
Abstract
Phase-change memory is regarded as the most appealing of the nonvolatile memory technologies, with attractive properties including scalability, bit alterability, and fast write/erase and read performance. Over the past decade, the technology has experienced rapid growth. Well-known semiconductor manufacturers such as IBM, Infineon, Samsung, and Macronix have spared no effort in the push to commercialize this technology. At the same time, many novel phase-change materials have been developed, such as typical Ge-Sb-Te alloys, Zn-Sb-Te alloys, and ZnO-\(\mathrm{Sb_{2}Te_{3}}\) nanocomposite.
New techniques such as ultrafast calorimetry are continuously emerging to better understand the crystallization kinetics of supercooled liquids for phase-change materials. In addition, phase-change materials are ideal functional materials for use in integrated photonic memory, which provides a new paradigm in all-photonic memory.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Over the past decade, the continuous size scaling of complementary metal-oxide-semiconductor () technology, along with its cost reduction, has driven the development of nonvolatile memory (NVM) to meet the growing demand for high-density digital information storage. The extent to which NVM has pervaded our day-to-day lives is truly remarkable. From the music on our MP3 players, to the photos on digital cameras, the stored email and text messages on smartphones, the documents we carry on our USB thumb drives, and the program codes that enable everything from our portable electronics to our cars, the NVM known as flash memory is ubiquitous. However, flash memory technology is inevitably approaching its fundamental limits. State-of-the-art NAND devices are restricted to \({\mathrm{12}}\,{\mathrm{nm}}\) node [44.1, 44.2] in size, where the node number refers to the minimum channel width of the CMOS.
The physical scaling of flash memory depends mainly on state-of-the-art photolithography techniques [44.3, 44.4], which are confined by the diffraction limit. Second, the scaling margin has also been shrinking, since the tunnel oxide layer inside flash memory needs to be thicker than \({\mathrm{8}}\,{\mathrm{nm}}\) in order to eliminate possible electron leakage [44.5]. Moreover, the coupling ratio between the floating gate and the control gate must be maintained at a value greater than 0.6 in order to control the conductive channel and prevent gate electron injection [44.6]. This can be achieved by wrapping the control gate around the floating gate to geometrically increase the gate coupling ratio. Obviously, there is not adequate space to contain such a wrapping structure as the downscaling process continues. Furthermore, the crosstalk effect between two adjacent cells will be strongly aggravated with future scaling; the electrons stored in one cell will have higher tunneling probability, which will adversely affect the performance of scaled devices [44.1].
To overcome the scaling limits of flash memory, more advanced storage technologies are being explored, giving rise to a series of new paradigms, including ferroelectric random-access memory (FRAM) [44.7], magnetic random-access memory (MRAM) [44.8], phase-change random-access memory () [44.9], and resistive random-access memory (RRAM) [44.10]. Among these, PRAM is arguably the most appealing. PRAM technology has the potential to provide inexpensive, high-speed, high-density, high-volume nonvolatile storage on an unprecedented scale. Working prototypes of PRAM chips have been tested by IBM, Infineon, Samsung, Macronix, and others. PRAM is regarded as a significant advance, and is likely to become one of the mainstream formats for semiconductor memory.
PRAM is based on the repeated switching of a phase-change material () between the amorphous and the crystalline states, inducing a large change in resistance. The amorphous, high-resistance state represents a binary 0, while the crystalline, low-resistance state represents a 1. Information is then stored in the phase of the material and is read by measuring the resistance of the PRAM cell. Programming a PRAM device involves the application of electric current, leading to temperature changes that either SET or RESET the cell, as shown schematically in Fig. 44.1. To SET a PCM cell to its low-resistance state, an electrical pulse is applied to heat the cell above the crystallization temperature \(T_{\text{c}}\) (but below the melting temperature \(T_{\text{m}}\)) of the phase-change material. The pulse is sustained for a period of sufficient length for the cell to transition to the crystalline state. On the other hand, to RESET the cell to its high-resistance amorphous state, a much larger electrical current is applied in order to increase the temperature above \(T_{\text{m}}\). After the material in the cell has melted, the pulse is abruptly cut off, causing the melted material to quench into the amorphous state. To READ the current state of a cell, a small current that does not perturb the cell state is applied to measure the resistance.


Currents and timings (not to scale) for SET, RESET, and READ operations on a PCM cell. After [44.11]
PCM is also the functional material widely used in rewritable optical media, such as CD-RW and DVD-RW. In those instances, the material's optical properties are manipulated rather than its electrical resistivity, as its refractive index also changes with the state of the material.
Generally speaking, several characteristics are important for the operation of the PCM, including rapid switching between the amorphous and crystalline phases, long data retention at high temperature, and a large difference in electrical resistivity or optical reflectivity between the amorphous and crystalline phases. However, trade-offs are necessary to balance each characteristic of PCM—for example, the contradictory relation between rapid phase change and long-term data retention. Long-term retention of data usually requires a material that is very stable in the amorphous state at high temperatures, which in turn implies that fast crystallization speed is not available. Furthermore, if the reliability of the PRAM cell is considered, then a small change in thickness during the amorphous-to-crystalline phase transition is needed to avoid the interface separation between the phase-change layer and the electrode. Since the thinner the phase-change layer, the smaller the change in thickness, a thinner phase-change layer is obviously of benefit for cell reliability. However, the thinner phase-change layer leads to a smaller difference in electrical resistivity or optical reflectivity, which will directly degrade the ON/OFF ratio for PRAM.
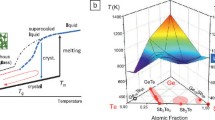
Tracing the development of PCM, it should be mentioned that in 1968, S.R. Ovshinsky of Energy Conversion Devices demonstrated very short reversible electrical switching phenomena in \(\mathrm{Te_{81}Ge_{15}Sb_{2}S_{2}}\) chalcogenide glasses as a potential memory technology [44.12]. Although material quality and power consumption issues prevented commercialization of the technology at that time, this work opened a new chapter in phase-change technology. The early phase-change materials used in optical storage comprised simple alloys based primarily on composition in the vicinity of the tellurium-germanium eutectic. In the early 1990s, Ge-Sb-Te alloys were reported as a second generation of high-speed phase-change materials [44.13]. These alloys have a stoichiometric composition along the GeTe-\(\mathrm{Sb_{2}Te_{3}}\) pseudo-binary line of the phase diagram. As indicated in Fig. 44.2, moving down this pseudo-line from \(\mathrm{Sb_{2}Te_{3}}\) to GeTe, the melting point and glass transition temperature of the material increases, crystallization speed decreases, and data retention increases [44.13]. Therefore, when a rapid phase change is required, a material with fast crystallization speed such as \(\mathrm{Sb_{2}Te_{3}}\) will be selected. However, because of its low activation energy, this material is not stable. On the other hand, a material such as GeTe with good amorphous stability has slow crystallization speed because of its high activation energy. Therefore, to balance thermal stability and crystallization speed, most important materials that have been explored, i. e., \(\mathrm{Ge_{2}Sb_{2}Te_{5}}\) (GST), \(\mathrm{GeSb_{2}Te_{4}}\), and \(\mathrm{GeSb_{4}Te_{7}}\), lie along the pseudo-line between \(\mathrm{Sb_{2}Te_{3}}\) and GeTe. These alloys are now also widely used in phase-change memory as well as for scientific research.


Phase diagram of the Ge-Sb-Te ternary alloy system. After [44.13]
In this chapter, we will first discuss the structure and crystallization behavior of ternary Ge-Sb-Te alloys, after which doped GST will be highlighted for the purpose of obtaining PRAM with lower power consumption and better data retention. We will then present the thermal and phase-change behavior for the novel doped Sb-Te alloys and some nanocomposites. The chapter also focuses on the recent hotspot in crystallization kinetics for GST materials by ultrafast calorimetry, which extends the heating rate to more than \({\mathrm{4\times 10^{4}}}\,{\mathrm{K{\,}s^{-1}}}\) and can provide more information on crystal growth in supercooled liquids. In addition, in recent years, phase-change materials have also been used in integrated photonic memory. In this case, both reading and writing of the memory can be performed with ultrashort optical pulses, because the guided light transfers the amorphous and crystallization phases of GST via its evanescent field interaction. This provides a pathway towards a new paradigm in all-photonic memory and nonconventional computing.
1 Conventional Ge-Sb-Te Phase-Change Films
Crystalline Ge-Sb-Te films have two possible configurations: a metastable face-centered cubic (fcc) and a stable hexagonal close-packed (hcp) lattice. Figure 44.3 depicts the structure of a typical cubic-phase Ge-Sb-Te alloy. The fcc state is preferred for discussion, because PRAM works in this crystalline state. As shown in the figure, the Te atoms occupy one lattice site, and Ge and Sb atoms randomly occupy the second lattice site of an atomic arrangement that closely resembles the rock-salt structure. There are a few noteworthy differences from an ideal rock-salt structure. In a material such as \(\mathrm{GeSb_{2}Te_{4}}\) or GST, there are a considerable number of vacancies, as schematically depicted in Fig. 44.3. For \(\mathrm{GeSb_{2}Te_{4}}\), for example, there are \({\mathrm{25}}\%\) vacancies on the Ge/Sb site, whereas the Te site is fully occupied.


Schematic image of the crystal structure of the rock-salt-like phase of, e. g., GST or \(\mathrm{GeSb_{2}Te_{4}}\). Te atoms occupy one sublattice of the crystal, and Ge atoms, Sb atoms, and vacancies randomly occupy the second sublattice. The typical nearest-neighbor spacing is close to \({\mathrm{3}}\,{\mathrm{\AA}}\). After [44.14]
The commonly studied phase-change alloys \(\mathrm{GeSb_{2}Te_{4}}\) and GST are found on the pseudo-binary GeTe-\(\mathrm{Sb_{2}Te_{3}}\) line, which is commonly believed to locate the most suitable Ge-Sb-Te-based compounds. The as-deposited films, which are deposited by magnetron sputtering, are amorphous, and the crystallization temperature increases along the \(\mathrm{GeSb_{2}Te_{4}}\)-\(\mathrm{Ge_{2}Sb_{2}Te_{4}}\) line, indicating higher overall stability with increasing a Ge content. The crystallization temperature of \(\mathrm{Ge_{2}SbTe_{4}}\) is similar to that of GST but higher than that of \(\mathrm{GeSb_{2}Te_{4}}\).
According to the resistance–temperature curve, phase-change materials upon amorphization exhibit a change in resistivity of several orders of magnitude. While the resistivity is low in the crystalline state, the amorphous system is highly resistive, as shown in Fig. 44.4.


