
Abstract
-
Stargardt disease (STGD) is one of the most common macular dystrophies in young adults. It progresses slowly. Its prevalence is about 1:8000–10,000.
-
Age of onset is a surrogate marker: The earlier the onset, the more severe the disease course. Onset usually occurs in childhood or early adolescence, at about 10–15 years of age.
-
Vision is between about 20/70 and 20/200.
-
The fundus shows a bull’s eye pattern or beaten-bronze appearance, with or without yellowish flecks (fundus flavimaculatus).
-
Fluorescein angiography may show dark choroid in about 80% of cases.
-
On fundus autofluorescence (FAF), newer flecks appear hyperautofluorescent (hyperAF); older ones become progressively more hypoAF with time. Some flecks are surrounded by a ring of decreased AF.
-
Peripapillary sparing is one the characteristics of Stargardt disease, but this area can be involved in about 2–7% of cases. The reason for this sparing is unclear; this area may be more resilient to the deleterious effect of ABCA4 gene mutation, and there might be a more favorable RPE photoreceptor ratio, resulting in less lipofuscin build-up, in the presence of a thicker overlying peripapillary retinal nerve fiber layer.
-
Patients with Stargardt disease should avoid bright light and excessive vitamin A.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
General Features
-
Stargardt disease (STGD) is one of the most common macular dystrophies in young adults. It progresses slowly. Its prevalence is about 1:8000–10,000.
-
Age of onset is a surrogate marker: The earlier the onset, the more severe the disease course. Onset usually occurs in childhood or early adolescence, at about 10–15 years of age.
-
Vision is between about 20/70 and 20/200.
-
The fundus shows a bull’s eye pattern or beaten-bronze appearance, with or without yellowish flecks (fundus flavimaculatus).
-
Fluorescein angiography may show dark choroid in about 80% of cases.
-
On fundus autofluorescence (FAF) , newer flecks appear hyperautofluorescent (hyperAF); older ones become progressively more hypoAF with time. Some flecks are surrounded by a ring of decreased AF.
-
Peripapillary sparing is one the characteristics of Stargardt disease, but this area can be involved in about 2–7% of cases. The reason for this sparing is unclear; this area may be more resilient to the deleterious effect of ABCA4 gene mutation, and there might be a more favorable RPE photoreceptor ratio, resulting in less lipofuscin build-up, in the presence of a thicker overlying peripapillary retinal nerve fiber layer.
-
Patients with Stargardt disease should avoid bright light and excessive vitamin A.
Clinical Stages and Groupings
Stages, Based on Fundus Appearance (Figs. 27.1, 27.2, 27.3, 27.4, 27.5 and 27.6)
-
STAGE I: Confined to macula , with beaten-metal appearance (discontinuous, as opposed to classic appearance); a discontinuous ring of flecks around fovea, about 1 disc diameter (DD). The electrooculogram (EOG) and electroretinogram (ERG) are normal.
-
STAGE II: Widespread flecks beyond temporal arcades and/or nasal to disc. Subnormal cone and rod response.
-
STAGE III: Re-absorption of flecks and widespread choriocapillaris (CC) atrophy. EOG shows subnormal ratio, and ERG shows cone/rod dysfunction.
-
STAGE IV: Further resorption of flecks, with extensive CC and RPE atrophy.

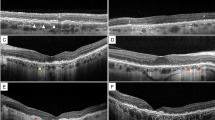
Stargardt disease (STGD) presenting as bull’s eye maculopathy (BEM), with no flecks. HypoAF on FAF. Optical coherence tomography (OCT) shows marked damage to photoreceptor layer and RPE (C4537delC; p.R107*)

STGD presenting as atrophic patch at the macula and flecks. Note sparing of the peripapillary area

STGD presenting as multiple flecks at the posterior pole, with a few atrophic patches at the macula

STGD presenting as a large atrophic patch at the macula with a few flecks, some of them encroaching on the peripapillary area (28:p.P1380L, 36:p.S1696N)

STGD presenting as a large RPE atrophic area in the macula with sparse flecks, some of them showing RPE atrophy as well

STGD presenting as BEM with minimal flecks; OCT shows marked thinning of the central retina. Note peripapillary sparing
Groupings, Based on Electroretinography (ERG)
-
GROUP I: Isolated abnormalities in the flash ERG (FERG) and pattern ERG (PERG); normal photopic/scotopic full-field (FF) ERG.
-
GROUP II: Abnormal FERG, PERG, and photopic FF ERG; normal scotopic FF ERG.
-
GROUP III: Abnormal FERG, PERG, and photopic/scotopic FF ERG.
Types, Based on FAF (Figs. 27.7, 27.8, 27.9, 27.10 and 27.11)
-
TYPE A: Central hypofluorescence, surrounded by a hyperfluorescent ring.
-
TYPE B: Only central hypofluorescence without surrounding hyperfluorescence.
-
TYPE C: No central hypofluorescence; instead, a speckled pattern of alternating hypofluorescence and hyperfluorescence.

