
Abstract
Gene therapy is a hopeful strategy for the treatment of retinal disorders with no effective treatment. Gene replacement therapy is the most widely used strategy to modulate the gene expression in clinical research of inherited or acquired ocular diseases. Viral vectors are at the forefront of translational gene therapy mainly due to their high efficacy; nevertheless, concerns regarding safety have fostered the progress of nonviral therapy. Nonviral systems are non-immunogenic and avoid the risk of insertional mutagenesis. Moreover, they can be easily produced at large scale and have the potential to deliver larger genetic payloads. However, vector engineering to attain tissue-selective targeting and/or regulate the extent of gene expression is a challenging issue of nonviral gene therapy. Subretinal or intravitreal injections are the best option for the success of gene delivery to the posterior segment of the eye, regardless of the type of vector used. Preclinical studies with nonviral vectors have shown encouraging results for the treatment of macular degeneration and some inherited retinal disorders such as X-linked retinoschisis, Stargardt disease, retinitis pigmentosa, and Leber congenital amaurosis. These recent advances point to nonviral gene therapy as a feasible therapeutic tool for retinal disorders.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Nonviral vectors
- Gene therapy
- Ocular diseases
- Solid lipid nanoparticles
- Liposomes
- Polymeric nanoparticles
Introduction
Ocular gene therapy is a hopeful approach to treat, cure, or prevent diseases changing the gene expression in the eyes . According to the European Medicines Agency (EMA), a gene therapy medicinal product means a biological medicinal product which fulfills the following two characteristics: (a) it contains an active substance which contains or consists of a recombinant nucleic acid used in or administered to human beings with a view to regulating, repairing, replacing, adding, or deleting a genetic sequence; (b) its therapeutic, prophylactic, or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains or to the product of genetic expression of this sequence [44]. The modulation of the gene expression to treat inherited or acquired pathological conditions can be addressed by introducing DNA, messenger RNA (mRNA), small interfering RNA (siRNA), microRNA, or oligonucleotides. Gene therapy based on the administration of DNA and mRNA acts by means of therapeutic protein supplementation, whereas the use of siRNA and microRNA provides a posttranslational gene silencing. An emerging field in the treatment of monogenic disorders is the genome editing, which corrects the disease by replacing a sequence of a defective gene by a healthy copy in order to restore the “wild-type” DNA, enabling the cell to produce what is needed to have optimal phenotypic outcome [32]. In vivo approach aimed at treating the mutations directly implies the use of sequence-specific endonucleases, such as meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effectors (TALENs), and CRISPR/Cas (clustered regularly interspaced short palindromic repeat (CRISPR)-associated) systems [104].
Among the organs targeted by gene therapy, the eye has been at the forefront of translational gene therapy largely due to appropriate disease targets and its suitable anatomic features. The main advantages of the eye when treated with gene therapy include the following: it has a well-defined anatomy, it is accessible, it is relatively immune privileged, it can be easily examined, and in the same subject, one eye can be used as the experimental target and the other one as a control [93]. These advantages have fostered research that has culminated in various gene therapy clinical trials for ocular diseases, most of them related to disorders of the retina, a major cause of severe vision impairment.
Various strategies can be applied in retinal gene therapy depending on the underlying disease. Gene replacement is employed for the treatment of disorders that are due to loss-of-function mutations, and it is based on the delivery of a correct copy of the defective gene without removal of the endogenous mutant one. Gene silencing inhibits the expression of a mutated gene via modification of mRNA, and it is applied to disorders caused by gain-of-function mutations [96]. In vivo retinal genome editing is under preclinical approach at the moment, although it is a very active field and advances at a rapid pace. Currently, in the database of Gene Therapy Clinical Trials Worldwide [50], 34 clinical trials restricted to “ocular diseases” are reported, with gene replacement therapy being the most widely used strategy.
The clinical application of gene therapy has been limited owing to many technical barriers. Among them, the development of safe and effective delivery vectors is a key challenge. Although viral vectors have substantially advanced the field of gene therapy thanks mainly to their high efficacy, concerns regarding safety are bringing interest in the progress of nonviral therapy. Viral vectors, apart from the immunological and oncogenic risk, present other disadvantages such as the limited gene size packaging capacity and production difficulties. Nonviral systems have the potential to overcome many of these drawbacks; they show generally a very low immunogenicity and avoid the risk of insertional mutagenesis. Moreover, they have also the potential to deliver larger genetic payloads, and their production is simpler, cheaper, and more reproducible than viral vectors [39, 89]. Nevertheless, despite the development of a wide variety of nonviral vectors, low transfection efficacy remains the main obstacle for the progress of these systems toward the clinic. In fact, 23 from the 34 clinical trials reported for ocular diseases use viruses as vectors. Different strategies and efforts are still ongoing in the field of nonviral vectors thanks to the advances in material sciences, including the design of new lipid and polymers useful for gene delivery, the rapid progress of nanotechnology, and the progress in nucleic acid chemistry [105].
Nonviral Vectors for Retinal Gene Therapy
Nonviral strategies based on physical methods (iontophoresis, electroporation, gene gun, nucleofection) have achieved considerable progress, but gene expression efficiency is still a limitation [20, 30, 79]. Chemical nonviral vectors are the most widely studied, including the nanoparticulated systems. Nanoparticles are exceedingly suitable for gene therapy because of their small size, ability to access the intracellular compartment, incredible surface-area-to-volume ratio, capacity to carry large payload, and minimal damage to cell membranes and cellular environment [77]. Another important advantage of nanoparticles is the capacity to transport different ligands such as antibodies, peptides, molecular sensors, and probes, among others, to target cells with high precision and specificity. Nonviral particles for gene therapy can be broadly divided into two groups depending on the material employed: lipidic systems, named lipoplexes, and polymeric systems, called polyplexes [108].
Lipidic Delivery Systems
The assembly of cationic lipids and nucleic acids through electrostatic interactions results in complexes named lipoplexes. However, cationic lipids confer excessive positive surface charge which has been shown to enable increased protein interactions and compromised distribution kinetics through rapid blood clearance as well as immune stimulation [47]. In order to minimize immunotoxicity of cationic lipids, lipid nanoparticles (LNP) have provided remarkable results in recent clinical trials. Among LNP, liposomes and solid lipid nanoparticles (SLNs) are the preferred ones to deliver nucleic acids.