Temperature dependence of the resistivity of a sputter-deposited \(\mathrm{GeSb_{2}Te_{4}}\) thin film. The temperatures of the phase transitions from the amorphous to the rock-salt state at \(\approx{\mathrm{150}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), and further transitions to the stable trigonal state, are indicated by the arrows. A significant resistivity change occurs only at the first phase transition. After [44.15]
The metastable crystalline phase of all the novel alloys shows the characteristic peaks of the rock-salt structure, with lattice parameters of around \({\mathrm{6}}\,{\mathrm{\AA}}\) (Fig. 44.5a-c and Table 44.1) and no evidence of phase separation in the metastable phase. The resistivity contrast of the new alloys is in the same range as the contrast of \(\mathrm{GeSb_{2}Te_{4}}\) and \(\mathrm{Ge_{2}Sb_{2}Te_{5}}\).


X-ray diffractograms () of (a) \(\mathrm{Ge_{1.5}Sb_{2}Te_{4}}\), (b) \(\mathrm{Ge_{2}Sb_{2}Te_{4}}\) and (c) \(\mathrm{Ge_{2}SbTe_{4}}\). The diffractograms show a metastable phase after crystallization from the as-deposited amorphous phase. The peaks have been identified and attributed to the metastable rock-salt structure. From [44.16]
Understanding the mechanism of the rapid reversible phase transition is important for improving the phase change performance. This mechanism has been discussed extensively during recent years. In 2004, for example, an umbrella-flip model was carried out to explain the rapid phase transition, as shown in Fig. 44.6a,b. Kolobov et al [44.17] used the (extended x-ray absorption fine structure) method to study the conventional GST, and concluded that GST does not have a rock-salt structure, but more likely consists of cubic symmetry that is randomly oriented in space. Laser-induced amorphization results in a dramatic shortening of the covalent bonds and obvious decrease in the relative displacement can be detected, which demonstrates a substantial increase in the degree of short-range ordering. This transition can be considered as an umbrella flip of Ge atoms from an octahedral position into a tetrahedral position without rupture of strong covalent bonds. In 2006, Kohara et al [44.18], using reverse Monte Carlo simulation with synchrotron-radiation x-ray diffraction data, found that the ring statistics of amorphous GST were dominated by four- and sixfold rings, analogous to the crystal phase, as shown in Fig. 44.7. They conclude that such unusual ring statistics for amorphous GST are the key for the fast phase transition speed of the material. In 2007, Akola and Jones [44.19] used density functional theory (DFT) to investigate the structure of GST. As shown in Fig. 44.8a,b, they found a long-range order among Te atoms and note that the crucial structural motif is a four-membered ring with alternating atoms of types A (Ge and Sb atom) and B (Te atom), which they called an ABAB square. They posit that the rapid amorphous-to-crystalline phase change is a reorientation of such disordered ABAB squares to form an ordered lattice. They determined that the vacancies in the amorphous GST provide the necessary space for the crystallization to take place, noting a vacancy concentration in GST of about \({\mathrm{11.8}}\%\).


Fragments of the local structure of GST around Ge atoms in the crystalline (a) and amorphous (b) states. After [44.17]


Schematic presentation of the possible ring size transformation in crystal–liquid–amorphous phase change and amorphous–crystal phase change in GST. Adapted from [44.18], with permission of AIP Publishing


ABAB squares and cubes in amorphous GST (Ge in red, Sb in blue, and Te in yellow). (a) Simulation box of amorphous GST with atoms and bonds of ABAB squares and cube highlighted. (b) Local environment of ABAB cube. Reprinted with permission from [44.19]. Copyright 2007 by American Physical Society


Structural transition from sixfold to fourfold in GeTe presented by Huang and Robertson in [44.20]
Huang and Robertson investigated structural transition for PCMs in five medium-range order [44.20]. They believe that many Ge sites in GeTe are distorted fourfold sites, indicating that the Ge atom is displaced along the (110) direction to complete the transition, as shown in Fig. 44.9. This is different from the umbrella-flip model as shown in Fig. 44.6a,b, where the Ge atoms move along the (111) direction corresponding to a change from an octahedral to tetrahedral structure in the crystalline-to-amorphous phase transition. Thus, the Ge atom is octahedrally rather than tetrahedrally coordinated, leading to a local Peierls-like distortion in the amorphous state. They also showed evidence to illustrate a large effect on the optical matrix elements, which are roughly twice as large for the resonantly bonded p state. Therefore, the large difference in optical contrast between amorphous and crystalline states in GeTe is attributed to the resonant bonds present in the crystalline phase. The concepts of Peierls-like distortion and resonant bonding in PCMs have become well accepted recently, and were discussed in detail by Jones [44.21].
2 Phase-Change Behaviors of Doped Ge\({}_{2}\)Sb\({}_{2}\)Te\({}_{5}\) Films
GST alloys have been proposed for use in PRAM due to their excellent properties with respect to thermal stability, cyclability, and crystallization speed. However, with the increasing need for low power consumption and good data retention, and the challenge in reducing the RESET current for PRAM applications, the performance of the commonly adopted GST still needs to be improved. In fact, many efforts have been made to improve performance by doping various elements, including N [44.22], O [44.23], Si [44.24], Bi [44.25], Ag [44.26], and Sn [44.27], into GST. The changes in properties as a function of doping mainly include increased resistivity of the phases, leading to smaller currents to write and erase, and accelerated crystallization speed or improved thermal stability for the amorphous phase due to increased crystallization activation energy.
Generally, appropriate doping enhances the thermal stability of the amorphous phase due to the formation of strong bonds in the glass network. For example, in O-doped GST film [44.28], the presence of nonstoichiometric Ge–O bonds and the \(\mathrm{Sb_{2}O_{3}}\) can be attributed to the suppression of crystallization and the enhancement of thermal stability. However, excess doping may deteriorate other phase transition properties, such as the speed of the phase change. For instance, the sheet resistance of W-GST drops more slowly with W doping of more than \({\mathrm{18}}\,{\mathrm{at.\%}}\), which implies that the excess W causes a decrease in the phase transition speed [44.29]. Excess doping can also lead to a phase separation that reduces the thermal stability. For example, crystallization was suppressed in film with \({\mathrm{16.8}}\,{\mathrm{at.\%}}\) incorporated oxygen, while the formation of a separate phase of \(\mathrm{Sb_{2}Te_{3}}\) was observed in films with more oxygen doping, indicating that the crystallization temperature was decreased with greater oxygen doping. We know that the GST film shows two types of phase transitions, i. e., metastable fcc and stable hcp. However, with proper doping, the second phase of hcp will always be inhibited. An ideal PCM possesses both high phase transition speed and amorphous thermal stability. However, a trade-off is required to balance these two properties in each PCM system. In the case of Bi- and Sn-doped GST [44.25], the balance of minimum time and maximum temperature for crystallization was obtained in the component with \({\mathrm{5.9}}\,{\mathrm{at.\%}}\) Bi and \({\mathrm{17.7}}\,{\mathrm{at.\%}}\) Sn, respectively. Therefore, the newer PCMs should be explored and characterized. Here, the crystallization behaviors, as well as the thermal and electrical properties, of Zn-doped GST will be highlighted for potential PRAM application.
Figure 44.10a-da–d shows x-ray diffraction results for the as-deposited films with different Zn doping concentrations annealed at 200, 250, and \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for \({\mathrm{3}}\,{\mathrm{min}}\), respectively. There are no diffraction peaks in Fig. 44.10a-da, confirming that all the as-deposited samples are amorphous. On the other hand, the diffraction peaks in the GST, \(\mathrm{Zn_{6.37}(GST)_{93.63}}\), and \(\mathrm{Zn_{8.13}(GST)_{91.87}}\) films annealed at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), as shown in Fig. 44.10a-db, correspond to fcc phase peaks in (200) and (220) orientations. Nevertheless, there are no fcc (200) and (220) diffraction peaks in \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) or \(\mathrm{Zn_{19.78}(GST)_{80.22}}\) films annealed at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). Diffraction peaks do appear with an increase in annealing temperature to \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) in the \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) and \(\mathrm{Zn_{19.78}(GST)_{80.22}}\) films, as shown in Fig. 44.10a-dc. Thus it can be clearly seen that the onset phase transition temperature of GST from the amorphous state to fcc structure increases with increased Zn doping. As shown in Fig. 44.10a-dd, the structures of \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) and \(\mathrm{Zn_{19.78}(GST)_{80.22}}\) thin films are kept at the fcc phase. However, the phase transition of fcc to hcp (hexagonal close-packed) is present in \(\mathrm{Zn_{6.37}(GST)_{93.63}}\) and \(\mathrm{Zn_{8.13}(GST)_{91.87}}\) when the annealing temperature was increased to \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). This implies that the high Zn dopant concentration in GST film introduces Zn atoms that serve as a center to restrain the fcc-to-hcp phase transition, resulting in a one-step crystallization process.


XRD patterns of GST and Zn-doped GST films: (a) as-deposited, (b) \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), (c) \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), and (d) \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) (f \(=\) fcc, h \(=\) hcp)
The temperature dependence of the sheet resistance (\(R\)–\(T\)) for the Zn-doped GST films with a heating rate of \({\mathrm{40}}\,{\mathrm{K{\,}min^{-1}}}\) are displayed in Fig. 44.11a. Clearly, the resistance of the films decreases slightly before their respective crystallization temperature (\(T_{\text{c}}\)) that exhibits an abrupt drop in sheet resistance, which implies the phase transition from amorphous to fcc crystalline structure. A second drop in the sheet resistance occurs in GST, \(\mathrm{Zn_{6.37}(GST)_{93.63}}\), and \(\mathrm{Zn_{8.13}(GST)_{91.87}}\) films, indicating the structural transition of fcc to hcp. Moreover, the \(T_{\text{c}}\) values increase with more Zn doping, and they are much higher than that of GST, which helps to improve the thermal stability of the films. An amorphous/crystalline resistance ratio larger than \(\mathrm{10^{5}}\) in the doped films provides a good signal-to-noise ratio for the reading operation in PCM applications. High crystalline resistance can also be obtained in these films, which helps to reduce the programming energy in the RESET operation.