An advanced case of STGD showing RPE atrophy over the macula and over all of the flecks as well. p.R18W and p.D2102E

STGD involving predominantly the macula, with sparse flecks in the periphery; OCT shows marked thinning of central retinal layers, including disruption of RPE layer (OS)

An advanced stage of STGD, with marked RPE atrophy, confluent at the posterior pole (3:p.C54Y,IVS14:c.2160+1G>C) (Stargardt Group 3)

STGD with just posterior involvement; the periphery is healthy. OCT shows marked thinning of the central retina. (Compare with Fig. 27.11, mother.) Two genetic variations: Cys54Tyr and Ala1038Val

STGD with involvement of the posterior pole and the periphery and marked atrophy of the RPE layer. (Compare with Fig. 27.10, son.) Two genetic variations: Cys54Tyr and Ala1038Val (Stargardt Group 3)
Classes, Based on Optical Coherence Tomography (OCT)
-
CLASS A: Flecks confined to outer segment (OS) layer
-
CLASS B: Flecks extend through junction of OS and inner segment (IS)
-
CLASS C: Protrude into outer nuclear layer (ONL)
-
CLASS D: Only in ONL
-
CLASS E: Drusen-like pigment epithelial detachment (PED). (Familial drusen shows echolucency in the center; flecks are echodense.)
Molecular Genetics
ABCA4 Gene (RIM Protein or ABCR)
Mutation in ABCA4 gene , which encodes an ATP-binding cassette (ABC) transporter protein, expressed in the outer segment (OS) of rods. ABCA4 protein transports potentially toxic substances out of photoreceptor cells. These substances are formed after phototransduction. Buildup of the toxic substance—lipofuscin—in the photoreceptor cells and surrounding cells causes cell death. Cytogenetic location: 1p22.1.
The genetics is extremely heterogenous and may be responsible for wide variations in clinical presentations. Mild reduction in ABCA4 activity results in about 95% of cases of STGD; moderate loss of activity results in cone-rod dystrophy (about 30–50% of cases); and complete loss results in retinitis pigmentosa (RP) with complete loss of rod and cone function (about 8% of cases of autosomal recessive RP).
Other Mutations
Other mutations, accounting for the remaining 5% of STGD cases, are in the dominant genes STGD4 and ELOVL4 and PRPH2. The ELOVL4 protein plays a role in making a group of fats called very long-chain fatty acids. Mutations in the ELOVL4 gene lead to the formation of ELOVL4 protein clumps in the cells, interfering with their activity and eventually leading to cell death. Cytogenetic location:6q14.1.
Genotype-Phenotype Correlation
-
c.5603A>T (p.Asn1868IIe) (Figs. 27.12, 27.13 and 27.14)
-
This is a hypomorphic allele that is expressed only when in trans with a deleterious mutation.
-
Phenotypes are late-onset (age about 36 years or later). About 85% of cases have foveal sparing and can masquerade as age-related macular degeneration (AMD).
-
Very few flecks are seen, which are somewhat larger, well-defined (a kind of “pizza sign”), and sparsely distributed.
-

STGD with predominant macular involvement; FAF shows central hypoAF surrounded by stippled AF (“pizza sign”) (arrows)

STGD caused by c.5603A>T (p.N1868I). Note the characteristic “pizza sign”
-
c.5882G>A (p.Gly1961Glu) (Fig. 27.15)
-
These patients exhibit mild disease expression, few specks, no confluence of flecks, and not much disease progression.
-
Age of onset is about 23 years, with foveal sparing in about 40% of cases.
-
Flecks are relatively large, well-defined, and sparsely distributed.
-
On OCT, these patients show focal loss of the ellipsoid zone (EZ) (Stage I), an optical gap (Stage II), and eventual collapse of the optical gap.
-

STGD caused by c.5603A>T (p.N1868I). Note foveal sparing on AF. This mutation accounts for foveal sparing in about 80% of cases, along with late onset and mild progression
-
Other ABCA4 alleles
-
These patients show innumerable flecks, which are confluent across the posterior pole and progressive; onset is about 18 years of age.
-
Foveal sparing, about 30%
-
-
Biallelic Null ABCA4
-
Age of onset about 10 years or earlier
-
Confluent flecks appear early and progress more rapidly, leading to end-stage atrophy.
-

STGD caused by c.5882G>A (p.G1961E) mutation. Note loss of foveal reflex, and central hypoAF surrounded by a hyperAF ring. OCT shows loss of the ellipsoid zone (EZ), causing an optical gap (arrow)
Suggested Reading
Burke TR, Tsang SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 2011;32:165–74.
Gelman R, Smith RT, Tsang SH. Diagnostic accuracy evaluation of visual acuity and fundus autofluorescence macular geographic atrophy area for the discrimination of Stargardt groups. Retina. 2016;36:159–601.
Greenstein VC, Nunez J, Lee W, Schuerch K, Fortune B, Tsang SH, et al. A comparison of en face optical coherence tomography and fundus autofluorescence in Stargardt disease. Invest Ophthalmol Vis Sci. 2017;58:5227–36.
Nõupuu K, Lee W, Zernant J, Tsang SH, Allikmets R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest Ophthalmol Vis Sci. 2014;55:7217–26.
Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017;101:25–30.
Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. 2017;54:404–12.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Tsang, S.H., Sharma, T. (2018). Stargardt Disease. In: Tsang, S., Sharma, T. (eds) Atlas of Inherited Retinal Diseases. Advances in Experimental Medicine and Biology, vol 1085. Springer, Cham. https://doi.org/10.1007/978-3-319-95046-4_27
Download citation
DOI: https://doi.org/10.1007/978-3-319-95046-4_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95045-7
Online ISBN: 978-3-319-95046-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)