Liposomes are spherical vesicles composed of an aqueous compartment surrounded by a phospholipid bilayer of natural or synthetic origin, with size that can range from 20 nm to a few microns. Due to their resemblance to biological membranes, liposomes show higher biocompatibility than polymeric vehicles, which contribute to better delivery systems. Technological factors, such as the lipid-to-nucleic acid ratio or total lipid concentration in the final complex, are determinant for efficient gene delivery. Liposomal encapsulation of nucleic acids has shown to be an effective method to transfect corneal cells, inner retinal layer, and retinal pigment epithelia (RPE) [74]. In order to improve the efficacy and site specificity, significant effort has been dedicated to modify the composition and chemical structure of liposomes [1]. Different compounds have been incorporated to their structure, such as protamine sulfate [69, 88], poly-ethylene glycol (PEG) [85], or Arg(R)-Gly(G)-Asp(D) motif peptides [25].
SLNs are considered one of the most effective lipid-based colloidal vehicles [93]. SLNs consist of an aqueous dispersion of a layer of surfactants surrounding a solid lipid core, with particle sizes ranging from 50 to 1000 nm. Like liposomes, SLNs are composed of well-tolerated physiological lipids, often approved in pharmaceutical preparations for human use. Moreover, they have demonstrated good stability and can be sterilized and lyophilized . To improve the capacity of transfection, a variety of ligands can be incorporated on the SLNs surface, including dextran [41], protamine [40], cell penetration peptides [37], chitosan [43], or hyaluronic acid (HA) [5]. These components provide a higher protection to the genetic material, favor the cell internalization, and/or improve the trafficking of the nucleic acids inside the cell [93]. SLNs loaded with different plasmids have been shown to transfect retinal cells after intraocular administration by different routes in rats [42] and in mice [6, 7]. Other lipid-based systems studied for gene delivery to the retina include niosomes [73, 80] and span-polyarginine nanoparticles [86].
Polymeric Delivery Systems
Several polymers have been assayed to prepare polymeric nanoparticles, either of synthetic nature, such as poly-lactic-co-glycolic acid (PLGA), poly-L-lysine (PLL), or polyethylenimine (PEI), or being readily available in nature, such as chitosan or cyclodextrins [81]. These materials have a great potential for gene delivery because they generally have good biocompatibility and biodegradability, both properties related to their chemical structure. Another advantage is that polymers allow for adequate vector size as well as structural modifications, which is an important strategy to increase the efficiency of the delivery process. PEI nanoparticles have emerged as a powerful tool for nonviral transfection mainly because PEI promotes the endosomal escape; this system has demonstrated capacity to deliver antisense oligonucleotides in vitro in rat retinal Müller glial cells and also in vivo after intravitreal administration [52, 59]. However, the cationic pDNA/PEI complexes have shown cytotoxicity on human RPE culture cells (ARPE-19) and strong aggregation in the vitreous body; high gene expression in the retina without such cytotoxicity after intravitreous administration was achieved by coating the PEI complexes with anionic polymers [65].
Polyesters, including poly(lactic) acid (PLA), poly(glycolic) acid (PGA), and their copolymer PLGA, have been also used for retinal nucleic acid delivery due to their ability to bind plasmids, their nontoxic features, and rapid internalization capacity [12, 23].
Polysaccharide -based nanoparticles are well suited for ocular gene delivery. HA and chitosan have been combined to obtain gene delivery nanoparticles (HA-CS-NP) for ocular applications [35]. The combination of HA-CS-NPs with cationic lipids has also been proposed as an effective nonviral vector for application in eye diseases [49].
Albumin [8], dendrimers [71], and PLL [70] are polymeric compounds also proposed as nucleic acid delivery systems for ocular applications. These compounds are able to protect the genetic material and to internalize them into the cell cytoplasm, increasing their presence in the nucleus.
Poly(2-(N,N-dimethylamino)ethyl methacrylate) (PDMAEMA) has been described as a very interesting polymer for gene therapy, and it is less toxic than PEI [106]. Recently, PDMAEMA, synthesized by reversible addition-fragmentation chain transfer in a defined-size polymer , has been able to direct gene expression in the RPE cell line D407 [15].
Barriers for Successful Nonviral Retinal Gene Therapy
The success of a treatment for a retinal disease based on gene therapy is greatly dependent on the selection of the most appropriate route of administration and the availability of a system efficiently internalized by the target cells. The ideal administration route will be the one that leads to the highest transfection rate for the targeted retinal cell type and the least risk of side effects. Topical administration is not currently an effective route to reach therapeutic concentrations of drugs in the back of the eye, especially in the case of large molecules such as the nucleic acids. Different periocular routes may be suitable for ocular administration including peribulbar, retrobulbar, posterior juxtascleral, sub-tenon, and subconjunctival, the most studied for gene delivery [60]. In a previous study in mice, after subconjunctival injection of RNA-loaded particles, most of them migrated toward the cornea, the targeted cells, although appreciable uptake by retinal cells was also observed [45]. In spite of the utility of the periocular routes, for a successful delivery of active molecules to the posterior segment of the eye, intravenous or intraocular administrations have been shown to be better options [11, 38]. The main barrier for drugs or genes injected systemically is the blood-retinal barrier, limited to large systemic doses of lipophilic molecules. Targeting strategies have led to gene expression in the inner retina and the RPE after intravenous administration in mice, but bioavailability is still a major limitation [107].
Injection of the vectors into the subretinal space allows the contact of the nucleic acids with photoreceptors (PR), outer retinal layers, and RPE cells. Studies in mice have shown that subretinal delivery is the most effective to transfect PR and RPE cells; in fact, this route is the most used for inherited retinal diseases [108]. However, subretinal administration is an invasive method, and there is a high risk of ocular damage, i.e., lesions in RPE, hemorrhages, retinal tears, sub- or preretinal fibrosis, and retinal detachment [16]. Drug delivery by intravitreal injection is relatively easy, high doses are possible, and it is already routinely used. Although intravitreal delivery is less invasive than subretinal administration, adverse events such as retinal detachment or endophthalmitis may occur [66]. Suprachoroidal administration, below the sclera and above choroid, is safer than subretinal route and delivers the drug close to RPE, although it has shown to be little effective. Touchard et al. [97] have developed a transfection method called suprachoroidal electrotransfer, which combines the administration of a nonviral plasmid DNA with the application of an electrical field. However, from a practical point of view, only subretinal and intravitreal administration provide a significant concentration of the therapeutic compound in the target tissue.