(a) Sheet resistance as a function of temperature for undoped and Zn-doped GST films. (b) The Arrhenius extrapolation at 10-year data retention for undoped and Zn-doped GST films
The data retention ability of GST and Zn-doped GST is shown in Fig. 44.11b. This can be extrapolated by the Arrhenius equation, \(t=\tau\exp(E_{\text{a}}/(k_{\text{B}}T))\), where \({\tau}\) is the proportional time coefficient, \(E_{\text{a}}\) is the activation energy for crystallization, and \(k_{\text{B}}\) is the Boltzmann constant. We determined \(t\) as the failure time when sheet resistance reaches half of its initial magnitude at a specific isothermal temperature, which is also indicated in Fig. 44.11b. Clearly, the 10-year data retention temperatures and \(E_{\text{a}}\) are all much higher than those of GST (\({\mathrm{88.9}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{2.98}}\,{\mathrm{eV}}\)), which means that the PCM based on Zn-doped GST can store the data for a longer time than that of GST. This indicates that Zn-doped GST (especially with high Zn dopant concentration) is an ideal PCM, with better amorphous stability, larger crystallization activation energy, and better 10-year data retention. Thus, we will focus on the GST film doped with a high Zn concentration, such as \(\mathrm{Zn_{15.16}(GST)_{84.84}}\), in the following section.
Figure 44.12 shows Raman spectra of GST and \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) annealed at 250 and \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). The vibration bands in \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed GST at 105 and \({\mathrm{155}}\,{\mathrm{cm^{-1}}}\) are attributed to the \(\mathrm{A_{1}}\) mode of \(\mathrm{GeTe_{4}}\) corner-sharing tetrahedral and Sb–Te vibrations in \(\mathrm{SbTe_{3}}\) units [44.30], respectively. The vibration band at \({\mathrm{155}}\,{\mathrm{cm^{-1}}}\) is significantly influenced by crystallization when the annealing temperature increases to \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), indicating a structural transformation from fcc to hcp. However, such a transition cannot be found in \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) film, indicating that the introduction of Zn into GST restrains the transition of fcc to hcp.


Raman spectra of GST and \(\mathrm{Zn_{15.16}(GST)_{84.84}}\) films annealed at 250 and \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)
3 Doped Sb-Te Films for Phase-Change Memory Applications
Compared with conventional GST films, another group of Sb-based materials, including Sb-Te, exhibits a fast growth-dominated crystallization process, and the set operation speed is faster. Therefore, Sb-based materials are also called fast-growth materials. While the fast-growth materials are obviously favored for high-speed reading and writing, the drawback of the materials is low amorphous phase stability and high media noise. An innovative approach for solving these problems is thus needed for the application of fast-growth materials in PCM devices. A preliminary investigation demonstrated that \(\mathrm{Sb_{2}Te_{3}}\) film doped with other metallic elements including Al [44.31, 44.32], Ag [44.33], and Ti [44.34] exhibits better data retention, higher crystalline resistance, and a larger amorphous/crystalline resistance ratio (\(\approx{\mathrm{10^{5}}}\)) during the crystallization process. \(\mathrm{Sb_{2}Te}\) alloys doped with Cu [44.35], Ti [44.36], and W [44.37] were reported to exhibit a high crystallization temperature and better data retention. However, the amorphous/crystalline resistance ratio or speed of crystallization in the alloys was found to degrade with increasing dopant concentrations. The doping of Ge [44.38], In [44.39], and Ag [44.40] into \(\mathrm{Sb_{7}Te_{3}}\) has been shown to increase the crystallization temperature, and has been frequently used for rewritable and recordable optical media such as DVD-RW and DVD+RW devices. However, these metal-doped \(\mathrm{Sb_{7}Te_{3}}\) materials are not good in all respects. For instance, Ag-doped \(\mathrm{Sb_{7}Te_{3}}\) always faces the problem of phase separation during the crystallization process, which can lead to poor endurance for PRAM applications. In order to increase the phase transition speed, researchers have tried to add the so-called fast-growth material Sb as much as possible to the phase-change material. Thus, Sb-Te alloys with high Sb content, including \(\mathrm{Sb_{3}Te}\) and \(\mathrm{Sb_{4}Te}\), have typically been considered for investigation. Doping of impurities such as Al [44.41] and Au [44.42] into \(\mathrm{Sb_{3}Te}\) can increase the crystallization temperature, data retention, and crystalline resistance, but other crystalline phases formed by these impurities will separate out during the crystallization process. Studies of Si-doped \(\mathrm{Sb_{4}Te}\) showed that Si doping significantly improved thermal stability, refined their grain size, and altered nucleation characteristics with an increase in silicon content. Investigation by in situ transmission electron microscopy (TEM), however, showed that the crystalline phase of Si-\(\mathrm{Sb_{4}Te}\) thin films could be indexed as a hexagonal Sb structure, and Si retained an amorphous state [44.43].
The above results have opened up an avenue of applications for metallic element-doped Sb-Te materials that have specific advantages compared with other phase-change materials. Here, Zn-doped Sb-Te films will be highlighted as a fast-transition and high-thermal-stability phase-change material for potential PRAM application.
Figure 44.13a shows the \(R\)–\(T\) curves of undoped and Zn-doped \(\mathrm{Sb_{4}Te}\) films. As we can see, the crystallization temperature (\(T_{\text{c}}\)) of the films increases with increasing Zn content. All of these doped films exhibit a higher \(T_{\text{c}}\) than \(\mathrm{Sb_{4}Te}\) (\({\mathrm{136}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)) and GST (\({\mathrm{168}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)). The \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film has the highest \(T_{\text{c}}\) at \({\mathrm{256}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and crystalline resistance (\(R_{\text{c}}\)) at \({\mathrm{780}}\,{\mathrm{\Upomega/\square}}\). The amorphous-phase-forming region of Zn-doped Sb-Te and other materials reported in the references are displayed in Fig. 44.13b. As we noted, the value of \(T_{\text{c}}\) is clearly increased with higher Zn concentration introduced into Sb-Te, including \(\mathrm{Sb_{2}Te_{3}}\), \(\mathrm{Sb_{2}Te}\), \(\mathrm{Sb_{7}Te_{3}}\), \(\mathrm{Sb_{3}Te}\), and \(\mathrm{Sb_{4}Te}\).


(a) Sheet resistance as a function of temperature for undoped and Zn-doped \(\mathrm{Sb_{4}Te}\) films. (b) Crystallization temperatures as a function of composition in the Zn-Sb-Te ternary amorphous-phase-forming region


The extrapolated data retention time of Zn-doped \(\mathrm{Sb_{4}Te}\) films at specified temperatures
By using the Arrhenius equation, the temperature for 10-year data retention can be extrapolated, and the fitting results are shown in Fig. 44.14. Clearly, the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film has the largest \(E_{\text{a}}\) of \({\mathrm{4.46}}\,{\mathrm{eV}}\) and the highest temperature for 10-year data retention of \({\mathrm{176.6}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), which meets automotive electronics criteria (10-year data retention temperature should be higher than \({\mathrm{120}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)).
A summary of the thermal and/or electrical parameters for Ge-Sb-Te, Ga-Sb-Te, and Zn-Sb-Te films and the respective undoped counterparts are listed in Table 44.2, which shows that the introduction of Zn can effectively increase the \(T_{\text{c}}\), \(E_{\text{a}}\), and 10-year data retention temperature. Compared with GST, \(\mathrm{Ga_{2}Te_{3}Sb_{5}}\), and other Zn-Sb-Te films, \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) possesses the best physical properties in terms of crystallization temperature, crystalline activation energy, and data retention, as well as crystalline resistance.


XRD patterns of undoped and Zn-doped \(\mathrm{Sb_{4}Te}\) films annealed at different temperatures for \({\mathrm{3}}\,{\mathrm{min}}\) in Ar atmosphere: (a) \(\mathrm{Sb_{4}Te}\), (b) \(\mathrm{Zn_{12.2}(Sb_{4}Te)_{87.8}}\), (c) \(\mathrm{Zn_{17.2}(Sb_{4}Te)_{82.8}}\), (d) \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\) and (e) \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\)
The XRD patterns of the as-deposited and annealed Zn-\(\mathrm{Sb_{4}Te}\) films are presented in Fig. 44.15a-e. In Fig. 44.15a-ea, the sharp crystalline diffraction peaks of \({\mathrm{150}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)- and \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Sb_{4}Te}\) films belong to the rhombohedral Sb-phase (JCPDS no. 35-0731). However, a new rhombohedral \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase begins to separate out (JCPDS no. 15-0874) in \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Sb_{4}Te}\) films. When the annealing temperature increases to \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), it is completely crystallized with the stable rhombohedral \(\mathrm{Sb_{2}Te_{3}}\) crystalline. Interestingly, there is no Sb or \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase in Zn-doped \(\mathrm{Sb_{4}Te}\) films. As can be seen in Fig. 44.15a-eb–e, only a hexagonal \(\mathrm{Sb_{2}Te}\) crystalline phase (JCPDS no. 80-1722) is present in the annealed Zn-\(\mathrm{Sb_{4}Te}\) films. In addition, the intensity of the diffraction peaks for the crystalline \(\mathrm{Sb_{2}Te}\) phase decreases with increasing Zn concentration, which implies that Zn doping can significantly suppress the growth of \(\mathrm{Sb_{2}Te}\) crystalline grains.
The crystalline phases of Sb-Te and Zn-Sb-Te films are listed in Table 44.3. It should be noted that the as-deposited \(\mathrm{Sb_{2}Te_{3}}\) is partially crystallized due to its poor thermal stability. Other as-deposited Zn-doped and undoped Sb-Te films, i. e., \(\mathrm{Sb_{2}Te}\), \(\mathrm{Sb_{3}Te}\), \(\mathrm{Sb_{4}Te}\), and \(\mathrm{Sb_{7}Te_{3}}\), are amorphous. Different crystalline phases are found in the films annealed at different temperatures. For instance, \(\mathrm{Sb_{2}Te}\) and \(\mathrm{Sb_{3}Te}\) films show a single \(\mathrm{Sb_{2}Te}\) crystalline phase, but \(\mathrm{Sb_{4}Te}\) and \(\mathrm{Sb_{7}Te_{3}}\) separate out Sb and \(\mathrm{Sb_{2}Te_{3}}\) crystalline phases with the increased annealing temperature. When Zn is doped in these Sb-Te films, however, it restrains the phase separation, and only a single \(\mathrm{Sb_{2}Te}\) crystalline phase can be detected in the above doped films (besides Zn-\(\mathrm{Sb_{2}Te_{3}}\)). Single \(\mathrm{Sb_{2}Te_{3}}\) crystalline phases are separated out from all the undoped and Zn-doped \(\mathrm{Sb_{2}Te_{3}}\) films in the crystallization process. All these results confirm that Zn can be used to stabilize the crystalline structure of Sb-Te and makes these phase-change materials more thermally stable.


