
Abstract
Nuclear factor-kappaB (NF-κB) is a signal-activated transcription factor complex with two functional subunits and controls the expression of >600 genes in the human genome. This protein complex was initially identified and described as a nuclear protein complex uniquely present in cells that transcribe immunoglobulin light chain genes. Hence, its pro-oncogenic functions were studied only in the context of hematologic malignancies. However, subsequent studies showed the presence of this complex in almost all cell types. Early studies of this complex suggested only pro-oncogenic role, which triggered massive interest in both academia and industry in developing drugs targeting this transcription factor complex. Because NF-κB activation requires multiple enzymatic steps, it is one of the few transcriptional regulators that are therapeutically targetable. Recent studies, however, have revealed both tumor suppressor and pro-oncogenic functions depending on the cellular context, stage of the disease, and concomitant genomic aberration in cancer cells. During initiation stage of tumorigenesis, NF-κB activates senescence program and prevents damaged cells from proliferating. During later stage, it promotes tumorigenesis by activating anti-apoptotic, pro-inflammatory, pro-metastatic, and tumor-immune suppressive machineries. Amplifications, mutations, and rare deletions in various components of NF-κB pathway are observed in multiple cancer types, and IKKε among them is a proven oncogene and is amplified in >30% of breast cancers. These complexities in NF-κB activities need to be taken into consideration while designing clinical trials with drugs with NF-κB inhibitory activity. Although specific inhibitors of NF-κB are yet to enter oncology clinic, NF-κB is one of the targets of several FDA-approved drugs including bortezomib used for treating multiple myeloma.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
NF-κB in Tumor Initiation
The potential role of NF-κB in cancer was first investigated in hematologic malignancies, particularly multiple myeloma and leukemia [1]. The focus subsequently shifted to solid tumors after discovery of its aberrant activity in breast cancers by others and us in 1998 [2]. Aberrant NF-κB activity was subsequently reported in prostate, bladder, lung, head and neck, and pancreatic cancers. Mechanistic studies, however, revealed dichotomous role of NF-κB in cancer. NF-κB has been shown to promote senescence and function as a master regulator of senescence-associated secretory phenotype (SASP) [3]. Soluble factors in SASPs reinforce senescence arrest, alter the microenvironment in the tumor, and trigger immune surveillance. In genetically engineered Kras-induced mouse model of pancreatic cancer, the p65 subunit of NF-κB triggers CXCL1 (part of SASP)-/CXCR2-dependent senescence and inhibits initial steps of carcinogenesis [4]. DNA damage also triggers NF-κB-dependent senescence. Switch to oncogenic function occurs once cells are immortalized. Other genomic aberrations in cancer cells will also determine tumor suppressor function of NF-κB. For example, in tumors where the expression of the anti-apoptotic BCL-2 is dependent on NF-κB, it functions as a survival factor. By contrast, in tumors with elevated BCL-2 independent of NF-κB, therapy-induced cell death requires NF-κB-mediated induction of SASPs [5]. In general, senescence is a double-edged sword in cancer because while senescent cells themselves rarely progress into cancer, SASPs from these cells can promote neoplastic progression of nearby preneoplastic cells by providing pro-inflammatory molecules and inducing epithelial-to-mesenchymal transition. Therefore, timing of NF-κB-induced senescence could have profound influence on cancer development. From the practical and clinical angle, this also poses challenge to ascertain whether concurrent presence of activated NF-κB (as determined by nuclear p65 and phosphorylated p65) and SASP is a good or bad prognostic marker.