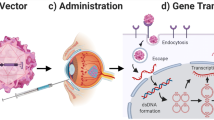
Once the vector reaches the retinal cells, it has to overcome several physical barriers, including cellular internalization, escape from endocytic vesicles, diffusion through the cytoplasm, and, in the case of DNA-based systems, the transport into the nucleus [39]. Fig. 8.1 shows a scheme with the cellular barriers that a nonviral vector has to overcome for a successful delivery of the genetic material.

Barriers that nonviral vectors need to overcome as nucleic acid delivery systems. (1) Binding to plasma membrane through electrostatic interactions between the cationic vectors and the negative charges of the plasma membrane or through interaction of ligands with specific receptors on the cell membrane. (2) Entry into the cell, mainly by endocytosis, which as ultimate step finishes in the degradative endolysosomes. (3) Escape of nucleic acids from the endosomes before degradation in lysosomal vesicles is crucial for effective transfection. Once released in the cytoplasm, RNA molecules will reach the intracellular target, whereas DNA molecules still have to bypass the nuclear membrane (4). In mitotic cells, DNA enters during interruption of the nuclear membrane, but in postmitotic cells, nuclear localization signals are needed to lead to active translocation by means of interaction with nuclear pore complexes (NPC)
Modifications on the particle and genetic materials are designed to overcome all these obstacles.
Cell Membrane
Once the nucleic acid delivery system reaches and binds to the surface of the target cell, the entrance into the cell is initiated. In the absence of any specific ligand on the surface of the vector, the attachment to the cell surface occurs through electrostatic interactions between the charges of the vector and the cell membrane [36], with endocytosis being the most frequent pathway for internalization. Multiple mechanisms of endocytosis have been described: phagocytosis, macropinocytosis, clathrin-dependent endocytosis, or clathrin-independent endocytosis, which include caveolae-mediated endocytosis, flotillin-dependent endocytosis, GRAF1-dependent endocytosis, Arf6-dependent endocytosis, or RhoA-dependent endocytosis [36, 90]. The predominant entry mechanism depends on the target cell and on the composition of the vector.
Internalization of the vectors may be improved by using cell-penetrating peptides (CPP). For instance, in a previous study, SLNs were decorated with SAP, a proline-rich peptide whose sequence is (VRLPPP)3 (made with three repetitive VRLPPP units; V = Val, R = Arg, L = Leu, and P = Pro) and that has demonstrated good translocation properties and is non-cytotoxic. SAP was able to improve the transfection efficacy of the SLNs in ARPE-19 cells because it induced a change in the dominant internalization mechanism , from clathrin endocytosis to caveolae/raft-dependent endocytosis, thereby decreasing the lysosomal pathway and consequently, reducing vector degradation [37]. In another study, a complex prepared with liposomes, protamine, and DNA was modified with the TAT protein of human immunodeficiency virus 1 (HIV-1), a cell-permeable peptide. This system resulted in efficient cell-specific delivery and a long-term expression of Rpe65 gene to mice lacking this gene; as a result, in vivo correction of blindness was detected [88].
The use of ligands for targeting to specific cells has also shown to improve the transfection effectiveness of nonviral vectors by means of the binding to cell surface receptors. For instance, the polysaccharide HA binds to the CD44 receptor, which is widely expressed in RPE cells, and it has been used to enhance the in vivo transfection efficiency of nanoparticles [7, 49, 103]. Cell internalization through the CD44 receptor avoids the degradation of the vector that occurs when other uptake mechanisms are involved, such as the lysosomal degradation after clathrin-mediated endocytosis.
Escape from Endosomes
Inside the cytoplasm, the release of the vector from either endosomes or lysosomes has been reported to be a major limitation for transfection. In the case of polyplexes, cationic protonable polymers can induce endosomal escape through the proton sponge effect, as mentioned above for PEI/DNA complexes. Vesicle-disturbing peptides conjugated to polyplexes may also facilitate the endosomal escape. Another strategy is the use of lysosomotropic agents, such as chloroquine, procaine, and spermidine, that promote pH buffering in endosomal vesicles [2]. Regarding lipoplexes, fusion of the cationic lipids has been proposed to facilitate not only the endosomal escape but also the DNA release [101].
Nuclear Envelope
Upon release in the cytoplasm, RNA molecules are available to reach the target and initiate the effect, but DNA has to bypass another important barrier, the nuclear membrane. When polyplexes are used as nonviral vectors, an incomplete polyplex dissociation in the nucleus has been proposed as a limiting step for efficient transfection [29]; in fact, polyplexes have been detected intact inside the nucleus, where they presumably undergo dissociation [17].
The entry into the nucleus is an important limiting step for transfection with DNA, as the nuclear membrane is a selective barrier to molecules bigger than 40 kDa, such as plasmids. In actively mitotic cells, the disruption of the nuclear membrane allows plasmids to enter into the nucleus; however, in postmitotic cells, like PR, the entry of large molecules depends on nuclear localization signals (NLS) . NLS sequences lead to active translocation through the nuclear envelope. The peptide protamine presents NLS sequences of six consecutive arginine residues, and it is frequently used as a component of nonviral vectors. In retinal cells, this peptide has shown to be able to significantly improve the transfection efficacy of nonviral vectors [40] and also in vivo after ocular administration [42, 88].
Decreasing the size of genetic material may help to increase nuclear internalization and transfection efficacy. In this sense, the minicircles, compact DNA vectors that lack a bacterial backbone, have led to superior levels and longer duration of gene expression with respect to full-length DNA plasmids [61].
Gene Expression
Plasmids delivered via nonviral vectors can be maintained episomally, thus avoiding the risk of insertional mutagenesis, although transient instead of stable transfection is usually achieved. Vector engineering to attain selective tissue targeting and/or regulation of the extent of gene expression is a challenging issue of retinal gene therapy that demands active research. Mammalian gene expression can be regulated by several elements such as enhancers, locus control regions, boundary elements, insulators, scaffold/matrix attachment regions (S/MARs), and CpG depletion [62, 82]. In this sense, the high load capacity of nonviral systems features an important advantage, allowing the inclusion of additional regulatory elements in order to target and to improve the level and long-term expression.