(a) The TEM BF micrograph and (b) SAED pattern of \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\) film annealed at \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\); (c) The TEM BF micrograph, (d) TEM DF micrograph, and (e) SAED pattern of \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film annealed at \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)
In order to obtain additional information on the structure of the crystalline film, transmission electron microscopy (TEM) was employed. The bright-field (BF) TEM micrograph and its corresponding selected-area electron diffraction (SAED) pattern of \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\) film are shown in Fig. 44.16a,b, and the BF TEM micrograph, corresponding SAED pattern, and dark-field (DF) TEM image of \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film are shown in Fig. 44.16c–e. The BF TEM micrographs and SAED patterns indicate uniform \(\mathrm{Sb_{2}Te}\) crystalline grains, several tens of nanometers in size, distributed in the Zn-\(\mathrm{Sb_{4}Te}\) films. In addition, the \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film has smaller crystalline grains and more continuous diffraction rings in the SAED pattern than the \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\) film. This implies that the crystalline grain size can be decreased with a higher Zn dopant concentration in the film, which is in line with the results estimated from the decreased line width and diffraction peak intensity of the XRD patterns. The DF TEM micrograph shown in Fig. 44.16e clearly demonstrates the uniform morphology, with bright areas (crystalline \(\mathrm{Sb_{2}Te}\) phase) embedded in dark areas (amorphous phase) in the \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film. The uniform distribution of the small crystalline grains is very beneficial for enhanced cycling reliability for PCM devices, which suggests that the \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film is the best candidate for PCM devices, with prolonged cyclability.


Power-time-effect diagrams of (a) \(\mathrm{Zn_{12.2}(Sb_{4}Te)_{87.8}}\), (b) \(\mathrm{Zn_{17.2}(Sb_{4}Te)_{82.8}}\), (c) \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\) and (d) \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) films. I and II indicate the amorphous and crystalline state, respectively
Phase transition speed is a key factor determining the switching speed of PCM devices. Here, a static tester with different laser pulse widths and power was employed to quantitatively obtain the phase transition speed of Zn-doped \(\mathrm{Sb_{4}Te}\) films. Figure 44.17a-da–d depicts the power-time-effect (PTE) diagrams for \(\mathrm{Zn_{12.2}(Sb_{4}Te)_{87.8}}\), \(\mathrm{Zn_{17.2}(Sb_{4}Te)_{82.8}}\), \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\), and \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) films, respectively. The optical contrast \({\Updelta}R\) is defined as \(\Updelta R=(R_{\text{after}}-R_{\text{before}})/R_{\text{before}}\) [44.25, 44.26], where \(R_{\text{before}}\) and \(R_{\text{after}}\) are the optical reflectivity before and after irradiation, respectively. This should not be confused with the resistance \(R\). The different colors indicate the magnitude of \(\Updelta R\) in various crystallization processes. Region I in the figures represents the amorphous state, with no change in reflectivity because of the short laser pulse width and/or low laser power, while region II represents the crystalline state, with significant change in reflectivity because of the wide laser pulse and/or high laser power. The maximum \({\Updelta}R\) (\(\Updelta R_{\text{max}}\)) value differs for each film, as follows: 0.26, 0.23, 0.20, and 0.19 for \(\mathrm{Zn_{12.2}(Sb_{4}Te)_{87.8}}\), \(\mathrm{Zn_{17.2}(Sb_{4}Te)_{82.8}}\), \(\mathrm{Zn_{20.2}(Sb_{4}Te)_{79.8}}\), and \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film, respectively.


Reflectivity changes in (a) \(\mathrm{Zn_{12.2}(Sb_{4}Te)_{87.8}}\) and (b) \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) films with different laser power and pulse width
Figure 44.18a,ba,b shows the normalized reflectivity evolutions induced by nano-laser pulse for \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) and \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) films. The initial low reflectivity of the curves in Fig. 44.18a,b indicates the amorphous state in the films. It increases gradually with increasing pulse duration for each curve (besides the lowermost curve in Fig. 44.18a,ba), indicating the crystallization process before achieving a homogeneous crystalline phase. As we can see, \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) begin to crystallize very quickly with a short pulse width; however, it is difficult for the \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) film because of its high crystallization threshold. For instance, no change can be found in the optical reflectivity for the \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) film, while a slight change can be observed in the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film, with laser power of \({\mathrm{15}}\,{\mathrm{mW}}\). Increasing the laser power to 30, 50, and \({\mathrm{70}}\,{\mathrm{mW}}\), onset crystallization times of 85, 60, and \({\mathrm{30}}\,{\mathrm{ns}}\), respectively, are detected for the \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) film, and 5, 15, and \({\mathrm{30}}\,{\mathrm{ns}}\) for the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film. The ending crystallization times for these two Zn-doped \(\mathrm{Sb_{4}Te}\) films with laser power of \({\mathrm{70}}\,{\mathrm{mW}}\) are 190 and \({\mathrm{164}}\,{\mathrm{ns}}\) for the \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) and \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) films, respectively. Obviously, both the onset and ending crystallization times for the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film are shorter than those for the \(\mathrm{Zn_{12.2}(\mathrm{Sb_{4}Te})_{87.8}}\) film. In addition, they are shorter than those for the GST (40 and \({\mathrm{280}}\,{\mathrm{ns}}\)) [44.50] and Sb-rich \(\mathrm{Zn_{30.2}(Sb_{7}Te_{3})_{69.8}}\) (10 and \({\mathrm{250}}\,{\mathrm{ns}}\)) films [44.49], indicating that the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film has the fastest crystallization speed among these reported phase-change materials.


The optical switching behavior over 50 cycles of operation on the \(\mathrm{Zn_{28.6}(Sb_{4}Te)_{71.4}}\) film
As discussed earlier, the uniform distribution of smaller \(\mathrm{Sb_{2}Te}\) crystalline grains in Zn-doped \(\mathrm{Sb_{4}Te}\) film is beneficial for enhancing cycling reliability. Therefore, here we employed static testing to investigate cycling reliability. Figure 44.19 illustrates the optical switching behavior over 50 cycles between the crystalline and amorphous phases for the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film. The optical contrast is positive when loaded on a pulse width of \({\mathrm{250}}\,{\mathrm{ns}}\) and laser power of \({\mathrm{35}}\,{\mathrm{mW}}\) (squares), indicating the crystalline state. On the other hand, the optical contrast is negative when loaded on a pulse width of \({\mathrm{150}}\,{\mathrm{ns}}\) and laser power of \({\mathrm{70}}\,{\mathrm{mW}}\) (circles), indicating the amorphous state. The difference in optical contrast between the amorphous and crystalline states remains almost constant during continuous laser application, which demonstrates that the \(\mathrm{Zn_{28.6}(\mathrm{Sb_{4}Te})_{71.4}}\) film has good cycling reliability.
4 Nanocomposite Films for Phase-Change Memory Applications
ZnO is a direct-band-gap (\({\mathrm{3.37}}\,{\mathrm{eV}}\)) semiconductor with excellent physical and chemical properties and thermodynamic stability at room temperature. It is an ideal material to be doped onto \(\mathrm{Sb_{2}Te_{3}}\) for fabrication of nanocomposite materials for potential PCM applications.
Figure 44.20 shows the \(R\)–\(T\) curves for GST, \(\mathrm{Sb_{2}Te_{3}}\), and ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films. As we see, there is no clear resistance drop in the as-deposited \(\mathrm{Sb_{2}Te_{3}}\) film due to the partial crystallization. The GST film exhibits two abrupt drops in sheet resistance at 168 and \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), indicating the transition of an amorphous to fcc structure and fcc to hcp structure, respectively. ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films show a sudden drop in resistance when the temperature is close to their specific crystallization temperature (\(T_{\text{c}}\)). The value of \(T_{\text{c}}\) is 212, 217, 223, 241, and \({\mathrm{275}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for \(\mathrm{(ZnO)_{0.62}(\mathrm{Sb_{2}Te_{3}})_{99.38}}\), \(\mathrm{(ZnO)_{0.82}(\mathrm{Sb_{2}Te_{3}})_{99.18}}\), \(\mathrm{(ZnO)_{1.69}(\mathrm{Sb_{2}Te_{3}})_{98.31}}\), \(\mathrm{(ZnO)_{5.22}(\mathrm{Sb_{2}Te_{3}})_{94.78}}\), and \(\mathrm{(ZnO)_{5.5}(\mathrm{Sb_{2}Te_{3}})_{94.5}}\), respectively. Clearly, the ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films have much higher \(T_{\text{c}}\) than GST and \(\mathrm{Sb_{2}Te_{3}}\), which implies that the introduction of ZnO into the \(\mathrm{Sb_{2}Te_{3}}\) film can significantly increase the amorphous thermal stability. However, the decrease in resistance becomes very sluggish in the \(\mathrm{(ZnO)_{5.5}(\mathrm{Sb_{2}Te_{3}})_{94.5}}\) film, indicating that the doping of ZnO is excessive and restrains the phase transition process.


\(R\)–\(T\) curve of ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films
The XRD patterns of undoped and ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films are shown in Fig. 44.21a-f. In Fig. 44.21a-fa, we note that there is one crystalline diffraction peak that corresponds to Te(110) in the XRD pattern of the as-deposited \(\mathrm{Sb_{2}Te_{3}}\) film, implying that it actually contains a crystalline Te phase. A rhombohedral \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase was found in \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{Sb_{2}Te_{3}}\) film, and the corresponding diffraction peak intensity was stronger when the annealing temperature was increased to 250, 300, and \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). Interestingly, a sharp diffraction peak located at \(2{\theta}=35.3^{\circ}\), which is attributed to the \(\mathrm{Sb_{2}O_{3}}\) crystalline phase, was found when the annealing temperature was increased to 300 and \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). It is believed that the poor thermal stability of the \(\mathrm{Sb_{2}Te_{3}}\) film results in this \(\mathrm{Sb_{2}O_{3}}\) phase separation.


