
Abstract
A common criticism of “prebiotic chemistry research” is that it is done with starting materials that are too pure, in experiments that are too directed, to get results that are too scripted, under conditions that could never have existed on Earth. Planetary scientists in particular remark that these experiments often arise simply because a chemist has a “cool idea” and then pursues it without considering external factors, especially geological and planetary context. A growing literature addresses this criticism and is reviewed here. We assume a model where RNA emerged spontaneously from a prebiotic environment on early Earth, giving the planet its first access to Darwinism. This “RNA First Hypothesis” is not driven by the intrinsic prebiotic accessibility; quite the contrary, RNA is a “prebiotic chemist’s nightmare.” However, by assuming models for the accretion of the Earth, the formation of the Moon, and the acquisition of Earth’s “late veneer,” a reasonable geological model can be envisioned to deliver the organic precursors needed to form the nucleobases and ribose of RNA. A geological model having an environment with dry arid land under a carbon dioxide atmosphere receiving effluent from serpentinizing igneous rocks allows their conversion to nucleosides and nucleoside phosphates. Mineral elements including boron and molybdenum prevent organic material from devolving to form “tars” along the way. And dehydration and activation allows the formation of oligomeric RNA that can be stabilized by adsorption on available minerals.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
3.1 Introduction
The chemical reactions that led to the first Darwinian systems on Earth did not occur in Pyrex glassware. Rather, they occurred in a geological context, in rock assemblages, basins, or other environments constructed from and bounded by mineral species. The precursors for those reactions also formed in geological contexts, in rock formations or, for simpler molecules, in an atmosphere above early Earth or in space. Even in space, however, organics may have been made on mineral grains that later came to Earth via meteorites.
A common criticism made of classical prebiotic chemistry by planetary scientists is that this geological context is too severely ignored (Sahai et al. 2016). Even non-geologists, like Robert Shapiro, have been harsh. For example, Shapiro, criticizing classical prebiotic chemistry performed by organic chemists, likened it to a golfer who himself having played 18 holes, believes that the ball could have propelled itself around the same course on its own without human intervention (Shapiro 2007). In this view, classical prebiotic chemistry examined compounds that were too pure, in experiments that were too contrived, to get results that were too scripted from environments that could never have actually existed on a planet.
Some of this criticism is unfair. Chemists have good reason to work with pure organic substances and to regularly intervene in the ongoing chemistry to isolate and purify reaction products before putting them into the next Pyrex flask for the next step. The products of every reaction must be analyzed for anything to be learned. That analysis cannot be done if a reaction generates dozens (or hundreds) of products, each in small amounts. Last, to directly use an unanalyzed product mixture as the starting point for another reaction sequence is, by hard experience, a recipe for disaster.
Further, chemists who do seek guidance from the planetary science community quickly discover that that guidance is generally sparse, often contradictory, and usually supported by few data (Schoonen and Smirnov 2016). While progress is being made in ways that we will describe below in some detail, the Hadean planetary environment remains too unconstrained to dictate specific environments where prebiotic chemistry must operate. Prebiotic chemists must choose among various models for the early Earth, constrain their experiments by that choice, and change those constraints if/when the preferred model changes.
For these reasons, most chemists feel justified when we follow John Sutherland who, after suggesting that the complicating reactivity of aminooxazole might be mitigated by having it sublime into an atmosphere and then “rain into” a separate environment that fortuitously contained a substantial amount of glyceraldehyde, wrote that “although the issue of temporally separated supplies of glycolaldehyde and glyceraldehyde remains a problem, a number of situations could have arisen that would result in the conditions of heating and progressive dehydration followed by cooling, rehydration and ultraviolet irradiation. Comparative assessment of these models is beyond the scope of this work” (Powner et al. 2009).
A reverse criticism is also possible. Few geologists are conversant with the reactivity of complex organic molecules. Thus, much prebiotic chemistry that emerges from geology-focused laboratories does little more than reduce carbon dioxide, create methane or ammonia, or create low molecular weight carboxylic acids such as acetic acid and straight chain fatty acids (Schoonen et al. 2004). Unfortunately, even if we stipulate that all of these reactions occurred in the Hadean, problems remain that make the origin of life seem (nearly) impossible. These problems arise from problematic reactivities of more complex organic molecules, not in the production of acetate or ammonia.
For example, Robert Hazen, a particularly daring geologist in this respect, went so far as to incubate pyruvic acid (CH3CO-COOH) with minerals at high pressures of carbon dioxide, expecting to see carbon fixation (Hazen 2005). He expressed surprise to see that he had created only a complex mixture of organic products (“tar”) that defied analysis. This surprise reflected a lack of experience with the reactivity of pyruvic acid (an “alpha-keto acid”), which is especially prone to form tars. The same lack of experience applies to proposals for a mineral-supported “reverse citric acid cycle,” which feature prominently in the “wish list” of many early Earth scientists (Russell and Martin 2004). This process also has molecules that share a propensity to form tars well before they fix CO2.
Such is the situation with multidisciplinary research in general (Benner et al. 2013). This review seeks to describe the current relation of the Hadean geological environment to prebiotic chemistry. To direct this discussion, we have chosen just one model for the origin of Darwinism on Earth: the “RNA First Model.” This model holds that Darwinism emerged on Earth through the creation of RNA molecules that carried information that could be replicated, with errors, with the errors themselves being replicable. This is the simplest combination of features needed for a system to use random variation followed by natural selection to increase its information to capture increasingly large fractions of the surrounding resources in competition with other Darwinian systems, as well as to compete with spontaneous degradation and devolution.
3.1.1 Decisions Must Be Made
Any model for the origin of life requires that we make decisions about what geology was available at the time that life emerged. These decisions cannot possibly rely on any statement of “proof.” Leaving aside the fact that proof is unknown in science for any interesting proposition, it is especially unknown for historical events (“What happened?”) that occurred over 4 billion years ago (Ga). A decade ago, too few constraints were available for either the geology or the time of life emerging to have much of a productive discussion.
The combinatorial explosion of parameter space does not even allow us to say that we are “agnostic” with respect to key decisions. Avoiding decisions leaves us with too little guidance among too many possibilities. Nor are we allowed to declare that we disavow “belief” in our scientific thought processes and therefore are “objective” in our consideration of “all options.” In fact, humans cannot be ideally objective or absent of belief; to claim otherwise simply offers an example of the Feynman observation that people are easy to fool, and the easiest person to fool is yourself (Feynman 1974).
Rather, productive science requires that the believing and nonobjective scientist first understands what those beliefs and biases are. Science then becomes the exercise of managing those. Self-management is preferred, but if that fails, science has a mechanism for the community to do the needed management. This allows science to succeed even though it is done by scientists.
3.1.2 The Logic Of Plausibility
We outline here what decisions guide our assembly of a prebiological geo-origins model. Here, we must manage the word “plausible,” which appears in phrases such as “this organic compound is prebiotically plausible.” “Plausible” in the field of origins has come to have a cultural definition, which in turn does little more than expressing the desire of a scientist to have the compound available in a prebiotic environment.
A survey of the contexts where “plausible” is used is beyond the scope of this review, but some examples are useful. For example, if a molecule can be seen by microwave spectroscopy of a nebula in our galaxy where stellar formation is active, then it is often said to be “prebiotically plausible.” This is the case for cyanoacetylene and glycolaldehyde, two relatively complex molecules in addition to primitives such as water, methane, hydrogen cyanide, and carbon dioxide.
Assuming that molecules found in interstellar gas clouds were available on a prebiotic Earth is not entirely defensible. As discussed below, during both the initial accretion of the Earth and in the subsequent Moon-forming event, essentially none of these molecules would have survived in any form more complex than their constituent atoms.
More defensible would be the view that organic materials are delivered to Earth by meteorites, including the carbonaceous chondrites that continue to fall on Earth. These organics have been the focus of much analysis. Further, they arrived continuously following the cataclysmic events in the natural history of Earth, did not experience extreme heating upon arrival, and could indeed have offered a reservoir of organic material for prebiotic synthesis. They may be used to define what was “plausibly” available by way of organic species on early Earth.
Concerned about the amounts and distribution of these, we take a narrower view. Here, we chose to consider an organic species as “plausible” only if it can be generated by Earth-based processes.
3.1.3 The Correspondence Principle
The Correspondence Principle in origins is analogous to the Correspondence Principle in physics (Van Vleck 1928), which holds that as one moves from the quantum to the macroscopic, the predictions of quantum mechanics must converge on the predictions of classical mechanics. In the field of bio-origins, the analogous Correspondence Principle starts with modern life as the end point. It then extrapolates modern biological chemistry back in time to ancient biological chemistry (Benner et al. 1989). That extrapolation, supported by paleogenetic experiments (Jermann et al. 1995; Benner et al. 2002; Benner 2017b), suggests that the last common ancestor of all life that we know had these features:
-
1.
It used RNA in key catalytic reactions (such as in the biosynthesis of proteins).
-
2.
It exploited RNA cofactors in a metabolically diverse metabolism that included carbon-carbon bond forming reactions, oxidation reactions, reduction reactions, methyl transfer reactions, and phosphoryl transfer reactions.
-
3.
It used triphosphates as activated species to drive reactions that are thermodynamically unfavorable in water.
The Correspondence Principle here would argue that the origin of life coming from unknown chemistry produced RNA, RNA cofactors, and triphosphates at the outset. This would allow the uninterrupted emergence of the molecules that we observe in modern life from the unknown model that we are building for its origins. Further, this Correspondence Principle holds that the form of RNA that originated is the same as the RNA present in the most distant ancestor whose RNA we can infer the structure of. According to the Correspondence Principle, the RNA that emerged from the origins contained bridging oxygens (not nitrogens) in the phosphate backbone, which was built from four nucleotides (not six or more) (Benner et al. 2016) that included thymine (not 2-thiothymine), and the backbone was linked by way of the 3′- and 5′-oxygens, inter alia.
For example, many consider replacing ribose by a four-carbon sugar such as threose, perhaps because it is easier to make prebiotically than ribose (Orgel 2000b; Herdewijn 2001; Ichida et al. 2005). Hud and others consider alternative hydrophobic organic molecules as “molecular midwives” that, because of their intrinsic hydrophobic nature, help assemble a stack of nucleotides (Hud et al. 2007). Some time ago, Orgel all but abandoned triphosphates as high-energy building blocks for RNA because they did not polymerize well, replacing the pyrophosphate leaving group with 2-methylimidazole (Inoue and Orgel 1982).