S/MAR sequences, which anchor chromatin to the nuclear matrix proteins during the interphase, were included in a plasmid containing the RPE65 gene [63]. The plasmid, administered encapsulated in PEGylated-PLL nanoparticles to the subretinal space of rpe65−/− mice, led to a long expression of the transgene related to the stability of the expression cassette, which was isolated intact 1 year postinjection [64]. Various systems for transgene integration have been developed to promote long-term expression, such as transposition systems based on the recombinases FC31 and Sleeping Beauty. Integrase from bacteriophage FC31 has also conferred genomic integration of plasmid DNA and has led to long-term expression in rat RPE cells after subretinal injection followed by electrotransfer [24]. A nonviral strategy based on the Sleeping Beauty transposon system also resulted in stable expression of pigment epithelium-derived factor (PEDF) in ARPE19 cells [57]. These findings have conducted to the evaluation of this strategy as a possible treatment of age-related macular degeneration associated to neovascularization [58].
Tissue-specific promoters for retinal cells have been used as a strategy to circumvent the lack of cell specificity of nonviral vectors. For instance, structural improvement of the Rs1h-deficient mice retina has been shown after successful delivery of a plasmid containing the gene RS1 under the control of a specific promotor for PR (murin opsin promoter, mOPS) formulated in SLNs [7]. Wang et al. used liposome-based vectors and different promoters that were able to achieve cell specificity for a variety of cell types: RPE cell specificity with vitelliform macular dystrophy (VMD2), rod cell specificity with mouse rhodopsin, cone cell specificity with red/green opsin, and ganglion cell specificity with thymocyte antigen promoters [100]. PEGylated liposomes containing an expression plasmid encoding bacterial galactosidase under the influence of either the simian virus (SV)40 promoter or the glial fibrillary acidic protein (GFAP) gene promoter have been used to target the cornea after intravenous administration [107].
The induction of the delivered gene expression only when it is needed is also a challenge. It can be achieved including inducible regulatory sequences in the promoter, which will be only active in the presence of specific environmental signals. In other cases, gene expression is regulated by drugs. For instance, an autogenous transgene regulatory system (ARES) is inducible by isopropyl β-d-1-thiogalactopyranoside (IPTG), which has no off-target effects in mammals. Sochor et al. used this system to control reversibly the luciferase expression in the murine retina after oral delivery of IPTG [92].
Nonviral Vectors in Gene Therapy for Retina and Posterior Segment Diseases
Many studies in animals have demonstrated the potential utility of gene therapy for the treatment of ocular diseases. As a result, translational clinical research has started. Nevertheless, up to date, the clinical trials reported for ocular diseases use viral vectors or naked genetic material, and nonviral vectors are still restricted to preclinical phases.
X-Linked Retinoschisis (XLRS)
XLRS is a retinal degenerative disease affecting young males, caused by mutations in the gene RS1 that encodes the secreted protein retinoschisin, with a prevalence of approximately 1:5000 to 1:25000 [91]. Disorganization of retinal layers and distinct abnormalities in the electroretinogram are hallmarks of the disease [94]. The splitting of retinal layers, with bilateral foveal schisis, is observed at early stages of the disease, and it results in cystic degeneration of the central retina. The progression and severity of XLRS is very variable leading to mild to severe loss in central vision. Currently, there is no cure for the schisis formation, and the treatment is focused on preserving the low vision.
Gene augmentation therapy may be an excellent therapeutic approach, due to the well-understood monogenic origin of this disease . In addition, retinoschisin is a secreted protein, and not only the transfected cells benefit from the replacement of the gene, since once secreted, the protein spreads from the site of expression. Novel therapies may be addressed in retinoschisin-deficient mice, which show close resemblance of the retinal phenotype with XLRS patients and represent an excellent disease model [18, 102]. Studies in the mouse model with viral vectors as delivery systems of RS1 emphasize the potential of gene therapy for XLRS and highlight the importance of careful design and optimization for specific, minimally invasive, and long-lasting gene therapy [56, 83, 84]. Recently, a preclinical dose escalation study of intravitreal RS1 gene delivery with viral vectors has been carried out [19]. Structural improvement was shown by reduction of retinal cavities 3–4 months after injection, and electroretinogram values were normalized at 3–4 months and 6–9 months postinjection, even when the production levels of retinoschisin were lower than in wild-type mice. A fully normal level of the protein expression seems not to be necessary for a therapeutic effect.
Nonviral vectors are a promising alternative to viral vectors for attempting the treatment of XLRS with gene therapy. Our research group has demonstrated the capacity of SLN loaded with a plasmid containing the RS1 gene to transfect a number of retinal layers after ocular injection to the mice model of XLRS and to induce the production of the therapeutic protein retinoschisin . As it is shown in Fig. 8.2, the production of retinoschisin led to a partial recovery of the structure of the retina [6, 7].

Microscopic images of retinas stained by Masson’s trichrome technique. Photographs show the differences in the retina structures of wild-type mice and retinoschisin-deficient mice untreated and treated with SLN-based vectors (HA-SLN and DX-SLN) 2 weeks after intravitreal administration. Green, connective tissue; dull green, muscle; dark blue, nuclei; pink, cytoplasm and muscle fibers. Images were captured at 20× magnification. Scale bar: 100 μm. (Reprinted from “Biomaterials, 90, Apaolaza et al., Copyright [7],” with permission from Elsevier). RPE Retinal Pigment Epithelium, PR Photoreceptors, ONL Outer Nuclear Layer, OPL Outer Plexiform Layer; INL Inner Nuclear Layer, IPL Inner Plexiform Layer, GC Ganglion Cell Layer
These studies showed successful gene transfer using lipid-based nanocarriers , with promising results that point to nonviral gene therapy as a feasible future therapeutic tool for posterior segment disorders.
