
Abstract
The chemical durability of silicate glasses has long been studied for many applications, in particular when glasses are subjected to environmental weathering and aqueous corrosion. Typical applications include optical instruments, glass vessels, radioactive waste confinement, and bone reparation. Glass corrosion involves ion exchange, water diffusion, network dissolution-recondensation, and secondary phase precipitation. These reactions may impact, among other things, the release of contaminants from waste glasses, and the glass mechanical, optical and catalytic properties. The glass corrosion mechanisms and alteration product formation have been well studied as a function of many environmental parameters (temperature, pH, water composition, etc.).
The present chapter describes the general phenomena behind glass corrosion and details glass dissolution in aqueous conditions on one hand and glass vapor hydration on the other hand. The latter phenomenon has not received the same level of attention in the literature relative to the corrosion in aqueous solutions. Research and development needs, in particular in complex systems such as radioactive waste geological repositories, are discussed in the conclusion of the chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Dissolution of Glass in Water
Glass–water interactions are studied to qualify glasses as materials for a variety of applications such as reaction vessels, optical instruments, building construction, cement enforcement, bone replacement, radioactive waste fixation, etc. Surface reactions are important in glass grinding, polishing, cleaning, coating, mechanical strengths, reflectance, and catalysis, amongst other areas.
Glasses are amorphous solids characterized at the molecular level by a nonperiodic network of different chemical elements. Glass network formers like Si, B, or Al stabilize the glass network by ion-covalent bridging bonds; glass network modifiers like the alkali ions break the ion-covalent bridging of the glass network by establishing nonbridging oxygen () bonds with largely ionic character. The ratio of NBO to bridging bonds is an indicator of the degree of polymerization of the glass network.
The interaction of glasses with fluids is studied by various test methods, exposing powdered or bulk glass samples to different kinds or stagnant or flowing aqueous media or to vapor phases. Glass–water interaction properties are characterized by the analyses of aqueous solutions for dissolved glass components or pH, by mass loss, or by solid alteration products on the glass surface.
Glasses react in aqueous solutions by a number of surface reactions, mainly hydrolysis of bridging bonds and ion exchange of NBO bond glass constituents. In the overall reaction mechanism between a fluid and the glass a number of coupled sequential and parallel partial reactions must be considered. The rate-limiting reaction step varies with time. It is possible to distinguish between congruent and incongruent dissolution. In the first case, all glass constituents are released into solution with release rate ratios corresponding to the composition of the glass. The glass dissolves homogeneously. In the second case, each glass constituent dissolves with a different rate. Those glass constituents dissolving faster than others are depleted from the glass surface. The surface becomes enriched in slowly releasing glass constituents. Glasses consisting of compounds with different solubilities have the tendency to dissolve incongruently. Incongruence can be a result of selective leaching or of a dissolution–reprecipitation mechanism. In the latter case clear phase boundaries are observed between the dissolving glass and a surface layer of reaction products.
A useful concept for data interpretation is the definition of the normalized mass loss \(NL\) (\(\mathrm{g{\,}m^{-2}}\)) of element \(i\)
where \(C_{i}\) is the concentration of element \(i\) in solution (\(\mathrm{g{\,}m^{-3}}\)), \(f_{i}\) is the fraction of an element \(i\) in the glass (unitless), and \(S/V\) is the ratio of the surface area to solution volume (\(\mathrm{m^{-1}}\)). Equal normalized elemental mass loss for two glass constituents signifies congruent release behavior of these two elements.
The principal partial reactions in the glass–water interaction mechanism are glass network hydration, ion exchange, and dissolution of the glass network, resulting in formation of often clearly structured altered surface layers. Transport processes of reactants (e. g., water) or products either in surface layers or in the fluid may become rate limiting under certain conditions.
Glass hydration and subsequent exchange of alkali ions from the glass with \(\mathrm{H^{+}}\) ions from solution on the one hand and glass network dissolution (often also termed glass corrosion, i. e., the detachment of network former elements from the glass) on the other are parallel reactions [12.1, 12.2, 12.3, 12.4], indicating that the fastest of these rates determines the overall reaction rate. The alkali–\(\mathrm{H^{+}}\) ion exchange process is often diffusion controlled, governed by the (inter)diffusion of alkali ions and \(\mathrm{H^{+}}\) ions. Alternatively, the rate-limiting reaction may be the diffusion of water molecules into the glass network [12.5, 12.6], i. e., the hydration of the glass network. This process can also be diffusion controlled. In the case of diffusion control, the rate of selective alkali release from glasses is initially highest and decreases with the square root of time until it becomes equal to the rate of network dissolution.
Data on soda lime glass provide some insight in the very early steps of the glass–water interaction: initially, the water diffusion–ion exchange reaction in 1 N \(\mathrm{H_{2}SO_{4}}\) proceeds with the square root of time [12.7]. The rate-limiting step was the rupture of an \(\mathrm{H{-}{}O}\) bond of inward-diffusing water molecules and subsequent hydrolysis of \(\mathrm{Si{-}{}O{-}{}Si}\) bonds to form two silanol groups. This reaction is a necessary precursor to Na/H ion exchange.
Glass network hydrolysis and/or dissolution becomes dominant for longer times. Under these conditions a steady state is established between network dissolution and ion exchange–water diffusion. The hydrolyzed glass constituents may either be dissolved in the solution or form secondary solid reaction products on the surface by a dissolution–precipitation mechanism. The steady state corresponds to the formation of a thin diffusion layer of constant thickness: the rate by which the layer moves into the glass equals the rate by which it is dissolved or altered at the interface with solution or with solid surface reaction products. For borosilicate glasses this dissolution layer is reported to be a few nm in thickness but more recent data obtained with the use of atom probe tomography () of high spatial resolution indicate that this layer, if it exists at all for borosilicate glass, is smaller than \({\mathrm{1}}\,{\mathrm{nm}}\) [12.6, 12.8].
For silicate glasses it has been shown [12.9] that there is a linear free energy relation between the corrosion rate of the glass and the thermodynamic stability of a hypothetical mechanical mixture of component oxides (\(\mathrm{SiO_{2}}+\mathrm{B_{2}O_{3}}+\mathrm{Al_{2}O_{3}}+\mathrm{Na_{2}O}+\dots\)), as shown for a vast variety of glass compositions such as window glasses, medieval church glasses, natural basalt glasses or nuclear waste form glasses [12.10].
The rates of reaction of glasses with aqueous solutions depend on the composition, pH, temperature, time, solution volume, etc. Dissolution rates \(r\) are minimal at neutral pH [12.11, 12.12] and increase as the solution becomes more acidic or alkaline. For silicate glasses, dissolution rates often decrease with increasing concentration of dissolved silicic acid. If accumulation of dissolved silicic acid in solution occurs, the dissolution rate decreases. It was shown for silicate glasses that this is due to a saturation effect of the solution with respect to dissolved silica, which in a simplified and approximate way can be written as [12.13, 12.14]
The saturation concentration of silicic acid, \(C_{\text{Si},\text{Saturation}}\), and the forward rate constant, \(k_{+}\), depend on pH, solution composition, glass composition, temperature, and ionic strength. The term saturation is used instead of equilibrium, since a true equilibrium between a glass and an aqueous solution cannot be achieved: the glass phase is thermodynamically instable with respect of a mixture of component oxides. A true equilibrium may be achieved under static test conditions between sparingly soluble dissolved glass constituents in solution and secondary reaction products.
At saturation of the solution with dissolved silica, the network dissolution rate decreases in some cases by up to a factor of \(\mathrm{1000}\) or more, but dissolution continues, either due to formation of secondary phases or due to continued alkali release by the ion exchange reaction.
Recently, it was shown that the rates of dissolution of a nuclear waste borosilicate glass decreased by a factor of \(\mathrm{150}\) with increasing Si concentration added to the contacting aqueous solution, but the decrease in rate is strongly nonlinear with respect to Si and the solution saturation state cannot be invoked in any simple way [12.15]. In addition, accumulation of other glass constituents such as Al in solution contributes to a reduction of reaction rates. For \({\mathrm{3}}\,{\mathrm{ppm}}\) Al in solution a reduction in dissolution rate by a factor \(\mathrm{45}\) was observed.
Under static conditions, glass dissolution rates depend strongly on the \(S/V\) ratio. This is due to (1) the fact that saturation is reached faster at high \(S/V\), and (2) that for alkali-rich glasses pH increases faster at high \(S/V\). At the lowest \(S/V\), \(NL\) values (see definition in (12.1)), corrosion rates, and corrosion layer thicknesses are at maximum and independent of \(S/V\). Two further reaction steps can be observed with higher \(S/V\) [12.16]: an intermediate step where the reaction slows down to a minimum, and at very high \(S/V\) where the reaction rate is augmented by secondary phase formation and/or pH excursion. For a similar soda lime composition under static dissolution conditions in nonbuffered solution, the pH rises with the progress of glass corrosion. The higher the temperature or the \(S/V\) ratio, the faster this pH rise occurs. If the pH is above 9, a rapid increase of corrosion rates occurs. This does not happen if the pH is buffered at neutral values, for example by boron release in case of borosilicate glasses. Soda lime glass normally has rather poor resistance in alkaline solutions. Consequently, significant staining of window glasses can be observed at the rain-exposed side of buildings, if constructions allow for contact with alkaline runoff water from concrete or cement walls.
For an understanding of the interaction of glasses with aqueous solutions, a detailed knowledge of surface chemistry is important. The zeta potential \((\zeta)\) (electrokinetical potential difference across phase boundaries between solids and liquids) as a characteristic parameter of the magnitude of surface charge at the solid–liquid interface was used to determine ionic effects on surface chemistry in the initial stages of basaltic glass dissolution [12.17]. Affinities of a large suite of ions to the basalt glass surfaces for up to \({\mathrm{12000}}\,{\mathrm{h}}\) were characterized by sorption isotherms at neutral pH. An initial decrease of zeta potential was explained by alkali and alkali earth ion release leading to formation of \(\mathrm{SiO^{-}}\) groups at the surface. Anions in solution increased the negative charge, whereas with time divalent cations, in particular \(\mathrm{Ca^{2+}}\) and \(\mathrm{Zn^{2+}}\), neutralized it. \(\mathrm{Al^{3+}}\) in solution led immediately to charge reversal. Far from equilibrium, there is a linkage between Si release rates and zeta potential. The anions \(\mathrm{F^{-}}\), \(\mathrm{C_{2}O_{4}^{2-}}\) and \(\mathrm{HPO_{4}^{2-}}\) increased Si release rates and had the most negative \(\zeta\) values, while \(\mathrm{Al^{3+}}\) and \(\mathrm{Zn^{2+}}\) had the most positive \(\zeta\) values linked to a strong decrease in dissolution rates. In the case of Al it is likely that \(\mathrm{Si{-}{}O{-}{}Al{-}{}O{-}{}Si}\) bonds are formed, reducing the rate of Si release. This may explain the above-mentioned reduction of glass dissolution rates with Al concentration and may contribute to the understanding of the nonlinear character of the saturation effect.
The stability of the glass network against dissolution, ion exchange, and water diffusion as well as saturation concentrations in water and the long-term corrosion mechanism, depends to a large extent on glass composition: increased concentrations of network modifiers like alkali ions in general decrease glass stability. Na-rich glasses showed stronger leach resistance than K-rich glasses, divalent cations stabilize the glass against ion exchange for compositions in the range \(\mathrm{20Na_{2}O}\cdot x\text{RO}\cdot(80-x)\mathrm{SiO_{2}}\) or 15(\(\mathrm{Na_{2}O}\) or \(\mathrm{K_{2}O})\cdot x\text{CaO}\cdot(75-x)\mathrm{SiO_{2}}\) (\(\mathrm{R}=\mathrm{Ca}\), Mg, Zn and \(x={\mathrm{0}},{\mathrm{5}},{\mathrm{10}},{\mathrm{15}}\,{\mathrm{mol\%}}\)) up to \(\text{R}={\mathrm{10}}\,{\mathrm{mol\%}}\) whereas higher contents of alkali earth ions (\({\mathrm{15}}\,{\mathrm{mol\%}}\)) lead to preferential alkali earth release. The partial replacement of Si by alkali earth ions decreases the corrosion resistance of alkali aluminum silicate glasses (\(\mathrm{18.5Na_{2}O\cdot 7.4Al_{2}O_{3}\cdot 74.1SiO_{2}}\)) at neutral pH and increases it at pH 10 [12.18]. The formation of \(\mathrm{Si{-}{}O{-}{}Al}\) bridges stabilizes the glass only if the hydrolytic stability of this bond structure is high. This is the case at neutral to alkaline pH, but not at acid pH. Hence, the stabilizing effect of Al depends also on solution chemistry. The stabilizing effects of Al and of divalent ions are not additive. If a glass composition is stabilized by Al addition, the further addition of alkali earth elements does not lead to further increase of corrosion resistance.
The corrosion resistance of glasses sometimes shows strong nonlinear behavior with variation in glass composition. For example, the addition of \(0{-}2\,{\mathrm{wt\%}}\) \(\mathrm{SiO_{2}}\) to a borosilicate nuclear waste glass leads only to a small increase in corrosion resistance, but a \({\mathrm{3}}\,{\mathrm{wt\%}}\) addition has shown a stabilizing effect by a factor of 10. A further increase in silica content had a negligible effect [12.19]. This abrupt change in corrosion resistance with small variations of glass composition would be unexpected with the thermodynamic solid solution theories for initial glass dissolution rates and may be explained by percolation theory, which also explains the shift from congruent to incongruent leach behavior of Si and B. The percolation model [12.20] distinguishes between fast reaction rates for breaking of \(\mathrm{Si{-}{}O{-}{}B}\) bonds and slow rates for \(\mathrm{Si{-}{}O{-}{}Si}\) bonds. According to the observations of the authors, the glasses fall into one of two categories that are markedly different in dissolution behavior, in terms of whether the \(\text{Si}/\text{B}\) ratio in the glass network exceeds a threshold value or not. The transition from one of the categories to the other is very sharp when the \(\text{Si}/\text{B}\) ratio is changed as the key parameter. The qualitative aspects of the experimental observation can be explained very well by a simple model combined with the site percolation theory.
2 Glass Corrosion in Aqueous Conditions
The chemical durability of glass refers to the resistance of the glass surface to chemical attack by fluids encountered during the service life of the glass. The largest scientific fields where chemical durability of glasses is of importance are related to the durability of the glass upon contact with aqueous fluids, in particular water, and the weathering of the glass surface against the atmosphere, in particular humidity. A large amount of information on the glass–water reaction collected over the past \(\mathrm{40}\) years has been summarized in the glass compendium [12.21] and numerous reviews [12.14, 12.22, 12.23, 12.24, 12.25, 12.26, 12.27, 12.28, 12.29, 12.30, 12.31]. In this section, we will present details on the various mechanisms responsible for glass corrosion in aqueous environments and the overall kinetics of the glass alteration process. Additionally, we will further explore the effects of durability on glass composition, fluid chemistry, and advanced materials characterization techniques that are being used to elucidate various processes responsible for glass alteration in aqueous fluids.
2.1 Mechanisms of Glass Alteration
The alteration rate of glass is not an inherent materials property. It depends on glass composition, structure, and surface states, as well as on environmental conditions such as the composition of the contacting solution (including pH and mineral saturation states), surface area of the glass exposed to the contacting solution, hydrodynamic conditions, temperature, stress, strain, vapor pressure, and so on. It is not possible to quantify the magnitude of each effect a priori for a given glass composition. Indeed, the overall mechanism of the glass–water interaction is a series of sequential and parallel reactions. The basic set of reactions may be applicable to most glass compositions.
Various partial reactions are responsible for the alteration of glass. The overall importance of various reactions is interpreted through responses to the glass alteration rate, selective leaching of various glass constituents, pH changes in solution, and careful examination of postmortem glass samples. Conceptual corrosion mechanisms and empirical rate laws may be constructed as a result of these observations. The nature of the rate-limiting reaction in the corrosion process is dependent on the glass composition and the environmental conditions under which the glass alteration proceeds. As a result of these processes, glass constituents are released into the contacting solution or remain at the glass surface and form an amorphous, porous, hydrated material known as a gel, or precipitate as secondary phases on the altered glass–solution interface.
Present understanding of corrosion has identified several coupled and parallel processes that control the rate of glass corrosion. These mechanisms include, but are not limited to, water diffusion [12.32, 12.33, 12.34, 12.35], ion exchange and interdiffusion between a water species and glass formers [12.36, 12.37, 12.4], hydrolysis of covalent and ionocovalent bonds, condensation of species at the glass surface, and precipitation of amorphous or crystalline minerals. Additionally, transport processes, such as access of reagents (e. g., water molecules) and removal of products (e. g., removal of glass constituents), may become rate limiting under certain conditions. These devitrification processes occur from the very first contact with water and continue, as far as is understood, ad infinitum. The rate-controlling process may change with time.
2.2 Kinetics of Glass Alteration
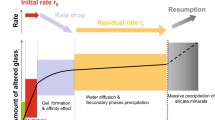
At this point, it is convenient to describe the various glass alteration processes in terms of their effect on the glass alteration rate on a glass sample that is initially placed in contact with dilute water. As the glass–water reaction proceeds, the extent of glass alteration can be divided into three regimes or stages (Fig. 12.1):
-
Stage I – Initial or forward rate, \(r_{0}\)
-
Stage II – Decreasing rate, \(r(t)\), and residual rate, \(r_{\mathrm{r}}\)
-
Stage III – Alteration renewal.