NF-κB in Tumor Progression/Metastasis
Overcoming senescence barrier either through inactivation of cell cycle inhibitors such as p16 or overexpression of cell cycle protein cyclin D1 and inactivation of p53 or through telomerase overexpression leads to cellular immortalization. In immortalized cells, NF-κB cooperates with other oncogenes such as RAS or functions downstream of oncogenes such as KRAS, mutant PIK3CA, activated EGFR, or ERBB2 to promote cancer progression. Cancer-promoting functions include upregulation of anti-apoptotic proteins such as BCL-2, BCL-XL, GADD45β, XIAP, cIAP1, and cIAP2; cell cycle proteins such as Cyclin D1; pro-invasion molecules such as MMP9; pro-metastatic molecules such as CXCR4; pro-inflammatory molecules such as IL-1α, IL-1β, IL-6, IL-8, TNFα, and COX2 and metabolic pathway genes such as GLUT3; and genes linked to epithelial-to-mesenchymal transition such as ZEB1 and ZEB2 [2, 6]. Additionally, there is evidence for NF-κB playing a significant role in self-renewal and maintenance of cancer stem cell phenotype, particularly in glioblastoma. NF-κB is essential for lung tumor development upon p53 mutation and KRAS (G12D) expression [7]. Inhibition of NF-κB sensitizes EGFR-mutant lung tumors to EGFR-targeted therapies. MLL fusion proteins that typically cause leukemia are dependent on NF-κB for transformation. NF-κB is also a central player in epigenetic switch that links chronic inflammation to cell transformation and subsequent metastasis [8].
Cancer Cell Non-autonomous Functions of NF-κB
Although initial studies on NF-κB were primarily focused on cell autonomous functions, several recent reports highlight its non-cell autonomous roles . NF-κB-inducible cytokines such as IL-6 and IL-8 not only alter tumor microenvironment by attracting different immune cells, but also these cytokines are associated with systemic effects of cancer. IL-6 is a major contributor to cancer cachexia. NF-κB itself contributes to cancer cachexia by blocking myogenic differentiation by affecting skeletal muscle transcription factor-microRNA circuitry and by repressing differentiation factor MyoD [9].
Recent studies have demonstrated a role for NF-κB in resistance to immune therapy. PD-L1, expressed mainly by cancer cells , plays a significant role in creating antitumor immunity. Therefore, several antibodies targeting PD-L1 have entered clinic. Although correlation between PD-L1 levels and response to therapy is yet to be established, NF-κB has been shown to increase PD-L1 at both transcriptional and posttranscriptional level. Pro-inflammatory cytokines such as TNFα induce the expression of COP9 signalosome 5 (CSN5), which deubiquitinates PD-L1 and stabilizes the protein.
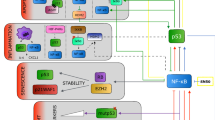
By increasing PD-L1 protein, NF-κB could reduce the effectiveness of PD-L1-targeting antibodies (Fig. 27.1) [10].

Canonical and noncanonical pathways of NF-κB activation. Steps associated with NF-κB activation in response to extracellular signals are shown. Different members of NF-κB and IκB family are listed. In mammals, the NF-κB family is composed of five related transcription factors: p50, p52, RelA (p65), c-Rel, and RelB. IKK complex is the main signaling hub that integrates extracellular/membrane-activated signals to nuclear events by activating NF-κB. Potential therapeutic agents that may reduce NF-κB activation or reduce the activities of downstream targets of NF-κB are also indicated. For example, ABX-IL8 may inhibit IL8-dependent signaling, and Plerixafar prevents CXCR4 activation by inhibiting CXCR4/CXCL12 interaction. Infliximab is a TNFα antagonist, which can block NF-κB activation by TNFα as well as block the effects of TNFα downstream of NF-κB activation
Current NF-κB-Targeted Therapies and Future Strategies for Treatment
Based on multiple functions ascribed to NF-κB in cancer and therapeutically targetable signaling cascades involved in NF-κB activation, there have been several attempts to develop drugs targeting NF-κB [11]. Commonly used drugs such as aspirin and food ingredient curcumin inhibit NF-κB. Several groups including us have shown anti-NF-κB activity of parthenolide, an active ingredient in the herb Tanacetum parthenium or feverfew used for migraine. Clinically, bortezomib used to treat multiple myeloma is a proteasome inhibitor that prevents IκB degradation and restricts NF-κB activation. However, for most of these drugs, NF-κB is one of their targets, and their clinical activity cannot be solely due to NF-κB inhibition. A highly specific NF-κB inhibitor is yet to enter clinic, although such an inhibitor may never be discovered. An ideal NF-κB inhibitor should dampen overactive NF-κB rather than completely eliminate its activity because basal NF-κB is necessary for normal function of the immune system and to prevent infection. Therefore, there is still considerable interest in both academia and industry to develop drugs that reduce but not eliminate NF-κB activity. Because of the role of NF-κB in upregulating anti-apoptotic proteins, these inhibitors will likely work as chemosensitizing agents rather than displaying single agent activity. However, chemosensitizing function NF-κB inhibitors needs to be tested with individual chemotherapeutic drugs because in certain instances, therapy-induced NF-κB, particularly therapies that promote replication stress, could augment cell death by activating extrinsic pathway of cell death through expression/activation of FAS-FASL-dependent cell death machinery [12].