Stargardt Disease
Stargardt disease is the most common inherited juvenile macular degeneration in humans, with a pattern of autosomal recessive inheritance [14]. The gene involved in Stargardt disease is named ABCA4, which encodes for a PR-specific all-trans-retinal transporter [4]. Due to a defective ABCA4 protein, vitamin A aldehyde forms deposits in RPE cells during the process of disk shedding and phagocytosis. Consequently, abnormal high levels of lipofuscin pigments accumulate in the RPE, triggering RPE cell death and causing secondary PR degeneration [78]. The impairment and loss of vision in Stargardt patients can be due to hundreds of mutations in the ABCA4 gene. The mutations in this gene are also responsible for other visual diseases such as cone-rod dystrophy and autosomal recessive retinitis pigmentosa [33, 72]. Heterozygous mutations in ABCA4 may lead to the development of age-related macular degeneration. At present, there is no cure for ABCA4-associated disease, and gene therapy has been proposed for Stargardt disease. Currently, a phase I/II clinical trial based on viral vectors for subretinal administration is underway [28]. The large size of the ABCA4 cDNA, a limitation for viral delivery, makes nonviral vectors as a suitable alternative. In a recent study [55], compacted DNA nanoparticles (8–10 nm in diameter) formulated with PEG-substituted PLL (CK30PEG) were used to inject ABCA4 to ABCA4-deficient mice by subretinal route. After administration, the expression of the transgene was detected for up to 8 months, and a significant correction of functional and structural Stargardt phenotypes was observed, including improved recovery of adaptation to darkness and decrease of lipofuscin granules.
Retinitis Pigmentosa
Retinitis pigmentosa is the most common subtype of retinal degeneration, responsible for loss of vision in one in 4000 people worldwide [87]. Defects in more than 60 genes have been identified in patients with retinitis pigmentosa, as autosomal dominant (30–40% of cases), autosomal recessive (50–60%), or X-linked (5–15%) forms. Notwithstanding, mutation in 30–35% of patients cannot be identified [87]. The features of the disease and its progression vary significantly among patients, but night blindness due to loss of rod PR in the early phase of the disease is very frequent. Over time, cone PR are also affected resulting in decreased central visual acuity. As a consequence, patients describe tunnel vision, which may result on complete blindness [13, 54].
Gene therapy for retinitis pigmentosa aims to slow down or stop the progress of retinal degeneration. Preclinical evaluation in animal models provides expectations for future clinical application. Due to the variety of genes involved in the disease, several animal models have been developed. One strategy to generate animal models that mimics the human autosomal recessive retinitis pigmentosa is directed to mutations in the genes encoding the two rod cyclic nucleotide-gated (CNG) channel subunits. Knockout of CNGB1 in mice results in a phenotype that recapitulates the principal pathology of retinitis pigmentosa patients [75]. Another animal model of retinitis pigmentosa is a dog deficient in a GTPase regulator-interacting protein 1 (RPGRIP1) [67]. Rhodopsin gene (RHO), which encodes the photosensitive pigment in rod PR, is another gene whose mutations (>100) have been identified in individuals with retinitis pigmentosa [87]. There has been an attempt to treat RHO-linked retinitis pigmentosa by means of a new strategy named “mutation-independent suppression and replacement,” which comprises both gene suppression and gene replacement [76]. The Royal College of Surgeons (RCS) rat is a widely studied animal model of retinal degeneration with a mutation in the MER proto-oncogene tyrosine kinase (Mertk) gene , and it serves as a model of an autosomal recessive form of retinal degeneration [34]. Both functional and structural retina preservations were achieved with gene replacement therapy in this model of Mertk-related retinitis pigmentosa. Based on this proof of concept, a phase I clinical trial of subretinally administered Mertk in a viral vector was conducted. Peak gains of greater than three lines of vision were observed in two of the six patients recruited in the trial [51].
The gene RDS, which encodes the retinal degeneration slow protein, is frequently associated to retinitis pigmentosa. A well-characterized animal model for RDS-associated retinitis pigmentosa (the Rds+/−mouse) is available to provide a valuable and readily accessible in vivo system for developing and testing gene therapy [31, 98]. RDS gene replacement mediated by nonviral vectors has been assayed in this model after subretinal administration. Cai et al. [21] developed DNA nanoparticles consisting of single molecules of DNA compacted with PEG-substituted lysine 30-mer peptide (CK30PEG10K). Vectors were administered by subretinal route, and RDS mRNA levels peaked at postinjection day 2–7 and remained elevated at the latest time point examined, 120 days after administration. A significant improvement in the outer segment structures was observed, rod function (measured by electroretinography) showed statistically significant improvement compared with controls, and cone function in nanoparticle-injected eyes reached the wild-type levels. More recently, span-polyarginine (SP-PA) nanoparticles were developed to mediate gene transfer in the subretinal space of a mouse model of retinitis pigmentosa carrying a point mutation (A216P) in the Prpf31 gene. SP-PA nanoparticles were able to efficiently transfect mice retinas with GFP and Prpf31 plasmid. Statistically significant improvement in visual acuity and retinal thickness was found in mice treated with the SP-PA- Prpf31 nanoplatform [86].
These findings confirm the potential of nonviral vector -mediated gene replacement as treatment of retinitis pigmentosa.
Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in people aged 50 years or older in the developed world. More than nine million Americans have AMD, and cases are expected to nearly double by 2050 to 17.8 million [26]. Depending on the histopathological characteristics, AMD can be classified into several categories: early, intermediate, and advanced AMD [99]. In early and intermediate AMD, only minimal visual acuity impairment occurs, but advanced AMD is the leading cause of blindness worldwide. AMD may be avascular or may be characterized by the subretinal invasion of choroidal vessels. Whereas avascular AMD is a slow progressing disorder, in which PR degeneration follows RPE cell degeneration, neovascularization-related AMD progresses rapidly to blindness following RPE cell degeneration.
Among the multiple factors that play a role in AMD, the strong genetic contribution is well documented [48]. In fact, gene therapy is considered one of the improved treatments under study for AMD [46]. In a phase I clinical trial, 28 patients with advanced neovascular AMD were treated using adenoviral vector-mediated intravitreal gene transfer of PEDF, which is an antiangiogenic cytokine. This therapeutic strategy appears to help arrest the growth of neovascular AMD [22]. As mentioned above, currently a nonviral strategy based on the Sleeping Beauty transposon system is also under preclinical evaluation to achieve stable expression of PEDF as treatment of AMD associated to neovascularization [58].