General schematic of the stages of glass–water reaction. The potential rate-limiting mechanism for each step is presented in the figure. Reprinted (adapted) from [12.38], with permission from Elsevier
It is important to note that in 2013 the international nuclear waste glass corrosion community reached agreement to refer to three stages rather than five stages [12.38]. The major difference between the three- and five-stage description is the condensing of mechanisms of interdiffusion (previously stage I), hydrolysis (previously stage II), and rate drop (previously stage III) into a single regime that is now referred to as stage I (initial or early stage of dissolution).
2.2.1 Stage I. Initial or Forward Rate
The initial stage (stage I) of the glass–water reaction consists of a set of coupled processes that includes: (1) ion exchange and subsequent interdiffusion that occurs via the selective removal of charge compensating cations (i. e., alkali and alkaline earth elements) by \(\mathrm{H^{+}}\) or \(\mathrm{H_{3}O^{+}}\), and (2) hydrolysis of the glass network, which occurs via protonation and rupture of bridging oxygen bonds \(\mathrm{Si{-}{}O{-}{}Si}\) or \(\mathrm{Si{-}{}O{-}{}Al}\). Rana and Douglas [12.39, 12.40] were amongst the first to recognize these steps in the reaction process. From experimental observations, they reasoned that because the release of sodium ions from the glass into solution exhibited square root time dependence, the initial dissolution behavior of the glass was controlled through a diffusion process. With increasing reaction progress, they noted that the square root time dependence persisted only at short time periods. As the reaction progressed, linear time dependence was observed, which they attributed to a different reaction. Stage I of glass alteration is regarded as the fastest stage in the glass dissolution process. It is limited first by diffusion-controlled ion release and then by hydrolysis. It leads to the release of principally cationic species and the development of a silica-like hydrated glass surface with a thickness of a few tens of nanometers.
Interdiffusion (ion exchange, water diffusion) refers to the exchange between alkali metals located at network terminal sites and protons in the solution. The interdiffusion process is defined by the exchange of \(\mathrm{H^{+}}\), \(\mathrm{H_{3}O^{+}}\), and/or \(\mathrm{H_{2}O}\) in the fluid phase for network-modifying cations in the glass. Interdiffusion has been observed experimentally for decades [12.39, 12.40].
The importance of the ion exchange process may be especially relevant at low temperature and in solutions that are near-saturated with respect to amorphous silica [\(\mathrm{SiO_{2}}\)(am)] [12.36, 12.37]. Additionally, Jantzen et al [12.41] concluded that the performance of a high-level waste glass buried in a field lysimeter for \(\mathrm{24}\) years was controlled by a diffusive process, thus highlighting the possible role of interdiffusion in the long-term durability of glass waste forms.
The overall chemical reaction describing the exchange of a cation, \(\mathrm{M^{+}}\), with a charged water species or molecular water, given here as a proton, is written as
where a horizontal bar represents one chemical bond. Equation (12.3) occurs in acidic conditions while (12.4) occurs in neutral conditions. The rate of the ion exchange reaction is controlled by the diffusion of a water species at the glass–water interface and the reaction results in an increase in the solution pH. The selective removal of alkali metals between the fluid and the glass, where the bulk silicate network is left intact, is referred to as leaching. Because the leaching process is interdiffusion controlled it follows Fick's second law of diffusion; both the alkali concentration in solution and depth of water penetration increase as a function of the square root of time.
The process of interdiffusion is followed by two simultaneous reactions: hydration and dissolution of the glass matrix. Hydrolysis involves a nucleophilic attack on a silicon atom by molecular water (12.5) or the hydroxyl ion (12.6). This reaction directly disrupts the connectivity of the silica network. During the reaction molecular water or the hydroxyl ion are consumed while silanol groups are formed. This reaction may be written in the following two ways
If the hydrolysis reaction occurs at a terminal silanol group it leads to the dissolution of the glass network and the release of orthosilicic acid (\(\mathrm{H_{4}SiO_{4}}\)) into the leachate. This is represented through the following reactions
where (12.7) occurs more rapidly in alkaline pH while (12.8) occurs at near neutral pH where the hydroxyl ion is less abundant. Equations (12.7) and (12.8) are known as network or matrix dissolution. If the matrix dissolution reaction occurs in the reverse direction it is referred to as the condensation reaction. The increase in the concentration of \(\mathrm{OH^{-}}\) accelerates network hydrolysis and also has the added effect that silica is more soluble at higher pH [12.42]. As network forming bonds are broken and the structure is hydrated, a gel-like structure is formed. If this reaction occurs in relatively dilute aqueous conditions, the release of glass constituents from the glass to the solution is congruent. The hydrolysis reaction is observed to be linear with time.
Several models have been developed to describe ion exchange and matrix dissolution behavior [12.36, 12.37, 12.43, 12.44]. Boksay et al [12.43, 12.44] explained the linear time dependence after the square root of time dependent behavior observed by Rana and Douglas by using a diffusion model that accounted for a continuously dissolving interface. Further development of this model by Doremus [12.4] allowed for the inward-diffusing and outward-diffusing species to have different diffusion coefficients, which was later confirmed by more accurate experimental data of elemental profiles [12.45, 12.46]. State-of-the art experimental studies dealing with diffusion in glass alteration still count on the models developed by Boksay and Doremus [12.47]. In addition to diffusion-controlled ion exchange reactions, models based on water diffusion have also been developed to explain long-term glass corrosion behavior [12.48, 12.49].
In summary, over the past \(\mathrm{30}\) years studies have focused on identifying the specific charge compensating cations (\(\mathrm{H^{+}}\) or \(\mathrm{H_{3}O^{+}}\)) responsible for the interdiffusion mechanism using a variety of molecular-scale techniques ([12.50] and references contained therein), such as pH stat titration experiments and surface profiling of corroded sodium borosilicate glass samples with Rutherford backscattering spectroscopy and elastic recoil detection analysis [12.51, 12.52, 12.6], isotopically labeled water (\(\mathrm{{}^{18}O}\) and deuterium) [12.36, 12.53], x-ray photoelectron spectroscopy [12.36], Fourier transform infrared () [12.54], x-ray reflectivity () studies [12.54], and quasielectric neutron scattering [12.55]. Efforts have also been made to isolate the alkali–alkali ion exchange and interdiffusion reaction from matrix dissolution [12.56, 12.57]. Although these studies have provided a wealth of information on the mechanism of ion exchange, they have not distinguished between \(\mathrm{H^{+}}\) or \(\mathrm{H_{3}O^{+}}\) as the charge compensation ion. However, it has been noted that the relative importance of the ion exchange depends on the diffusion coefficient and the corrosion rate.
2.2.2 Stage II. Decreasing Rate and Residual Rate
The initial dissolution regime is followed by a slowing of the dissolution rate. The onset of this regime is caused by saturation of the solution with respect to Si (affinity-controlled process) and the growth of one or several alteration layers that forms between the contact solution and the pristine glass. The composition and role of this alteration layer will be explained in more depth in Sect. 12.2.4. The slowdown regime is characterized by incongruent dissolution of glass components. Though the effects of an affinity and gel layer protection processes are well noted in the literature, a consensus does not exist on the treatment of observed results. Here, we will first give an overview of the underlying principles of an affinity-based theory, followed by an explanation of the passivating gel layer, and how the two theories may both play a role in the glass alteration process.
A theory to kinetically examine the slowdown of the reaction progress of minerals in water was proposed in the early 1980s [12.58]. It is based on the concept that there is a thermodynamic driving force controlling mineral corrosion. Affinity is a term that is used in irreversible thermodynamics and transition state theory denoting the deviation from equilibrium [12.58, 12.59]. Close to equilibrium the reaction rate decreases proportionally to decreasing affinity as described in (12.2) [12.58, 12.60, 12.61].
Equation (12.2) was proposed by Grambow [12.61] where it was postulated that the hydrolysis of silica in the glass, leading to the buildup of silica (in the form of orthosilicic acid, \(\mathrm{H_{4}SiO_{4}}\)) in the leachate, is the rate-limiting step. This equation is called Grambow's first-order law and it shows the essential role of silica in the dissolution process of glasses. This assumes that there is an intrinsic leachate (\(\mathrm{H_{4}SiO_{4}}\)) saturation constant \(C^{*}_{\text{Si},\text{Saturation}}\) that exists for the glass. This saturation value is dependent only on the glass composition, temperature, and pH. The second part of this equation is more representative of the slowdown process and considers activities of silica. Transition state theory is only valid for elementary reactions. Using a first-order dissolution rate law is not valid for a system, such as glass, where numerous coupled reactions occur and a rate-limiting elementary reaction is undefined. The value of \(C^{*}_{\text{Si},\text{Saturation}}\) is not intrinsic to a given glass composition, but depends on how the equilibrium conditions were obtained, such as environmental conditions and the exposure history of the glass [12.61]. A pristine glass coupon placed in a saturated leachate is altered at a high rate until a protective gel layer has formed [12.30]. This leads to a postulation that the reduction in the reaction rate is due to the formation of the gel layer at the surface (see below). The chemical affinity term only accounts for a rate decrease of a factor of five, and not by the observed factor of \(\mathrm{10^{4}}\).
In experiments, the silica concentration eventually reaches a saturation value and decreases the corrosion rate by three to four orders of magnitude, however corrosion continues, due either to the formation of secondary phases [12.62, 12.63] or continued alkali release by the ion exchange reaction [12.36]. An effect of saturation has also been reported for phosphate and some borate glasses [12.64]. It is important to note that in the case of glass, the dissolution rate, although low, does not become zero because silicate glasses cannot achieve thermodynamic equilibrium in water. Thermodynamic equilibrium is defined as the same solid phase dissolving into solution precipitating from solution at the exact same rate. Glass, which is a high-temperature phase, cannot precipitate from aqueous solution under these conditions and, therefore, the corrosion glass surface forms an amorphous hydrated layer.
Another process that is presented in the literature to account for the decreasing alteration rate of glass with reaction progress is attributed to the protective gel reaction layer that forms a diffusion barrier between the pristine glass and surrounding solution. Redensification of the gel may be one of the reasons for a decrease in the alteration rate. The role of the gel density on the alteration rate has been studied elsewhere [12.65]. Protective gel layers formed in silica-rich solutions are formed because the degree of silanol group condensation produces very dense gel layers. Reactive mass transfer of silica across the gel layer is an important rate-limiting step but it is not related to affinity. The rate of mass transfer is not a thermodynamic value but rather a constant, which may vary from experiment to experiment. Further description of these alteration layers is provided in Sect. 12.2.4, Surface Modification.
The altered layer (i. e., amorphous hydrated surface layer, also known as gel layer, and crystalline reaction products) represents a complex region, both physically and chemically, located between two distinct boundaries: the pristine glass surface at the innermost interface and the aqueous solution at the outermost interface. The thickness, chemical composition, and pore structure of the altered layer changes as a function of glass composition, pH, temperature, and bulk fluid composition. In addition, the thickness and stability of the altered layer are affected by the formation of crystalline reaction products, whether in residual rate or alteration rate renewal regimes. Altered layers have been identified on basaltic glasses [12.50, 12.66], mineral analogue glasses [12.67], medieval stained glasses [12.68], and sodium borosilicate glasses [12.69].
For completeness, an alternative conceptual model is also highlighted that has been proposed within the geochemical community. The alternative conceptual model, referred to as the dissolution–reprecipitation mechanism, suggests congruent dissolution of the glass, which is spatially and temporally coupled to amorphous silica precipitation at an inward-moving reaction interface [12.70, 12.8]. In comparison to the leaching mechanism, which suggests the altered layer forms by the repolymerization of partially bound elements at the glass–fluid interface after the selective removal of certain elements, the dissolution–reprecipitation model suggest that all bonds are broken at the glass–fluid interface followed by the immediate precipitation of an altered layer from a supersaturated thin film of water at the glass–fluid interface.
To conclude, the inarguable trend is observed that glass dissolution rates decrease with time. This decrease is due to affinity effects and the gel layer. Many arguments given to explain an observation for one effect do not necessarily exclude the role of the other. Being able to quantify the effects of the two theories would aide greatly in their understanding. Regardless, treatment of these two concepts as related to the slowdown in the rate is only important if this particular mechanism controls the long-term alteration rate.
Following the slowdown regime is the onset of the so-called residual rate. In terms of nuclear waste disposal, this is the most important regime because the release of radioactive elements into the surrounding environment will be controlled by this mechanism for thousands of years. A value for the rate of glass corrosion in deionized water at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for a large number of simplified waste glasses is on the order of magnitude of \({\mathrm{10^{-4}}}\,{\mathrm{g{\,}m^{-2}{\,}d^{-1}}}\) [12.71]. The mechanism controlling glass corrosion during the residual rate is not completely understood but it is assumed that affinity, the reactive diffusion of mobile elements in the glass towards the solution from the penetration of water into the glass, and the precipitation of secondary phases at the glass surface play important roles [12.72]. Though much attention is paid to affinity and gel layer contributions to the slowdown regime, the role of ion exchange and water diffusion, which are prevalent at the beginning of glass alteration, may not be overlooked once all other processes have slowed [12.36]. The effects of all these processes on the residual rate are difficult to identify.
2.2.3 Stage III. Reaction Rate Acceleration
As the glass dissolution reaction progresses, the solution becomes saturated and secondary minerals begin to form, principally at the surface–solution interface. The alteration phases are often a clay mineral, such as a smectite or chlorite [12.73]. The precipitation kinetics associated with these phases can be complex, but in general, the rate of secondary phase growth increases in response to the increase in magnitude of supersaturation [12.74, 12.75]. Depending on the type of alteration phase, the glass–water reaction can increase from the residual rate and return to a rate consistent with the saturation and pH conditions observed during stage II (e. g., stage III – alteration rate renewal). This type of behavior has been observed in accelerated weathering tests.
The stage III rate acceleration has been attributed mostly to the precipitation of zeolitic phases [12.26, 12.76, 12.77, 12.78, 12.79], but the process through which the effect occurs remains poorly understood. The mechanism by which phase precipitation and glass dissolution are coupled must be known to incorporate the effect into the waste glass degradation model. We note again that an increase in rate has only been observed in certain conditions and glass compositions [12.80, 12.81]. High-level waste glasses [12.77, 12.79] have exhibited this phenomenon, and so have a few representative low-activity waste () glasses [12.82]. Static experiments have suggested that aluminum may play a significant role in stage III behavior, with Al concentrations in solution decreasing abruptly as the alteration rate increases [12.61, 12.77, 12.83]. On the other hand, Si concentrations in solution have been shown to increase during the same period [12.83], which was wrongly assumed to be inconsistent with precipitation of aluminosilicate phases ([12.84] and references contained therein). Strachan and Neeway [12.78] recently identified an increase in Si concentrations to be possible because of a decrease in the rate-limiting \(\mathrm{H_{4}SiO_{4}}\) activity that occurs near pH 9 with an increase in the negatively charged \(\mathrm{H_{3}SiO_{4}^{-}}\) activity causing a net increase in Si concentrations. A recent experiment by Fournier et al [12.85] showed no effect on the alteration rate of the intermediate activity glass, CSD-B (\({\mathrm{50.32}}\,{\mathrm{wt\%}}\) \(\mathrm{SiO_{2}}\), \({\mathrm{14.44}}\,{\mathrm{wt\%}}\) \(\mathrm{B_{2}O_{3}}\), \({\mathrm{12.58}}\,{\mathrm{wt\%}}\) \(\mathrm{Na_{2}O}\), \({\mathrm{8.70}}\,{\mathrm{wt\%}}\) \(\mathrm{Al_{2}O_{3}}\), \({\mathrm{13.96}}\,{\mathrm{wt\%}}\) others), when analcime grains were added to the experiment as a seed for nucleation, but that the rate accelerated when the silicon-rich zeolite \(\mathrm{(NaAlO_{2})_{7}(SiO_{2})_{9}}\) was added to a separate system under the same experimental conditions. The large difference in effect with small compositional difference suggests the conditions required to initiate stage III are complex.
2.3 Dissolution Rate Measurement Methods
Rates for the different kinetic stages can be measured using a variety of durability tests [12.86]. Stage I dissolution rates are measured with tests where the solution is relatively dilute and affinity or a protective alteration layer has not been formed. Two common test methods are used to measure stage I, or forward rates: the single-pass flow through () method or the ASTM C1220 Test. The SPFT test uses a flow-through system with an influent solution of controlled compositions and steady-state release rates of important elements are achieved. The duration required for the reaction to achieve a steady state can last from a few days to several months depending on the temperature and pH of the system. The ASTM C1220 uses a glass monolith in a large volume of water. The test duration can be from a few hours to a few days but, compared to the SPFT, it has the disadvantage that species released from the glass build up in solution. This causes local pH fluctuations and the risk that the reaction affinity increases. In general, the forward rates of glass dissolution varies between \(\mathrm{0.1}\) and \({\mathrm{10}}\,{\mathrm{g{\,}m^{-2}{\,}d^{-1}}}\) at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) at pH \(\mathrm{9}\) measured at \({\mathrm{23}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\).
The stage II dissolution rate or residual rate is generally calculated using a static system or a system with low renewal rates (less than a few mL per day). Solution aliquots are periodically taken and the change in solution species concentration with time is used to calculate a rate. Because the solution concentrations are relatively high and the release rates are low, an accurate measurement of rates is difficult to achieve. Residual rates are often reported between \(\mathrm{10^{-4}}\) and \({\mathrm{10^{-3}}}\,{\mathrm{g{\,}m^{-2}{\,}d^{-1}}}\) at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), i. e., several orders of magnitude lower than the forward rate.
On the other hand, the stage III rate is often difficult to measure, as prediction of the onset of this regime has not been achieved. Recent experiments by Fournier et al [12.85] have seeded tests with zeolitic phases that cause resumption in the glass alteration rate. This technique should provide a method to better quantify stage III rates because it removes the latency period between the residual rate and resumption.
For all of these tests, it is important to know the surface area of the reacting sample. The representative surface area for glass dissolution has been the subject of many studies [12.15, 12.87]. In addition, the solution volume must be known in order to interpret the results and compare to other studies. Careful controls of experimental temperature and, if necessary, pH are also needed.
2.4 Effects of Glass Composition on Corrosion Kinetics
The bulk glass composition affects the rate of chemical and structural alteration that occurs at the surface of the glass. This, in turn, determines the composition and physical characteristics of the glass alteration layers [12.88]. The chemical nature and concentration of species in the glass are both important. It should be noted that the effects of different glass components are not additive so it is difficult to predict the corrosion rates of multicomponent glass from the corrosion rates of simple glasses. Therefore, the chemical durability of a given glass must be experimentally determined. Nevertheless, some qualitative information on the relative durability of a given glass composition is possible.
From a structural point of view, the addition of various species to a silicate disrupts and weakens the intermolecular bonds, which destabilizes the solid by increasing the free energy of the material. Addition of alkali ions reduces the melting point and corrosion resistance of silica due to the formation of ionic nonbridging oxygen bonds. On the other hand, alkaline earth cations can increase the crosslink density of the glass network. Intermediate elements such as Al and B can function either as network formers or decrease the connectivity of the network. Because of the various roles and complexity of the various species we will provide some examples of how specific species and elemental groups affect the overall chemical durability of various glasses.
2.4.1 Surface Modification
This section gives an overview of the morphology of the altered layer, the behavior of the leached elements, and an explanation of important secondary precipitates that may be formed as a result of glass alteration. The gel layer differs from the pristine glass in that it is porous, silica rich, and depleted in soluble glass constituents such as boron and alkali elements. It is also enriched in hydrolysable elements such as Fe, Zr, lanthanides, and actinides [12.72]. This altered layer is also seen in basaltic mineral dissolution but is referred to as palagonite. This gel layer is strongly dependent on several parameters such as the composition of the altered glass, the leachate chemistry, the length of experiment, and the temperature.
After the first contact of a corrosive fluid with the glass and even in presence of humid air, the initial surface modification is glass hydration over a few nanometers. The quantity of water entering the altered glass depends, among other parameters, on space and strain considerations in the glass network [12.89]. Molecular water may result from inwards diffusion of water accompanying \(\mathrm{H^{+}}\) ions or from the reaction [12.90, 12.91] (12.9).
If sufficient glass network bonds are hydrolyzed, a transformed layer [12.92] (gel) is formed, with a clear phase boundary to the glass. Gel layers are porous [12.93], containing molecular water [12.94, 12.95] and allowing for high ionic mobility [12.96] as well as high water mobility. Studying glass dissolution at pH 0 and \({\mathrm{150}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and using \(\mathrm{{}^{18}O}\) (measured by time-of-flight secondary ion mass spectrometry) and \(\mathrm{{}^{26}Mg}\) tracers (measured by laser ablation inductively coupled plasma mass spectrometry), it was shown that the gel layer is formed by a dissolution–precipitation mechanism, without evidence at \(\mathrm{\upmu{}m}\) space resolution of interdiffusion-controlled ion exchange at the glass–gel reaction front [12.70]. Recently it was shown for a borosilicate nuclear waste glass at \({\mathrm{50}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and neutral pH that also at nm scale a clear phase boundary exists between the pristine glass and the gel layer [12.8]. Imaging with (scanning) transmission electron microscopy () with a high-angle annular dark-field () detector allows discrimination of the altered zone from the pristine glass. The width of the altered zone increases, from \(35{-}40\,{\mathrm{nm}}\) after \({\mathrm{4}}\,{\mathrm{d}}\), \(50{-}60\,{\mathrm{nm}}\) at one month, to \(50{-}70\,{\mathrm{nm}}\) at three months, and to \(70{-}90\,{\mathrm{nm}}\) after seven months. The glass–altered zone interface is always distinct and very sharp at nm to sub-nm scale (Fig. 12.2).