In many cases, changing the target structure away from the RNA suggested by the Correspondence Principle creates something of a “mission creep.” For example, the Szostak laboratory, seeking to finally solve the half-century old problem of template directed “uncatalyzed” synthesis of RNA, moved from the methylimidazole of Orgel to an aminoimidazole leaving group (Li et al. 2017). They also replaced the 3′-oxygen of modern ribose by a 3′-nitrogen (Izgu et al. 2016) and got better performance from 2-thiothymidine instead of standard thymidine (Zhang et al. 2013). Some spectacular examples of success have emerged. However, much of the work seeking prebiotic routes to the modern RNA building blocks must be set aside, as 3′-amino-2-thiothymine ribonucleoside requires a different prebiotic synthesis “concept” than the ones that we have been working so hard to create.
Thus, targeting the exact structure of modern RNA is useful simply because it is a clear target. For example, considering some of the most productive collaborations in this field, the Sutherland laboratory, supported by Simons philanthropy, attempts to find ways to make nucleosides such as cytidine and thymidine, not thiothymidine, with oxygens on the 2′-, 3′-, and 5′- positions, not nitrogens, and with ribose as the sugar, not threose. Analogous statements can be made by the recent work of Carell, which generated adenosine attached to ribose by N-9, exactly as found in the RNA extrapolated for the most ancient life on Earth, not some other purine attached in some other way (Becker et al. 2016). Both laboratories consider their chemistry to be successful when it delivers these exact molecules.
In this review, we embrace the Correspondence Principle, in part because alternatives are being heavily explored by other laboratories, in part because we appreciate the clarity of the goal. Our model must deliver RNA building blocks exactly as they are found in modern life, put phosphates onto the correct positions, and condense to give the natural RNA, preferably initially, but certainly as an end product.
3.1.4 The “RNA First Hypothesis”
So, why is RNA our target? In part, it is because of the results cited above that arise if we extrapolate modern molecular biology back in time to infer the molecular biology of the last common ancestor of all life on Earth. However, the RNA First Hypothesis (RFH) was introduced by Alex Rich (1962) for other reasons. Rich saw this as a way to manage two problems in particular:
-
(a)
The “chicken or egg” problem, from terran molecular biology: Proteins are needed to make nucleic acids, while nucleic acids are needed to make proteins.
-
(b)
A lack of molecular mechanisms by which proteins might transfer their information to descendant proteins.
Rich proposed that if RNA had catalytic power, an RNA system could pass its information to descendants by RNA catalysis. This is the strong version of the “RNA World Hypothesis,” which holds only that an earlier episode of Darwinism on Earth (not necessarily the first) used RNA as its only genetically encoded component of biological catalysis. This hypothesis is supported by the ribosome and RNA cofactors, inter alia. In 1989, the Benner lab built a model for a metabolism of the last riboorganism based on an analysis of modern biochemistry (Benner et al. 1989).
The discovery by Cech, Altman, and others that RNA could catalyze the splicing of introns and the cleavage of pre-tRNA molecules strengthened this model (Cech and Bass 1986; Guerrier-Takada et al. 1983). RNA can catalyze reactions. Further, the structure of the ribosome showed what had been suspected from “wet” biochemistry years earlier: The machine that makes proteins across the known biosphere uses RNA as the catalyst (Cech 2000).
We have remarked on the challenges of obtaining a single biopolymer that can do both genetics and catalysis (Benner 2017a). A catalytic molecule must fold, so as to surround its substrates and its transition state. A genetic molecule should not fold, so that it can template the formation of its complement. A catalytic molecule should change its behavior rapidly with small changes in sequence so that it can effectively search sequence space. A genetic molecule should not change its behavior with changes in sequence, so as to not disrupt the biophysics that allow faithful transmission of genetic information. A catalytic molecule should have many building blocks, so that they can catalyze many different kinds of reactions. A genetic biopolymer should have few building blocks, as this allows for the highest fidelity of information transfer.
RNA is a biopolymer that appears to make an effective compromise between these competing molecular attributes. Proteins, by way of contrast, do not. Accordingly, RNA is a reasonable target biopolymer to first support Darwinism. This, in turn, allows us to reserve for later the prebiotic synthesis of amino acids and peptides. This reservation has strategic value, as amino acids and peptides are both more stable than RNA and RNA components, are easier to form abiotically, and are therefore far less challenging (and less interesting) targets.
3.1.5 What Makes a Model Persuasive
Accordingly, our goal is to construct a model for a process that converts compounds that were likely to have been available on early Earth, in environments that are likely to have been available on early Earth, and to support this by experimental work. We next must ask what we should consider as a “solution to the problem. When will these experiments end (Galison 1987)? What form must a proposed model have to be convincing to the communities that the problem has been “solved”?
This is, of course, a matter of culture, not logic. Here, however, we must also make a choice about what metrics we will use for progress. In this work, we have chosen the metric to be the number of steps that require human intervention as the process is performed in the laboratory, which translates into the continuity of the process. Of course, for the analogous process to work as a solution in the natural environment, no intervention at all is allowed.
3.2 The Planetary History
3.2.1 Choices Made Concerning Planetary History
Any model for the origin of life in a geological context requires that we make choices about what geology was available at the time that life emerged. A decade ago, too few constraints were available for either the geology of early Earth or the time when life emerged to have a productive discussion. However, today, the situation is changed. We rely on the preponderance of evidence to make these choices. We do not, because we cannot, rely on any concept of “proof.” Rather, we simply recognize that our view of the availability of prebiotic resources is quite different depending on those choices. It is preferable to make them explicitly than implicitly, writing out the evidence that we rely upon to make those choices.
3.2.1.1 Deciding What Cosmogenic Factors to Build into the Model
Several cosmogenic events are of special importance when we consider the possible geological context on early Earth where RNA might have emerged. We consider these individually.
3.2.1.1.1 Initial Accretion
It is generally accepted that the Earth was formed stepwise, with material from the pre-solar cloud of gas gravitationally collapsing to form “embryonic” planets having approximately the size of the Moon or, later, Mars, which then gravitationally accreted. While the specific material that formed the Earth can no longer be analyzed as such, meteorites that (until recently) escaped accretion are generally used as proxies for this material.
Interestingly, the Earth and its Moon both have isotopic compositions that are indistinguishable from enstatite meteorites (Javoy et al. 2010; Qin et al. 2010; Warren 2011; Zhang et al. 2012; Dauphas et al. 2015; Dauphas and Schauble 2016; Young et al. 2016). Enstatites are very reduced, with their iron being present as a metal or as a sulfide; essentially no iron oxide is present. Enstatites are named for their dominant mineral enstatite (MgSiO3), which is the end member of a series of pyroxene silicate minerals that grade to ferrosilite (FeSiO3). In a ratio of 9:1, this is a dominant mineral of the Earth’s mantle, including its peridotite, which features prominently in discussions of the origins of life. It is also observed with forsterite (Mg2SiO4) in spectra obtained from outside of the solar system (Molster et al. 2001).
These isotopic similarities between enstatites and Earth include 17O, which is the same in both the Earth and the Moon. However, these similarities also extend to isotopes that record different stages of the accretion of the Earth, including the isotopes of lithophilic (“rock loving”) elements such as oxygen, calcium, titanium, and neodymium; the moderately siderophilic (“iron loving”) elements chromium, nickel, and molybdenum; and strongly siderophilic elements such as ruthenium. This isotopic similarity suggests that the material that accreted to create the Earth was always dominated by “E-type” enstatite chondrites. These contributed perhaps half of the accreting material in the first 60% of the accretion, with the remaining accretion being essentially all enstatites.
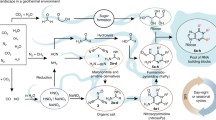
This model has the advantage of allowing the impactor that formed the Moon to have essentially the same isotopic ratio as the proto-Earth that it impacted. This, in turn, relaxes the constraints of models for lunar formation (Fig. 3.1).

Asteroid “21 Lutetia” appears to be an E-type chondrite of the type that dominated Earth’s early accretion. This image was obtained by ESA’s Rosetta mission. Source: ESA 2010 MPS for OSIRIS Team MPS/UPD/LAM/IAA/RSSD/INTA/UPM/DASP/IDA—https://www.flickr.com/photos/europeanspaceagency/4781143008/
3.2.1.1.2 Formation of the Moon
Many lines of evidence suggest that the Moon was formed by the impact on a growing Earth of a Mars-sized body that led to substantial vaporization of the rocks. The Moon congealed from this vapor. Because the vaporization was planetwide, little doubt exists that the origin of life must postdate the formation of the Moon. Analysis of the surface of the Moon, which has been preserved in composition (ignoring the time during which it was reworked by continuing impacts), is likely to give us as well an idea of the surface composition of the Earth immediately after the Moon was formed. Especially important are the potassium-rare earth element-phosphate (KREEP) species on the Moon.
The redox state (in geologist terms, the oxygen “fugacity” of the Hadean mantle) has been constrained by the analysis of zircon (ZrSiO4) crystals that have survived from the Hadean. Trail and his coworkers measured the ratio of Ce3+/Ce4+ in these zircons (Trail et al. 2011). They found that the mantle in which they were formed had an oxidation state similar to the oxidation state of the modern Earth. This corresponds to a fugacity near that of the fayalite-quartz-magnetite (FQM) redox state. This represents essentially complete loss of Fe0 to the core by this time. Other evidence suggests that surface water was also available deep in the Hadean.
3.2.1.1.3 The “Late Veneer” (LV)
When the metallic iron sank to form the core of the Earth, it took the siderophilic elements with it, leaving behind a crust and mantle that were depleted in these elements relative to chondrites (Becker 2006). Thus, the mantle and the crust are far more oxygenated than the core, are dominated by silicate, and have a higher redox potential, as measured by the fugacity of oxygen.
However, a puzzle arises because the abundances of siderophilic metals in the mantle and crust are much greater than predicted by experimental data that measure the partitioning of siderophilic elements between liquid iron and silicate (Mann et al. 2012; Rubie et al. 2015a, b). Indeed, the relative amounts of these siderophilic elements in the terrestrial and Martian mantels are nearly the same as those in chondrites (Brasser and Mojzsis 2017).