Leber Congenital Amaurosis
Leber congenital amaurosis (LCA) is an autosomal recessive disease resulting from mutation in at least 15 genes [3]. Prevalence of LCA is 1 in 35,000 newborn of all blind children [95]. Patients with this severe retinal disease suffer from a marked impairment of the visual acuity at birth or during the first 6 months of life, sensory poorly reactive pupils, and severely diminished or non-detectable electroretinogram activity [95].
There is no successful treatment for LCA, but four independent clinical trials have been carried out for human RPE65-associated LCA [9, 10, 27, 68]. The RPE65 gene encodes for all-trans-retinyl-ester hydrolase, which is a 65 KDa enzyme that in RPE is critical for the production of 11-cis-retinal. This compound is transported to the PR where it binds to apo-rhodopsin; the apo-rhodopsin-11-cis-retinal complex reacts with a photon to produce a change in membrane potential, which generates a nerve signal that travels to the visual cortex for image formation and recognition. Deficiency of all-trans-retinyl-hydrolase due to mutation in RPE65 gene happens in about 6% of LCA cases in humans [95]. In the most recent clinical trial for RPE65-LCA, a phase III clinical trial, patients that received subretinally injections of the RPE65 gene in a viral vector have shown successful improvement of the sensitivity to light and functional vision [53].
RPE65 gene has been also formulated in nonviral vectors composed of liposomes and protamine. Efficient cell-specific delivery and long-term expression of the RPE65 gene in mice lacking RPE65 protein led to in vivo correction of blindness [88]. In order to obtain long expression of the RPE65 gene with nonviral vectors, the S/MAR sequence was included in the corresponding plasmid. As mentioned above, this strategy led to detection of the expression cassette 1 year after subretinal injection to mice lacking the RPE65 gene [63].
Conclusions
Ocular gene therapy is a hopeful approach to treat, cure, or prevent diseases by modulating gene expression in the retina and in the posterior segment of the eye. Human clinical trials are beginning to show encouraging results, although nonviral vector engineering to attain tissue-selective targeting and/or regulate the extent of gene expression is still a challenge. Preclinical studies with nonviral vectors have shown encouraging results for the treatment of some ocular diseases, such as macular degeneration, and some inherited retinal disorders including X-linked retinoschisis, Stargardt disease, retinitis pigmentosa, and Leber congenital amaurosis. These recent advances point to nonviral gene therapy as a feasible therapeutic tool for retinal disorders.
References
Abul-Hassan K, et al. Optimization of non-viral gene transfer to human primary retinal pigment epithelial cells. Curr Eye Res. 2000;20:361–6.
Aied A, et al. Polymer gene delivery: overcoming the obstacles. Drug Discov Today. 2013;18:1090–8.
Al-Saikhan FI. The gene therapy revolution in ophthalmology. Saudi J Ophthalmol. 2013;27:107–11.
Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46.
Apaolaza PS, et al. A novel gene therapy vector based on hyaluronic acid and solid lipid nanoparticles for ocular diseases. Int J Pharm. 2014;465:413–26.
Apaolaza PS, et al. Solid lipid nanoparticle-based vectors intended for the treatment of X-linked juvenile retinoschisis by gene therapy: in vivo approaches in Rs1h-deficient mouse model. J Control Release. 2015;217:273–83.
Apaolaza PS, et al. Structural recovery of the retina in a retinoschisin-deficient mouse after gene replacement therapy by solid lipid nanoparticles. Biomaterials. 2016;90:40–9.
Arnedo A, et al. Albumin nanoparticles improved the stability, nuclear accumulation and anticytomegaloviral activity of a phosphodiester oligonucleotide. J Control Release. 2004;94:217–27.
Bainbridge JW, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–9.
Banin E, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther. 2010;21:749–1757.
Battaglia L, et al. Application of lipid nanoparticles to ocular drug delivery. Expert Opin Drug Deliv. 2016;13:1743–57.
Bejjani RA, et al. Nanoparticles for gene delivery to retinal pigment epithelial cells. Mol Vis. 2005;11:124–32.
Berger W, et al. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75.
Binley K, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of StarGen for Stargardt disease. Invest Ophthalmol Vis Sci. 2013;54:4061–71.
Bitoque DB, et al. Efficiency of RAFT-synthesized PDMAEMA in gene transfer to the retina. J Tissue Eng Regen Med. 2017;11:265–75.
Bloquel C, et al. Potential ocular therapeutic avenues. Adv Drug Deliv Rev. 2006;58:1224–42.
Breunig M, et al. Gene delivery with low molecular weight linear polyethylenimines. J Gene Med. 2005;7:1287–98.
Bush RA, et al. Convergence of human genetics and animal studies: gene therapy for X-linked retinoschisis. Cold Spring Harb Perspect Med. 2015;5:a017368.
Bush RA, et al. Preclinical dose-escalation study of intravitreal AAV-RS1 gene therapy in a mouse model of X-linked Retinoschisis: dose-dependent expression and improved retinal structure and function. Hum Gene Ther. 2016;27:376–89.
Cai X, et al. Nanoparticle applications in ocular gene therapy. Vis Res. 2008;48:319–24.
Cai X, et al. Gene delivery to mitotic and postmitotic photoreceptors via compacted DNA nanoparticles results in improved phenotype in a mouse model of retinitis pigmentosa. FASEB J. 2010;24:1178–91.
Campochiaro PA, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial. Hum Gene Ther. 2006;17:167–76.
Carrasquillo KG, et al. Controlled delivery of the anti-VEGF aptamer EYE001 with poly(lactic-co-glycolic)acid microspheres. Invest Ophthalmol Vis Sci. 2003;44:290–9.
Chalberg TW, et al. phiC31 integrase confers genomic integration and long-term transgene expression in rat retina. Invest Ophthalmol Vis Sci. 2005;46:2140–6.
Chen CW, et al. Efficient downregulation of VEGF in retinal pigment epithelial cells by integrin ligand-labeled liposome-mediated siRNA delivery. Int J Nanomedicine. 2013;8:2613–27.
Cheung LK, Eaton A. Age-related macular degeneration. Pharmacotherapy. 2013;33:838–55.
Cideciyan AV, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112–7.