Three-dimensional (), reconstruction of one-month altered glass by atom probe tomography (APT), showing chemical maps for B and Ca, where each dot represents a single atom. The altered zone (or layer) is strongly depleted in B and Ca, and the chemical interfaces delimiting the pristine glass–altered zone boundary are almost atomically sharp. Reproduced from [12.8]
These data are clear evidence for dissolution–precipitation [12.8] as a mechanism of gel layer formation, but it does not preclude a diffusion controlled process [12.38]. Indeed, the precipitated gel layer may constitute a diffusion barrier for water and dissolved silica. As a matter of fact, a thorough review by Gin et al [12.23] highlighted that the alteration layer mechanism formation is strongly linked to the solution saturation conditions. They added that neither a simple interdiffusion model nor the interfacial dissolution–precipitation model can explain the observed elemental profiles within the alteration layer. More generally, far-from-dilute and close-to-saturation conditions must be differentiated.
The reacted glass may also become entirely dissolved (congruent dissolution, as occurs with some potassium silicate glasses) but in many cases the reacted glass surface remains at the surface in altered form and becomes enriched with sparingly soluble glass constituents. This is particularly true if the glass contains large quantities of heavy metals.
Often surface layers with a layered structure are formed: next to the pristine glass a hydration layer is found. There is typically a smooth transition between the pristine and the hydrated glass. Going outwards, the hydrated glass is followed by an amorphous and highly porous gel layer, which may also contain microcrystalline phases, precursors of a crystalline phase. There is often a clear phase boundary between the hydrated glass and the gel layer. This gel layer is depleted in alkali elements and B, but enriched in Si, Al, transition and heavy metal contents [12.63, 12.97]. Metal ions from solution (e. g., Mg) also become incorporated into the gel layer. The interface between the diffusion layer and the gel layer often has the character of a phase boundary. The thickness of the gel layer is a function of solution chemistry, fluid flow rates and surface-to-volume ratio. Particularly dense gel layers are formed in static dissolution tests at high \(S/V\) ratios and gel layers of very low density are formed in dissolution tests in deionized water at high water flow rates (Soxhlet test). Using x-ray reflectometry, Rébiscoul et al [12.98] identified four stages of gel layer densification for the nuclear waste glass SON68:
-
Stage 1: Creation of thin dealkalized interdiffusion layers (\({\mathrm{18}}\,{\mathrm{nm}}\)) with \(\rho={\mathrm{1.9}}\,{\mathrm{g{\,}cm^{-3}}}\)
-
Stage 2: Beginning of alteration by the creation of a thin low-density gel layer (\({\mathrm{24}}\,{\mathrm{nm}}\)) with \(\rho={\mathrm{1.5}}\,{\mathrm{g{\,}cm^{-3}}}\) on top of the dealkalized layer
-
Stage 3: Decrease of alteration rate and densification of the gel layer of \({\mathrm{35}}\,{\mathrm{nm}}\) \((\rho={\mathrm{1.8}}\,{\mathrm{g{\,}cm^{-3}}})\)
-
Stage 4: High reaction progress with a gel density of \(\rho={\mathrm{1.8}}\,{\mathrm{g{\,}cm^{-3}}}\).
In long-term experiments in silica-saturated alkaline solutions, very dense dealkalized layers have been observed \((\rho={\mathrm{1.9}}\,{\mathrm{g{\,}cm^{-3}}})\) [12.99]. Gel layers are rather stable reaction products [12.100], whose dissolution rate may be smaller than that of the glass. Depending on solubility limits in solution, sparingly soluble glass components precipitate as pure phase or a solid solution in the gel layer or at the interface of the gel layer and the solution. Constituents from solution may become incorporated in the precipitates. Provided thermodynamic data are available, the composition and type of precipitated phases can be estimated, using simple solid phases in geochemical reaction path models [12.101, 12.102, 12.103, 12.104, 12.105]. A paragenetic sequence of secondary phases during silicate glass corrosion is metal(hydr)oxides \(\rightarrow\) clays \(\rightarrow\) zeolites \(\rightarrow\) \(\mathrm{SiO_{2}}\). Clays are among the most abundant crystalline alteration products both of natural glasses as well as manmade glasses. Their composition depends both on glass and solution composition. Zeolites are formed mainly at alkaline pH (\(8{-}14\)). Under alkaline conditions calcium-silicate hydrate () phases are formed in Ca-rich glasses, and under acid to neutral conditions sulfates, such as \(\mathrm{BaSO_{4}}\), or molybdates may be observed.
The coordination number of various elements is quite different in the surface layer when compared to the pristine glass. \(\mathrm{Mn^{2+}}\) exists in the dry glass in tetrahedral coordination, whereas the coordination is octahedral in the surface layer [12.106]. For \(\mathrm{Sr^{2+}}\) an increase of coordination number from six to seven and for equatorial coordination of uranyl ions from four to five [12.107] is observed.
There was an intensive debate over more than \(\mathrm{30}\) years as to whether saturation effects in solution (affinity rate law) [12.49] or protective gel layers [12.14] are responsible for the slowdown of reaction rates with time under static leaching conditions. The temporal decrease of corrosion rates was explained [12.108, 12.88] by a hypothetical protective effect but other authors [12.109] have shown that in many instances this is not the case, and corrosion rates often decrease due to affinity effects in solution. However, particularly in Mg-rich solutions, a protective effect was confirmed for borosilicate glass [12.109, 12.110], soda lime glasses [12.111], natural tektites [12.112] or bioactive glasses [12.112]. Using x-ray reflectometry [12.113], three different steps are observed:
-
An interdiffusion step
-
The formation of a homogeneous gel
-
An electronic density gradient forms (porosity and/or composition) within the gels at long reaction times. This density gradient was suggested to explain the protective properties of the gel.
The effect of \(S/V\) and of water flow rate on surface layer properties and dissolution of various compositions of sodium borosilicate glasses has been simulated using Monte Carlo techniques, both for static and flow-through conditions. In static conditions, dissolution and condensation reactions are calculated to lead to the formation of a protective layer composed of polymerized Si sites. At high water flow rates, the dissolving glass exhibits a thin alteration layer and fast congruent dissolution; while at low flow rates, a protective layer is formed as in static conditions leading to near-saturation conditions in the vicinity of the pristine glass and lower reaction rates. A similar impact of gel layer densification on slowdown of reaction rates was observed in a combined experimental/Monte Carlo study.
Today we know that both protective surface layer formation and affinity effects contribute to a slowdown of glass dissolution rates. The above-mentioned densification of the gel layer has a protective effect, since in dense layers the mass transfer of silica dissolved from the glass is very slow. Hence, due to the mass transfer resistance between the bulk solution and the water in the pores of the gel layer at the gel–glass interface, the affinity term does not apply to saturation in bulk solution but only to saturation in the pores of the gel close to the pristine glass. Also, the affinity rate law would suggest an end to the glass–water reaction, once saturation in bulk solution is encountered. This end of reaction is never observed and the reaction continues with a very small rate, probably controlled by diffusion of water into the glass or driven by the slow crystallization of secondary alteration products.
2.4.2 Gel Layer Structure
Common physical properties of the gel layer are that it does not detach from the pristine glass and it is isovolumetric, i. e., the remaining material fills the same space as the pristine glass from which it is derived. These properties are observable after \({\mathrm{30}}\,{\mathrm{d}}\) for SON68, a simulant waste glass based on the R7T7 radioactive glass produced in France (\({\mathrm{45.85}}\,{\mathrm{wt\%}}\) \(\mathrm{SiO_{2}}\), \({\mathrm{14.14}}\,{\mathrm{wt\%}}\) \(\mathrm{B_{2}O_{3}}\), \({\mathrm{10.22}}\,{\mathrm{wt\%}}\) \(\mathrm{Na_{2}O}\), \({\mathrm{5.00}}\,{\mathrm{wt\%}}\) \(\mathrm{Al_{2}O_{3}}\), \({\mathrm{24.79}}\,{\mathrm{wt\%}}\) others) [12.114] and after \(\mathrm{1800}\) years for a Roman glass [12.115, 12.116, 12.117]. This gel layer, which has a density \(<{\mathrm{1.9}}\,{\mathrm{g{\,}cm^{-3}}}\) (compared to an SON68 density of \({\mathrm{2.7}}\,{\mathrm{g{\,}cm^{-3}}}\)) shows an increased density when solution concentrations of silica increase with reaction progress [12.98].
An altered SON68 glass surface generated from the alteration experiments leads to several different identifiable zones as can be seen in Fig. 12.3. It should be noted that this is the classical description of glass corrosion and as new techniques and experimental results become available more layers and the role of those layers in controlling glass corrosion continues to evolve.