One solution to this puzzle is the assumption that after core formation was complete [~4.45 Ga (Allègre et al. 2008)], small-scale accretion [an additional 1% (weight)] delivered more siderophilic elements to the crust under conditions that did not give them the opportunity to descend into the iron core. This “late veneer” period remains controversial (Willbold et al. 2011, 2015; Touboul et al. 2012, 2014; Puchtel et al. 2014; cf. Righter et al. 2015) but is supported by the preponderance of evidence.
The amount of the siderophilic elements that made it to the Moon appears to be smaller, perhaps only 0.02 to 0.035 weight percent (Day and Walker 2015; Kruijer et al. 2015). This means that the Earth, relative to the Moon, got 1950 ± 650 times more of these siderophilic elements than would be expected based simply on their relative gravitational cross sections.
To explain this, various authors have suggested that the late veneer came from a very few, mostly large, later impacts, ranging in size from the asteroid 4 Vesta (535 km diameter) to our Moon. An impactor of this size would itself have had an iron core, with its siderophilic elements separated from its surface silicate shell.
Thus, as long as the iron core of the impactor did not hit the Earth “square on” (where its core would simply have joined Earth’s), the impact would have fragmented the impactor into smaller particles, shearing the core of the impactor into fragments of molten iron that would descent “in a hail” to the surface of the Earth (Brasser et al. 2016; Genda et al. 2017), together with its siderophilic elements. This accounts for the amounts of siderophilic elements “polluting” Earth’s mantle/crust and the relative paucity of these on the Moon. A glancing blow at 45 °C is, of course, at the center of the probability distribution of impact angles.
Why is this important to prebiotic chemistry? The delivery of molten iron to the Earth’s atmosphere after the core had formed, the initially accreted Fe0 had been lost to the core, the mantle had achieved a FQM redox state, and the oceans had formed, would give a pulse of reducing power in the atmosphere of the early Earth. The reaction between iron and water produces dihydrogen, and Genda et al. (2017) estimate that 90 bars of H2 would be produced for an impactor having the size of the Moon. Ultraviolet radiation from a young Sun would erode that H2 over a period of ~200 million years (Genda et al. 2017; Hamano et al. 2013).
This model appears to be supported by the preponderance of evidence, and we can constrain our prebiotic chemistry models using it. It has the prebiotically important consequence of creating an atmosphere that, at least for a time, is more reducing than the mantle and crust, and more reducing than the volcanic gasses that might emerge from the mantle and crust at a FQM redox state. This planetary thermodynamic disequilibrium can be exploited. An atmosphere with substantial amounts of H2 (and, therefore, some methane and ammonia) is a productive source of hydrogen cyanide (HCN), cyanamide (HNCNH), cyanoacetylene (HCCCN), and other species that we will incorporate into our model for the prebiotic generation of RNA. At the same time, it allows minerals to have higher oxidation states, including silicates, borates, phosphates, and molybdates.
This productive opportunity, however, is short-lived (if 200 million years is seen as a short period of time). For prebiotic chemistry to exploit it, it must occur within less than 200 million years after the impact that created the late veneer.
3.2.1.1.4 The Late Heavy Bombardment (LHB)
There is little doubt that as time went forward, the number of impacts on the Earth decreased as its local region of the solar system was “clean swept” by the Earth itself. What remains unclear is whether this first-order exponential decreasing of impacts was interrupted by a spike in impacts 3.9 to 4.1 Ga. Originally proposed in 1974 based on an analysis of lunar rocks collected by the Apollo mission (Tera et al. 1974), this “late heavy bombardment” (LHB) has been revisited often in the past four decades (Kring and Cohen 2002; Morbidelli et al. 2012; Marchi et al. 2013).
A LHB is troubling for prebiotic chemists, as it implies that the prebiotic sphere had less time to generate life than the full lifespan of the Earth. If the late bombardment of the Earth was heavy enough, it could have sterilized the planet (Maher and Stevenson 1988). Failing that, a late heavy bombardment would have forced the proto-biosphere into refugia that constrain the nature and diversity of its metabolism (Abramov and Mojzsis 2009).
This problem becomes only worse as carbon isotope ratios indicative of life are found in increasingly more ancient strata. Indeed, if the most ancient life-indicating 12C/13C ratios are accepted at face value at the dates as early as 4.1 Ga (Mojzsis et al. 1996; Rosing 1999; Bell et al. 2015), life would have effectively had to arise on Earth instantly after any late heavy bombardment, and possibly because of it.
However, a critical review of the literature finds that the evidence for the late heavy bombardment is equivocal, at best. The originally proposed “terminal lunar cataclysm” was based on 18 samples collected by the Apollo mission from a lunar highland where the U-Pb fractionation and rubidium-strontium isochrones dated them at ca. 3.9 Ga. These samples were interpreted as having come from “widely separated areas,” therefore implying multiple large impacts well after the formation of the Moon.
This view was immediately contradicted (Hartmann 1975). The contradiction noted that the few samples were not from widely separated areas. Further, it was noted that older lunar rocks would be hard to find based on the apparent history of cratering on the Moon. More recently, Chapman et al. reviewed this and other evidence, pointing out that the samples were likely to be strongly biased against older examples (Chapman et al. 2007).
Nevertheless, the late heavy bombardment remains a staple in the thinking of prebiotic chemists whose focus lies outside of geology. For example, the Sutherland group references it as contributing to several features of their prebiotic organic chemical models (Sutherland 2016).
Here, we choose a model that holds that the number and intensity of impacts declined smoothly over time, without any noticeable paucity of those impacts before 4.1 Ga, and no spike in those in the 4.1–3.9 Ga. In part, this is because of an absence of data to contradict the more reasonable model of monotonic decline. In particular, we find the analysis of Boehnken and Harrison (2016) sufficiently persuasive for us to discount the 40Ar/39Ar “plateau” dates; we see it likely that diffusion of this noble gas under repeated reworking of the lunar surface would create the “illusion” of a late bombardment. These 40Ar/39Ar data stand behind a number of current arguments for the LHB. Here, Moon-derived meteorites, which are presumed to come from all around the Moon, were exploited (Cohen et al. 2000; Kring and Cohen 2002; Marchi et al. 2013).
3.2.2 The Geological Consequence of the Model Derived from These Choices
We then turn to some interrelated questions that are addressed by this model for the early Earth that have relevance to prebiotic processes for the formation of RNA:
-
(A)
What was the global inventory of free water on Earth relative to the global topography? Specifically, did they permit dry land?
-
(B)
Related to this, what was the degree and nature mixing of materials within and between the crust and mantle, perhaps different from modern plate tectonics?
-
(C)
Related to this, what was the redox potential of the global crust and the global mantle, and the consequent composition and redox state of volcanic gasses and magma?
-
(D)
Related to this, what was the redox potential of the global atmosphere?
-
(E)
Related to this, what mineral species were available globally?
All of these questions have time-dependent answers; the states of the atmosphere, crust, mantle, topography, and other parameters were all likely different 4.5 Ga than 3.5 Ga, when life presumably had become a historical fact. However, because they ask about global averages, they are not complex questions. They are difficult to answer only because theoretical models for planetary accretion that might allow us to work “forward in time” are poorly constrained, physical records from the Hadean are largely lost, and “backwards in time” approaches from the biosphere become ambiguous well before the first living organism can be modeled.
Several classical views provide answers to these questions. The first argues that the inventory of water on Earth was high enough to threaten a flooded planet, especially if plate tectonics and other global processes had not created a substantial topography. Here, the classical answer held that plate tectonics took hundreds of millions of years to begin and may not have begun in the modern sense of the term until quite late, perhaps even requiring billions of years to get started (Condie 2018). This encourages the possibility that the early Earth was a “waterworld.” This, in turn, threatens the loss by dilution into a global ocean of any progress making RNA under prebiotic conditions.
Here again, we rely on data from crystals of Hadean zircon. Zircon is an exceptionally durable mineral that retains its primary chemistry with respect to most of its elemental constituents and isotopes from the time of its crystallization (Cherniak et al. 1997). This includes uranium and thorium, which are easily incorporated into the zircon crystal, while their radioactive daughter product lead atoms are not. This allows relatively precise dating of zircon crystals.
These zircons retain also the oxygen isotopes from the reservoir in which they are formed, and inclusions. Together, they suggest that the Earth did contain at least some dry land. Our model uses this dry land as a key locale to form RNA.
3.2.3 What Organic Species Are Available from the Atmosphere?
Dating back again to the time of Stanley Miller, many scientists have identified experimental conditions in which prebiotically interesting small organic molecules might be obtained by spark discharge, silent electrical discharge (Löb 1913), ultraviolet irradiation, or other energy sources. These are summarized in Table 3.1.
These small organics are believed to be useful to create RNA precursors. For example, purine and pyrimidine nucleobases may be formed from hydrogen cyanide (HCN) (Sekimoto and Takayama 2012) and NH4CN (Miyakawa et al. 2002), cyanoacetylene (Sanchez et al. 1966; Ferris et al. 1968), and cyanamide (NCNH2) (Sanchez and Orgel 1970; Levy et al. 1999) [but see critical evaluation in Shapiro (1995, 1999)]. These, in turn, are formed by lightning or UV in atmospheres containing CO2, N2, and H2O (Yuasa et al. 1984; Bada et al. 2016). Indeed, a Georgia Tech group developed an undergraduate laboratory teaching recipe for making prebiotic adenine from these (using diaminomaleonitrile instead of HCN) (Anumukonda et al. 2011).
Many modern models for the prebiotic origins of RNA components rely on these molecules. For example, Sutherland’s model for the prebiotic synthesis of cytidine requires cyanoacetylene (Powner et al. 2009). The Saladino model for the prebiotic formation of nucleobases depends heavily on the production of substantial amounts of formamide, which in turn requires large amounts of HCN to be formed (Saladino et al. 2003, 2011). Our model relies on formamide and/or urea as a solvent/reaction environment to solve the “water problem,” which reflects the instability of many bonds in RNA with respect to hydrolysis in water (Neveu et al. 2013).
The amounts of small organic molecules produced depend very much on the extent to which the atmosphere is reduced. For example, the production of HCN drops rapidly as the overall oxidation potential of the gas mixture increases (Pearce et al. 2017; Pearce and Pudritz 2015). Thus, the amounts of HCN required for various of these models are simply not available if the atmosphere has a redox potential that corresponds to the FQM redox potential of a mantle. We chose to manage this problem by relying on the model outlined above for the formation of the late veneer, which allows that atmosphere to be reducing enough to enable these RNA precursors to be formed in it.