XRD patterns of as-deposited and annealed ZnO-\(\mathrm{Sb_{2}Te_{3}}\) films: (a) \(\mathrm{Sb_{2}Te_{3}}\), (b) \(\mathrm{(ZnO)_{0.62}(Sb_{2}Te_{3})_{99.38}}\), (c) \(\mathrm{(ZnO)_{0.82}(Sb_{2}Te_{3})_{99.18}}\), (d) \(\mathrm{(ZnO)_{1.69}(Sb_{2}Te_{3})_{98.31}}\), (e) \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\), and (f) \(\mathrm{(ZnO)_{5.5}(Sb_{2}Te_{3})_{94.5}}\)
Figure 44.21a-fb–f shows the XRD patterns of the as-deposited and annealed ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films. We can see that the \(\mathrm{Sb_{2}Te_{3}}\) crystalline phases begin to separate out in
films. The \(\mathrm{Sb_{2}O_{3}}\) crystalline phase appears at \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for the \(\mathrm{(ZnO)_{0.62}(Sb_{2}Te_{3})_{99.38}}\), annealed \(\mathrm{(ZnO)_{0.82}(Sb_{2}Te_{3})_{99.18}}\), and \(\mathrm{(ZnO)_{1.69}(Sb_{2}Te_{3})_{98.31}}\) films. However, the \(\mathrm{Sb_{2}O_{3}}\) crystalline phase is almost suppressed in the \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) and \(\mathrm{(ZnO)_{5.5}(Sb_{2}Te_{3})_{94.5}}\) films, even with the annealing temperature increased to \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). This is evidence that the thermal stability can be significantly improved and phase separation suppressed with increased ZnO doping. In addition, broader line width and lower diffraction peak intensity can be found in Fig. 44.21a-fe,f, indicating that the growth of crystalline grains is restrained with increased ZnO doping.
We also performed Raman spectroscopy to study the structure of the undoped and ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films, and the results are shown in Fig. 44.22a-f. As seen in Fig. 44.22a-fa, three vibration peaks ascribed to the \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase can be found at 65 (peak A), \({\mathrm{110}}\,{\mathrm{cm^{-1}}}\) (peak B), and \({\mathrm{165}}\,{\mathrm{cm^{-1}}}\) (peak C) in the \(\mathrm{Sb_{2}Te_{3}}\) target and films [44.50, 44.51]. They become sharper with increasing annealing temperature as more of the \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase is separated out. However, two new vibration peaks at \({\mathrm{140}}\,{\mathrm{cm^{-1}}}\) (peak D) and \({\mathrm{123}}\,{\mathrm{cm^{-1}}}\) (peak E) in the \(\mathrm{Sb_{2}Te_{3}}\) films are detected when the annealing temperature is increased to \({\mathrm{350}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). These two vibration peaks are ascribed to the vibrations of the \(\mathrm{Sb_{2}O_{3}}\) phases [44.52]. The Raman spectra of the as-deposited and annealed \(\mathrm{(ZnO)_{0.62}(Sb_{2}Te_{3})_{99.38}}\) and \(\mathrm{(ZnO)_{0.82}(Sb_{2}Te_{3})_{99.18}}\) films are displayed in Fig. 44.22a-fb,c. Peak C can be found in these two films, but it is much weaker than in the undoped \(\mathrm{Sb_{2}Te_{3}}\) film, indicating that the \(\mathrm{Sb_{2}Te_{3}}\) crystalline phase is significantly restrained by a small amount of ZnO doping. Peaks E and D appear in the Raman spectra of the \({\mathrm{250}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{(ZnO)_{0.62}(Sb_{2}Te_{3})_{99.38}}\) and \(\mathrm{(ZnO)_{0.82}(Sb_{2}Te_{3})_{99.18}}\) films, which confirms that the \(\mathrm{Sb_{2}O_{3}}\) phases were crystallized in the films. As depicted in Fig. 44.22a-fd–f, some common features of Raman spectra for three other ZnO-doped films can be concluded as follows. Peak C appears from the overlapping broad band from 100 to \({\mathrm{180}}\,{\mathrm{cm^{-1}}}\) with increasing annealing temperature, indicating that the crystalline \(\mathrm{Sb_{2}Te_{3}}\) phases can be separated out at high temperature. However, peaks D and E are not so obviously detected in these Raman spectra. All these results support the conclusions summarized from XRD, i. e., high ZnO dopant content can significantly restrain the formation of the \(\mathrm{Sb_{2}O_{3}}\) phase and improve the thermal stability for ZnO-doped \(\mathrm{Sb_{2}Te_{3}}\) films.


Raman spectra of as-deposited and annealed ZnO-\(\mathrm{Sb_{2}Te_{3}}\) films: (a) \(\mathrm{Sb_{2}Te_{3}}\), (b) \(\mathrm{(ZnO)_{0.62}(Sb_{2}Te_{3})_{99.38}}\), (c) \(\mathrm{(ZnO)_{0.82}(Sb_{2}Te_{3})_{99.18}}\), (d) \(\mathrm{(ZnO)_{1.69}(Sb_{2}Te_{3})_{98.31}}\), (e) \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\), and (f) \(\mathrm{(ZnO)_{5.5}(Sb_{2}Te_{3})_{94.5}}\)
The BF and DF TEM micrographs, SAED patterns, and high-resolution TEM (HRTEM) images for the \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)-annealed \(\mathrm{(ZnO)_{5.2}(Sb_{2}Te_{3})_{94.8}}\) film are shown in Fig. 44.23a–d, respectively. From the BF and DF TEM micrographs, the uniform crystalline grains of \(10{-}20\,{\mathrm{nm}}\) in size can be found in the annealed films, which is in good agreement with the estimated XRD results. These nanocrystals are surrounded by amorphous ZnO that exists in the dark area in the DF TEM micrograph. The SAED pattern and HRTEM image indicate that these nanocrystals in the \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film are \(\mathrm{Sb_{2}Te_{3}}\) phases, and no other crystalline phase exists in the film. This is in line with the results estimated from XRD and Raman spectroscopy. Based on these analyses, we believe that the nanocomposite structure with mixed amorphous ZnO and crystalline \(\mathrm{Sb_{2}Te_{3}}\) phases in the \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film results in enhanced thermal stability and reduced operating power.


(a) The TEM BF micrograph, (b) TEM DF micrograph, and (c) SAED pattern of \(\mathrm{(ZnO)_{5.2}(Sb_{2}Te_{3})_{94.8}}\) film annealed at \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\); (d) the HRTEM images of the same film
A static tester using pulsed laser irradiation was employed to determine the phase transition speed for optimal \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film. The phase transition speed can be obtained by observing the change in optical contrast (\({\Updelta}R\)), which is defined as \({\Updelta}R=(R_{\text{after}}-R_{\text{before}})/R_{\text{before}}\), where \(R_{\text{before}}\) and \(R_{\text{after}}\) are the reflectivity before and after irradiation, respectively. Pulse-width-dependent \({\Updelta}R\) with different laser power levels and the PTE diagram for the \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film are displayed in Fig. 44.24a,b. \({\Updelta}R\) increases with increasing laser pulse width and/or laser power, but it does not change with increasing pulse width when the laser power is \({\mathrm{5}}\,{\mathrm{mW}}\), as the applied laser energy is not sufficient to induce film crystallization (region I in the PTE pattern). When laser power is increased, crystallization occurs and \({\Updelta}R\) increases (region II in the PTE pattern). Researchers have reported onset crystallization times for GST of 300, 150, 80, and \({\mathrm{40}}\,{\mathrm{ns}}\) with laser power of 10, 30, 50, and \({\mathrm{70}}\,{\mathrm{mW}}\), respectively [44.49]. For \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film, onset of crystallization occurred in 130, 40, 20, and \({\mathrm{10}}\,{\mathrm{ns}}\) with the same power, which would indicate that the crystallization speed of the \(\mathrm{(ZnO)_{5.22}(Sb_{2}Te_{3})_{94.78}}\) film is faster than that of GST.


(a) Reflectivity changes of \(\mathrm{(ZnO)_{5.2}(Sb_{2}Te_{3})_{94.8}}\) film with different laser power and pulse width; (b) PTE (power-time-effect) diagram of \(\mathrm{(ZnO)_{5.2}(Sb_{2}Te_{3})_{94.8}}\) film
5 Crystallization Kinetics Studied by Ultrafast Calorimetry for Phase-Change Materials
Phase-change materials (PCMs) demonstrate promising performance for next-generation memory technology, with fast, reversible switching speeds between amorphous and crystalline phases, and large optical and electrical contrast. However, the kinetics of switching must be better understood for optimization of PCMs and phase-change devices, especially with regard to the crystallization kinetics, which has aroused much interest over the years in the form of experiments [44.53, 44.54, 44.55, 44.56, 44.57] and molecular dynamics simulations [44.58, 44.59].
In recent decades, due to the limitations in instrumentation, a majority of experimental investigations of the crystallization kinetics of PCMs have focused primarily on the relatively low-temperature region. For real-world PCM applications, however, crystallization always occurs at higher temperatures, with ultrafast optical or electrical pulses. Fortunately, a new type of calorimetry, ultrafast differential scanning calorimetry (ultrafast DSC), has been introduced, which extends the heating rate to more than \({\mathrm{4\times 10^{4}}}\,{\mathrm{K{\,}s^{-1}}}\).