Different alteration layers formed during glass alteration. Adapted from [12.118]
The layers, described in order from the bulk glass to the solution are:
- 1.
The first layer is the reaction or diffusion zone, which is adjacent to the pristine glass. This is the region where the main alteration of the pristine glass into the resulting product takes place. The layer has a constant thickness when ion exchange is equal to the rate of matrix dissolution [12.43, 12.44]. At a steady state, the two steps are coupled and the slower matrix dissolution step controls the rate of alteration [12.118]. Formation of the layer is parallel to the surface and dissolution pits and preferential leaching zones are not observed [12.115]. It is prudent to note that a study by Hellmann et al [12.119] using an approach combining high-resolution transmission electron microscopy () and energy-filtered transmission electron microscopy () with \(\mathrm{\AA{}}\) to nm spatial resolution on a labradorite feldspar altered at acid pH has shown that this reaction zone may not be present and that dissolution follows immediate interfacial reprecipitation.
- 2.
The second layer is the gel layer, which makes up the majority of the spatial extent of altered layer. The gel formation is the result of restructuring through hydrolysis and condensation. While the glass is being restructured some of the silicon is released from this gel and becomes a component of the leachate. The layer shows the strong presence of phyllosilicates [12.114, 12.120] and the crystallinity degree of secondary minerals found in this layer tends to increase with time [12.121].
- 3.
The last layer consists of precipitates found on the glass surface. These precipitates may be crystalline or amorphous in form. The components of the precipitates come from either leached glass elements or the surrounding environment. Sorption and precipitation processes may also occur within the high surface gel layer leading to complex surface layer chemistries. The gel may also contain crystallized phases precipitated from solution, which are mainly composed of phyllosilicates or rare earth element phosphates and amorphous products [12.122].
The behavior and partitioning of different elements as a function of alteration conditions is very diverse. Here we attempt to show the different behaviors of hydrogen, the alkali metals, the alkali earth metals, aluminum, the transition metals, and the rare earth metals during the course of glass alteration.
Due to the interdiffusion process the altered layer possesses an elevated concentration of hydrogen, which is usually found in several forms such as hydroxyls, water, or as the hydronium ion terminating silicate sites. The concentration of hydronium ions is especially elevated in the surface phyllosilicates. If the leachate contains NaCl, a decrease in the penetration of hydrogen is observed due to the diminution of the gradient of chemical potential at the solid–liquid interface, which hampers or stops the diffusion of Na from the glass to the solution [12.123].
The gel layer is lacking in Li, Cs, and Na compared to the pristine glass but these elements are found in higher concentrations in surface phyllosilicate phases as opposed to the gel layer [12.121, 12.124, 12.46]. The concentrations of these alkalis in the phyllosilicates remains lower than that in the pristine glass except for the case of Cs whose phyllosilicate concentration is higher than its original concentration in the pristine glass due to reversible incorporation into the phyllosilicates phase [12.121, 12.124]. Incorporation of alkalis into solid surface phases can be explained by their high energy of hydration where incorporation of larger alkalis is more likely due to the smaller absolute value of the free energy of hydration as one moves down the periodic table [12.125].
Boron is depleted in the gel layer and is not found in abundance in the phyllosilicate layer. In fact, boron does not show preferential retention in any of the altered layers and is soluble in solution, thus it is often used as a tracer for dissolution kinetics [12.13, 12.15, 12.33].
Alkali earth metals have been shown to behave differently in the gel in comparison with their alkali counterparts. To begin with, they are relatively less depleted in the gel layer [12.126]. This is most likely due to retention in the gel and phyllosilicate layers. One reason proposed for this is that \(\mathrm{Ca^{2+}}\) assures the electroneutrality of the \(\mathrm{ZrO_{6}^{2-}}\) [12.127]. Ba and Sr are strongly partitioned in the surface products while Ca shows enrichment at the interface between the gel layer and the phyllosilicates, corresponding to calcium and rare earth element phosphates [12.121, 12.128]. One more point of interest was investigated by Curti et al [12.129] who showed that the replacement of Mg by Ca leads to slower nucleation of clay minerals and thus slows the corrosion rate. This is due to the formation of phyllosilicates even below amorphous silica saturation and at a pH close to \(\mathrm{10}\). More recently, Debure et al [12.130] have further studied the kinetics and thermodynamics of Mg-phyllosilicate phases and their effect on glass corrosion.
Another important element that is present at \({\mathrm{5}}\,{\mathrm{wt\%}}\) in SON68 is aluminum. After glass alteration this element is held in the phyllosilicate layer and is enriched in the gel layer in comparison to the pristine glass. This high concentration shows that the aluminum is released from the glass network during glass dissolution but is then reprecipitated, as seen by its low concentration in the leachate. Despite being a trivalent compound, it has been shown that Al always conserves its \(4+\) coordination and replaces \(\mathrm{SiO_{4}}\) through the negatively charged species \(\mathrm{AlO_{4}^{-}}\) [12.131]. This means that other elements such as Na, Cs, and Ca that are associated to nonbridging oxygens may be necessary to carry out the charge balance [12.132]. Aluminum may also be found in zeolite phases which, when formed during glass alteration, may be detrimental to the stability of the glass because it may lead to a resumption in higher alteration rates as already explained in this work.
Other transition metals of general interest in waste glass corrosion are Mo, Zn, Ni, Fe, Zr, and Cr, which are also shown to have high concentrations in the surface precipitates. Mo shows a depth profile similar to those found with the alkalis, thus it is often used as a dissolution tracer. This is due to isolated \(\mathrm{MoO_{4}^{2-}}\) species being encapsulated within cavities formed in the glass network and thus easily released during glass dissolution [12.133]. Zn and Ni behavior shows enrichment in the crystalline surface layers while being less concentrated in the amorphous gel. This differs from Fe, Zr, and Cr, which are relatively enriched throughout the gel layer.
Zr is an important analogue for trivalent actinides [12.129] and is found to have a six-fold coordination, inserted in the silicate network through \(\mathrm{Si{-}{}O{-}{}Zr}\) bonds [12.134]. This structure is also conserved in the gel layer [12.135]. A recent study has stressed the importance of Zr in the dissolution behavior of borosilicate glasses [12.65]. For a glass composition containing greater than \({\mathrm{4}}\,{\mathrm{wt\%}}\) \(\mathrm{ZrO_{2}}\), the authors showed that substituting the soluble oxide, \(\mathrm{ZrO_{2}}\), for silica prevents reorganization of the gel layer and inhibits a pore closure mechanism, thus leading to a larger corrosion rate. The densification of the gel layer slows glass alteration kinetics by reducing the diffusion coefficient of water. This study also highlights the importance of gel structural formation in glass dissolution kinetics.
Rare earth elements () are important in nuclear waste glass studies because their chemical behavior in nonradioactive reference glasses is used to predict the behavior of the actinides in radioactive glasses such as R7T7, which corresponds to the active version of SON68 glass (Sect. 12.2.4, Gel Layer Structure). After glass alteration, REEs have been shown to be relatively concentrated in the region between the gel–phyllosilicate interface [12.121]. The mechanisms of retention of actinides and REE are mainly sorption and precipitation [12.136, 12.137]. REEs can either be located in an interlayer position and in exchangeable sites of the argillaceous minerals [12.138], in REE molybdate phases (powellite), or REE hydroxide phases during leaching in acidic brines [12.139].
Nevertheless, in the context of radioactive waste geological disposal, an important contribution of iron from the corrosion of overpacks used to encapsulate the primary waste package (glass and the stainless steel container) may indirectly affect the glass dissolution and alteration products formation. Hence, iron corrosion products (magnetite: \(\mathrm{Fe_{3}O_{4}}\), siderite: \(\mathrm{FeCO_{3}}\) and iron sulfide: FeS, \(\mathrm{FeS_{2}}\)), and iron silicates could affect the glass dissolution kinetics and long-term behavior via silica consumption [12.117, 12.140, 12.141, 12.142].
3 Glass Vapor Hydration
Glass vapor phase hydration can be defined as the process of altering the chemical and/or physical properties of surface by means of exposure to water vapor in contrast to exposure to liquid water, which leads to elemental dissolution and leaching. Vapor hydration proceeds by reaction with the glass and a tiny water layer condensed on the surface, whose thickness mainly depends on the relative humidity and temperature. The \(S/V\) ratio is then very high (\(\approx{\mathrm{10^{8}}}\,{\mathrm{m^{-1}}}\) in comparison to the ratios obtained in leaching experiments of \(10{-}E5\,{\mathrm{m^{-1}}}\)), which could lead to high pH values of the water film unfavorable for silica-rich glass chemical durability [12.143]. Thereby, chemical saturation and high pH values in the limited water film are quickly achieved, leading to alteration product formation including silica- and/or alumina-consuming phases, e. g., zeolites and calcium silicate hydrates [12.143, 12.144, 12.145, 12.146]. Bates et al [12.147, 12.148] indicated that the glass hydration kinetics follow an Arrhenius relationship, thus suggesting that vapor hydration could be used to accelerate glass alteration and product formation [12.147, 12.148].
Hydration of natural glasses have long been studied as a method of dating of natural samples of geological and archeological importance, in particular obsidians [12.149, 12.150]. The dating technique relies on extrapolation of the results of hydration experiments conducted at elevated temperatures to environmental low temperature using an Arrhenius relationship [12.151, 12.152]. These studies, used to determine the age of the obsidian samples, coupled the hydration layer thickness with the reaction time using dating techniques usual for archaeology. These studies obtained hydration kinetics and thus led to the identification of temperature and relative humidity as the main parameters controlling the glass hydration. Though most of the archeological studies have been done with obsidian, other glass compositions have been studied [12.153, 12.154].
The interest in studying glass vapor hydration has been growing since the early 1980s due to the research and development needs in relation to geological disposal of glass as a matrix of nuclear waste confinement [12.147, 12.148, 12.155, 12.156, 12.157, 12.158, 12.159, 12.160, 12.161, 12.162, 12.163]. These nuclear waste glasses arise from the reprocessing of spent nuclear fuels and the vitrification of fission products and minor actinides, e. g., high-level waste () [12.164, 12.165, 12.33, 12.47]. The first hydration studies with nuclear waste glasses included inactive simulant and radioactive glasses [12.147]. The first studies with the reference simulated nuclear waste glass SON68 were conducted in mid-1990s but the need for an improved understanding of the foreseen high-level waste repository highlighted the importance of studying the glass hydration in unsaturated conditions. In fact, the multibarriers concept developed by many countries, such as France, includes the primary waste package with the glass poured in a stainless steel container, a thick carbon-steel overpack separated from the host rock (Callovo-Oxfordian argillite) by a carbon steel liner and a cementitious material [12.166, 12.167]. Nevertheless, the metallic liner is expected to produce a large amount of hydrogen via anoxic corrosion in the presence of humidity, which contributes to keeping unsaturated conditions for thousands of years [12.141].
3.1 Hydration of Natural Glasses
Interest in hydration of glasses was first driven by Friedman and Smith [12.149] who developed a new dating method from artifacts made from obsidian. The method relies on the measurement of the hydration layer (birefringent layer) in connection with the estimated temperature to estimate the time at which the glass first contacted the water. Friedman and Smith [12.149] described the rate of layer formation as
where \(l\) is the thickness of the hydration layer, \(t\) is the hydration time, and \(k\) is the proportionality constant, which describes the temperature dependence of the process. The rate dependence was then shown to be a function of square root of time, indicating a diffusion-controlled hydration process. The inward diffusion of molecular water is the rate-determining step for the obsidian hydration [12.168, 12.169, 12.170].
The dating method was then developed by many authors including [12.171, 12.172, 12.173, 12.174, 12.175, 12.176]. At the beginning of this new methodology scientists thought that the glass hydration rate was independent of the relative humidity (). Thus, experimental work with obsidian at different RH and temperatures up to \({\mathrm{245}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) showed no dependency between the hydration rate and the RH [12.151]. However, additional experimental work conducted at lower relative humidities and temperature confirmed that the RH has a strong effect on glass hydration rate [12.177]. The authors conducted hydration experiments with obsidian at different RH (\(\mathrm{60}\), \(\mathrm{90}\), \(\mathrm{95}\) and \({\mathrm{100}}\%\)) and temperatures (\(\mathrm{150}\), \(\mathrm{160}\) and \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)). The authors found that the hydration rate increased by \({\mathrm{25}}\%\) between \(\mathrm{60}\) and \({\mathrm{100}}\%\) RH, with the majority of the rate increase between \(\mathrm{90}\) and \({\mathrm{100}}\%\). Indeed, the water sorption isotherm for obsidian showed a doubling of the amount of sorbed water between \(\mathrm{90}\) and \({\mathrm{100}}\%\) RH. They attributed the effect of RH to the chemical potential difference between water sorbed on the obsidian surface and intrinsic water in the obsidian. Finally, Mazer et al [12.177] insisted on the fact that a successful use of obsidian-hydration dating requires a careful estimation of RH and temperature at the given site. Furthermore, the importance of temperature effects on obsidian hydration dating was stressed by Stevenson et al [12.178] and Lynch and Stevenson [12.179]. The authors recommended temperature controls, more subsurface glass samples to date, and when possible confirmation dates by techniques such as radiocarbon dating. More recently a review by Liritzis and Laskaris [12.180] on \(\mathrm{50}\) years of obsidian hydration dating in archeology indicated that the empirical dating method developed by Friedman and Smith [12.149] still produces satisfactory results but fails to produce a reliable chronometer. The use of new techniques such as secondary ion mass spectrometry () helped to accurately measure the hydration profile. Modeling via equations describing the diffusion profile helped to obtain accurate ages with promising results. Stevenson and Novak [12.181] proposed to use structural water measurements by infrared spectroscopy to derive Arrhenius constants and thus archeological hydration rates at ambient temperature.
3.2 Hydration of Commercial Glasses
A detailed investigation on hydration of commercial glasses was published by Moriya and Nogami [12.182, 12.183] with a particular emphasis on water speciation in the hydrated glass and the role of oxides on hydration rates.
Bates et al [12.147] applied first the vapor hydration methodology to nuclear waste glasses as a part of a safety analysis of a disposal site. These glasses originate from the vitrification of high-level wastes (e. g., fission products and minor actinides) arising from the reprocessing of nuclear spent fuel [12.184, 12.185, 12.186, 12.38]. A consensus within the scientific community is agreed on concerning high-level nuclear waste management and consists of disposal of packages in deep geological formations. For safety case purposes, intended to justify that the disposal system is acceptably safe, safety scenarios are considered including normal and altered scenarios. These scenarios describe the phenomenological evolution of the site including thermal, hydraulic, radiological, mechanical and chemical evolution of the site. In all cases it is expected that the nuclear glass will be in contact with water vapor for a more or less long term. As a consequence, glass hydration constitutes an important issue to study before complete saturation of the disposal site. The site undersaturation conditions are expected to occur and last for several thousands of years due to hydrogen generation upon steel canisters and overpack corrosion.
The essential early work on nuclear waste glasses hydration was done during the 1980s and early 1990s by Bates and collaborators. Bates et al [12.147] proposed the methodology to perform the hydration tests and presented the first results of glass hydration. Then the authors examined the relevance of vapor hydration of nuclear glasses to geological disposal [12.155] and the behavior of stressed glasses in humid environments [12.156]. Abrajano et al [12.157] investigated the comparison between rates of hydration in liquid and vapor environments as well as the mechanisms of vapor hydration of natural and nuclear waste glasses. The sorption of water onto obsidian and a nuclear waste glass and the effect of relative humidity were studied by Ebert et al [12.159]. In another study Ebert et al [12.160] examined the role of the \(S/V\) ratio on the reaction. Wronkiewicz et al [12.162, 12.163] examined the effect of radiation exposure on glass alteration in a steam environment. Gong et al [12.187] published the first hydration work with the French reference nuclear glass SON68 with particular interest in the hydration layer structure and composition.
Studies of water diffusion and speciation in hydrated glasses were conducted Tomozawa et al during the 1980s and 1990s [12.188, 12.189, 12.190].
Later, during the 2010s hydration works on nuclear glasses gained interest notably for the French radioactive waste management program by Abdelouas et al In this case the experimental conditions were well controlled to ensure undersaturation conditions during the run [12.145, 12.146].
3.3 Experimental Procedures and Parameters
3.3.1 Procedures
Nearly all the hydration experiments were conducted at saturated water vapor conditions. In general, two experimental setups have been used to study glass vapor hydration. The first one consists of placing the glass specimens (rods, chips, discs) inside an environmental chamber that allows control of the temperature and relative humidity [12.161]. The second setup consists of hanging the glass specimen on the top of the water volume in a sealed reactor heated in an oven. Positioning the glass inside the reactor is an important issue to solve in order to prevent water condensation on the glass surface. Water condensation can distort the results and conclusions on the role of relative humidity. Hence, two methods were developed to position the glass in the reactor and they are:
- 1.
The glass suspension by platinum or Teflon wire. The major disadvantage of this technique is the risk of it falling either by tilting its attachment or wire breakage during the run. The Teflon wires have the disadvantage of being fragile with ionizing radiation in comparison with platinum wires. Thus, they cannot be used in radioactive environments (experiments with radioactive glass or experiments with external radiation source such as gamma sources) [12.147, 12.148, 12.158, 12.159, 12.161, 12.162, 12.163, 12.191, 12.192]. Platinum wires are much more resistant to ionizing radiation but have a disadvantage of favoring the nucleation of secondary phases [12.193, 12.194, 12.195, 12.196, 12.197, 12.198]. The feedback from the experimental works recommended the horizontal suspension of the glass specimens to minimize water condensation on the glass surface upon cooling [12.199].
- 2.
The vertical suspension of the glass specimen on a Teflon bracket [12.145, 12.146, 12.200, 12.201, 12.202, 12.203, 12.204]. This method allows suspension of the glass on the top of a solution with two major advantages:
-
Maximize the contact between the glass and the atmosphere
-
Avoid the falling down of the glass into the solution.
-
The protocol established by Argonne National Laboratory (ANL) [12.162] for the vapor phase hydration consists of suspending of the glass specimen horizontally or vertically by a Teflon or platinum wire above the solution (Fig. 12.4). The hydration is conducted inside a \({\mathrm{22}}\,{\mathrm{mL}}\) stainless steel autoclave with a Teflon liner. The temperature is then controlled by placing the autoclave in an oven. The protocol established by SUBATECH laboratory [12.146] uses a Teflon liner placed in a stainless autoclave (Fig. 12.5). The glass specimen is deposited on a holder, to the vertical, above a volume of solution. The autoclave is itself placed in a relatively thick aluminum overpack to homogenize the temperature during glass hydration.


Experimental apparatus for glass vapor hydration developed by Pacific Northwest National Laboratory (PNNL), USA. Adapted from [12.162]


Experimental apparatus for glass vapor hydration developed by SUBATECH Laboratory, France. Adapted from [12.200]
3.3.2 Experimental Parameters
3.3.2.1 The Relative Humidity
During the vapor hydration experiments the water layer absorbed on the glass surface is supposed to be at equilibrium with the atmosphere and does not condense and drip. This condition is a priori fulfilled in the case of water below saturation conditions (\(<{\mathrm{100}}\%\) RH). Under saturation conditions (\({\mathrm{100}}\%\) RH) water condensation may occur and lead to glass leaching instead of hydration and thus modify the glass alteration kinetics and secondary phase formation. The risk of water condensation can be reduced by using a small volume of water, just above the volume necessary to reach water saturation in the reactor at the given temperature. The occurrence or absence of condensation or dripping may also be established by measuring the pH of the solution used for the steam supply water from the atmosphere of the reactor; runoff generally giving rise to very high pH (\(\text{pH}> {\mathrm{11}}\)). This measure allows identification of experiments with abnormalities. However, experiments in saturated atmospheres remain unfavorable for long-term studies.
Two methodologies were developed to study vapor hydration:
- 1.
The alteration in dry steam, using a small volume of water insufficient to saturate the reactor atmosphere in water [12.157, 12.161, 12.205]. The main difficulties associated with this method are the exact determination of free reactor volume accessible to steam, perfectly guaranteeing the seal can work with a constant relative humidity, and minimizing the risk of change of the relative humidity over alteration.
- 2.
The use of saline solutions accordingly to control the relative humidity [12.206]. The use of NaCl solutions allows the control of water activity and thus control of the relative humidity [12.145, 12.146, 12.200, 12.201, 12.207].
3.3.2.2 The Experimental Procedure for Hydration Experiments
Prior to hydration experiments the glass specimens are polished in many ways using:
- 1.
Polishing sandpaper Grit 600 according to Pacific Northwest National Laboratory protocol [12.131, 12.147, 12.158, 12.160, 12.162, 12.191, 12.192, 12.193, 12.194, 12.195, 12.196, 12.199, 12.205, 12.208, 12.209].
- 2.
Polishing with sandpaper Grit 600 and a finish at \(1{-}6\,{\mathrm{\upmu{}m}}\) [12.210].
- 3.
Polishing sandpaper of \({\mathrm{3}}\,{\mathrm{\upmu{}m}}\) according to SUBATECH laboratory protocol [12.146, 12.200, 12.201, 12.202, 12.203, 12.207].
In the case of hydration experiments in reactors, a predetermined volume of solution, from \(0.025{-}8\,{\mathrm{mL}}\) depending on the reactor vessel volume and setup protocol, is added to supply the reactor with water vapor and the glass specimen is positioned above the water. The reaction system (autoclave—PNNL protocol—or Teflon reactor, autoclave and overpack—SUBATECH protocol) is then placed in an oven at the temperature selected for the desired time. After retrieval from the oven the reactor may be either cooled in water or ice [12.147, 12.148, 12.158, 12.160, 12.191, 12.194, 12.196, 12.209, 12.211] (PNNL protocol) in order to promote the condensation of water on the reactor walls and minimize dripping on the glass surface, or is left to cool down to room temperature [12.146, 12.200, 12.201, 12.202, 12.203, 12.204] (SUBATECH protocol), to avoid a too rapid condensation. For studies in environmental chambers, the chamber directly imposes the hydration conditions selected.
3.3.2.3 Water Sorption on Silicate Glasses
A major study on water sorption on obsidian and the SRL \(\mathrm{165}\) borosilicate nuclear waste glass (\({\mathrm{51.6}}\,{\mathrm{wt\%}}\) \(\mathrm{SiO_{2}}\), \({\mathrm{11.7}}\,{\mathrm{wt\%}}\) \(\mathrm{Fe_{2}O_{3}}\), \({\mathrm{10.8}}\,{\mathrm{wt\%}}\) \(\mathrm{Na_{2}O}\), \({\mathrm{6.8}}\,{\mathrm{wt\%}}\) \(\mathrm{B_{2}O_{3}}\), \({\mathrm{4.8}}\,{\mathrm{wt\%}}\) \(\mathrm{Li_{2}O}\), \({\mathrm{4.3}}\,{\mathrm{wt\%}}\) \(\mathrm{Al_{2}O_{3}}\), \({\mathrm{2.8}}\,{\mathrm{wt\%}}\) \(\mathrm{Mn_{2}O_{3}}\), \({\mathrm{1.6}}\,{\mathrm{wt\%}}\) CaO, \({\mathrm{5.6}}\,{\mathrm{wt\%}}\) others) was conducted by Ebert et al [12.160]. The authors collected sorption isotherms for water at \({\mathrm{23}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and found results similar to those of water on simple silicate and other oxides (Fig. 12.6). The authors used the t-plot method to convert the amount of adsorbed water into number of monolayers according to the Hagymassy et al method. They described the sorption as occurring primarily at silanol sites and the sorption to other sites remaining very low. Then, the water forms clusters around the silanol sites, which could cover about \({\mathrm{70}}\%\) of the glass surface starting at \({\mathrm{3}}\%\) RH. Next, with increasing relative humidity, the water clusters start to coalesce until they generate a water film above \({\mathrm{95}}\%\) RH (Fig. 12.6). Ebert et al [12.160] examined water sorption on obsidian and identified three steps as follows:
- 1.
Water sorption on the surface. At this stage the water sorption is much greater than water diffusion rate so the water layer could be considered to remain at equilibrium with the vapor phase throughout the experimental time.
- 2.
Water condensation in the porous layer.
- 3.
Water diffusion in the glass bulk to form the hydrated layer (Fig. 12.7).
The water ingress rate in the hydration phase 3 is similar to data extrapolated from high-temperature experiments with obsidian. The same trend of water sorption on obsidian was found with experiments conducted at \({\mathrm{58}}\%\) RH. The water sorption and diffusion in the glass matrix are controlled by the difference in chemical potential between water sorbed on the glass and the water in the gas phase, which corresponds to the water available for the reaction.