3.2.4 Local Variation in Mineral Species Available from the Mantle and Crust
As discussed below, valuable minerals for our models for the prebiotic synthesis of RNA are obtained from a mantle having an FQM fugacity. In some cases, as with silicate and borate, the oxidation state comes naturally; elemental silicon and elemental boron are not likely to be found in any mineralogical environment other than iron metal. In some cases, the useful oxidation state seems accessible even in a more reducing environment. This includes ferrous iron (Fe2+) and phosphate, although a lower oxidation state of phosphorus is key to its mobilization by the mechanism proposed by Matthew Pasek (see below).
Global statements about planetary geology are not sufficient, however. More complex and, unfortunately, likely more relevant to the origins of life are questions that ask about the potential for early Earth to have diverse local environments. Even the simplest models for the emergence of RNA involve multiple models that have differing access to water, different redox potentials, and locally distinctive minerals. For example, the discontinuous model for the synthesis of RNA (Neveu et al. 2013) proposes:
-
1.
To create simple organic molecules (formaldehyde, hydrogen cyanide) in an atmosphere containing at least some methane, substantial carbon dioxide, and water.
-
2.
Then, have them rain into another environment, where they are transformed by percolation through a different environment, alkaline basalts containing olivine, igneous tourmaline, and igneous apatites (Piccoli and Candela 2002), followed by runoff water carrying their product to a different environment.
-
3.
Evaporate basins under an atmosphere rich in carbon dioxide and volcanic gasses, where borate, phosphate, and sulfate minerals are present.
Sleep pointed out that this change does not create a Shapiro-objectionable requirement for human intervention (Sleep et al. 2011). Except for the atmosphere, environments (2) and (3) are known on modern Earth near each other geographically.
Further, material easily moves from one environment to the other by gravity, even on modern Earth. For example, water percolating through rocks that contain olivine and tourmaline in the Sierra Nevada creates serpentinization, producing as much as 9.1 μM/kg per hour (200 °C, 300 bar) or 0.3 mmol/kg total inorganic carbon (Jones et al. 2010). The water flows out of these rocks into Death Valley, where borate from eroding tourmalines gives abundant borate minerals in the evaporite. Such a complex environment also appears to have been present on Mars, where remnants of evaporite minerals include gypsum facie containing borate minerals (http://www.manyworlds.space/index.php/(2016/12/14/with-the-discovery-of-boron-on-mars-the-package-of-chemicals-needed-for-life-is-complete/). If we can establish that such diversity in environments could have been present during the Hadean, the prebiotic chemist can go to work.
The possibility of proximal mineralogical diversity is key to prebiotic chemistry. Molecular systems do not see a planet. Indeed, nine orders of magnitude separate the nanometer dimensions of a molecule from the meter dimensions of a typical rock formation. However, no matter how difficult it is to model “global” properties of the Hadean, modeling local environments is worse. That modeling must begin with a picture of the Hadean Earth as a whole, suffer all of its ambiguities, and then tackle spatial variation, a large task indeed.
3.2.5 When Is a Mineral Considered Impossible
As interesting as a discussion about what minerals might be present is a discussion that guides us to expect that certain minerals might be excluded from the early Earth environment. For example, Hazen et al. (2008) have proposed various natural histories of mineral evolution. The idea is driven in part by empirical “mineral counting” in facie of different ages with various attempts at mitigating “preservation bias” (the fact that some minerals are harder, less soluble in water, or more likely to survive eons of geological time), sampling bias, or other factors that make it appear as if fewer minerals were present long ago than today. It is also driven by theoretical models that identify a dozen preplanetary refractory mineral species (“ur-minerals”) that might have been present on early Earth.
These authors have divided Earth’s history into three eras and ten stages of “mineral evolution.” Each has been proposed to have seen significant changes in the planet’s near-surface mineralogy, including increases in the number of mineral species; shifts in the distribution of those species; systemic changes in major, minor, and trace element and isotopic compositions of minerals; and the appearance of new mineral grain sizes, textures, and morphologies. These changes include changes that have been affected by the biosphere, including its production of atmospheric dioxygen.
We consider the likelihood for an early environment to contain specific minerals in the discussion below, each time that mineral is proposed to be part of a process that mitigates problems in the prebiotic chemistry of RNA. Interestingly, at the 2017 ISSOL meeting (the XVIII International Conference on the Origins of Life), Hazen introduced the “Benner rule” to mitigate the severity by which specific mineral species might be rejected through their incompatibility with the broader picture of mineral evolution. In this rule, Hazen noted that if Benner, or some other prebiotic chemist, found exceptional value in the reactivity of a mineral that was excluded by a global view of mineral evolution, he would consider revising that view. This implies that, given sufficient reasons to look for it, the mineral will actually be found.
3.3 Classical Literature on Minerals
Given these choices about the natural history of Earth, its atmospheric history, and the history of its crust and mantle, what can minerals do to help the prebiotic chemistry research based on the Correspondence Principle and the RNA First Model for the origins of Darwinism on Earth?
In 1947, two distinguished authors mentioned that clays might have been involved in early prebiotic chemistry on Earth as concentrators and catalysts. Unfortunately, these were both “false starts.” Thus, Bernal’s mention of clay was casual (Bernal 1951) and he never developed the suggestion. Goldschmidt, the founder of geochemistry, prepared a more elaborate analysis independently, published in 1952 after being edited by N. W. Pirie (Goldschmidt 1952). The article is prescient, mentioning the geological ubiquity of carbon dioxide, the chirality of quartz, and the possible prebiotic relevance of micas, carbonates, and clays. Unfortunately, Goldschmidt died before he could develop his insights.
Indeed, experiments from the classical period of prebiotic chemistry rarely began with a mineralogical context. Instead, these experiments focused on the organic molecules that might have emerged from the atmosphere. Löb (1913) examined an atmosphere model whose composition contained CO2. Miller (1953) was more influenced by the composition of the atmosphere of Jupiter and therefore examined more reducing environments. Oró provided an extensive review of these classical experiments (Oró 1965), all without any detailed discussion of minerals.
3.3.1 Classical Prebiotic Chemistry Involving Minerals: Clays
The first modern effort to bring geology into prebiotic chemistry also began with clays, but in roles more exotic than as concentrators and catalysts. Cairns-Smith mused on the challenges of obtaining biopolymers out of prebiotic soups, and commented favorably on the self-organizing properties of minerals. He then suggested that clays might themselves support Darwinism, replicating themselves. In this replication, the defects in their structures would hold information that is transmitted to descendant clays, with errors, where those errors could be further transmitted. This, he suggested, would allow a mineral species to evolve without a genetic biopolymer (Cairns-Smith 1977, 1982, 2005).
The Cairns-Smith approach proved, again unfortunately, to be a false start. No working system has yet emerged where minerals in two or three dimensions replace linear biopolymers to support Darwinism. However, it is not clear that much effort has actually been devoted to explore this possibility. Nevertheless, Cairns-Smith’s introduction of the term “genetic takeover” has had considerable impact, for example, on heroes in the field like Albert Eschenmoser (1997). The view, of course, requires renunciation of the Correspondence Principle.
James Ferris returned to explore the less exotic possible roles of clays in prebiotic chemistry in the decades to follow. His focus was montmorillonite [(Na,Ca)0.33(Al,Mg)2(Si4O10)(OH)2·nH2O]. This clay arises from the aqueous erosion of igneous rocks containing feldspars and therefore might have occurred on early Earth. Perhaps the most interesting outcome of his work was the observation that montmorillonite catalyzes the assembly of RNA building blocks having a 5′-phosphate activated by being pre-conjugated to an imidazole. Again, this imidazole was not the triphosphate that the Correspondence Principle would lead us to prefer. However, it is among the best examples for the formation of long RNA molecules without direction from a template (Ferris and Ertem 1992, 1993; Ertem and Ferris 1996, 1997; Ferris 2005).
Clay has allowed the coupling of RNA building blocks in other experiments. For example, Burcar et al. (2015) observed clay-based polymerization of RNA building blocks in the presence of a water-soluble carbodiimide reagent (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) that creates the imidazolide in water.
Montmorillonite has been explored by the prebiotic community as an adsorbent. For example, montmorillonite can bind to and adsorb macromolecular RNA, including catalytic RNA, protecting it from degrading agents and in some cases even enhancing its catalytic activity (Biondi et al. 2007a, b; Stephenson et al. 2016). The interactions between clays and liposomes have been studied (Hanczyc et al. 2003). Adsorption onto clays may protect other biomolecules (Ertem et al. 2017), including peptides (Lahav et al. 1978).
3.3.2 Classical Prebiotic Chemistry Involving Minerals: Pyrites and Other Sulfides
A separate line of research involving minerals in classical prebiotic chemistry focused on various transition metal sulfide minerals, whose redox potential allows them to transform organic molecules. This was popularized in the 1990s by Günter Wächtershäuser with a focus on pyrite. Here, ferrous ion (in the +2 oxidation state, e.g., FeS) reacts with hydrogen sulfide to form pyrite (FeS2, with iron in its +4 oxidation state) as it delivers electrons that can reduce organic species (Wächtershäuser 1988a, b, 1990a, b, 1993; Blöchl et al. 1992).
Huber carried this concept further by considering how this process on iron-nickel sulfide surfaces might activate amino acids to form peptides (Huber and Wächtershäuser 1997, 1998). Russell and his associates took this concept further, proposing that sulfide mineral assemblages near ocean floor hydrothermal vents might be venues for the origin of Darwinism (Russell et al. 1994; Russell and Hall 1997).
For the most part, experimental work with pyrites and other sulfides has delivered only simple compounds, such as acetate, methane, and ammonia (Brandes et al. 1998). However, many of these models, theoretically elaborated, lead to a “metabolism first” view for the origins of life. Here, rather than seeking to generate a genetic biopolymer by prebiotic chemistry, they propose to get a compositional form of Darwinism working through the reactions of assemblies of molecules (Markovitch and Lancet 2014). This work continues in prebiotic chemical research in the current millennium (Huber et al. 2003, 2012; Cody et al. 2000, 2001, 2004; Cody 2004, 2005).
At one level, we have no quarrel with the “metabolism first” concept. Even models that require a biopolymer to give biology its first access to standard Darwinism require a preceding set of connected organic transformations to generate the precursors for that biopolymer. The borate-molybdate formose process (see below) may be one example (Kim et al. 2011). However, as discussed below, such systems often “leak,” losing material nonproductively to give (again) tar. Further, it is difficult to conceive real chemical systems that are, by “compositional inheritance,” able to evolve to capture more resources to the exclusion of other systems (Anet 2004).