(a) Ultrafast heating of amorphous \(\mathrm{Ge_{2}Sb_{2}Te_{5}}\) (GST). Here, the DSC traces are labeled with heating rate in \(\mathrm{K{\,}s^{-1}}\). The inset shows a close-up of the exothermic peaks at the lowest heating rates; (b) Kissinger plot for crystallization of GST. The data of Park et al [44.55] are from conventional DSC (\(\Phi=0.08{-}0.33\,{\mathrm{K\mskip 3.0mus^{-1}}}\)) on amorphous GST films. The data of Friedrich et al [44.60] (\(\Phi=0.08{-}0.33\,{\mathrm{K\mskip 3.0mus^{-1}}}\)) and of Choi et al [44.61] (\(\Phi=0.05{-}8.33\,{\mathrm{K\mskip 3.0mus^{-1}}}\)) are from electrical resistance measurements. The red curve represents the temperature dependence of the crystal growth rate \(U\) in GST, related to the dashed line through the data by numerical modeling of DSC curves. The kinetic coefficient or limiting velocity for crystal growth \(U_{\text{kin}}\) (green line) is related to \(U\) using (44.2). Adapted from [44.62]
In 2012, Orava et al first applied ultrafast DSC to the conventional PCM \(\mathrm{Ge_{2}Sb_{2}Te_{5}}\) [44.62]. As shown in Fig. 44.25a, the crystallization peaks are readily detectable in these DSC traces, and the corresponding temperature, \(T_{\text{p}}\), increases with the increase in the heating rate. According to the Kissinger equation [44.63]
where \(\Phi\) is the heating rate, \(T_{\text{p}}\) is the crystallization temperature, \(Q\) is the activation energy for crystallization, \(R\) is the gas constant, and \(A\) is a constant, at relatively low heating rates, the value of \(Q\) will always be a constant, and the relationship between \(\ln(\Phi/T_{\text{p}}^{2})\) and \(T_{\text{p}}\) would then be straightforward. Therefore, strict Arrhenius behavior with a linear Kissinger plot in crystallization is generally obtained in a conventional low-heating-rate test. However, as depicted in Fig. 44.25b, the Kissinger plot of GST based on ultrafast DSC is obviously curved, and the effective activation energy \(Q\) decreases at higher temperature, as expected for a fragile liquid. The red curve represents the temperature dependence of the crystal growth rate \(U\) in GST, related to the dashed line through the data by numerical modeling of DSC curves. The kinetic coefficient or limiting velocity for crystal growth \(U_{\text{kin}}\) is related to \(U\). The real crystal growth rate \(U\) can be assumed as [44.64]
with \(R\) the gas constant and \({\Updelta}G\) the driving force for crystallization. For \({\Updelta}G\), here, the equation of Thompson and Spaepen was used [44.65]. It is likely to be an approximation for which
where \({\Updelta}H_{\text{m}}\) is the latent heat of melting (\({\Updelta}H_{\text{m}}\) is \({\mathrm{12.13}}\,{\mathrm{kJ/mol}}\) for GST), \(T_{\text{m}}\) is the melting temperature (\({\mathrm{900}}\,{\mathrm{K}}\) for GST), and \({\Updelta}T\) is the supercooling (\(T_{m}-T\)).
According to classical nucleation theory [44.66], the crystal growth rate
so there is a relationship between the kinetic coefficient for crystal growth \(U_{\text{kin}}\) and diffusivity \(D\) that can be written as \(U_{\text{kin}}\propto D\). A Stokes–Einstein relation between \(D\) and viscosity \({\eta}\) is \(D\propto{T}/{\eta}\). Therefore, the relationship between \(U_{\text{kin}}\) and \({\eta}\) can be obtained and described as \(U_{\text{kin}}\propto{T}/{\eta}\). Thus, the \(U_{\text{kin}}\) term can be evaluated based on the Cohen and Grest expression for the viscosity of glass-forming liquids, which is [44.67]
where \(A\), \(B\), \(C\), and \(T_{0}\), are all adjusted parameters. However, the Stokes–Einstein relation between \(D\) and \({\eta}\) can be broken in the low-temperature range, such as around glass transition temperature (\(T_{\text{g}}\)), so the decoupling in \(U_{\text{kin}}\) and \({\eta}\) possibly breaks down on cooling towards \(T_{\text{g}}\). It has been expressed by Ediger et al [44.64] as a scaling \(U_{\text{kin}}\propto{T}/{\eta^{\xi}}\), where \({\xi}<1\). They found that a similar decoupling occurred with GST. With the definition of fragility,
a large \(m\) (\(=90\)) can be obtained, which indicates an obvious non-Arrhenius behavior, as shown in Fig. 44.25a.
After \(U_{\text{kin}}\) is evaluated over the entire temperature range down to \(T_{\text{g}}\), and the thermodynamic effects are incorporated (44.3), the temperature dependence of the crystal growth rate can be represented as shown in Fig. 44.26. As expected for supercooled liquids [44.64, 44.68], the interplay of kinetics and thermodynamics yields a maximum in \(U\). This maximum is close to \(T_{\text{m}}\), and for GST the maximum is at \(0.76T_{\text{m}}\). The arrow in this figure indicates that ultrafast DSC extends conventional measurements and can characterize the temperature dependence of the crystallization rate of GST up to at least \(0.72T_{\text{m}}\) (\(1.70T_{\text{g}}\)), where the crystal growth rate approaches its maximum.


Crystal growth rate in supercooled liquid GST from \(T_{\text{g}}\) to \(T_{\text{m}}\). The green line in Fig. 44.25b is based on (44.4) and gives the temperature dependence of the kinetic coefficient for crystal growth \(U_{\text{kin}}\). The crystal growth rate \(U\) is obtained using (44.2). The ranges are shown (in brown), where the temperature dependence of \(U\) can be obtained from DSC measurements. After [44.62]
In 2015, Orava et al studied the crystallization kinetics for another PCM, Ag-In-\(\mathrm{Sb_{2}Te}\) (AIST), based on ultrafast DSC [44.69]. For this material, however, the Cohen and Grest model for viscosity may be not suitable. The authors believed that there was a fragile-to-strong crossover on cooling the liquid, and applied the generalized Mauro–Yue–Ellison–Gupta–Allan () expression to study the crystallization kinetics. The generalized MYEGA equation from metallic glass-forming systems can be described as [44.70]
\(W_{1}\), \(W_{2}\), \(C_{1}\), and \(C_{2}\) are adjusted parameters, and \(\eta_{\infty}\) is the viscosity at infinite temperature.
Months later, Chen et al employed this ultrafast DSC to study the crystallization kinetics of a growth-dominated PCM, \(\mathrm{Ge_{7}Sb_{93}}\) [44.71]. Two expressions of viscosity fitting have been used in this material: Cohen and Grest, and Salinga and Mauro. The Salinga and Mauro equation is not shown here, but can be obtained from [44.71]. According to the results, the Salinga and Mauro model for viscosity and growth rate appears more appropriate for this \(\mathrm{Ge_{7}Sb_{93}}\) material. This was also confirmed in a GeTe material [44.72].
It may be noted that both nucleation and crystal growth are determinants of crystallization speed, but the role of nucleation has not been discussed here. As described by Sebastian et al [44.73], nucleation is expected to become less important, and crystallization would govern crystal growth, at the technologically relevant nanometer length scale and nanosecond timescale. This is particularly true in the melt-quenched amorphous state, where a large population of nuclei already exist and a crystal/amorphous interface is present [44.74]. Therefore, we have emphasized the crystal growth rather than the nucleation for the crystallization kinetics of PCMs.
In summary, we have achieved important progress regarding the crystallization kinetics of PCMs at higher temperatures by employing ultrafast DSC. In this way, a large range of heating rates (from 10 to \({\mathrm{40000}}\,{\mathrm{K{\,}s^{-1}}}\)) can be applied, whereby crystallization becomes available over a relatively wide temperature range. The decoupling of \(U_{\text{kin}}\) and \({\eta}\), and their marked non-Arrhenius temperature dependence, shows that conventional DSC crystallization measurements near \(T_{\text{g}}\) have limited relevance for the fast crystallization occurring in PCMs. Nevertheless, the specific model that can be employed for PCM viscosity fitting is not uniform, whether Cohen and Grest, generalized MYEGA, Salinga and Mauro, or any others. In the future, greater attention should be paid to solving this critical argument.
6 Phase-Change Materials for Applicationsin Integrated Photonic Memory
With the large contrast in optical properties such as refractive index between amorphous and crystalline states, the use of phase-change materials opens up a new dimension for integrated photonic memory. Pernice and Bhaskaran [44.75] have theoretically proposed photonic memory devices using phase-change materials, as shown in Fig. 44.27a. A microring resonator was used to coat a short section of GST. The nearby nanophotonic waveguides propagated the visible control light through evanescent coupling to the microring resonator. GST is optically triggered by the transition between the amorphous and crystalline phases, which changes the transmission characteristics of the microring resonator. The phase status can be read by sending a probe beam, which serves as a digital data bit. Numerical analysis indicates that the crystallization of GST can be induced by a \({\mathrm{600}}\,{\mathrm{fs}}\) optical pulse with power of \({\mathrm{5.4}}\,{\mathrm{pJ}}\), and reversible amorphization can be achieved by an optical pulse with higher power. Moreover, multilevel recording can be realized in such integrated photonic platforms.


(a) Schematic overview of the proposed memory element, and the cross-sectional view of the coupling region showing the control port and the GST-covered freestanding waveguide section. Reprinted from [44.75], with the permission of AIP Publishing. (b) A scanning electron microscope image of a fabricated GST optical memory device, from [44.76]. (c) Two-level and eight-level operations realized by precise control of the volume ratio of the amorphous and crystalline phase. From [44.77]
Rios et al [44.78] experimentally demonstrated a hybrid nanophotonic circuit for implementing tunable photonic devices based on nanoscale GST junctions. A \({\mathrm{10}}\,{\mathrm{nm}}\)-thick GST layer was deposited on a section of racetrack resonators, Mach–Zehnder interferometers, and balanced splitters. A thin layer of indium tin oxide was capped on top of the GST to prevent oxidation. The transmission spectra of the devices were strongly affected by the complex propagation constant and attenuation coefficient of the waveguide mode, which was dependent on the phase state of the GST. Using racetrack resonators as memory elements, the authors were able to detect optical properties including the \(Q\)-factor, central wavelength of the peak resonances, and the extinction ratio, and to track the state of the GST. This work provides a feasibility study for optically tunable photonic circuits that can potentially be switched on in a picosecond timescale.
Using optical near-field effects, GST can be employed to realize bit storage of up to eight levels in integrated nonvolatile photonic memory [44.76], as shown in Fig. 44.27b. By precisely controlling the intensity of the writing pulse with power from 372 to \({\mathrm{601}}\,{\mathrm{pJ}}\), the ratio of crystalline and amorphous states can be modulated in a controlled manner to realize eight different transmittance levels, as shown in Fig. 44.27c. In addition, the transmittance can arbitrary reach any of eight levels in the case of selective erasure of pulse intensity. The 5-\(\mathrm{{\upmu}m}\)-long GST devices can be erased and written under an \({\mathrm{100}}\,{\mathrm{ns}}\) optical pulse with power of \({\mathrm{533}}\,{\mathrm{pJ}}\). In a more extreme situation with a \({\mathrm{1}}\,{\mathrm{{\upmu}m}}\)-long GST device, a short \({\mathrm{10}}\,{\mathrm{ns}}\) optical pulse is sufficient for operation at a power energy of only \({\mathrm{13.4}}\,{\mathrm{pJ}}\). GST-based photonic memory has been found to be comparable to cutting-edge electronic PCM memory in terms of energy and speed [44.77]. To demonstrate multi-bit access capability, wavelength selectively operated through a single waveguide was coupled to three ring resonators with embedded GST elements (\(1{\times}{\mathrm{1}}\,{\mathrm{{\upmu}m^{2}}}\)), as shown in Fig. 44.28a. The three ring resonators with different ring radii were separated from the central waveguide by a gap of \({\mathrm{300}}\,{\mathrm{nm}}\). The bit status was able to be primarily retrieved by the transmittance change of the GST devices. Using three cavity modes with different wavelengths of 1560.1, 1561.5, and \({\mathrm{1563.35}}\,{\mathrm{nm}}\), a single pulse of \({\mathrm{10}}\,{\mathrm{ns}}\) and a train of consecutive \({\mathrm{50}}\,{\mathrm{ns}}\) pulses were employed to complete write and erase operations, respectively, as shown in Fig. 44.28b.