Water sorption isotherms on the SRL \(\mathrm{165}\) borosilicate nuclear waste glass at \({\mathrm{23}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) (data from [12.160]). Data were obtained with experiments using increasing or decreasing humidity techniques


Water sorption expressed as mass gain (\(\mathrm{\upmu{}g}\)) of obsidian hydrated at \({\mathrm{23}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{84}}\%\) RH (data from [12.160])
3.3.2.4 Methodology for Measurement of Hydration Layer Thickness
The absence of leachate makes the determination of hydration rate limited to glass analysis. Hence, the hydration progress could be studied by layer thickness measurements [12.146, 12.147, 12.148, 12.149, 12.151, 12.157, 12.205, 12.207, 12.210, 12.211, 12.212, 12.213, 12.214, 12.215], or by pristine glass thickness measurements [12.192, 12.193, 12.194, 12.195, 12.196, 12.197, 12.199, 12.209].
The first method that was developed to determine the thickness of the hydration layer consists of the observation of cross sections under the light microscope and is based on the difference in refractive index between the pristine glass and hydrated glass (Fig. 12.8a) [12.194]. Observations under the scanning electron microscope () allow the measurement of layer thickness at the submicrometer level (Fig. 12.8b). The hydration profile can also be observed and analyzed by transmission electron microscopy () (Fig. 12.8c) or by time-of-flight secondary ions mass spectrometry (-) (Fig. 12.8d). Figure 12.8d shows the leaching of boron from the glass surface, which is parallel to enrichment in \(\mathrm{{}^{18}O}\) from the spiked water entering the glass. SEM, TEM and SIMS studies allow chemical analysis and structural analysis (TEM) of the hydration layer. Abdelouas et al [12.201] used nuclear reaction analysis () to study hydrogen profiles in a hydrated borosilicate glass. They determined the hydration layer thickness based on water penetration in the glass structure.
Another methodology based on Fourier transform infrared spectroscopy (FTIR) has been developed earlier by Davis and Tomozawa [12.216] and later for nuclear borosilicate glasses by Ferrand et al [12.33] to study water speciation and hydration thickness in the glass. An example of FTIR spectra obtained for the SON68 borosilicate hydrated in vapor at \({\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) is given in Fig. 12.9. The band assignments were based on the literature [12.216, 12.217, 12.218, 12.33]. Efimov et al [12.217] and Navarra et al [12.218] studied the hydration of silicate glasses by FTIR. The authors assigned bands to different water species (free-bound molecular water, Silanol-SiOH):
-
\(3595{-}3605\,{\mathrm{cm^{-1}}}\) attributed to OH stretching mode in SiOH.
-
\(515{-}3518\,{\mathrm{cm^{-1}}}\) attributed to OH stretching mode in the bound water silanol groups.
-
\(3400{-}3415\,{\mathrm{cm^{-1}}}\) attributed to symmetrical stretching OH mode in the free water molecule.
-
\(170{-}3185\,{\mathrm{cm^{-1}}}\) attributed to OH stretching mode in bound water silanol groups.
-
\({\mathrm{700}}\,{\mathrm{cm^{-1}}}\) attributed to silica matrix.
The hydration thickness could then be inferred from SEM or TEM observations. Hence, Neeway [12.200] found a correspondence between the thickness of hydration layer measured by SEM or TEM and the SiOH absorbance at different experimental conditions obtained by FTIR, which allowed him to obtain hydration kinetics as a function of time of exposure. Neeway et al [12.146] assigned an absorbance unit for SiOH of \(0.09{-}1\,{\mathrm{\upmu{}m}}\) of the hydrated layer for the SON68 glass hydrated at various temperatures and RH. Abdelouas et al [12.201] studied the vapor hydration of the simplified nuclear waste borosilicate International Simple Glass () glass hydration at \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and various RH. Using NRA data, they found a correspondence average of \({\mathrm{0.11}}\pm{\mathrm{0.02}}\) absorbance unit for SiOH per micrometer of alteration layer. This method is particularly used to study the glass hydration kinetics.


(a) Hydrated HLP-09 low-activity waste glass at \({\mathrm{300}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for \({\mathrm{3}}\,{\mathrm{d}}\), adapted from [12.194], ©PNNL, US Department of Energy, showing a white alteration layer surrounding the pristine glass. (b) SEM picture of SON68 glass hydrated at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{92}}\%\) RH. (c) TEM thin section of SON68 glass hydrated at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{92}}\%\) RH. (d) ToF-SIMS profile of boron and \(\mathrm{{}^{18}O}\)/\(\mathrm{{}^{16}O}\) isotopic ratio for SON68 glass hydrated at \({\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{95}}\%\) RH for \({\mathrm{600}}\,{\mathrm{d}}\). The depth profile is about \({\mathrm{5}}\,{\mathrm{\upmu{}m}}\)


Experimental and deconvoluted FTIR spectra of SON68 glass hydrated at \({\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{95}}\%\) RH
3.3.2.5 Effect of Time on Glass Hydration
Most of the hydration experiments were conducted at high temperature and in the short term. In this case the hydration follows a linear law. An example is given in Fig. 12.10 where we found that water ingress rate in the SON68 and ISG glasses follows a linear trend. In the case of the ISG glass the fitting of the NRA hydration profiles with Fick's second law allowed us to infer the hydrogen diffusion coefficients of \(2{-}7E-21\,{\mathrm{m^{2}\mskip 3.0mus^{-1}}}\).
However, experiments at low temperatures and conducted in the long term (more than a year) showed inflection of the hydration curve for borosilicate nuclear waste glasses [12.202, 12.207] (Fig. 12.11). Aït Chaou et al [12.207] calculated, based on FTIR measurement, the short-term (the first \({\mathrm{35}}\,{\mathrm{d}}\)) and the long-term hydration rates (corresponding to the period starting from the inflection of the glass hydration until the end of the experiment). They found that the long-term rates are one order of magnitude lower than the initial rates. The drop of hydration with time is still not well understood.


The growth of the SiOH peak at \({\mathrm{3595}}\,{\mathrm{cm^{-1}}}\) for the SON68 and ISG glasses hydrated at different temperatures and relative humidities


3.3.2.6 Effect of Temperature on Glass Hydration
The temperature plays an important role in glass hydration (Fig. 12.12). The study of the role of temperature on the glass hydration allowed the proposition of an Arrhenius relationship to permit extrapolating data from high temperatures to low temperatures. Many authors proposed the Arrhenius model for different glasses including natural glasses (obsidian) [12.151], for the SRL type of glass [12.147, 12.148], and for the French SON68 glass [12.146, 12.200, 12.202]. Typical activation energies \((E_{\mathrm{a}})\) obtained by Neeway et al [12.146] for temperatures between \(\mathrm{125}\) and \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{92}}\%\) RH are \(E_{\mathrm{a}}=43{-}47\,{\mathrm{kJ\mskip 3.0mumol^{-1}}}\), and \(E_{\mathrm{a}}={\mathrm{22.7}}\,{\mathrm{kJ{\,}mol^{-1}}}\) for temperatures of \(35{-}125\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{95}}\%\) RH [12.202].


The growth of the SiOH peak at \({\mathrm{3595}}\,{\mathrm{cm^{-1}}}\) for the SON68 glass hydrated at different temperatures. Adapted from [12.202]
3.3.2.7 Effect of the Relative Humidity on the Glass Hydration
As clearly shown by Ebert et al [12.160], increasing the relative humidity results in an increase in the number of layers of water in contact with glass. This leads to higher rate of water condensation and ingress in the glass matrix. An example can be seen in Fig. 12.13 for the SON68 glass hydrated at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). This dependence of hydration rate of the relative humidity has been studied by several authors for natural and commercial glasses [12.145, 12.146, 12.161, 12.182, 12.183, 12.200, 12.201, 12.202, 12.205, 12.207, 12.219].
Moriya and Nogami [12.182, 12.183] pioneered a study on commercial glasses hydration in a steam atmosphere. They particularly investigated the role of alkalis (Na), alkaline earths (Ca, Mg) and alumina on glass hydration at \(100{-}180\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for \(3{-}72\,{\mathrm{h}}\). They showed that the hydration rate was larger in the glass containing more Na in the \(\mathrm{Na_{2}O}\)-\(\mathrm{SiO_{2}}\) system and decreased by the substitution of \(\mathrm{Na_{2}O}\) by CaO, MgO or by the addition of \(\mathrm{Al_{2}O_{3}}\). Partial substitution of \(\mathrm{Na_{2}O}\) in the \(\mathrm{30Na_{2}O\cdot 70SiO_{2}}\) system by MgO (\(\mathrm{20Na_{2}O\cdot 10MgO\cdot 70SiO_{2}}\)) led to the decrease of the hydration rate by a factor of ten. The hydration rate doubled for the \(\mathrm{25Na_{2}O\cdot 5MgO\cdot 70SiO_{2}}\) system in comparison to the \(\mathrm{25Na_{2}O\cdot 5CaO\cdot 70SiO_{2}}\) system at \({\mathrm{140}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{18}}\,{\mathrm{h}}\). The hydration rate of the \(\mathrm{20Na_{2}O\cdot 10MgO\cdot 70SiO_{2}}\) glass increased by a factor of seven at \({\mathrm{160}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) when the water vapor pressure doubled.
The effect of relative humidity on obsidian hydration was thoroughly studied by Friedman et al [12.219]. The relative humidity varied from \(28{-}100\%\) by using a liquid and solid mixture of different chloride salts (KCl or \(\mathrm{MgCl_{2}}\)) at \(\mathrm{40}\) and \({\mathrm{70}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). The authors found that the hydration rate at \({\mathrm{75}}\%\) RH is generally double that at \({\mathrm{28}}\%\) both at \(\mathrm{40}\) and \({\mathrm{70}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\). In fact, in most environments, the RH of the soil atmosphere approaches \({\mathrm{100}}\%\) below \({\mathrm{10}}\,{\mathrm{cm}}\), and \({\mathrm{25}}\,{\mathrm{cm}}\) in extremely dry desert areas. They suggested that obsidian hydration ages of surface samples usually approximate those derived from buried samples.
Abdelouas et al [12.201] conducted hydration experiments with the ISG borosilicate glass at \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and relative humidities of \(60{-}98\%\). They found that the hydration occurred even at \({\mathrm{60}}\%\) RH and the hydration rate is typically proportional to the number of water monolayers, which is a function of the relative humidity (Fig. 12.5) [12.160].


The growth of the SiOH peak at \({\mathrm{3595}}\,{\mathrm{cm^{-1}}}\) for the SON68 glass hydrated at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and different relative humidity. Adapted from [12.202]
3.3.2.8 Effect of pH on the Glass Hydration
A study of the role of pH on glass hydration has been conducted by Aït Chaou et al [12.145]. The authors investigated the SON68 glass hydration at \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{98}}\%\) relative humidity and different gas atmospheres ensuring a pH control. Thus, gas–water equilibrium calculations with the geochemical code PHREEQC [12.221] at \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) give pH estimations as follow: pH \(\mathrm{4.5}\) under \(\mathrm{CO_{2}}\), pH \(\mathrm{4.9}\) under \(\mathrm{H_{2}S}\), pH \(\mathrm{5.7}\) under Ar, and \(\mathrm{9.1}\) under \(\mathrm{NH_{3}}\) [12.145, 12.220]. The results shown in Fig. 12.14 clearly highlight the role of pH in glass hydration. Hence, under acidic atmospheres the dominant process is the interdiffusion of \(\mathrm{H_{3}O^{+}}\)/alkalis, which leads to the formation of a hydrated gel. Under basic conditions network glass dissolution, due to high \(\mathrm{OH^{-}}\) activity, strongly enhances the glass alteration. The SEM observations of the altered glass confirm the slight alteration of the glass under a \(\mathrm{CO_{2}}\) atmosphere with the formation of a clay-like layer (Fig. 12.15a,ba). The strong alteration of the SON68 glass under an ammonia atmosphere allowed the precipitation of analcime, which often precipitates under high reaction progress (the quantity of dissolved glass per unit of volume) (Fig. 12.15a,bb).


The growth of the SiOH peak at \({\mathrm{3595}}\,{\mathrm{cm^{-1}}}\) for the SON68 glass hydrated at \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{98}}\%\) relative humidity under acidic (\(\mathrm{CO_{2}}\) equilibrated water vapor) and alkaline (\(\mathrm{NH_{3}}\) equilibrated water vapor) conditions. Adapted from [12.220]


SEM photos of SON68 glass hydrated under a (a) \(\mathrm{CO_{2}}\) atmosphere (initial pH \(\mathrm{4.9}\)) and (b) \(\mathrm{NH_{3}}\) atmosphere (initial pH \(\mathrm{9.1}\))
3.3.2.9 Effect of Ionizing Radiation on the Glass Vapor Hydration
Ionizing radiation is known to damage solid waste forms including crystalline ceramics and glass matrices [12.222, 12.223, 12.224, 12.225, 12.226]. In addition, in contact with water vapor, alpha and gamma radiation may produce nitric acid via nitrogen radiolysis, organic acids and radicals (\(\mathrm{H_{2}O^{\bullet}}\) and \(\mathrm{O_{2}^{-}}\)). These may decrease the pH and consequently increase the glass hydration via the increase of the water diffusion coefficient in the glass matrix [12.33]. To infer the role of ionizing radiation on the glass hydration rates experiments were conducted with actinides (for alpha effect) or \(\mathrm{{}^{99}Tc}\)-doped (for beta effect) and external gamma irradiation [12.162, 12.163]. Other works focused on secondary phase formation and the behavior of vapor hydrated glasses in liquid water [12.155, 12.156, 12.158, 12.160, 12.227] or the behavior of radionuclides during vapor hydration tests [12.228, 12.229].
Wronkiewicz et al [12.162, 12.163] studied the SRL defense waste glasses (SRL \(\mathrm{131}\), SRL \(\mathrm{165}\) and SRL \(\mathrm{200}\)) doped with \({\mathrm{851}}\,{\mathrm{kBq{\,}g^{-1}}}\) of \(\mathrm{{}^{99}Tc}\) and actinides (\(\mathrm{{}^{237}Np}\), \(\mathrm{{}^{238}U}\), \(\mathrm{{}^{239}Pu}\), \(\mathrm{{}^{241}Am}\)) and subjected them to gamma external irradiation with a dose rate of \({\mathrm{35}}\,{\mathrm{Gy{\,}h^{-1}}}\). These glasses have, in addition to minor elements and nuclides, average composition in oxide \(\mathrm{wt\%}\) of \(\mathrm{SiO_{2}}\): \(43.8{-}52.9\%\), \(\mathrm{Na_{2}O}\): \(10.8{-}17.5\%\), \(\mathrm{Fe_{2}O_{3}}\): \(4.8{-}12.7\%\), \(\mathrm{B_{2}O_{3}}\): \(6.8{-}9.7\%\) and \(\mathrm{Li_{2}O}\): \(3{-}4.7\%\). After \({\mathrm{56}}\,{\mathrm{d}}\) of hydration at \({\mathrm{150}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) the authors showed an increase of glass hydration of the doped glasses with a factor of \(10{-}15\) times for SRL \(\mathrm{200}\) and four times for the SRL \(\mathrm{131}\), in comparison with the nondoped glasses. They attributed the increase to water vapor acidification.
McKeown et al [12.228] and Buechele et al [12.215] studied the hydration of \(\mathrm{{}^{99}Tc}\)-doped glasses at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for up to \({\mathrm{30}}\,{\mathrm{d}}\). They showed that \(\mathrm{{}^{99}Tc}\), initially present in the glass as pertechnetate \(\mathrm{TcO_{4}^{-}}\), was reduced in contact with water vapor, thus increasing its stability in the alteration layer.
Leaching experiments conducted with two types of defense nuclear waste glass (SRL \(\mathrm{211}\) and \(\mathrm{131}\)) and a prototype commercial glass (PNL \(\mathrm{76}\)-\(\mathrm{68}\)) were conducted at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) in deionized water for \({\mathrm{28}}\,{\mathrm{d}}\) by Bates et al [12.155, 12.156]. These glasses were first hydrated at high temperature \((> {\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}})\) and \({\mathrm{100}}\%\) RH for a few days. It was found that the leaching rates of actinides from the hydrated glass subjected to leaching in water are higher than those from pristine glass directly leached in water.
3.3.2.10 Hydration Layer and Secondary Phases
The low amount of water content in vapor hydration tests leads to a high surface area of glass to volume of water ratio (). As a consequence, strong saturation conditions with respect secondary phases are reached in the water condensed on the glass surface.
Transmission and scanning electron microscopy (SEM, TEM) studies allowed identification of a complex gel-like layer underneath well-crystallized secondary phases, in particular at high temperature \((> {\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}})\) [12.146, 12.147, 12.148, 12.155, 12.156, 12.161, 12.162, 12.163, 12.200, 12.205, 12.207, 12.211]. At lower temperatures (\(35{-}125\,{\mathrm{{}^{\circ}\mathrm{C}}}\)) the main alteration product, in addition to a hydrated gel-like layer, is a clay-type mineral. Hence, in Fig. 12.16a-fa we can observe that at \({\mathrm{35}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and after \({\mathrm{654}}\,{\mathrm{d}}\) of hydration the only detected product is a clay-type phase. The same product has been identified at \(\mathrm{50}\) and \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) (not shown in the figure) and at \({\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) but only after \({\mathrm{154}}\,{\mathrm{d}}\) (Fig. 12.16a-fb). The same product formed at higher temperatures (Fig. 12.16a-fc) in addition to analcime and Ca-silicate hydrates (tobermorite, gyrolite) (Fig. 12.16a-fd,e,f). It is clear that the temperature plays an important role in the formation of silica-consuming minerals (analcime, Ca-silicate hydrates), thus leading to the increase of glass alteration. Rare earth elements released from the glass precipitate with phosphate (apatite) and molybdate (powellite). A summary of secondary phases identified during vapor hydration of nuclear glasses is given in Table 12.1.