We have chosen not to pursue a “metabolism first” model in the work reviewed here. Nevertheless, we continue to be interested in “metabolism first” models that suggest actionable chemistry with specific organic molecules that might create “compositional inheritance.” We have frequently offered to synthesize these molecules and deliver them to anyone wishing to test the actionable idea. We renew this offer here.
3.3.3 Classical Prebiotic Chemistry Involving Minerals: Silicates, Oxides, and Others
In a third independent line of work, classical prebiotic chemistry proposed roles in the origin of life for silica-containing minerals. Quartz (SiO2), the basic silica mineral, was especially interesting because it forms chiral crystals. Adsorption of prebiotic molecules onto these was suggested as possibly being the origin of chirality in biomolecular chemistry, a suggestion mentioned by Goldschmidt (Bonner et al. 1974, 1975; Evgenii and Wolfram 2000).
The surfaces of more complex silica minerals have been suggested to be useful as containers, concentrators, activators, and stabilizers. This includes feldspars (Parsons et al. 1998).
Zeolites, with their large water content and large pores, have also been discussed (Smith 1998; Smith et al. 1999).
In still another line of research, the serpentinization of iron magnesium silicates is central to many models for early Earth chemistry, both on the land and beneath the ocean. Analogous to the Wächtershäuser pyrite formation, this serpentinization provides reducing power in many models for the reduction of CO2 and the formation of ammonia from a variety of oxidized nitrogen species (Berndt et al. 1996; McCollom and Seewald 2001; Summers and Chang 1993). Mineral oxides and hydroxides have also been discussed, again as concentrators and catalysts (Holm et al. 1993; Weber 1995).
3.4 How to Tackle the RNA First Problem Using Mineralogy
Several excellent reviews capture the state of affairs at the start of the current millennium (Lahav 1994; Orgel 2000a; Schoonen et al. 2004). Minerals do well at providing small organic molecules, but few that are needed to manage the problems of the RNA First Hypothesis for the origins of Darwinism.
Indeed, at a Gordon Conference on the origin of life in 2002, Stanley Miller remarked that, in his view, very little progress had been made in this direction. This was, of course, just a few years after he had published a study measuring the instability of carbohydrates, an instability that had driven him to conclude that “stability considerations preclude the use of ribose and other sugars as prebiotic reagents …. It follows that ribose and other sugars were not components of the first genetic material…” (Larralde et al. 1995). This study dampened enormously the enthusiasm of the community for the “RNA first” hypothesis for the origin of life on Earth.
This contrasts with the enthusiasm that dominated the field just 25 years earlier. For example, in 1969, Burton et al., citing Calvin, declared that “the successful ‘chance’ synthesis of nearly all the biologically important monomeric substances (amino acids, sugars, fatty acids, even purines and pyrimidines) has been accomplished” [italics added] (Calvin and Calvin 1964; Burton et al. 1969).
Instead, the situation seemed grim at the start of the new millennium. Nowhere is this stated more directly than by Bregestovski (2015). After noting that the model is the “most accepted and widespread contemporary scenario of prebiotic evolution that led to the emergence of the first cells on our planet,” Bregestovski remarked that the hypothesis “suffers from a number of insurmountable [italics added] problems of chemical and informational nature”. Among these, he included the
-
(a)
unreliability of the synthesis of starting components;
-
(b)
catastrophic increasing instability of the polynucleotide molecules as they elongate;
-
(c)
exceedingly low probability of meaningful sequences;
-
(d)
lack of the mechanism that would generate membrane-bound vesicles able to divide regularly and permeable to the nitrogenous bases and other RNA components;
-
(e)
absence of driving forces for the transition from the ‘RNA world’ to the much more complex ‘DNA-RNA world.’
The conclusion was captured in the title of the paper: “RNA World, a highly improbable scenario of the origin and early evolution of life on Earth.” Or, as Joyce and Orgel remarked in 1999, RNA is “the prebiotic chemist’s nightmare” (Joyce and Orgel 1999).
3.4.1 Paradoxes
One way to direct research in a field that approaches “big questions” (Benner 2009, 2013; Malaterre 2013) does not ask a historical question (“How did life arise in the solar system?”), but rather asks a “contrary to fact” question: “What generally accepted facts and theories suggest that it is impossible for life to have arisen in our solar system?” This framework focuses attention on places where existing geologic, chemical, and biological theory seems to rule out the possibility of life emerging, despite the fact that it evidently did. In doing so, a focus on apparent “paradoxes” redirects activity away from the approach where a chemist has what he/she thinks is a “cool idea” to make an RNA precursor, runs some experiments to show that it works in Pyrex, and then constructs an argument to explain why it might be “prebiotically plausible.”
The “RNA First” Model (Rich 1962; Benner et al. 1989) has many such apparent “paradoxes.” We list some of these below, ranking these in an “even if” order.
3.4.1.1 The Tar Paradox
As an observation well-established by chemists and non-chemists alike, organic molecules devolve to give complex mixtures if given energy and left to themselves. These products are “tars” or “asphalts,” better suited for paving roads than supporting Darwinism. Any scenario to create building blocks for RNA requires a way to allow organic material to escape this devolution. Here, we ask whether minerals can help.
3.4.1.2 The Phosphate Paradox
Even if precursors of RNA can escape devolution, one component of RNA seems geologically scarce according to generally accepted facts: phosphate. Free phosphate is likely scarce in alkaline serpentinizing prebiotic environments that contain calcium (Bach et al. 2006; Sleep et al. 2004, Andreani et al. 2013), as calcium and phosphate precipitate as insoluble minerals (e.g., Ca5(PO4)3(OH), hydroxyapatite). Yet some models for the prebiotic formation of RNA precursors require phosphate concentrations as high as 96 g/L (1 M) (Powner et al. 2009), since these models use phosphate as a buffer as well as a precursor to manage undesired reactivity of organic species that, absent large amounts of dissolved phosphate, would devolve to give tar. Can minerals help?
3.4.1.3 The Water Paradox
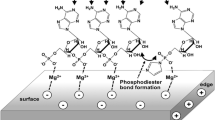
Even if organic precursors for RNA escape devolution, and even if phosphate is available to prevent this, many bonds in RNA are thermodynamically unstable in water (Fig. 3.2). This is also paradoxical, as the repeating phosphate backbone charge in RNA makes the molecule likely to perform only in water. The water-sensitive bonds might be made using high-energy dehydrating reagents, like carbodiimides. However, these themselves are unstable in water. Thus, any scenario for origins based on the RNA First Hypothesis must manage the apparent contradiction where life needs a solvent (water) that is inherently corrosive to the Darwinian biopolymer that is also needed. Can minerals help?

Red bonds in this generic RNA structure are thermodynamically unstable in water. They can be formed in water from “activated” species (e.g., the phosphates may be activated as triphosphates or imidazolides). However, these activated species must also be unstable in water
3.4.1.4 The Chirality Problem
Theory suggests that to be evolvable, the building blocks of a biopolymer must be homochiral, to allow the semicontinuous changes in the behavior of a biopolymer as it moves across a sequence landscape required for Darwinism (Benner 2017b). Illustrating with proteins, changing an amino acid from tryptophan to arginine (for example) already influences the physical properties of the biopolymer (hence, the “semi”). However, semicontinuous change is impossible if, in addition to changing the biophysical properties of a side chain, the change also switches the orientation of the side chain. Unfortunately, even if a prebiotic chemistry makes building blocks, not tar, with phosphates, it rarely generates them with common chirality. Can minerals help?
3.4.1.5 The Information-Need Paradox
The amount of information required for a chemical system to gain access to replication with imperfections that are themselves replicable is difficult to estimate. However, by any current theory, biopolymers that might support Darwinism seem to be too long to have arisen spontaneously from the amounts of building blocks that might have escaped devolution in water. If a biopolymer is assumed to be needed for Darwinism, some way is needed to obtain high enough concentrations of building blocks to allow thermodynamically tolerable assembly of those biopolymers with adequate amounts of information. Can minerals help?
3.4.1.6 The Biopolymer Stability Problem
Even if organic precursors avoid the tar problem, even if they gain access to phosphate to form the linking bonds, even if the phosphate linkers in RNA can be made by dehydration, even if the nucleoside groups linked all have the same chirality, and even if enough can be gathered to give an RNA system that supports Darwinism (Mutschler et al. 2015), the RNA must survive long enough to initiate Darwinism. Unfortunately, RNA is not very stable, especially at high Mg++ where experimental studies of catalytic RNA suggest that its power is the strongest. Can minerals help?
Clearly, if minerals are to help, they have their job set out for them.
3.4.2 Minerals as a Source of Prebiotic Components
We begin with a very simple problem: How might the phosphate “mineral” components of RNA have been extracted from a geological environment? Available phosphate is required as a linking group in RNA. Phosphate is also found in many RNA cofactors. And at least one model for the formation of an RNA precursor, from Sutherland, requires large amounts of dissolved phosphate to manage the “tar” paradox (Powner et al. 2009).
An abundant source of geological phosphate is apatite, a calcium phosphate that has a fluoride, chloride, or hydroxide group filling a spot in the structure to give chlorapatite, fluorapatite, and hydroxylapatite. Apatite has the general formula A5(XO4)3Z, where Z = Cl, Fl, or OH (and, less commonly, Br and I), and A represents Ca2+ or various cations that can substitute for Ca2+ in the structure (e.g., Sr2+, Pb2+, Ba2+, Mg2+, Mn2+, Fe2+, rare earth elements (REE)3+, Eu2+, Cd2+, and Na+). Hydroxyapatite is, of course, widely used in contemporary biology; it is a principal component of vertebrate bone.
Apatite is found in many environments. Igneous apatite is seen in mafic igneous rocks (i.e., basalts), but also in felsic rocks (i.e., andesite and rhyolite) and the ultramafic rocks of the Earth’s crust and mantle. It is also found on the Moon with whitlockite (Ca9(MgFe)(PO4)6PO3OH), in meteorites (Fuchs 1969), and on Mars (Santos et al. 2013).