Clinical-Trials.gov. Phase I/IIa study of SAR422459 in patients with Stargardt’s macular degeneration. StarGen, Clinical-Trialsgov number, NCT01367444. https://clinicaltrials.gov/ct2/show/NCT01367444. Last query: 26 June 2017.
Cohen RN, et al. Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection. J Control Release. 2009;135:166–74.
Conley SM, Naash MI. Nanoparticles for retinal gene therapy. Prog Retin Eye Res. 2010;29:376–97.
Connell G, et al. Photoreceptor peripherin is the normal product of the gene responsible for retinal degenera-tion in the rds mouse. Proc Natl Acad Sci U S A. 1991;88:723–6.
Cox DB, et al. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–31.
Cremers FP, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in Stargardts disease ABCR. Hum Mol Gen. 1998;7:355–62.
D’Cruz PM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51.
de la Fuente M, et al. Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Invest Ophthalmol Vis Sci. 2008;49:2016–24.
del Pozo-Rodríguez A, et al. Solid lipid nanoparticles for retinal gene therapy: transfection and intracellular trafficking in RPE cells. Int J Pharm. 2008;360:177–83.
del Pozo-Rodríguez A, et al. A proline-rich peptide improves cell transfection of solid lipid nanoparticle-based non-viral vectors. J Control Release. 2009;133:52–9.
del Pozo-Rodríguez A, et al. Lipid nanoparticles as drug/gene delivery systems to the retina. J Ocul Pharmacol Ther. 2013;29:173–88.
del Pozo-Rodríguez A, et al. Applications of lipid nanoparticles in gene therapy. Eur J Pharm Biopharm. 2016;109:184–93.
Delgado D, et al. Understanding the mechanism of protamine in solid lipid nanoparticle-based lipofection: the importance of the entry pathway. Eur J Pharm Biopharm. 2011;79:495–502.
Delgado D, et al. Dextran-protamine-solid lipid nanoparticles as a non-viral vector for gene therapy: in vitro characterization and in vivo transfection after intravenous administration to mice. Int J Pharm. 2012a;425:35–43.
Delgado D, et al. Dextran and protamine-based solid lipid nanoparticles as potential vectors for the treatment of X-linked juvenile retinoschisis. Hum Gene Ther. 2012b;23:345–55.
Delgado D, et al. New gene delivery system based on oligochitosan and solid lipid nanoparticles: ‘in vitro’ and ‘in vivo’ evaluation. Eur J Pharm Sci. 2013;50:484–91.
European Medicines Agency. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. Draft. EMA/CAT/80183/2014, 2015.
Feng L, et al. Ocular delivery of pRNA nanoparticles: distribution and clearance after subconjunctival injection. Pharm Res. 2014;31:1046–58.
Feret A, et al. Macular degeneration: types, causes, and possible interventions. Geriatr Nurs. 2007;28:387–92.
Foged C. siRNA delivery with lipid-based systems: promises and pitfalls. Curr Top Med Chem. 2012;12:97–107.
Fritsche LG, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45:433–9.
Gan L, et al. Hyaluronan-modified core-shell liponanoparticles targeting CD44-positive retinal pigment epithelium cells via intravitreal injection. Biomaterials. 2013;34:5978–87.
Gene Therapy Clin Trials Worldwide. Provided by the Journal of GeneMedicine. Jon Wiley and Sons Ltd., 2017. http://www.abedia.com/wiley/indications.php. Updated Apr 2017; last query: 26 June 2017.
Ghazi NG, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular sub-retinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135:327–43.
Gomes dos Santos AL, et al. Oligonucleotide-polyethylenimine complexes targeting retinal cells: structural analysis and application to anti-TGFbeta-2 therapy. Pharm Res. 2006;23:770–81.
Hafler BP. Clinical progress in inherited retinal degenerations: gene therapy clinical trials and advances in genetic sequencing. Retina. 2017;37:417–23.
Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40.
Han Z, et al. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J Clin Invest. 2012;122:3221–6.
Janssen A, et al. Effect of late-stage therapy on disease progression in AAV-mediated rescue of photoreceptor cells in the retinoschisin deficient mouse. Mol Ther. 2008;16:1010–7.
Johnen S, et al. Sleeping Beauty transposon-mediated transfection of retinal and iris pigment epithelial cells. Invest Ophthalmol Vis Sci. 2012;53:4787–96.
Hudecek M, et al. Going non-viral: the sleeping beauty transposon system breaks on through to the clinical side. Crit Rev Biochem Mol Biol. 2017;52:355–80.
Horbinski C, et al. Polyethyleneimine-mediated transfection of cultured postmitotic neurons from rat sympathetic ganglia and adult human retina. BMC Neurosci. 2001;2:2.
Kaur IP, Kakkar S. Nanotherapy for posterior eye diseases. J Control Release. 2014;193:100–12.
Kay MA, et al. A robust system for production of minicircle DNA vectors. Nature Biotech. 2010;28:1287–9.
Koirala A, et al. A review of therapeutic prospects of non-viral gene therapy in the retinal pigment epithelium. Biomaterials. 2013a;34:7158–67.
Koirala A, et al. S/MAR-containing DNA nanoparticles promote persistent RPE gene expression and improvement in RPE65-associated LCA. Hum Mol Genet. 2013b;22:1632–42.
Koirala A, et al. Episomal maintenance of S/MAR-containing non-viral vectors for RPE-based diseases. Adv Exp Med Biol. 2014;801:703–9.
Kurosaki T, et al. Ocular gene delivery systems using ternary complexes of plasmid DNA, polyethylenimine, and anionic polymers. Biol Pharm Bull. 2013;36:96–101.
Ladas ID, et al. Safety of repeat intravitreal injections of bevacizumab versus ranibizumab: our experience after 2,000 injections. Retina. 2009;29:313–8.
Lhériteau E, et al. Successful gene therapy in the RPGRIP1-deficient dog: a large model of cone-rod dystrophy. Mol Ther. 2014;22:265–77.
Maguire AM, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–8.
Mannermaa E, et al. Long-lasting secretion of transgene product from differentiated and filter-grown retinal pigment epithelial cells after nonviral gene transfer. Curr Eye Res. 2005;30:345–53.