Several of downstream targets of NF-κB can be targeted therapeutically (Table 27.1). In fact, drugs targeting TNF, IL-1, IL-6, IL-8, CXCR4, and PD-L1 are already in clinical use and can be exploited to treat cancers in which NF-κB pathway is essential for cancer cell survival.
Summary
Although NF-κB was discovered more than 30 years ago, its regulation and function still remain at the forefront of research not only from the oncology point of view but also with other diseases such as autoimmune disorders, microbial infections, and neurodegenerative diseases. While constitutive activation of NF-κB has been reported in advanced stages of many cancers, causes of this activation vary widely and may involve both genomic and non-genomic events. Despite early-stage-specific tumor suppressor role described for this transcription factor complex, this observation should not hinder clinical development of NF-κB inhibitors because NF-κB has already transitioned to oncogenic role at the time of clinical manifestation of the disease. Thus, studies focusing on NF-κB, both at regulatory and functional level, will continue to unlock mysteries surrounding cancer progression and potentially to new cancer therapies. Reliable biomarkers of constitutive NF-κB activity in cancer are yet to be identified because of close link between NF-κB activation and inflammatory process. Although drugs that directly target NF-κB are yet to enter clinic, several drugs that inhibit the function of proteins overexpressed as a consequence of increased NF-κB activity in cancer are showing promising results.
Abbreviations
- COX2:
-
Cyclooxygenase 2
- CSN5:
-
COP9 signalosome 5
- CXCL:
-
Chemokine (C-X-C) ligand
- CXCR:
-
Chemokine (C-X-C) receptor
- EGFR:
-
Epidermal growth factor receptor
- ERBB2:
-
ERB-B2 receptor tyrosine kinase 2
- FAS:
-
Fas cell death receptor
- FASL:
-
FAS ligand
- IAP:
-
Inhibitor of apoptosis
- IKK:
-
IκB kinase
- IL:
-
Interleukin
- IκB:
-
Inhibitor of kappaB
- MLL:
-
Mixed lineage leukemia
- MMP:
-
Matrix metalloproteinase
- MyoD:
-
Myogenic differentiation 1
- NF-κB:
-
Nuclear factor-kappaB
- PD-L1:
-
Programmed death ligand 1
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- SASP:
-
Senescence-associated secretory phenotype
- TNF:
-
Tumor necrosis factor
- ZEB:
-
Zinc finger E-box-binding homeobox
References
Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34.
Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–10.
Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012;24(4):835–45.
Lesina M, Wormann SM, Morton J, Diakopoulos KN, Korneeva O, Wimmer M, et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J Clin Invest. 2016;126(8):2919–32.
Klein U, Ghosh S. The two faces of NF-kappaB signaling in cancer development and therapy. Cancer Cell. 2011;20(5):556–8.
Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 2006;13(5):759–72.
Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462(7269):104–7.
Kuo HP, Wang Z, Lee DF, Iwasaki M, Duque-Afonso J, Wong SH, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-kappaB. Cancer Cell. 2013;24(4):423–37.
Bakkar N, Guttridge DC. NF-kappaB signaling: a tale of two pathways in skeletal myogenesis. Physiol Rev. 2010;90(2):495–511.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–39.
DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400.
Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12(2):121–32.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nakshatri, H. (2019). NF-κB Signaling Pathways in Carcinogenesis. In: Badve, S., Kumar, G. (eds) Predictive Biomarkers in Oncology. Springer, Cham. https://doi.org/10.1007/978-3-319-95228-4_27
Download citation
DOI: https://doi.org/10.1007/978-3-319-95228-4_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95227-7
Online ISBN: 978-3-319-95228-4
eBook Packages: MedicineMedicine (R0)