The presence of the calcium phosphates on the Moon makes them likely to have been available to prebiotic chemistry early in the Hadean. This is also consistent with the behavior of apatite in igneous environments. Although apatite has low abundance overall reflecting the low abundance of phosphate relative to silicate and aluminate, it is enriched in igneous melts due to its low solubility and the limited capacity of common rock-forming minerals to accept phosphate into their structures (Piccoli and Candela 2002 op. cit.).
Thus, the “phosphate problem” exists in prebiotic chemistry not because phosphate rocks are likely absent from the Hadean, but rather because phosphate is generally seen to be difficult to extract from phosphate minerals, such as apatite. However, many phosphate minerals have low solubility. Even magnesium phosphate, a common source of phosphate in biochemical laboratories, has low solubility (2.27 × 10−3 g/L at 20 °C). Likewise, most transition metal phosphates have low solubility, as exemplified by ferrous phosphate (the mineral vivianite), copper phosphate (the mineral libenthite), and zinc phosphate (the mineral hopeite). Thus, calcium alone is not the only challenge to models that use high concentrations of dissolved phosphate to manage other paradoxes in the RNA First Model; strontium, barium, and all transition metals, including ferrous iron, create problems.
Multiple attempts were made in the prebiotic chemistry of the last millennium to mobilize phosphate from phosphate minerals, usually apatite. These generally failed. For this reason, in much of the classical literature, discussed below, apatite is seen primarily as useful for adsorbing and concentrating prebiotically interesting molecules, including prebiotic nucleoside phosphates. However, several solutions have been proposed to “the phosphate problem” in the new millennium.
3.4.2.1 Solutions to the Phosphate Problem: Acidification
While many phosphate salts are quite insoluble in water, hydrogen phosphates tend to be more soluble. This is, of course, well-known to those who experience dental disease, where bacteria secreting acid slowly dissolve tooth enamel calcium phosphate. Thus, calcium dihydrogen phosphate Ca(H2PO4)2 dissolves in water to ~ 18 g/L at 20 °C, contrasting with the estimated <0.02 g/L for apatite itself. Lowering the pH escapes the calcium precipitation “trap,” although it needs not rescue models that require one molar phosphate at pH 7.0 [the conditions used in Powner et al. (2009)].
The principal challenge in exploiting acidity as a way of mobilizing phosphate from insoluble phosphate minerals is to find a source of acid. An atmosphere of 100 bar CO2, considered possible for early Earth, would create a pH of ~4 in pure water oceans, absent buffering. This is sufficiently acidic to convert phosphate into mobile dihydrogen phosphate. However, the challenge comes in the next step. For phosphate ester bond formation to be thermodynamically accessible, the water must be evaporated. If the acidity comes from carbonic acid, evaporation would cause the carbonic acid to revert to carbon dioxide and water, effectively removing a proton in the process of raising the pH.
Volcanoes offer alternative sources of acidity, depending on the redox state of the crust and mantle. If the oxygen fugacity is sufficiently high (general geological question C), SO2 would be prominent among volcanic gasses. Sulfur dioxide reacts with water to produce sulfurous acid, which is acidic enough to also dissolve phosphate. Further, sulfurous acid would more easily remain behind upon evaporation than carbonic acid.
3.4.2.2 Solutions to the Phosphate Problem: Change the Redox State
In a very influential insight, Pasek pointed out some time ago that the low oxidation state of phosphorus in the meteoritic mineral schreibersite ((Fe,Ni)3P) could, upon corrosion, deliver phosphite and other low oxidation forms of phosphorus to an aquifer (Pasek 2016). Pasek estimated that 1–10% of the accessible phosphorus in the Hadean would have been in the form of schreibersite contributed by impact (general geological question E) (Gull et al. 2015). Phosphite minerals are generally more soluble than the corresponding phosphates (compare barium hydrogen phosphate and barium hydrogen phosphite at 0.1 and 6.9 g/L, respectively, at 20 °C). Further, phosphorus in the +3 oxidation state easily exchanges ligands to transiently form esters that could be stabilized by the subsequent oxidation of phosphorus +3 to phosphorus +5.
The Pasek model requires multiple geological microenvironments in proximity, of course. The first involves aqueous corrosion of low oxidation state phosphorus to give phosphite. The last requires oxidation. Minerals might provide the source of oxidation. For example, the manganese mineral pyrolusite (MnO2) has manganese at its +4 oxidation state, which might serve as an oxidant. If the volcanic gasses have the correct oxygen fugacity, elemental sulfur (S8) may also serve as an oxidant, giving the thiophosphate analog of RNA. Indeed, in solid-phase commercial DNA synthesis commonly used in the biotechnology industry, phosphorus in its 3+ oxidation state is converted to thiophosphate using elemental sulfur.
The challenge here is that in classical DNA synthesis, a phosphite triester is the intermediate. A phosphate diester, in contrast, does not have the same tautomeric form. Rather, it exists as RO-P(=O)(H)-OR. This does not behave well, making it a problematic intermediate in the assembly of oligonucleotides.
3.4.2.3 Solutions to the Phosphate Problem: Change the Solvent
The insolubility of phosphate minerals is, of course, discussed with reference to water as the solvent. Accordingly, one solution is to change the solvent. Some years back, we explored the ability of apatite to dissolve in formamide, without much success. However, more recently, the Hud group explored mixtures of semi-aqueous solvents in the hope of finding some that would mobilize the phosphate from apatite (Burcar et al. 2016).
Particularly successful were mixtures that combined urea with ammonium formate and water in a molar ratio of 1:2:4. Ammonium formate was proposed to be the end product of hydrolysis of HCN by way of formamide (Bada et al. 2016). Urea is produced in reactions (McCollom 2013), when ammonium cyanide (NH4CN) is exposed to sunlight (Lohrmann 1972), or via hydrolysis of cyanamide (HNCNH), which may be formed by electrical discharge in somewhat reducing N2-containing atmospheres (Fiore and Strazewski 2016). A small amount of formamide is apparently made upon heating the mixtures containing ammonium formate. Hud suggested that the composition of the mixture was stabilized around a eutectic point by evaporation of water and ammonium formate and/or loss of urea by precipitation.
Burcar et al. then tested the ability of both soluble phosphates (such as sodium phosphate) and insoluble phosphates (such as apatite) to donate phosphate to nucleosides in these mixtures. Perhaps as expected, the soluble phosphates gave mixtures of phosphorylated nucleosides at 65 °C over 19 days. No phosphorylation was seen with hydroxyapatite as a donor. However, when the temperature was raised, apatite also generated small amounts of phosphorylated products. The analogous phosphorylation was seen with glycerol as the phosphate acceptor. Further, addition of magnesium sulfate and free phosphate, at concentrations suggested to be comparable to those that dissolved apatite generated when treated with acidic heated surface waters, improved phosphorylation yields (Grosjean et al. 1995; Feely et al. 1998; Hedenquist et al. 2000).
While the reaction conditions here remain to be explored in greater depth, it appears as if the substantial part of the success is not due to changing the solvent per se, but rather due to the lowering of the pH in these mixtures, the mechanism outlined above. Thus, Burcar et al. report the formation of both struvite (MgNH4PO4• 6H2O) and newberyite (Mg(HPO4)· 3H2O), minerals reflecting a lower pH.
3.4.2.4 Solutions to the Phosphate Problem: Use a Realistically Complex Mineralogical Background
The mechanism of these reactions notwithstanding, Hud’s results show that mineral complexity in a geological environment has an impact on the outcome of organic reactions. Although today’s oxygen-containing atmosphere makes the surface and near-surface environments quite different in many respects for what could have been possible during the Hadean, Earth has plenty of environments to explore natural mineral complexity in natural planetary environments. Unfortunately, the literature is poorly indexed. Accordingly, a degree of random reading is necessary to come across, serendipitously, mineral assemblages that are interesting enough to lead to experimental work.
For example, random reading came across a report of terran evaporite formations that contained together in one locale gypsum (calcium sulfate, CaSO4•2H2O), boracite (Mg3(B7O13)Cl), and lüneburgite (Mg3B2(PO4)2(OH)6•8H2O) (Müller and Fabricius 1978). The occurrence in natural evaporites of a mineral containing calcium, but combined with sulfate (gypsum) rather than phosphate, and a mineral containing phosphate, but combined with magnesium and boron (lüneburgite) rather than calcium, suggested that phosphate in nature need not be irrevocably tied up with calcium, even in mineral environments rich in calcium, if borate is present. We therefore asked whether borophosphate evaporite mineral assemblages could address the “phosphate problem” by segregating calcium and phosphate. da Silva and Holm have addressed the larger possible prebiotic relevance of borophosphate minerals in general (da Silva and Holm 2014).
As with the Hud laboratory, we began with experiments that showed that hydroxyapatite does not dissolve to any measurable extent in formamide, despite formamide being an excellent solvent for many organic solutes. Parallel experiments showed that synthetic apatite does not phosphorylate nucleosides in urea under standard conditions.
We then asked whether lüneburgite itself could act as a phosphorylating agent in nonaqueous conditions, needed for phosphate esters to be formed thermodynamically. Synthetic lüneburgite was made by mixing and evaporating boric acid, phosphoric acid, sulfuric acid, calcium oxide, and magnesium oxide in the needed ratios. The resulting material was analyzed by powder X-ray diffraction. Absent calcium, lüneburgite and (with excess magnesium) periclase (MgO) were formed. However, if calcium was added, powder X-ray diffraction found that lüneburgite and gypsum segregated as separate minerals. This spontaneous removal of phosphate from calcium in the presence of borate, but not in the absence of borate, reproduced in the laboratory what was observed in natural evaporites. This segregation may be understood as a consequence of the relative solubilities of the mineral species involved.
But is phosphate more accessible from lüneburgite than from hydroxyapatite when nucleosides are available to be phosphorylated? Here, the affinity of borate for the diol units of ribose and ribonucleosides suggested that ribonucleoside phosphorylation targets might themselves extract the borate from lüneburgite, with the accompanying release of phosphate that then can be used. Ribonucleosides have 2′,3′-diol units that can bind borate.
Simple NMR experiments showed this to be true. While the amount of inorganic phosphate released to pure water from lüneburgite alone was very small, uridine nucleoside released phosphate in increasing amounts. By extracting the borate from the mineral, uridine made the lüneburgite phosphate soluble, even in the presence of calcium-containing gypsum.