(a) Scanning electron microscope micrograph of the device. Light is coupled in and out of the on-chip circuitry by means of focusing grating couplers at the lower left and right; (b) The changes in transmission for an individual element for write/erase operations. Adapted from [44.76]
In this section, we have summarized recent advances in photonic memory based on phase-change materials. These materials are characterized by high contrast between the crystalline and amorphous phases of optical properties such as refractive index. A striking and functional feature of phase transition is high speed and scalability, and the high thermal stability of two states satisfies a key requirement for truly nonvolatile memory. Compatibility with CMOS technology vastly expands their capability into photonic devices. With these attributes, PCMs are ideal candidates for optical memory.
7 Summary
PRAM is an appealing memory technology, and its functional material is a key component of the memory cell. Recent decades have seen considerable progress in the development of phase-change materials for memory devices with lower power consumption and better data retention. However, a challenge remains in understanding a basic set of scientific concepts, i. e., how to understand the rapid structural change when the phase-change materials undergo amorphous to crystallization transition. In this chapter, we have presented the current knowledge about the structural and crystallization behavior of ternary Ge-Sb-Te alloys, and highlighted the use of Zn-doped \(\mathrm{Ge_{2}Sb_{2}Te_{5}}\) to enhance thermal stability in an amorphous state and increase crystalline resistance. The thermal and phase-change behavior for the novel Zn-Sb-Te alloys and ZnO-\(\mathrm{Sb_{2}Te_{3}}\) nanocomposite was further discussed. We also focused on recent scientific research on crystallization kinetics for phase-change materials by ultrafast calorimetry. Based on this new measurement technology, we can extend our knowledge to the process of crystal growth under supercooled liquid for phase-change materials. Finally, we have discussed recent progress involving the use of PCMs in integrated photonic memory, although much additional work is needed to further reduce the power required for writing and erasure. Nevertheless, this provides a pathway towards a new paradigm in all-photonic memory.
References
S.K. Lai: Brief history of ETOX™ NOR flash memory, J. Nanosci. Nanotechnol. 12(10), 7597–7603 (2012)
Y. Fujisaki: Overview of emerging semiconductor non-volatile memories, IEICE Electron. Express 9(10), 908–925 (2012)
H. Lan, H. Liu: UV-nanoimprint lithography: Structure, materials and fabrication of flexible molds, J. Nanosci. Nanotechnol. 13(5), 3145–3172 (2013)
H.J. Borg, R.V. Woudenberg: Trends in optical recording, J. Magn. Magn. Mater. 193(1–3), 519–525 (1999)
C.-Y. Lu: Future prospects of NAND flash memory technology-the evolution from floating gate to charge trapping to 3D stacking, J. Nanosc. Nanotechnol. 12(10), 7604–7618 (2012)
C. Miccoli, C.M. Compagnoni, L. Chiavarone, S. Beltrami, A.L. Lacaita, A.S. Spinelli, A. Visconti: Reliability characterization issues for nanoscale flash memories: A case study on 45-nm NOR devices, IEEE Trans. Dev. Mater. Reliab. 13(2), 362–369 (2013)
J.F. Scott: Ferroelectric Memories (Springer, Berlin, Heidelberg 2000)
S.S.P. Parkin, K.P. Roche, M.G. Samant, P.M. Rice, R.B. Beyers, R.E. Scheuerlein, E.J. O'Sullivan, S.L. Brown, J. Bucchigano, A. D. W., Y. Lu, M. Rooks, P.L. Trouilloud, R.A. Wanner, W.J. Gallagher: Exchange-biased magnetic tunnel junctions and application to nonvolatile magnetic random access memory, J. Appl. Phys. 85, 5828 (1999)
S. Raoux, G.W. Burr, M.J. Breitwisch, C.T. Rettner, Y.C. Chen, R.M. Shelby, M. Salinga, D. Krebs, S.-H. Chen, H.-L. Lung, C.H. Lam: Phase-change random access memory: A scalable technology, IBM J. Res. Dev. 52(4.5), 465–479 (2008)
H. Akinaga, H. Shima: Resistive random access memory (ReRAM) based on metal oxides, Proc. IEEE 98(12), 2237–2251 (2010)
G.W. Burr, M.J. Breitwisch, M. Franceschini, D. Garetto, K. Gopalakrishnan, B. Jachson, B. Kurdi, C. Lam, L.A. Lastras, A. Padilla, B. Rajendran, S. Raoux, R.S. Shenoy: Phase change memory technology, J. Vac. Sci. Technol. B 28(2), 223–262 (2010)
S.R. Ovshinsky: Reversible electrical switching phenomena in disordered structures, Phys. Rev. Lett. 21, 1450–1453 (1968)
X.S. Miao, L.P. Shi, H.K. Lee, J.M. Li, R. Zhao, P.K. Tan, T.C. Chong: Temperature dependence of phase-change random access memory cell, Jpn. J. Appl. Phys. 45, 3955 (2006)
M. Wuttig, N. Yamada: Phase-change materials for rewriteable data storage, Nat. Mater. 6, 824–832 (2007)
W. Welnic, M. Wuttig: Reversible switching in phase-change materials, Mater. Today 11(6), 20–27 (2008)
M. Wuttig, D. Lusebrink, D. Wamangi, W. Welnic, M. Gilleßen, R. Dronskowski: The role of vacancies and local distortions in the design of phase-change mateirals, Nat. Mater. 6, 122–128 (2007)
A.V. Kolobov, P. Fons, A.I. Frenkel, A.L. Ankudinov, J. Tominaga, T. Uruga: Understanding the phase-change mechanism of rewritable optical media, Nat. Mater. 3(10), 703–708 (2004)
S. Kohara, K. Kato, S. Kimura, H. Tanaka, T. Usuki, K. Suzuya, Y. Tanaka: Structural basis for the fast phase change of Ge2Sb2Te5: Ring statistics analogy between the crystal and amorphous states, Appl. Phys. Lett. 89(20), 201910 (2006)
J. Akola, R.O. Jones: Structural phase transitions on the nanoscale: The crucial pattern in the phase-change materials Ge2Sb2Te5 and GeTe, Phys. Rev. B 76(23), 235201 (2007)
B. Huang, J. Robertson: Bonding origin of optical contrast in phase-change memory materials, Phys. Rev. B 81(8), 081204 (2010)
R.O. Jones: Bonding in phase change materials: Concepts and misconceptions, J. Phys. Condens. Matter 30(15), 153001 (2018)
Y.K. Kim, K. Jeong, M.H. Cho, U. Hwang, H.S. Jeong, K. Kim: Changes in the electronic structures and optical band gap of Ge2Sb2Te5 and N-doped Ge2Sb2Te5 during phase transition, Appl. Phys. Lett. 90, 171920 (2007)
S. Privitera, E. Rimini, R. Zonca: Amorphous-to-crystal transition of nitrogen- and oxygen-doped Ge2Sb2Te5 films studied by in situ resistance measurements, Appl. Phys. Lett. 85, 3044 (2004)
B.W. Qiao, J. Feng, Y.F. Lai, Y. Ling, Y.Y. Lin, T. Tang, B.C. Ca, B. Chen: Effects of Si doping on the structural and electrical properties of Ge2Sb2Te5 films for phase change random access memory, Appl. Surf. Sci. 252(24), 8404–8409 (2006)
T.J. Park, S.Y. Choi, M.J. Kang: Phase transition characteristics of Bi/Sn doped Ge2Sb2Te5 thin film for PRAM application, Thin Solid Films 515(12), 5049–5053 (2007)
K.H. Song, S.W. Kim, J.H. Seo, H.Y. Lee: Influence of the additive Ag for crystallization of amorphous Ge-Sb-Te thin films, Thin Solid Films 517(14), 3958–3962 (2009)
N. Bai, F.R. Liu, X.X. Han, Z. Zhu, F. Liu, X. Lin, N.X. Sun: Effect of the Sn dopant on the crystallization of amorphous Ge2Sb2Te5 films induced by an excimer laser, Opt. Laser Technol. 74, 11–15 (2015)
M.H. Jang, S.J. Park, D.H. Lim, M.-H. Cho, K.H. Do, D.-H. Ko, H.C. Sohn: Phase change behavior in oxygen-incorporated Ge2Sb2Te5 films, Appl. Phys. Lett. 95(1), 012102 (2009)
X. Zhou, L. Wu, Z. Song, F. Rao, M. Zhu, C. Peng, D. Yao, S. Song, B. Liu, S. Feng: Carbon-doped Ge2Sb2Te5 phase change material: A candidate for high-density phase change memory application, Appl. Phys. Lett. 101(14), 202 (2012)
P. Němec, A. Moreac, V. Nazabal, M. Pavlišta, J. Přikryl, M. Frumar: Ge-Sb-Te thin films deposited by pulsed laser: An ellipsometry and Raman scattering spectroscopy study, J. Appl. Phys. 106, 103509 (2009)
F. Wei, L. Wang, T. Kong, L. Shi, R. Huang, J. Zhang, G. Cheng: Amorphous thermal stability of Al-doped Sb2Te3 films for phase-change memory application, Appl. Phys. Lett. 103(18), 181908 (2013)
C. Peng, L.C. Wu, Z.T. Song, F. Rao, M. Zhu, X.L. Li, B. Liu, L.M. Cheng, S.L. Feng, P.X. Yang, J.H. Chu: Performance improvement of Sb2Te3 phase change material by Al doping, Appl. Surf. Sci. 257(24), 10667–10670 (2011)
J. Xu, B. Liu, Z. Song, S. Feng, B. Chen: Crystallization and C-RAM application of Ag-doped Sb2Te3 material, Mater. Sci. Eng. B 127(2/3), 228–232 (2006)
Y.J. Chen, B. Zhang, Q.Q. Ding, Q.S. Deng, Y. Chen, Z.T. Song, J.X. Li, Z. Zhang, X.D. Han: Microstructure evolution and crystallography of the phase-change material TiSbTe films annealed in situ, J. Alloy. Compd. 678, 85–92 (2016)