Männistö M, et al. Structure-activity relationships of poly(L-lysines): effects of pegylation and molecular shape on physicochemical and biological properties in gene delivery. J Control Release. 2002;83:169–82.
Marano RJ, et al. Inhibition of in vitro VEGF expression and choroidal neovascularization by synthetic dendrimer peptide mediated delivery of a sense oligonucleotide. Exp Eye Res. 2004;79:525–35.
Martinez-Mir A, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease ABCR. Nat Genet. 1998;18:11–2.
Mashal M, et al. Retinal gene delivery enhancement by lycopene incorporation into cationic niosomes based on DOTMA and polysorbate 60. J Control Release. 2017;254:55–64.
Masuda I, et al. Gene transfer with liposomes to the intraocular tissues by different routes of administration. Invest Ophthalmol Vis Sci. 1996;37:1914–20.
Michalakis S, et al. Gene therapy restores vision and delays degeneration in the CNGB1(−/−) mouse model of retinitis pigmentosa. Adv Exp Med Biol. 2014;801:733–9.
Millington-Ward S, et al. Strategems in vitro for gene therapies directed to dominant mutations. Hum Mol Genet. 1997;6:1415–26.
Mohan RR, et al. Corneal gene therapy: basic science and translational perspective. Ocul Surf. 2013;11:150–64.
Molday RS, Zhang K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res. 2010;49:476–92.
Naik R, et al. Gene delivery to the retina: focus on non-viral approaches. Drug Discov Today. 2009;14:306–15.
Ojeda E, et al. The in fluence of the polar head-group of synthetic cationic lipids on the transfection efficiency mediated by niosomes in rat retina and brain. Biomaterials. 2016;77:267–79.
Oliveira C, et al. Recent advances in characterization of nonviral vectors for delivery of nucleic acids: impact on their biological performance. Expert Opin Drug Deliv. 2015;12:27–39.
Oliveira AV, et al. Non-viral strategies for ocular gene delivery. Mater Sci Eng C Mater Biol Appl. 2017;77:1275–89.
Ou J, et al. Synaptic pathology and therapeutic repair in adult retinoschisis mouse by AAV-RS1 transfer. J Clin Invest. 2015;125:2891–903.
Park TK, et al. Intravitreal delivery of AAV8 retinoschisin results in cell type-specific gene expression and retinal rescue in the Rs1-KO mouse. Gene Ther. 2009;16:916–26.
Peeters L, et al. Vitreous: a barrier to nonviral ocular gene therapy. Invest Ophthalmol Vis Sci. 2005;46:3553–61.
Pensado A, et al. Span poly-L-arginine nanoparticles are efficient non-viral vectors for PRPF31 gene delivery: an approach of gene therapy to treat retinitis pigmentosa. Nanomedicine. 2016;12:2251–60.
Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol. 2014;8:127–36.
Rajala A, et al. Nanoparticle-assisted targeted delivery of eye-specific genes to eyes significantly improves the vision of blind mice in vivo. Nano Lett. 2014;14:5257–63.
Rodríguez-Gascón A, et al. Vaginal gene therapy. Adv Drug Deliv Rev. 2015;92:71–83.
Ruiz de Garibay AP, et al. Role of endocytic uptake in transfection efficiency of solid lipid nanoparticles-based nonviral vectors. J Gene Med. 2013;15:427–40.
Sikkink SK, et al. X-linked retinoschisis: an update. J Med Genet. 2007;44:225–32.
Sochor MA, et al. An autogenously regulated expression system for gene therapeutic ocular applications. Sci Report. 2015;5:17105.
Solinís MA, et al. Treatment of ocular disorders by gene therapy. Eur J Pharm Biopharm. 2015;95:331–42.
Tantri A, et al. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49:214–30.
Thumann G. Prospectives for gene therapy of retinal degenerations. Curr Genomics. 2012;13:350–62.
Trapani I, et al. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108–28.
Touchard E, et al. Suprachoroidal electrotransfer: a nonviral gene delivery method to transfect the choroid and the retina without detaching the retina. Mol Ther. 2012;20:1559–70.
Travis GH, et al. The retinal degeneration slow (rds) gene product is a photoreceptor discmembrane-associated glycoprotein. Neuron. 1991;6:61–70.
Van Lookeren Campagne M, et al. Mechanisms of age-related macular degeneration and therapeutic opportunities. J Pathol. 2014;232:151–64.
Wang Y, et al. Cell-specific promoters enable lipid-based nanoparticles to deliver genes to specific cells of the retina in vivo. Theranostics. 2016;6:1514–27.
Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116:255–64.
Weber BH, et al. Inactivation of the murine X-linked juvenile retinoschisis gene, Rs1h, suggests a role of retinoschisin in retinal cell layer organization and synaptic structure. Proc Natl Acad Sci U S A. 2002;99:6222–7.
Yamada Y, et al. Hyaluronic acid controls the uptake pathway and intracellular trafficking of an octaarginine-modified gene vector in CD44 positive- and CD44 negative-cells. Biomaterials. 2015;52:189–98.
Yanik M, et al. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog Retin Eye Res. 2017;56:1–18.
Yin H, et al. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541–55.
Zhang Y, et al. Design and biophysical characterization of bioresponsive degradable poly(dimethylaminoethyl methacrylate) based polymers for in vitro DNA transfection. Biomacromolecules. 2012;13:313–22.
Zhu C, et al. Widespread expression of an exogenous gene in the eye after intravenous administration. Invest Ophthalmol Vis Sci. 2002;43:3075–80.
Zulliger R, et al. Non-viral therapeutic approaches to ocular diseases: an overview and future directions. J Control Release. 2015;219:471–87.
Acknowledgments
This work was supported by the University of the Basque Country UPV/EHU (PPG17/65 and GIU17/032). J Torrecilla thanks UPV/EHU for her research grant.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
del Pozo-Rodríguez, A., Torrecilla, J., Rodríguez-Gascón, A., Solinís, M.Á. (2018). Nonviral Delivery Systems for Gene Therapy for Retina and Posterior Segment Disease. In: Patel, J., Sutariya, V., Kanwar, J., Pathak, Y. (eds) Drug Delivery for the Retina and Posterior Segment Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-95807-1_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-95807-1_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95806-4
Online ISBN: 978-3-319-95807-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)