However, release of phosphate to water does not address the thermodynamic instability of phosphate esters there. Thus, we asked whether lüneburgite itself could phosphorylate nucleosides in nonaqueous media. This proved to be the case in urea, when the water was evaporated at 85 °C for 16 h. This simulated desert was then flooded to give uridine and uridine-5′-phosphate (85:15) with few side products. The same was seen for the other three ribonucleosides G, A, and C.
Interestingly, borate extracted from lüneburgite by ribonucleosides to be phosphorylated also mitigated a part of the “tar problem” in the reaction. Without borate from lüneburgite (instead using sodium dihydrogen phosphate), complex phosphorylation product mixtures were found. With lüneburgite, by complexing the 2′- and 3′- oxygens, borate directs the phosphorylation reaction to the 5′-hydroxyl group, preventing complex product formation (Kim et al. 2016).
3.4.2.5 Borate Minerals to Mitigate the “Tar Problem”
Whether any of these offered solutions to the phosphate problem are acceptable is a cultural statement. However, the phosphate problem is “easy,” in that the phosphate unit is stable; it will not decompose. Nucleobases are similar, since they are aromatic ring systems whose kernels are stable under many conditions, although their amino functional groups can be lost by hydrolysis (Levy and Miller 1998).
However, the third entity, the ribose carbohydrate, is an archetypal source of tar. Indeed, as noted above, Stanley Miller concluded from the instability of carbohydrates that neither ribose nor any other sugar could be components of the first genetic material (Larralde et al. 1995).
Ribose (a 5-carbon sugar, or pentose C5H10O5) is not a problem because it cannot be formed. On the contrary, it has been known for a half century that ribose can be formed from formaldehyde under alkaline conditions (Appayee and Breslow 2014). Ribose and other carbohydrates (with general formulae CnH2nOn) are “polymers” of formaldehyde (HCHO, C1H2O1), which almost certainly form in prebiotic atmospheres containing CO2, H2O, and electrical discharge and/or ultraviolet radiation (Cleaves 2008). Thus, HCHO can be the sole starting material to form many carbohydrates in a process known as the “formose reaction” (Butlerov 1861). This requires Ca(OH)2 (pH of ~12.5) at elevated temperatures (65–80 °C).
Further, glycolaldehyde (C2H4O2) is formed by electrical discharge in moist atmospheres, albeit in smaller amounts than HCHO (Löb 1913; Harman et al. 2013). This “C2” species reacts with HCHO (a “C1 species”) to give glyceraldehyde (a “C3” species, C3H6O3). This C3 species can react with C2 glycolaldehyde to give C5 species, including ribose (Ricardo et al. 2004). For formose cycling (Fig. 3.3), glycolaldehyde is a “catalyst,” as one glycolaldehyde can fix many HCHO molecules. These reactions proceed on the hour time scale at pH ~10.5, somewhat milder conditions than for the formose reaction. Thus, the classical literature viewed the formose process as a solution to the problem of prebiotic carbohydrate formation (Dyson 1985).

Formose-like processes make useful carbohydrate. However, under conditions where they are formed, these react further, making the unmoderated process useless as a prebiotic source for ribose, the “R” in “RNA.” The Benner lab has synthesized many of these intermediates, including with 13C isotopic labels to trace reactions in this process (Benner and Kim 2015). Sedimentary minerals containing borate (e.g., kernite, ulexite, colemanite) constrain the complexity to the box by binding adjacent -OH groups in carbohydrate intermediates, giving the branched pentose C5b as a metastable product as its borate complex
Rather, ribose becomes a problem because it then goes on to react further under the conditions where it was formed to give complex mixtures of products containing only small amounts of ribose (Decker et al. 1982). As shown in Fig. 3.3, the reactions that can occur with carbohydrates are many and represent the carbohydrate version of the tar paradox. Thus, even if the formose process is manually supervised, it gives tar. And if it is unsupervised, anything productive reacts further to give saccharinic acids, furans, and other “tars”, especially after HCHO is depleted.
How does naturally complex mineralogy mitigate this problem? Here, as with the phosphorylation with lüneburgite described above, the ability of mineral borate to coordinate to the adjacent hydroxyl groups of organic species offers a solution. In a series of experiments, including those where intermediates in Fig. 3.3 were prepared with 13C substitution, Kim et al. dissected the formose processes (Kim et al. 2011). This dissection showed that minerals containing borate (e.g., kernite, ulexite, and colemanite) mitigate the intrinsic instability of ribose in several ways. First, for the reaction between glycolaldehyde and glyceraldehyde (i.e., C3 + C2 ➔ C5), borate stabilizes the ribose formed as the preferred species under the conditions where it was formed. The same can be happening if glyceraldehyde is made in situ from glycolaldehyde and formaldehyde (i.e., C1 + C2 ➔ C3). This is because ribose, in its cyclic hemiacetal form, presents two adjacent hydroxyl groups in a pre-organized conformation, allowing the resulting borate complex to be stabilized by an internal hydrogen bond. Indeed, the complex between ribose and borate is as stable as for any pentose.
This represents a partial solution to the carbohydrate “tar” problem with ≥25 mM borate. This work prompted Stephenson et al. to seek such concentrations of borate in clay of a Mars meteorite; these were found (Stephenson et al. 2013). These results, in turn, prompted the Mars Science Laboratory to seek borate in Gale Crater on Mars, where it was found associated with gypsum {http://www.manyworlds.space/index.php/2016/12/14/with-the-discovery-of-boron-on-mars-the-package-of-chemicals-needed-for-life-is-complete/}. This association is, of course, exactly the same as discussed above with lüneburgite.
However, as HCHO is produced in large excess over glycolaldehyde, the net stoichiometry of this reaction (2 glycolaldehydes +1 HCHO = only 1 ribose) implies that much of the HCHO generated in a prebiotic atmosphere will be wasted. Interestingly, experiments in our laboratory then found that borate can improve the stoichiometry (Neveu et al. 2013). This can be seen by extracting the portion of Fig. 3.3 contained within the magenta box. Here, just one molecule of glycolaldehyde presented with an excess of HCHO generates branched pentose C5b in a C2 + C1 + C1 + C1 ➔ C5 process.
At 50 mM (and higher) concentrations of borate, the C5b branched pentose is quite stable as its borate complex; indeed, unless Mars holds life to eat it, we expect that a pentose-borate complex will be found today on the surface of Mars as an organic mineral, where it will be continuously formed from HCHO formed by ultraviolet irradiation of the contemporary Martian atmosphere and delivered onto eroding basaltic rocks having igneous borate-containing tourmaline.
This stoichiometry can be improved, as C5b lacks an acidic proton adjacent to its C=O unit, making it unable to enolize. Kim showed that C5b can suffer a retroaldol reaction to give glycolaldehyde and glyceraldehyde (C5b ➔ C2 + C3, the vertical dotted arrows in Fig. 3.3). The resulting glycolaldehyde and glyceraldehyde molecules can then repeat the cycle, adding a total of five more HCHO molecules to give two C5b molecules. At the end of the next cycle, one glycolaldehyde has fixed eight HCHO molecules. Repeating the cycle, as many as 17 HCHO molecules can be fixed by a single molecule of glycolaldehyde.
But were borate minerals present on early Earth? For sure, borate minerals do not appear in Hazen’s list of “early minerals.” However, borates have now been found in 3.8 Ga Isua formations, and work in the Trail laboratory with Hadean zircons may yield information on this shortly.
3.4.2.6 Molybdate Chemistry and the Influence of Redox States
Unfortunately, although the pentose has the same collection of atoms as ribose, it is not ribose. Further, the retroaldol reaction fragments of C5b to give glycolaldehyde and glyceraldehyde place these two compounds “at risk” of further reaction into an unproductive manifold. Thus, the glycolaldehyde and glyceraldehyde molecules need not cooperatively react with formaldehyde to give ribose. Instead, two glycolaldehyde molecules might react with each other to create a 4-carbon sugar; two glyceraldehyde molecules might react with each other to create a 6-carbon sugar. Further, to allow the retroaldol reaction to occur requires that the concentration of borate (relative to C5b) drops to release C5b. This risks the loss of carbon to “tar” at high pH by leakage from the cycle.
One solution is to find mechanisms that would rearrange the atoms in C5b to give ribose without fragmenting the molecule. This would be done by carbon and hydride shifts that occur while the entire molecule is kept together and does not allow pieces to move apart. Can mineral species help do this?
Again, the answer is “yes.” Specifically, molybdenum will in its +6 oxidation state (molybdate, Mo6+) catalyze the rearrangement of C5b at pH 6–7 to give linear pentoses, including ribose, stereospecifically (C5b is either erythro or threo), without formation of “tar” (Fig. 3.4) (Petrus et al. 2001). Again, this requires movement of material from one environment to another. Here, once C5b is formed at high pH (~10) in aqueous media, it must move in the aquifer into an arid basin beneath an atmosphere rich in CO2. Here, the pH necessarily drops. The stability of the complex between C5b and borate drops as well as the pH drops. At pH 6–7, the rates of the tar-forming reactions of carbohydrates are quite slow, making stabilization by borate no longer necessary.

Mo(6+) catalyzes the stereospecific interconversion of pentoses (the Bilik reaction) at neutral pH, without risk of tar formation, giving linear carbohydrates. At this lower pH, only the most stable borate complexes form
But is molybdate accessible on early Earth? Molybdate minerals do not appear in Hazen’s list of “early minerals.” However, recent experiments suggest that molybdate minerals were easily present, given our knowledge of the fugacity in the mantle at times deep in the Hadean (general geological question C). Again, silicate melts at different oxygen fugacities have been studied experimentally in the laboratory, and the amount of Mo in either 4+ or 6+ oxidation states measured. Molybdenum is shown in quartz melts to transition from Mo+4 to Mo+6 one log unit just below the iron-wustite buffer (Fig. 3.5) (Borisov 2016). Trail’s work with zircons containing cerium has the terran mantle near the FMQ redox state, where Mo+6 > > Mo+4. Even Mars, more reducing overall than the Earth, may have had Mo+6. The fO2 of ALH 84001 [4.1 Ga by Lu-Hf dating (Lapen et al. 2010)] has been estimated to be at a higher oxidation state (~ FMQ −2.7) (Righter et al. 2008), where Mo(6+) dominates.

Molybdenum average valence as a function of oxygen fugacity relative to the iron-wüstite buffer. Experiments were done at 16 and 24 GPa (Danielson et al. 2011)
Thus, Mo6+ seems to be likely to have been present to prebiotic chemistry in analogous arid regions on Hadean Earth. Here, with borate minerals, it would have been available to mitigate the tar problem associated with carbohydrates.