SEM photos of SON68 glass hydrated at (a) \({\mathrm{35}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{95}}\%\) RH for \({\mathrm{654}}\,{\mathrm{d}}\), (b) \({\mathrm{125}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{92}}\%\) RH for \({\mathrm{154}}\,{\mathrm{d}}\), (c,d) \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{95}}\%\) RH for \({\mathrm{99}}\,{\mathrm{d}}\), (e) \({\mathrm{175}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{98}}\%\) RH for \({\mathrm{99}}\,{\mathrm{d}}\), and (f) \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\), \({\mathrm{92}}\%\) RH for \({\mathrm{57}}\,{\mathrm{d}}\)
Extensive analytical electron microscopy studies were conducted by Gong et al [12.211] with the SON68 glass hydrated in saturated water vapor at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) for up to \({\mathrm{1021}}\,{\mathrm{d}}\). The authors found that the surface layer thickness increased with increasing reaction time. A complex alteration layer formed with six distinctive zones. The alteration layer is mainly composed of an A-domain containing well-crystallized fibrous smectite aggregates and a B-domain consisting of poorly crystallized regions containing smectite and a \(\mathrm{ZrO_{2}}\)-rich amorphous silica matrix. Chemical segregation occurs in A- and B-domains with transition metal elements such as Zn, Cr, Ni, Mn, and Fe concentrated in A-domain, and rare earth elements, Mo and Zr are mostly retained in the B-domain. Based on the structural studies of the alteration layer a mechanism of formation was proposed and consists of the nucleation of smectite crystallites at the early stages of the water vapor–glass interaction. Continuous restructuring of the hydrated glass matrix, followed by partitioning and migration of elements, leads to the growth of smectite crystallites to form the A-domain. The migration of alkalis and alkaline earths leads to the precipitation of well-crystallized secondary phases such as analcime and Ca-silicate hydrates. Experiments with SON68 and CSDB glasses conducted in nonsaturated water conditions (\({\mathrm{92}}\%\)) showed less complex layers than those formed in saturated water vapor by Gong et al [12.211]. Typical alteration layers can be seen in Fig. 12.17a-d. The amorphous hydrated layer underneath the external crystalline layer is relatively homogeneous in terms of morphology and elemental composition at high (\({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)) and low temperature (\({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\)). The low activity of water did not allow a significant transport of heavy elements except alkalis. Dissolution figures observed in Fig. 12.17a-dc denote the probable high pH in the condensed water.