Y. Lu, S. Song, Z. Song, F. Rao, L. Wu, M. Zhu, B. Liu, D. Yao: Investigation of CuSb4Te2 alloy for high-speed phase change random access memory applications, Appl. Phys. Lett. 100(19), 193114 (2012)
M. Zhu, L. Wu, Z. Song, F. Rao, D. Cai, C. Peng, X. Zhou, K. Ren, S. Song, B. Liu, S. Feng: Ti10Sb60Te30 for phase change memory with high-temperature data retention and rapid crystallization speed, Appl. Phys. Lett. 100(12), 122101 (2012)
C. Peng, L. Wu, F. Rao, Z. Song, P. Yang, H. Song, K. Ren, X. Lin, M. Zhu, B. Liu, J. Chu: W-Sb-Te phase-change material: A candidate for the trade-off between programming speed and data retention, Appl. Phys. Lett. 101(12), 122108 (2012)
Y.C. Her, Y.S. Hsu: Optical properties and crystallization characteristics of Ge-doped Sb70Te30 phase change recording film, Jpn. J. Appl. Phys. 42, 804 (2003)
Y.S. Hsu, Y.C. Her, S.T. Cheng, S.Y. Tsai: Thermal-and laser-induced order-disorder switching of In-doped fast-growth Sb70Te30 phase-change recording films, IEEE Trans. Magn. 43(2), 936–938 (2007)
Y.S. Hsu, Y.C. Her, S.T. Cheng, S.Y. Tsai: Thermal-and laser-induced order-disorder switching of Ag-doped fast-growth Sb70Te30 phase-change recording films, Jpn. J. Appl. Phys. 46(6S), 3945 (2007)
C. Peng, Z. Song, F. Rao, L. Wu, M. Zhu, H. Song, B. Liu, X. Zhou, D. Yao, P. Yang, J. Chu: Al1.3Sb3Te material for phase change memory application, Appl. Phys. Lett. 99(4), 043105 (2011)
F. Wang, T. Zhang, C. Liu, Z. Song, L. Wu, B. Liu, S. Feng, B. Chen: Au doped Sb3Te phase-change material for C-RAM device, Appl. Surf. Sci. 254(8), 2281–2284 (2008)
Y. Cheng, Z. Song, Y. Gu, S. Song, F. Rao, L. Wu, B. Liu, S. Feng: Influence of silicon on the thermally-induced crystallization process of Si-Sb4Te phase change materials, Appl. Phys. Lett. 99(26), 261914 (2011)
G.X. Wang, Q.H. Nie, X. Shen, R.P. Wang, L.C. Wu, J. Fu, T.F. Xu, S.X. Dai: Phase change behaviors of Zn-doped Ge2Sb2Te5 films, Appl. Phys. Lett. 101, 051906 (2012)
K.F. Kao, C.M. Lee, M.J. Chen, M.J. Tsai, T.S. Chin: Ga2Te3Sb5 – A candidate for fast and ultralong retention phase-change memory, Adv. Mater. 21(17), 169509 (2009)
G.X. Wang, X. Shen, Y.G. Lu, S.X. Dai, Q.H. Nie, T.F. Xu: Understanding the role of Zn in improving the phase change behaviors of Sb2Te3 films, Thin Solid Films 585, 57–65 (2015)
X. Shen, G.X. Wang, R.P. Wang, S.X. Dai, L.C. Wu, Y.M. Chen, T.F. Xu, Q.H. Nie: Enhanced thermal stability and electrical behavior of Zn-doped Sb2Te films for phase change memory application, Appl. Phys. Lett. 102, 131902 (2013)
G.X. Wang, X. Shen, Q.H. Nie, T.F. Xu, S.X. Dai, Y.G. Lu, Y.M. Chen, J.J. Li: Characterization of physical properties for Zn-doped Sb3Te films, Appl. Phys. Express 6, 095801 (2013)
G.X. Wang, X. Shen, Q.H. Nie, R.P. Wang, L.C. Wu, Y.G. Lu, S.X. Dai, T.F. Xu, Y.M. Chen: Improved phase-change characteristics of Zn-doped amorphous Sb7Te3 films for high-speed and low-power phase change memory, Appl. Phys. Lett. 103, 031914 (2013)
K.M.F. Shahil, M.Z. Hossain, V. Goyal, A.A. Balandin: Micro-Raman spectroscopy of mechanically exfoliated few-quintuple layers of Bi2Te3, Bi2Se3, and Sb2Te3 materials, J. Appl. Phys. 111(5), 054305 (2012)
J.S. Wei, H. Yuan, F.X. Gan: Crystallization mechanism and course of the Ge2Sb2Te5 thin films under focused pulse laser, J. Inorg. Mater. 17(6), 1245–1252 (2002)
D.W. Zeng, C.S. Xie, B.L. Zhu, W.L. Song: Characteristics of Sb2O3 nanoparticles synthesized from antimony by vapor condensation method, Mater. Lett. 58(3/4), 312–315 (2004)
J. Rocca, M. Erazu, M. Fontana, B. Arcondo: Crystallization process on amorphous GeTeSb samples near to eutectic point Ge15Te85, J. Non-Cryst. Solids 355, 2068–2073 (2009)
J. Coombs, A. Jongenelis, W. van Es-Spiekman, B. Jacobs: Laser-induced crystallization phenomena in GeTe-based alloys. I. Characterization of nucleation and growth, J. Appl. Phys. 78, 4906–4917 (1995)
J. Park, M.R. Kim, W.S. Choi, H. Seo, C. Yeon: Characterization of amorphous phases of Ge2Sb2Te5 phase-change optical recording material on their crystallization behavior, Jpn. J. Appl. Phys. 38, 4775 (1999)
J. Kalb, F. Spaepen, M. Wuttig: Atomic force microscopy measurements of crystal nucleation and growth rates in thin films of amorphous Te alloys, Appl. Phys. Lett. 84, 5240–5242 (2004)
S. Raoux, K. Virwani, C. Cabral Jr, L. Krusin-Elbaum, J.L. Jordan-Sweet, M. Hitzbleck, M. Salinga, A. Madan, T.L. Pinto: Phase transitions in Ge-Sb phase change materials, J. Appl. Phys. 105(6), 064918 (2009)
T. Matsunaga, J. Akola, S. Kohara, T. Honma, K. Kobayashi, E. Ikenaga, R.O. Jones, N. Yamada, M. Takata, R. Kojima: From local structure to nanosecond recrystallization dynamics in AgInSbTe phase-change materials, Nat. Mater. 10, 129–134 (2011)
J. Hegedüs, S. Elliott: Microscopic origin of the fast crystallization ability of Ge-Sb-Te phase-change memory materials, Nat. Mater. 7, 399–405 (2008)
I. Friedrich, V. Weidenhof, W. Njoroge, P. Franz, M. Wutting: Structural transformations of Ge2Sb2Te5 films studied by electrical resistance measurements, J. Appl. Phys. 87, 4130–4134 (2000)
Y. Choi, M. Jung, Y.-K. Lee: Effect of heating rate on the activation energy for crystallization of amorphous Ge2Sb2Te5 thin film, Electrochem. Solid-State Lett. 12, F17–F19 (2009)
J. Orava, A. Greer, B. Gholipour, D. Hewak, C. Smith: Characterization of supercooled liquid Ge2Sb2Te5 and its crystallization by ultrafast-heating calorimetry, Nat. Mater. 11, 279–283 (2012)
H.E. Kissinger: Reaction kinetics in differential thermal analysis, Anal. Chem. 29, 1702–1706 (1957)
M. Ediger, P. Harrowell, L. Yu: Crystal growth kinetics exhibit a fragility-dependent decoupling from viscosity, J. Chem. Phys. 128, 034709 (2008)
C.V. Thompson, F. Spaepen: On the approximation of the free energy change on crystallization, Acta Metall. 27, 1855–1859 (1979)
S. Raoux, D. Ielmini: Phase change materials and their application to nonvolatile memories, Chem. Rev. 110, 240–267 (2009)
M.H. Cohen, G. Grest: Liquid-glass transition, a free-volume approach, Phys. Rev. B 26, 6313 (1982)
M.L.F. Nascimento, E. Dutra Zanotto: Does viscosity describe the kinetic barrier for crystal growth from the liquids to the glass transition?, J. Chem. Phys. 133, 174701 (2010)
J. Orava, D.W. Hewak, A.L. Greer: Fragile-to-strong crossover in supercooled liquid Ag-In-Sb-Te studied by ultrafast calorimetry, Adv. Funct. Mater. 25, 4851–4858 (2015)
C. Zhang, L. Hu, Y. Yue, J.C. Mauro: Fragile-to-strong transition in metallic glass-forming liquids, J. Chem. Phys. 133, 014508 (2010)
B. Chen, J. Momand, P.A. Vermeulen, B.J. Kooi: Crystallization kinetics of supercooled liquid Ge–Sb based on ultrafast calorimetry, Cryst. Growth Des. 16, 242 (2015)
Y. Chen, G. Wang, L. Song, X. Shen, J. Wang, J. Huo, R. Wang, T. Xu, S. Dai, Q. Nie: Unraveling the crystallization kinetics of supercooled liquid GeTe by ultrafast calorimetry, Cryst. Growth Des. 17, 3687 (2017)
A. Sebastian, M. Le Gallo, D. Krebs: Crystal growth within a phase change memory cell, Nat. Commun. 5, 4314 (2014)
B.-S. Lee, K. Darmawikarta, S. Raoux, Y.-H. Shih, Y. Zhu, S.G. Bishop, J.R. Abelson: Distribution of nanoscale nuclei in the amorphous dome of a phase change random access memory, Appl. Phys. Lett. 104, 071907 (2014)
W.H.P. Pernice, H. Bhaskaran: Photonic non-volatile memories using phase change materials, Appl. Phys. Lett. 101(17), 171101 (2012)
C. Ríos, M. Stegmaier, P. Hosseini, D. Wang, T. Scherer, C.D. Wright, H. Bhaskaran, W.H.P. Pernice: Integrated all-photonic non-volatile multi-level memory, Nat. Photon. 9(9), 725–732 (2015)
E. Kuramochi, M. Notomi: Optical memory: Phase-change memory, Nat. Photon. 9(11), 712–714 (2015)
C. Rios, P. Hosseini, C.D. Wright, H. Bhaskaran, W.H.P. Pernice: On-chip photonic memory elements employing phase-change materials, Adv. Mater. 26(9), 1372–1377 (2013)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter
Cite this chapter
Shen, X., Chen, Y., Wang, G., Lv, Y. (2019). Phase-Change Memory and Optical Data Storage. In: Musgraves, J.D., Hu, J., Calvez, L. (eds) Springer Handbook of Glass. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-319-93728-1_44
Download citation
DOI: https://doi.org/10.1007/978-3-319-93728-1_44
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93726-7
Online ISBN: 978-3-319-93728-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)