TEM micrographs showing the alteration layer of SON68 glass hydrated at \({\mathrm{200}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{92}}\%\) for \({\mathrm{57}}\,{\mathrm{d}}\) (a,b) and CSDB glass hydrated at \({\mathrm{90}}\,{\mathrm{{}^{\circ}\mathrm{C}}}\) and \({\mathrm{92}}\%\) for \({\mathrm{365}}\,{\mathrm{d}}\) (c,d)
4 Conclusion
Glass corrosion in aqueous media has thoroughly been discussed throughout the chapter in terms of corrosion mechanisms including ion exchange, water diffusion, and network dissolution–recondensation, but also in terms of dissolution kinetics and alteration product formation. Environmental parameters have also been discussed and their impact on glass corrosion developed, in particular for radioactive waste repositories. The scientific knowledge acquired during the last \(\mathrm{30}\) years in this field allowed the prediction of the glass dissolution for long durations via geochemical modeling. However, the evolution of repository concepts, such as the introduction of new materials, has necessarily led to new R&D needs required to improve the predictive models. These future needs include a better characterization of the alteration products (e. g., iron silicates formed on the glass–iron container interface) and their sorption capacities with regard radionuclides released by the glass. For the glass vapor hydration R&D needs are more important and include the understanding of the underlying hydration mechanisms, the evaluation of the influence of irradiation on glass hydration and the interactions of the multibarrier materials in the vapor phase.
References
H. Schröder: Über die Angreifbarkeit des Glases durch Lösungen mit pH-Werten nahe 7, Glastech. Ber. 26, 91–97 (1953)
K. Zagar, A. Schillmöller: Über die physikalischchemischen Vorgänge bei der Wasserauslaugung von Glasoberflächen, Glastech. Ber. 33, 109–116 (1960)
R.W. Douglas, T.M. El Shamy: Reactions of glasses with aqueous solutions, J. Am. Ceram. Soc. 50, 1–8 (1967)
R.H. Doremus: Interdiffusion of hydrogen and alkali ions in a glass surface, J. Non-Cryst. Solids 19, 137–144 (1975)
R.M.J. Smets, T.P.A. Lommen: The leaching of sodium aluminosilicate glasses studied by secondary ion mass spectrometry, Phys. Chem. Glasses 23, 83–87 (1982)
B.C. Bunker, G.W. Arnold, E.K. Beauchamp, D.E. Day: Mechanisms for alkali leaching in mixed Na-K-silicate glasses, J. Non-Cryst. Solids 58, 295–322 (1983)
P. March, F. Rauch: Leaching studies of soda-lime-silica glass using deuterium- and oxygen-18 enriched solutions, Glastech. Ber. 63, 154–162 (1990)
R. Hellmann, S. Cotte, E. Cadel, S. Malladi, L.S. Karlsson, S. Lozano-Perez, M. Cabié, A. Seyeux: Nanometre-scale evidence for interfacial dissolution–reprecipitation control of silicate glass corrosion, Nat. Mater. 14, 307–311 (2015)
A. Paul: Chemical durability of glasses: A thermodynamic approach, J. Mater. Sci. 13, 2246–2268 (1979)
J. Plodinec, C.M. Jantzen, G.G. Wicks: A Thermodynamic Approach to Prediction of the Stability of Proposed Radwaste Glasses (Savannah River Laboratory, Savannah 1982), DP-MS-82-66
K.G. Knauss, T.J. Wolery: Dependence of albite dissolution kinetics on pH and time at 25 °C and 70 °C, Geochim. Cosmochim. Acta 50, 2481–2497 (1986)
T. Advocat: Les mécanismes de corrosion en phase aqueuse du verre nucléaire R7T7. Approche Experimentale, Essai de modélisation Thermodynamique et Cinetique, Ph.D. Thesis (Université Louis Pasteur, Strasbourg 1991)
B. Grambow: Influence of saturation on the leaching of borosilicate nuclear waste glasses, Glastech. Ber. 56K, 566–571 (1983)
A.A. Barkatt, P.B. Macedo, B.C. Gibson, C.J. Montrose: Modeling of waste form performance and system release, Mater. Res. Soc. Symp. Proc. 44, 3–13 (1985)
J.P. Icenhower, C.I. Steefel: Experimentally determined dissolution kinetics of SON68 glass at 90 °C over a silica saturation interval: Evidence against a linear rate law, J. Nucl. Mater. 439, 137–147 (2013)
W.L. Ebert, J.K. Bates: A comparison of glass reaction at high and low glass surface/solution volume, Nucl. Technol. 104, 372–384 (1993)
S. Dultz, H. Behrens, G. Helsch, J. Deubener: Electrolyte effects on surface chemistry of basaltic glass in the initial stages of dissolution, Chem. Geol. 426, 71–84 (2016)
I.O. Isard, W. Müller: Influence of alkaline earth ions on the corrosion of glasses, Phys. Chem. Glasses 27, 55–58 (1986)
X. Feng, I.L. Pegg, A.A. Barkatt, P.B. Barkatt, P.B. Macedo, S.J. Cucinell, S. Lai: Correlation between composition effects on glass durability and the structural role of the constituent oxides, Nucl. Technol. 85, 334–345 (1989)
M. Kinoshita, M. Harada, Y. Sato, Y. Hariguchi: Percolation phenomenon for dissolution of sodium borosilicate glasses in aqueous solutions, J. Am. Ceram. Soc. 74, 783–787 (1991)
J.K. Bates, C.R. Bradley, E.C. Buck, J.C. Cunnane, W.L. Ebert, X. Feng, J.J. Mazer, D.J. Wronkiewicz, J. Sproull, W.L. Bourcier, B.P. McGrail, M.K. Altenhofen: High-Level Waste Borosilicate Glass: A Compendium of Corrosion Characteristics (US Department of Energy, Washington DC 1994), DOE-EM-0177
W.L. Bourcier: Overview of chemical modeling of nuclear waste glass dissolution, Mater. Res. Soc. Symp. Proc. 212, 3–18 (1991)
S. Gin, L. Neill, M. Fournier, P. Frugier, T. Ducasse, M. Tribet, A. Abdelouas, B. Parruzot, J. Neeway, N. Wall: The controversial role of inter-diffusion in glass alteration, Chem. Geol. 440, 115–123 (2016)
B.C. Bunker: Waste glass leaching: Chemistry and kinetics, Mater. Res. Soc. Symp. Proc. 84, 493–507 (1987)
L.L. Hench, D.E. Clark, A.B. Harker: Review nuclear waste solids, J. Mater. Sci. 21, 1457–1478 (1986)
D.M. Strachan, T.L. Croak: Compositional effects on long-term dissolution of borosilicate glass, J. Non-Cryst. Solids 272, 22–33 (2000)
P. Van Iseghem, M. Aerstens, S. Gin, D. Deneele, B. Grambow, D.M. Strachan, B.P. McGrail, G.G. Wicks: GLAMOR—Or how we achieved a common understanding on the decrease of glass dissolution kinetics. In: Ceramic Transactions, Vol. 207, ed. by A. Cosi, T. Ohji (American Ceramic Society, Westerville 2009) pp. 115–126
A.K. Varshneya: Fundamentals of Inorganic Glasses (Academic Press, San Diego 1994)
E.Y. Vernaz, J.L. Dussossoy: Current state of knowledge of nuclear waste glass corrosion mechanisms: The case of R7T7 glass, Appl. Geochem. 1, 13–22 (1992)
E.Y. Vernaz, S. Gin, C. Jegou, I. Ribet: Present understanding of R7T7 glass alteration kinetics and their impact on long-term behavior modeling, J. Nucl. Mater. 298, 27–36 (2001)
L. Werme, I.K. Bjorner, G. Bart, U. Zwicky, B. Grambow, W. Lutze, R. Ewing, C. Magrabi: Chemical corrosion of highly radioactive borosilicate waste glass under simulated repository conditions, J. Mater. Res. 5, 1130–1146 (1990)
R.H. Doremus: Diffusion-controlled reaction of water with glass, J. Non-Cryst. Solids 55, 143–147 (1983)
K. Ferrand, A. Abdelouas, B. Grambow: Water diffusion in the simulated French nuclear waste glass SON68 contacting silica rich solutions: Experimental and modeling, J. Nucl. Mater. 355, 54–67 (2006)
D. Rébiscoul, F. Rieutord, F. Né, P. Frugier, R. Cubitt, S. Gin: Water penetration mechanisms in nuclear glasses by x-ray and neutron reflectometry, J. Non-Cryst. Solids 353, 2221–2230 (2007)
H. Tomozawa, M. Tomozawa: Diffusion of water into a borosilicate glass, J. Non-Cryst. Solids 109, 311–317 (1989)
B.P. McGrail, J.P. Icenhower, D.K. Shuh, P. Liu, J.G. Darab, D.R. Baer, S. Thevuthasen, V. Shutthanandan, M.H. Engelhard, C.H. Booth, P. Nachimuthu: The structure of Na2O-Al2O3-SiO2 glass: Impact on sodium ion exchange in H2O and D2O, J. Non-Cryst. Solids 296, 10–26 (2001)
M.I. Ojovan, A. Pankov, W.E. Lee: The ion exchange phase in corrosion of nuclear waste glasses, J. Nucl. Mater. 358, 57–68 (2006)
S. Gin, A. Abdelouas, L.J. Criscenti, W.L. Ebert, K. Ferrand, T. Geisler, M.T. Harrison, Y. Inagaki, S. Mitsui, K.T. Mueller, J.C. Marra, C.G. Pantano, E.M. Pierce, J.V. Ryan, J.M. Schofield, C.I. Steefel, J.D. Vienna: An international initiative on long-term behavior of high-level nuclear waste glass, Mater. Today 16, 243–248 (2013)
M.A. Rana, R.W. Douglas: The reaction between glass and water. Part 2. Discussion for the result, Phys. Chem. Glasses 2, 196–204 (1961)
M.A. Rana, R.W. Douglas: The reaction between glass and water. Part I. Experimental methods and observations, Phys. Chem. Glasses 2, 179–195 (1961)
C.M. Jantzen, D.I. Kaplan, N.E. Bibler, D.K. Peeler, M.J. Plodinec: Performance of a buried radioactive high level waste (HLW) glass after 24 years, J. Nucl. Mater. 378, 244–256 (2008)
R.K. Iler: The Chemistry of Silica (Wiley, New York 1979)
Z.G. Boksay, G. Bouquet, S. Dobos: The kinetics of the formation of leached layers on glass surfaces, Phys. Chem. Glasses 8, 140–144 (1967)
Z.G. Boksay, G. Bouquet, S. Dobos: The kinetics of the formation of leached layers on glass surfaces, Phys. Chem. Glasses 9(2), 69–71 (1968)
W.A. Lanford: 15N hydrogen profiling: Scientific applications, Nucl. Instrum. Methods 149, 1–8 (1978)
W.A. Lanford: Hydration of soda-lime glass, J. Non-Cryst. Solids 33, 249–266 (1979)
S. Gin, J.V. Ryan, D.K. Schreiber, J.J. Neeway, M. Cabié: Contribution of atom-probe tomography to a better understanding of glass alteration mechanisms: Application to a nuclear glass specimen altered 25 years in a granitic environment, Chem. Geol. 349–350, 99–109 (2013)
B.C. Bunker: Molecular mechanisms for corrosion of silica and silicate glasses, J. Non-Cryst. Solids 179, 300–308 (1994)
B. Grambow, R. Müller: First-order dissolution rate law and the role of surface layers in glass performance assessment, J. Nucl. Mater. 298, 112–124 (2001)
W.H. Casey, B.C. Bunker: Leaching of mineral and glass surfaces during dissolution. In: Mineral-Water Interface Geochemistry. Reviews in Mineralogy, ed. by M.F. Hochella, A.F. White (Mineralogical Society of America, Washington DC 1990) pp. 397–424
B.C. Bunker, G.W. Arnold, E.E. Day, P.J. Bray: The effect of molecular structure on borosilicate glass leaching, J. Non-Cryst. Solids 87, 226–253 (1986)
B.C. Bunker, D.R. Tallant, T.J. Headley, G.L. Turner, R.J. Kirkpatrick: The structure of leached sodium borosilicate glass, Phys. Chem. Glasses 29, 106–120 (1988)
L.R. Pederson, D.R. Baer, G.L. McVay, M.H. Engelhard: Reaction of soda lime silicate glass in isotopically labelled water, J. Non-Cryst. Solids 86, 369–380 (1986)
D. Rébiscoul, F. Bruguier, V. Magnin, S. Gin: Impact of soda-lime borosilicate glass composition on water penetration and water structure at the first time of alteration, J. Non-Cryst. Solids 358, 2951–2960 (2012)
D. Rébiscoul, J. Cambedouzou, I. Matar Briman, M. Cabié, H.P. Brau, O. Diat: Water dynamics in nanoporous alteration layer coming from glass alteration: An experimental approach, J. Phys. Chem. C 119, 15982–15993 (2015)
J.J. Neeway, S. Kerisit, S. Gin, Z. Wang, Z. Zhu, J.V. Ryan: Low-temperature lithium diffusion in boroaluminosilicate glasses, J. Non-Cryst. Solids 405, 83–90 (2014)
J.J. Neeway, S.N. Kerisit, J. Liu, Z. Zhu, B. Riley, J.V. Ryan: Ion-exchange interdiffusion model with potential application to long-term nuclear waste glass performance, J. Phys. Chem. C 120, 9374–9384 (2016)
P. Åagaard, H.C. Helgeson: Thermodynamic and kinetic constraints on reaction rates among minerals and aqueous solutions I. Theoretical considerations, Am. J. Sci. 282, 237–285 (1982)
J.D. Rimstidt, H.L. Barnes: The kinetics of silica-water reactions, Geochim. Cosmochim. Acta 44, 1683–1699 (1980)
T. Advocat, J.L. Crovisier, B. Fritz, E. Vernaz: Thermokinetic model of borosilicate glass dissolution: Contextural affinity, Mater. Res. Soc. Symp. Proc. 176, 241–248 (1990)
B. Grambow: A general rate equation for nuclear waste glass corrosion, Mater. Res. Soc. Symp. Proc. 44, 15–27 (1985)
S. Gin, J.P. Mestre: SON68 nuclear glass alteration kinetics between pH 7 and pH 11.5, J. Nucl. Mater. 295, 83–96 (2001)
A. Abdelouas, J.L. Crovisier, W. Lutze, R. Müller, W. Bernotat: Structure and chemical-properties of surface-layers developed on R7T7 simulated nuclear waste glass altered in brine at 190 °C, Eur. J. Mineral. 7, 1101–1113 (1995)
W. Lutze, R.C. Ewing: Radioactive Waste Forms for the Future (North-Holland, New York 1988)
C. Cailleteau, F. Angeli, F. Devreux, S. Gin, J. Jestin, P. Jollivet, O. Spalla: Insight into silicate-glass corrosion mechanisms, Nat. Mater. 7, 978–983 (2008)
D. Wolff-Boenisch, S.R. Gislason, E.H. Oelkers: The effect of fluoride on the dissolution rates of natural glasses at pH 4 and 25 °C, Geochim. Cosmochim. Acta 68, 4571–4582 (2004)
J.P. Hamilton, S.L. Brantley, C.G. Pantano, L.J. Criscenti, J.D. Kubicki: Dissolution of nepheline, jadeite and albite glasses: Toward better models for aluminosilicate dissolution, Geochim. Cosmochim. Acta 65, 3683–3702 (2001)
J. Sterpenich, G. Libourel: Water diffusion in silicate glasses under natural weathering conditions: Evidence from buried medieval stained glasses, J. Non-Cryst. Solids 352, 5446–5451 (2006)
C. Lenting, T. Geisler, A. Gerdes, E. Kooijman, E.E. Scherer, A. Zeh: The behavior of the Hf isotope system in radiation-damaged zircon during experimental hydrothermal alteration, Am. Mineral. 95, 1343–1348 (2010)
T. Geisler, A. Janssen, D. Scheiter, T. Stephan, J. Berndt, A. Putnis: Aqueous corrosion of borosilicate glass under acidic conditions: A new corrosion mechanism, J. Non-Cryst. Solids 356, 1458–1465 (2010)
S. Gin, X. Beaudoux, F. Angéli, C. Jégou, N. Godon: Effect of composition on the short-term and long-term dissolution rates of ten borosilicate glasses of increasing complexity from 3 to 30 oxides, J. Non-Cryst. Solids 358, 2559–2570 (2012)
P. Frugier, S. Gin, Y. Minet, T. Chave, B. Bonin, N. Godon, J.E. Lartigue, P. Jollivet, A. Ayral, L. De Windt, G. Santarini: SON68 nuclear glass dissolution kinetics: Current state of knowledge and basis of the new GRAAL model, J. Nucl. Mater. 380, 8–21 (2008)
E.M. Pierce, B.P. McGrail, P.F. Martin, J. Marra, B.W. Arey, K.N. Geiszler: Accelerated weathering of high-level and plutonium-bearing lanthanide borosilicate waste glasses under hydraulically unsaturated conditions, Appl. Geochem. 22, 1841–1859 (2007)
K.L. Nagy, A.C. Lasaga: Simultaneous precipitation kinetics of kaolinite and gibbsite at 80 °C and pH 3, Geochim. Cosmochim. Acta 57, 4329–4335 (1993)
K.L. Nagy: Dissolution and precipitation kinetics of sheet silicates. In: Chemical Weathering Rates of Silicate Minerals, ed. by A.F. White, S.L. Brantley (Mineralogical Society of America, Washington DC 1995) pp. 173–234
W.L. Ebert: Solution Replacement Tests with SON-68 Glass (Argonne National Laboratory, Lemont 2012), FCRD-SWF-2012-0001222
S. Ribet, S. Gin: Role of neoformed phases on the mechanisms controlling the resumption of SON68 glass alteration in alkaline media, J. Nucl. Mater. 324, 152–164 (2004)
D.M. Strachan, J.J. Neeway: Effects of alteration product precipitation on glass dissolution, Appl. Geochem. 45, 144–157 (2014)
P. Van Iseghem, B. Grambow: The long-term corrosion and modelling of two simulated Belgian beference high-level waste glasses, Mater. Res. Soc. Symp. Proc. 112, 631–639 (1988)
W.L. Ebert, J.A. Fortner, C.L. Crawford, J.C. Marra: Stage 3 Dissolution tests with AFCI glass (US Department of Energy, Washington DC 2012), FCRD-SWF-2012-000204
M. Fournier, S. Gin, P. Frugier: Resumption of nuclear glass alteration: State of the art, J. Nucl. Mater. 448, 348–363 (2014)
B.P. McGrail, W.L. Ebert, D.H. Bacon, D.M. Strachan: A Strategy to Conduct an Analysis of the Long-Term Performance of Low-Activity Waste Glass in a Shallow Subsurface Disposal System at Hanford (Pacific Northwest National Laboratory, Richland 1998), PNNL-11834
A. Barkatt, A. Olszowka, W. Sousanpour, M.A. Adel-Hadadi, R. Adiga, A. Baraktt, G.S. Marbury, S. Li: Leach rate excursions in borosilicate glasses: Effects of glass and leachant composition, Mater. Res. Soc. Symp. Proc. 212, 65–76 (1991)
J.D. Vienna, J.V. Ryan, S. Gin, Y. Inagaki: Current understanding and remaining challenges in modeling long-term degradation of borosilicate nuclear waste glasses, Int. J. Appl. Glass Sci. 4, 283–294 (2013)
M. Fournier, P. Frugier, S. Gin: Effect of zeolite formation on borosilicate glass dissolution kinetics, Procedia Earth Plan. Sci. 7, 264–267 (2013)
D.M. Strachan: Glass dissolution: Testing and modeling for long-term behavior, J. Nucl. Mater. 298, 69–77 (2001)
M. Fournier, A. Ull, E. Nicoleau, Y. Inagaki, M. Odorico, P. Frugier, S. Gin: Glass dissolution rate measurement and calculation revisited, J. Nucl. Mater. 476, 140–154 (2016)
L.L. Hench, D.E. Clark: Physical chemistry of glass surfaces, J. Non-Cryst. Solids 28, 83–105 (1978)
H. Scholze: Glass-water interactions, J. Non-Cryst. Solids 102, 1–10 (1988)
H. Scholze (Ed.): Glas – Natur, Struktur und Eigenschaften, 2nd edn. (Springer, Berlin 1977)
D.R. Baer, L.R. Pederson, G.L. McVay: Glass reactivity in aqueous solutions, J. Vac. Sci. Technol. A2, 738–743 (1984)
H. Schnatter, H. Doremus, W.A. Lanford: Hydrogen analysis of soda-lime silicate glass, J. Non-Cryst. Solids 102, 11–18 (1988)
B.C. Bunker, T.J. Headly, D.C. Douglas: Gel structures in leached alkali silicate glass, Mater. Res. Soc. Symp. Proc. 32, 41–46 (1984)
R.D. Aines, H.C. Weed, J.K. Bates: Hydrogen speciation in hydrated layers on nuclear waste glass, Mater. Res. Soc. Symp. Proc. 84, 547–558 (1987)
F.M. Ernsberger: Electrical transport of protons in solids, Glastech. Ber. 56K, 963–968 (1983)
R.H. Doremus, Y. Mehrotra, W.A. Lanford, C. Burman: Reaction of water with glass: Influence of a transformed surface layer, J. Mater. Sci. 18, 612–622 (1983)
A. Abdelouas, J.L. Crovisier, J. Caurel, E. Vernaz: Analyse par microscopie électronique à transmission des produits de l'altération hydrothermale du verre nucléaire R7T7, C. R. Acad. Sci. Paris 317, 1333–1340 (1993)
D. Rébiscoul, A. Van der Lee, F. Rieutord, F. Ne, O. Spalla, A. El-Mansouri, P. Frugier, A. Ayral, S. Gin: Morphological evolution of alteration layers formed during nuclear glass alteration: New evidence of a gel as a diffusive barrier, J. Nucl. Mater. 326, 9–18 (2004)
K. Ferrand: Effet de la diffusion d'eau et de la radiolyse alpha et gamma sur la corrosion des verres type SON68 en solutions aqueuses riches en silicium, Ph.D. Thesis (Univ. Nantes, Nantes 2004)
S. Ricol: Etude du gel d'altération des verres nucléaires et synthèse de gels modèles, Ph.D. Thesis (Univ. Pierre and Marie Curie, Paris 1995)
L.N. Plumme: Geochemical Modeling: A Comparison of Forward and Inverse Method (National Water Well Assoc, Worthington 1984) pp. 149–177
G. Berger, J. Schott, M. Loubet: Fundamental processes controlling the first stage of alteration of a basalt glass by seawater: An experimental study between 200 °C and 320 °C, Earth Planet. Sci. Lett. 84, 431–445 (1987)
J.L. Crovisier, T. Advocat, J.C. Petit, B. Fritz: Alteration of basaltic glass in Iceland as a natural analogue for nuclear waste glasses: Geochemical modelling with DISSOL, Mater. Res. Soc. Symp. Proc. 127, 57–64 (1989)
L. Michaux, E. Mouche, J.C. Petit, B. Fritz: Geochemical modeling of the long-term dissolution behavior of the French nuclear glass R7T7, Appl. Geochem. Suppl. 1, 41–54 (1992)
B. Grambow: Geochemical modeling of the reaction between glass and aqueous solution. In: 85th Am. Ceram. Soc. Annu. Meeting (American Chemical Society, Columbus 1984) pp. 474–481
S.C. Kohn, J.M. Charnock, C.M.B. Henderson, G.N. Greaves: The structural environments of trace elements in dry hydrous silicate glasses: A manganese and strontium K-edge x-ray absorption spectroscopy study, Contrib. Mineral. Petrol. 105, 359–368 (1990)
N.T. Barrett, G.M. Antonini, G.N. Greaves, F.R. Thornley, A. Manara: Grazing incidence fluorescence EXAFS and near edge spectroscopy for corroded glass surfaces, J. Phys. Colloq. C8 12(47), C8-879–882 (1986)
G. Malow: The mechanisms for hydrothermal leaching of nuclear waste glasses: Properties and evaluation of surface layers, Mater. Res. Soc. Symp. Proc. 11, 25–36 (1982)
B. Grambow, D.M. Strachan: Leach testing of waste glasses under near-saturation conditions, Mater. Res. Soc. Symp. Proc. 26, 623–634 (1984)
A. Barkatt, E.E. Saad, R.B. Adiga, W. Sousanpour, A. Barkatt, M.A. Adel-Hadai, J.A. O'Keefe, S. Alterescu: Leaching of natural and nuclear waste glasses in sea water, Appl. Geochem. 4, 593–603 (1989)
J.C. Sang, R.F. Jakubik, A. Barkatt, E. Saad: The interaction of solutes with silicate glass and its effect on dissolution rates, J. Non-Cryst. Solids 167, 158–171 (1994)
H. Attassi, J.L. Crovisier, A. Mosser, D. Muster: Rôle protecteur de la couche d'altération formée à la surface du verre bioactif KGS altéré dans le plasma artificiel, C. R. Acad. Sci. Paris 318(II), 935–939 (1994), Protective effect of the alteration layer formed on the KGS bioactive glass altered in artificial plasma
D. Rébiscoul, A. van der Lee, P. Frugier, A. Ayral, S. Gin: X-ray reflectometry characterization of SON68 glass alteration films, J. Non-Cryst. Solids 325, 113–123 (2003)
N. Donzel, S. Gin, F. Augereau, M. Ramonda: Study of gel development during SON68 glass alteration using atomic force microscopy. Comparison with two simplified glasses, J. Nucl. Mater. 317, 83–92 (2003)
A. Verney-Carron, S. Gin, G. Libourel: Archaeological analogs and the future of nuclear waste glass, J. Nucl. Mater. 406, 365–370 (2010)
A. Verney-Carron, S. Gin, G. Libourel: A fractured Roman glass block altered for 1800 years in seawater: Analogy with nuclear waste glass in a deep geological repository, Geochim. Cosmochim. Acta 72, 5372–5385 (2008)
C. Mayant, B. Grambow, A. Abdelouas, S. Ribet, S. Leclercq: Surface site density, silicic acid retention and transport properties of compacted magnetite powder, Phys. Chem. Earth 33, 991–999 (2008)
D.E. Clark, R.L. Schulz, G.G. Wicks, A.R. Lodding: Waste glass alteration processes, surface layer evolution and rate limiting steps, Mater. Res. Soc. Symp. Proc. 333, 107–122 (1994)
R. Hellmann, J.M. Penisson, R.L. Hervig, J.H. Thomassin, M.F. Abrioux: An EFTEM/HRTEM high-resolution study of the near surface of labradorite feldspar altered at acid pH: Evidence for interfacial dissolution-reprecipitation, Phys. Chem. Miner. 30, 192–197 (2003)
T.A. Abrajano, J.K. Bates, A.B. Woodland, J.P. Bradley, W.L. Bourcier: Secondary phase formation during nuclear waste-glass dissolution, Clays Clay Miner. 38, 537–548 (1990)
N. Valle, A. Verney-Carron, J. Sterpenich, G. Libourel, E. Deloule, P. Jollivet: Elemental and isotopic (29Si and 18O) tracing of glass alteration mechanisms, Geochim. Cosmochim. Acta 74, 3412–3431 (2010)
A. Abdelouas, J.L. Crovisier, W. Lutze, B. Fritz, A. Mosser, R. Müller: Formation of hydrotalcite-like compounds during R7T7 nuclear waste glass and basaltic glass alteration, Clays Clay Miner. 42, 526–533 (1994)
J.C. Petit, M.C. Magonthier, J.C. Dran, G.D. Mea: Long-term dissolution rate of nuclear glasses in confined environments: Does a residual chemical affinity exist?, J. Mater. Sci. 25, 3048–3052 (1990)
Y. Inagaki, A. Shinkai, K. Idemistu, T. Arima, H. Yoshikawa, M. Yui: Aqueous alteration of Japanese simulated waste glass P0798: Effects of alteration-phase formation on alteration rate and cesium retention, J. Nucl. Mater. 354, 171–184 (2006)
G. Berger, J. Schott, C. Guy: Behavior of Li, Rb and Cs during basalt glass and olivine dissolution and chlorite, smectite and zeolite precipitation from seawater: Experimental investigations and modelization between 50 and 300 °C, Chem. Geol. 71, 297–312 (1988)
A. Lodding, P. Van Iseghem: In-depth distributions of elements in leached layers on two HLW waste glasses after burial in clay; step-scan by SIMS, J. Nucl. Mater. 298, 197–202 (2001)
O. Deruelle, O. Spalla, P. Barboux, J. Lambard: Growth and ripening of porous layers in water altered glasses, J. Non-Cryst. Solids 261, 237–251 (2000)
S. Gin, C. Jegou, E. Vernaz: Use of orthophosphate complexing agents to investigate mechanisms limiting the alteration kinetics of French SON68 nuclear glass, Appl. Geochem. 15, 1505–1525 (2000)
E. Curti, J.L. Crovisier, G. Morvan, A.M. Karpoff: Long-term corrosion of two nuclear waste reference glasses (MW and SON68): A kinetic and mineral alteration study, Appl. Geochem. 21, 1152–1168 (2006)
M. Debure, L. De Windt, P. Frugier, S. Gin, P. Vieillard: Mineralogy and thermodynamic properties of magnesium phyllosilicates formed during the alteration of a simplified nuclear glass, J. Nucl. Mater. 475, 255–265 (2016)
P.C. Burns, R.A. Olson, R.J. Finch, J.M. Hanchar, Y. Thibault: KNa3(UO2)2(Si4O10)2(H2O)4, a new compound formed during vapor hydration of an actinide-bearing borosilicate waste glass, J. Nucl. Mater. 278, 290–300 (2000)
F. Angeli, J.M. Delaye, T. Charpentier, J.C. Petit, D. Ghaleb, P. Faucon: Influence of glass chemical composition on the Na–O bond distance: A 23Na 3Q-MAS NMR and molecular dynamics study, J. Non-Cryst. Solids 276, 132–144 (2000)
R.J. Hand, R.J. Short, S. Morgan, N.C. Hyatt, G. Mobus, W.E. Lee: Molybdenum in glasses containing vitrified nuclear waste, Eur. J. Glass Sci. Technol. Part A 43, 121–124 (2005)
L. Galoisy, E. Pélegrin, M.A. Arrio, P. Ildefonse, G. Calas, D. Ghaleb, C. Fillet, F. Pacaud: Evidence for 6-coordinated zirconium in inactive nuclear waste glasses, J. Am. Ceram. Soc. 82, 2219–2224 (1999)
F. Angeli, T. Charpentier, M. Gaillard, P. Jollivet: Influence of zirconium on the structure of pristine and leached soda-lime borosilicate glasses: Towards a quantitative approach by 17O MQMAS NMR, J. Non-Cryst. Solids 354, 3713–3722 (2008)
T. Advocat, P. Jollivet, J.L. Crovisier, M. del Nero: Long-term alteration mechanisms in water for SON68 radioactive borosilicate glass, J. Nucl. Mater. 298, 55–62 (2001)
O. Menard, T. Advocat, J.P. Ambrosi, A. Michard: Behaviour of actinides (Th, U, Np, and Pu) and rare earths (La, Ce, and Nd) during aqueous leaching of a nuclear glass under geological disposal conditions, Appl. Geochem. 13, 105–126 (1998)
O. Vidal, M.C. Magonthier, V. Joanny, M. Creach: Partitioning of La between solid and solution during the ageing of SiAlFeLaCa gels under simulated near-field conditions of nuclear waste disposal, Appl. Geochem. 10, 269–184 (1995)
B. Grambow, R. Müller, A. Rother, W. Lutze: Release of rare earth elements and uranium from glass in low pH brines, Radiochim. Acta 52/53, 501–506 (1991)
H. El Hajj, A. Abdelouas, Y. El Mendili, G. Karakurt, B. Grambow, C. Martin: Corrosion of carbon steel under sequential aerobic-anaerobic environmental conditions, Corros. Sci. 76, 432–440 (2013)
Y. El Mendili, A. Abdelouas, A. Aït Chaou, J.F. Bardeau, M. Schlegel: Carbon steel corrosion in clay-rich environment, Corros. Sci. 88, 56–65 (2014)
Y. El Mendili, A. Abdelouas, G. Karakurt, A. Aït Chaou, R. Essehli, J.F. Bardeau, J.M. Greneche: The effect of temperature on carbon steel corrosion under geological conditions, Appl. Geochem. 52, 76–85 (2015)
J.K. Bates, M.J. Steindler, B. Tani, F.J. Purcell: The hydration alteration of a commercial nuclear waste glass, Chem. Geol. 51, 79–87 (1985)
M. Fournier, P. Frugier, S. Gin: Resumption of alteration at high temperature and pH: Rates measurements and comparison with initial rates, Procedia Mater. Sci. 7, 202–208 (2014)
A. Aït Chaou, A. Abdelouas, Y. El Mendili, R. Bouakkaz, C. Martin: The French SON68 glass vapor hydration under different atmospheres, Procedia Mater. Sci. 7, 179–185 (2014)
J. Neeway, A. Abdelouas, B. Grambow, S. Schumacher, C. Martin, M. Kogawa, S. Utsunomiya, S. Gin, P. Frugier: Vapor hydration of SON68 glass from 90 °C to 200 °C: A kinetic study and corrosion products investigation, J. Non-Cryst. Solids 358, 2897–2905 (2012)
J.K. Bates, L.J. Jardine, M.J. Steindler: Hydration aging of nuclear waste glass, Science 218, 51–54 (1982)
J.K. Bates, L.J. Jardine, M.J. Steindler: The Hydration Process of Nuclear-Waste: An Interim Report, Vol. ANL-82-11 (Argonne National Laboratory, Argonne 1982)
I. Friedman, R.L. Smith: A new dating method using obsidian: Part I, The development of the method, Am. Antiq. 25, 476–493 (1960)
I. Friedman, R.L. Smith, W.D. Long: Hydration of natural glass and formation of perlite, Geol. Soc. Am. Bull. 77, 323–328 (1966)
I. Friedman, W. Long: Hydration rate of obsidian, Science 191, 347–352 (1976)
J.W. Michels, I.S.T. Tsong, G.A. Smith: Experimentally derived hydration rates in obsidian dating, Archaeometry 25, 107–117 (1983)
M. Morgenstein, J. Riley: Hydration-rind dating of basaltic glass: A new method for archaeological chronologies, Asian Perspect 17, 145–159 (1975)
M. Morgenstein, P. Rosendahl: Basaltic glass hydration dating in Hawaiian archaeology. In: Advances in Obsidian Glass Studies, ed. by R.E. Taylor (Noyes, New Jersey 1976) pp. 141–164
J.K. Bates, M.G. Seitz, M.J. Steindler: The relevance of vapor phase hydration aging to nuclear waste isolation, Nucl. Chem. Waste Manag. 5, 63–73 (1984)
J.K. Bates, M.J. Steindler, P.L. McDaniel: Hydration of stressed nuclear waste glass, Mater. Lett. 2, 296–300 (1984)
T.A. Abrajano, J.K. Bates, C.D. Byers: Aqueous corrosion of natural and nuclear waste glasses I, J. Non-Cryst. Solids 84, 251–257 (1986)
W.L. Ebert, J.K. Bates: The reaction of synthetic nuclear waste glass in steam and hydrothermal solution, Mater. Res. Soc. Symp. Proc. 176, 339–346 (1989)
W.L. Ebert, R.F. Hoburg, J.K. Bates: The sorption of water on obsidian and a nuclear waste glass, Phys. Chem. Glasses 32, 133–137 (1990)
W.L. Ebert, J.K. Bates, W.L. Bourcier: The hydration of borosilicate waste glass in liquid water and steam at 200 °C, Waste Manag. 11, 205–221 (1991)
B.M. Biwer, J.K. Bates, T.A. Abrajano, J.P. Bradley: Comparison of the layer structure of vapor phase and leached SRL glass by use of AEM, Mater. Res. Soc. Symp. Proc. 176, 255–263 (1989)
D.J. Wronkiewicz, L.M. Wang, J.K. Bates, B.S. Tani: Effect of radiation exposure on glass alteration in a steam environment, Mater. Res. Soc. Symp. Proc. 294, 183–190 (1993)
D.J. Wronkiewicz, C.R. Bradley, J.K. Bates, L.M. Wang: Effects of radiation exposure on SRL 131 composition glass in a steam environment, Mater. Res. Soc. Symp. Proc. 333, 259–267 (1994)
A. Abdelouas, J.L. Crovisier, W. Lutze, B. Grambow, J.C. Dran, R. Müller: Surface layers on a borosilicate nuclear waste glass corroded in MgCl2 solution, J. Nucl. Mater. 240, 100–111 (1997)
P. Jollivet, S. Gin, S. Schumacher: Forward dissolution rate of silicate glasses of nuclear interest in clay-equilibrated groundwater, Chem. Geol. 330/331, 207–217 (2012)
Andra: Andra research on the geological disposal of high-level long-lived radioactive waste: Results and perspectives (National Agency for Radioactive Waste Management, Châtenay-Malabry 2005) p. 40 265 AV
A. Abdelouas, B. Grambow: Aquatic chemistry of long-lived mobile fission and activation products in the context of deep geological disposal. In: Radionuclide Behavior in the Natural Environment: Science, Impacts and Lessons for the Nuclear Industry, ed. by C. Poinssot, H. Geckeis (Woodhead, Cambridge 2012) pp. 70–102
R.H. Doremus: Exchange and diffusion of ions in glass, J. Phys. Chem. 68, 2212–2018 (1964)
R.R. Lee, D.A. Leich, T.A. Tombrello: Obsidian hydration profile measurements using an nuclear reaction technique, Nature 250, 44–47 (1974)
J.K. Bates, T.A. Abrajano Jr., W.L. Ebert, J.J. Mazer, T.J. Gerding: Experimental hydration studies of natural and synthetic glasses, Mater. Res. Soc. Symp. Proc. 123, 237–244 (1988)
J.W. Michels: Archeology and dating by hydration of obsidian, Science 158, 211–214 (1967)
L. Johnson Jr.: Obsidian hydration rate for the Klamath basin of California and Oregon, Science 165, 1354–1356 (1969)
F.J. Findlow, V. Bennett, J. Ericson, S. DeAltley: A new obsidian hydration rate for certain obsidians in American southwest, Am. Antiq. 40, 344–348 (1975)
I. Friedman, F.W. Trembour: Obsidian: The dating stone, Am. Sci. 66, 44–52 (1978)
I. Friedman, F.W. Trembour: Obsidian hydration dating update, Am. Antiq. 48, 544–547 (1983)
F.W. Trembour, I. Friedman: The present status of obsidian hydration dating. In: Quaternary Dating Methods, ed. by W. Mahaney (Elsevier, Amsterdam 1984) pp. 141–151
J.J. Mazer, C.M. Stevenson, W.L. Ebert, J.K. Bates: The experimental hydration of obsidian as a s function of relative humidity and temperature, Am. Antiq. 56, 504–513 (1991)
C.M. Stevenson, M.D. Dinsmore, B.E. Scheetz: An inter-laboratory comparison of hydration rim measurement, Int. Assoc. Obsidian Stud. Newsl. 1, 7–14 (1989)
T.F. Lynch, C.M. Stevenson: Obsidian hydration dating and temperature controls in the Punta Negra region of Northern Chile, Quarternary Res. 37, 117–124 (1992)
I. Liritzis, N. Laskaris: Fifty years of obsidian hydration dating in archaeology, J. Non-Cryst. Solids 357, 2011–2023 (2011)
C.M. Stevenson, S.W. Noak: Obsidian hydration dating by infrared spectroscopy: Method and calibration, J. Archaeol. Sci. 38, 1716–1726 (2011)
Y. Moriya, M. Nogami: Hydration of silicate glass in steam atmosphere, J. Non-Cryst. Solids 38, 667–672 (1980)
Y. Moriya, M. Nogami: Hydration of silicate glass in steam atmosphere, J. Non-Cryst. Solids 39, 667–672 (1980)
B. Grambow: Nuclear waste glasses – How durable?, Elements 2, 357–364 (2006)
J. Neeway, A. Abdelouas, B. Grambow, S. Schumacher: Dissolution mechanism of the SON68 reference nuclear waste glass: New data in dynamic system in silica saturation conditions, J. Nucl. Mater. 415, 31–37 (2011)
E. Vernaz, J. Bruezière: History of nuclear waste glass in France, Procedia Mater. Sci. 7, 3–9 (2014)
W.L. Gong, R.C. Ewing, L.M. Wang, E. Vernaz, J.K. Bates, W.L. Ebert: Secondary phase formation and the microstructural evolution of surface layers during vapor phase alteration of the French SON68 nuclear waste glass at 200 °C, Mater. Res. Soc. Symp. Proc. 412, 197–204 (1996)
H. Li, M. Tomozawa: Effects of water in simulated borosilicate-based nuclear waste glasses on their properties, J. Non-Cryst. Solids 195, 188–198 (1996)
K.M. Davis, A. Agarwal, M. Tomozawa, K. Hirao: Quantitative infrared spectroscopic measurement of hydroxyl concentrations in silica glass, J. Non-Cryst. Solids 203, 27–36 (1996)
A. Agarwal, K.M. Davis, M. Tomozawa: A simple IR spectroscopic method for determining fictive temperature of silica glasses, J. Non-Cryst. Solids 185, 191–198 (1995)
X. Feng, P.R. Hrma, J.H. Westsik Jr., N.R. Brown, M.J. Schweiger, H. Li, J.D. Vienna, G. Chen, G.F. Piepel, D.E. Smith, B.P. McGrail, S.E. Palmer, D. Kim, Y. Peng, W.K. Hahn, A.J. Bakel, W.L. Ebert, D.K. Peeler, C. Chang: Glass Optimization for Vitrification of Hanford Site Low Level Tank Waste (Pacific Northwest National Laboratory, Richland 1996), PNNL-10918
D. Kim, M.J. Schweiger, C.P. Rodriguez, W.C. Lepry, J.B. Lang, J.V. Crum, J.D. Vienna, F.C. Johnson, J.C. Marra, D.K. Peeler: Formulation and Characterization of Waste Glasses with Varying Processing Temperature (Pacific Northwest National Laboratory, Richland 2011), PNNL-20774
J.D. Vienna, A. Jiricka, B.P. McGrail, B.M. Jorgensen, D.E. Smith, B.R. Allen, J.C. Marra, D.K. Peeler, K.G. Brown, I.A. Reamer, W.L. Ebert: Hanford Immobilized LAW Product Acceptance: Initial Tanks Focus Area Testing Data Package (Pacific Northwest National Laboratory, Richland 2000), PNNL-13101
R.I. Schulz, T.H. Lorier, D.K. Peeler, K.G. Brown, I.A. Reamer, J.D. Vienna, A. Jiricka, B.M. Jorgensen, D.E. Smith: Hanford Immobilized LAW Product Acceptance: Tanks Focus Area Testing Data Package II (Pacific Northwest National Laboratory, Richland 2000), PNNL-13344
B.P. McGrail, J.P. Icenhower, P.F. Martin, H.T. Schaef, M.J. O'Hara, E.A. Rodriguez, J.L. Steele: Waste Form Release Data Package for the 2001 Immobilized Low-Activity Waste Performance Assessment (Pacific Northwest National Laboratory, Richland 2001), PNNL-13043
T.H. Lorier: Establishment of Vapor Hydration Test (VHT) Capabilities at the Savannah River Technology Center (Savannah River Technology Center, Savannah 2002), WSRC-2002-00572
P.R. Hrma, D.S. Kim, J.D. Vienna, J. Matyas, D.E. Smith, M.J. Schweiger, J.D. Yeager: Testing of Large-Scale ICV Glasses with Hanford LAW Simulant (Pacific Northwest National Laboratory, Richland 2005), PNNL-15107
G.J. Sevigny, M.L. Kimura, C.M. Fischer, M.J. Schweiger, C.P. Rodriguez, D. Kim, B.J. Riley: Iron Phosphate Glass-Containing Hanford Waste Simulant (Pacific Northwest National Laboratory, Richland 2012), PNNL-20670
A. Jiricka, J.D. Vienna, P. Hrma, D.M. Strachan: The effect of experimental conditions and evaluation techniques on the alteration of low activity glasses by vapor hydration, J. Non-Cryst. Solids 292, 25–43 (2001)
J. Neeway: The Alteration of the SON68 Reference Waste Glass in Silica Saturated Conditions and in the Presence of Water Vapor, Ph.D. Thesis (Univ. Nantes, Nantes 2011)
A. Abdelouas, Y. El Mendili, A. Aït Chaou, G. Karakurt, C. Hartnack: A preliminary investigation of the ISG glass vapor hydration, Int. J. Appl. Glass Sci. 4, 307–316 (2013)
R. Bouakkaz: Altération aqueuse et hydratation en phase vapeur du verre SON68 à basse température (35-90 °C), Ph.D. Thesis (École des Mines de Nantes, Nantes 2014)
R. Bouakkaz, A. Abdelouas, A. Aït Chaou, Y. El Mendili: A borosilicate glass hydration at low temperature, J. Chem. Pharm. Res. 7, 332–337 (2015)
P. Jollivet, J.L. Chouchan, J.P. Mestre, C. Marcou: Caractérisation du premier échantillon de verre altéré en phase vapeur au LCLT, Compte Rendu d'Essai (CEA Marcoule, Bagnols-sur-Cèze 2014)
T.A. Abrajano, J.K. Bates, J.J. Mazer: Aqueous corrosion of natural and nuclear waste glasses II, J. Non-Cryst. Solids 108, 269–288 (1989)
K.S. Pitzer, D.J. Bradley, P.S.Z. Rogers, J.C. Peiper: Thermodynamic of high temperature brines. In: American Society for Testing and Materials Symposium (Lawrence Berkeley Laboratory, Berkeley 1979), LBL-8973
A. Aït Chaou, A. Abdelouas, Y. El Mendili, R. Bouakkaz, S. Utsunomiya, C. Martin, X. Bourbon: Vapor hydration of a simulated borosilicate nuclear waste glass in unsaturated conditions at 50 °C and 90 °C, RSC Adv. 5, 64538–64549 (2015)
J.K. Bates, M.J. Steindler, B. Tani, F.G. Purcell: The hydration alteration of a commercial nuclear waste glass, Chem. Geol. 51, 79–87 (1985)
D.S. Kim, J.D. Vienna, P.R. Hrma, M.J. Schweiger, J. Matyas, J.V. Crum, D.E. Smith, G.J. Sevigny, W.C. Buchmiller, J.S. Tixier Jr, J.D. Yeager, K.B. Belew: Development and Testing of ICV Glasses for Hanford LAW (Pacific Northwest National Laboratory, Richland 2003), PNNL-14351
J.S. Luo, W.L. Ebert, J.J. Mazer, J.K. Bates: Simulation of natural corrosion by vapor hydration test: Seven-year results, Mater. Res. Soc. Symp. Proc. 465, 157–163 (1997)
W.L. Gong, L.M. Wang, R.C. Ewing, E. Vernaz, J.K. Bates, W.L. Ebert: Analytical electron microscopy study of surface layers formed on the French SON68 nuclear waste glass during vapor hydration at 200 °C, J. Nucl. Mater. 254, 249–265 (1998)
L.R. Riciputi, J.M. Elam, L.M. Anovitz, D.R. Cole: Obsidian diffusion dating by secondary ion mass spectrometry: A test using results from Mound 65, Chalco, Mexico, J. Archaeol. Sci. 29, 1055–1075 (2001)
I. Liritzis: SIMS-SS, a new obsidian hydration dating method: Analysis and theoretical principles, Archeometry 48, 533–547 (2005)
A.A. Kruger, S.K. Cooler, I. Joseph, I.L. Pegg, G.F. Piepel, H. Gan, I. Muller: ILAW PCT, VHT, Viscosity and Electrical Conductivity Model Development (Office of River Protection, Richland 2007), ORP-56502
A.C. Buechele, D.A. McKeown, W.W. Lukens, D.K. Shuh, I.L. Pegg: Tc and Re behavior in borosilicate waste glass vapor hydration tests II, J. Nucl. Mater. 429, 159–165 (2012)
K.M. Davis, M. Tomozawa: An infrared spectroscopic study of water-related species in silica glasses, J. Non-Cryst. Solids 201, 177–198 (1996)
A.M. Efimov, V.G. Pogareva, A.V. Shashkin: Water-related bands in the IR absorption spectra of silicate glasses, J. Non-Cryst. Solids 332, 93–114 (2003)
G. Navarra, I. Iliopoulos, V. Militello, S.G. Rotolo, M. Leone: OH-related infrared absorption bands in oxide glasses, J. Non-Cryst. Solids 351, 1796–1800 (2005)
I. Friedman, F.W. Trembour, G.I. Smith, F.L. Smith: Is Obsidian hydration dating affected by relative humidity?, Quaternary Res. 41, 185–190 (1994)
A. Aït Chaou, A. Abdelouas, Y. El Mendili, C. Martin: The role of pH in the vapor hydration at 175 °C of the French SON68 glass, Appl. Geochem. 76, 22–35 (2017)
D.L. Parkhurst, C.A.J. Appelo: User's Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations, Water-Resources Investig., Vol. Report 99-4259 (USGS, Denver 1999)
R.C. Ewing, W.J. Weber, F.W. Clinard Jr.: Radiation effects in nuclear waste forms for high-level radioactive waste, Prog. Nucl. Energy 29, 63–127 (1995)
K. Sun, L.M. Wang, R.C. Ewing, W.J. Weber: Electron irradiation induced phase separation in a sodium borosilicate glass, Nucl. Instrum. Methods Phys. Res. B 218, 368–374 (2004)
T. Suzuki-Muresan, J. Vandenborre, A. Abdelouas, B. Grambow, S. Utsunomiya: Studies of (Cs,Ba)-hollandite dissolution under gamma irradiation at 95 °C and at pH 2.5, 4.4 and 8.6, J. Nucl. Mater. 419, 281–290 (2011)
A. Abdelouas, S. Utsunomiya, T. Suzuki, B. Grambow, T. Advocat, F. Bart, R.C. Ewing: Effects of ionizing radiation on the hollandite structure-type: Ba0.85Cs0.26Al1.35Fe0.77Ti5.90O16, Am. Mineral. 93, 241–247 (2008)
G. Karakurt, A. Abdelouas, J.P. Guin, M. Nivard, T. Sauvage, M. Paris, J.F. Bardeau: Understanding of the mechanical and structural changes induced by alpha particles and heavy ions in the French simulated nuclear waste glass, J. Nucl. Mater. 475, 243–254 (2016)
J.K. Bates, W.L. Ebert, T.J. Gerding: Vapor hydration and subsequent leaching of transuranic-containing SRL and WV glasses. In: High Level Radioactive Waste Management 1990 (American Society of Civil Engineers, New York 1989) pp. 1095–1102
D.A. McKeown, A.C. Buechele, W.W. Lukens, D.K. Shuh, I.L. Pegg: Tc and Re behavior in borosilicate waste glass vapor hydration tests, Environ. Sci. Technol. 41, 431–436 (2007)
A.C. Buechele, S.T. Lai, I.L. Peg: Alteration phases on high-sodium waste glasses after short- and long-term hydration, Ceram. Trans. 107, 251–259 (2000)
W.L. Ebert, A.J. Bakel, N.R. Brown: Measurement of the glass dissolution rate in the presence of alteration phases. In: Proc. Int. Conf. Nucl. Hazard. Waste Manag., Spectrum‘96 (American Nuclear Society, La Grange Park 1996) pp. 569–575
J.K. Bates, M.J. Steindler: Alteration of nuclear waste glass by hydration, Mater. Res. Soc. Symp. Proc. 15, 83–90 (1982)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter
Cite this chapter
Abdelouas, A., Neeway, J., Grambow, B. (2019). Chemical Durability of Glasses. In: Musgraves, J.D., Hu, J., Calvez, L. (eds) Springer Handbook of Glass. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-319-93728-1_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-93728-1_12
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93726-7
Online ISBN: 978-3-319-93728-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)