3.4.3 Minerals as Reservoirs of Organic Materials
We have mentioned that borate-ribose and other species might be organic minerals that serve as reservoirs of unstable compounds, stabilizing them and concentrating them for later use. Organic minerals are scarce today on the surface of modern Earth, as they are easily food for organisms in the biosphere. Nevertheless, several are known where they survive rapid consumption by having stable mineral salts and low energy value.
For example, when graphitic carbon comes in contact with the modern dioxygen-containing atmosphere, it slowly is oxidized to a core benzenehexacarboxylic acid (mellitic acid) that is metastable with respect to further oxidation. Its aluminum salt is quite stable and appears as the mineral mellite.
Mellite has played a role in the exploration of Mars. Given that meteorites containing graphitic carbon fall to Mars continuously, the 1976 Viking mission that visited and explored Mars fully expected to find some organic material. The instrument designed to find it, however, looked for mass spectrometry signatures, volatile material that would emerge from the Martian soil on heating to temperatures as high as 400 °C. The volatiles from these organic materials were then passed in a stream of dihydrogen gas into a chromatography column; dihydrogen was then removed by a palladium separator and the residual material was injected into a mass spectrometer. The unexpected outcome was that no organic molecules were seen at all.
To explain the missing Mars organics, the immediate hypothesis was that photolysis of water by the ultraviolet light that penetrates to the surface of Mars would lead to hydroxyl radicals that “combust” the graphitic carbon. However, Benner et al. (2000) suggested an alternative explanation, based on the expectation that the graphitic carbon arriving by a meteorite every day to Mars would also be oxidized to the metastable mineral mellite (Boily et al. 2000a, b). Experiments showed that mellite was sufficiently stable so as not to yield substantial amounts of product upon heating under the Viking conditions.
Subsequently, the Mars Polar Lander discovered evidence of perchlorate salts in the Martian soil. Upon heating to 400 °C in the presence of perchlorate, organic matter combust, even in the form of mellite. The combustion products include chlorohydrocarbons; interestingly, these were seen by the Viking 1976 mass spectrometer.
Still other organic minerals on the modern Earth survive due to a combination of instabibility and lack of metabolic value. Materials are present where the already high level of oxidation makes them metastable with respect to further oxidation. Best known among these are minerals that contain oxalate, which is C2H2O4. Oxalate forms an insoluble salt with calcium; in medicine, such salts are commonly found in kidney stones. Further, just one oxidation state away from carbon dioxide, oxalate is not a valuable resource to a microbe even if it were able to metabolize it.
Of course, in the prebiotic world, organic minerals can survive without fear of being eaten by microorganisms. There, organic minerals become possible reservoirs, allowing the accumulation of organic species that would otherwise be transformed to tars over geological periods of time.
3.4.3.1 Organic Carbohydrate Minerals
Carbohydrates are the archetypal examples of organic molecules that react with each other to give unproductive products. This creates problems for prebiotic chemists.
For example, the 2009 model of Powner and Sutherland to form pyrimidine nucleoside phosphates requires large amounts of the C2 carbohydrate glycolaldehyde to await encounter with a large amount of cyanamide to form aminooxazole. Glycolaldehyde needs not wait for cyanamide to arrive, since it can react with itself to form threose and erythrose, and then tar (if the reaction is not stopped by human intervention) (Kim et al. 2011). Next, to manage the reactivity of resulting aminooxazole, Sutherland suggested that it sublimes to condense at a distant location that had substantial amounts of glyceraldehyde (Powner et al. 2009). Glyceraldehyde also reacts with itself (Kim et al. 2011) and may not wait for the arrival of aminooxazole before doing so.
Organic minerals may be exploited as “safe harbors” to allow carbohydrates to accumulate in unreactive form. Borate-ribose (Fig. 3.6) is one such mineral. Here, the stability of the complex arises because ribose forms a cyclic hemiacetal that presents three hydroxyl groups on the same side of the molecule. Borate coordinates tightly to these pre-organized cis-diols, with the five-ring complex stabilized by an internal hydrogen bond.

By making the geo-surrounding more complex by including volcanic SO2, bisulfite addition products (R-(CHOH)-SO3−) form with aldehydes, stabilizing these as organic minerals to give reservoirs of carbohydrate adducts for later use. The HCHO bisulfite addition product slowly bleeds HCHO into an alkali mixture of evolving carbohydrates, preventing many classes of reactions that destroy these (Fig. 3.7), while never allowing HCHO to reach levels where Cannizzaro destruction dominates. Thus, Hadean volcanoes mitigate a problem that Powner manages with an organic partner (Islam et al. 2017). Note how bisulfite addition to ribose competes with hemiacetal formation (right)
Smaller carbohydrates such as glycolaldehyde and glyceraldehyde are not as fortunate. Because these species cannot form cyclic hemiacetals that pre-organize cis-diols to present to borate for complexation, their borate complexes are less stable.
Even HCHO is a problem. It is likely to be formed in large amounts in a post-veneer atmosphere (Cleaves 2008). However, if present in sufficient concentrations at high pH, HCHO reacts with itself in the Cannizzaro reaction at a rate that scales with the square of its concentration to yield formate and methanol, neither having much prebiotic value (although MeOH might be a solvent and antifreeze).
To stabilize aldehydes, Powner recently suggested that aminothiazole might react with glycolaldehyde and glyceraldehyde to form imines that form stable crystalline minerals (Islam et al. 2017) (Fig. 3.6). However, mineral species may offer a better solution, if we make a geo-environment more realistically complex. For example, SO2 is expected to emerge from Hadean volcanoes under the model for planetary history that we have chosen. SO2 reacts reversibly with HCHO to give hydroxymethanesulfonate (HMS), which crystallizes in an evaporite. HMS is stable against self-reaction but, upon dissolution, slowly releases HCHO into a mixture, where it can react.
A constant source of low HCHO is an effective way to prevent carbohydrates in alkali from becoming “tar.” Here, any enediolate formed is trapped by HCHO (Kim et al. 2011) before it can react with another higher carbohydrate, or suffer beta-elimination, or form saccharinic acids (Fig. 3.7). This is especially true with borate, which moderates formation of enediolates.

Low levels of HCHO, slowly bled into a mixture from hydroxymethylsulfonate, manage the intrinsic propensity of carbohydrates to form “tars.” This arises from the ability of HCHO to capture enediol(ate)s before they have a chance to do undesired reactions (Kim et al. 2011)
3.4.3.2 Organic Minerals Incorporating the Nucleobases
Nucleobases likewise are species that are conceivably formed from hydrogen cyanide, cyanoacetylene, and other small molecules created in a sufficiently reducing Hadean atmosphere. However, their reaction with water generates hydrolysis products. An organic mineral that stabilizes them would be useful. Here, very little work has been done to explore the situation. However, complexes between purines and ferrous iron are known (Richter and Fischer 2003). These two might combine to create organic minerals to allow the accumulation of nucleobases in metastable forms.
Here, it is important to appreciate how choices made in our geological modeling influence how prebiotic chemistry gets driven. For example, Pearce and Pudritz (2015) are troubled from the fact that a Hadean atmosphere dominated by carbon dioxide, dinitrogen, sulfur dioxide, and water would not be very efficient at producing organic molecules. Pearce chose instead to assume that the Hadean atmosphere and lithosphere were in equilibrium with each other, and chose to have purines delivered by meteorite. This made their abundances very low, with the abundance of adenine in a warm little pond being called “negligible.” This was the case even if seepage from those ponds was turned off (Pearce et al. 2017).
We exploited recent models for the formation of the late veneer to manage the problem arising from the inefficient formation of purines in oxidized atmospheres. However, various organic minerals might be considered. For example, nucleobases form complexes with various transition metals. These may allow both their concentration and their stabilization.
3.4.3.3 Organic Minerals Incorporating Cyanide
Hydrogen cyanide is a key prebiotic material, serving as a precursor for nucleobases, amino acids and, in some models, even carbohydrates. It is formed best in reducing atmospheres, including those that may have existed above the Earth for about 200 million years following the late veneer. The formation of adenine is especially well studied. However, like formaldehyde, it is difficult to get HCN in high concentrations without itself reacting. The most prominent self-reaction product is the hydrogen cyanide polymer, which is even today poorly defined at a structural level.
Before the end of the last millennium, Keefe, Orgel, and others had noted the incompatibility between hydrogen cyanide and ferrous iron (Keefe and Miller 1996), which was likely to be the dominant oxidation state of iron in a prebiotic ocean, especially under the atmosphere proposed to have been formed during the late veneer. Cyanide reacts with ferrous iron to form ferrous ferrocyanide, well-known as the dye “Prussian blue.”
Sutherland recently offered an intriguing proposal that Prussian blue might be a reservoir for hydrogen cyanide, extending the concept to transition metal complexes with other quite unstable prebiotic materials, notably cyanoacetylene (Sutherland 2016). Williams and his group have pointed out that under a reducing atmosphere, ferrous iron (Fe2+) might be a replacement for Mg2+, which is commonly used today under an oxygen-rich atmosphere where iron is scarce due to the poor solubility of many ferric Fe3+ salts (Okafor et al. 2017).
This raises the general question of the incompatibility of mineral species and various needed prebiotic organic species. The incompatibility between ferrous iron and cyanide is easily resolved without the formation of Prussian blue. Indeed, depending on the concentration of dissolved ferrous iron, the hydrolysis of cyanide to give formamide and ammonium formate is faster than the formation of Prussian blue. Conversely, Prussian blue will hydrolyze to give ammonium formate. As noted above in the Hud work involving nucleoside phosphorylation, mixtures of formamide and ammonium formate are useful for other prebiotic chemistry problems. Further, as Saladino has shown with his colleagues (Saladino et al. 2003), formamide is as good as a precursor for many nucleobases as hydrogen cyanide. Further, formamide itself is a useful solvent for the discontinuous synthesis model for RNA prebiotic chemistry. This model uses formamide as a solvent to manage the water paradox in the assembly of nucleoside to nucleoside phosphates in the presence of borate minerals and urea.
3.4.4 Minerals as a Way to Stabilize Prebiotic Species That Manage to Be Formed
3.4.4.1 Adsorption onto Phosphate Minerals
Organic minerals allow the storage, one hopes in stable form, of organic species in bulk. They also require the existence of arid desert-like environments (general geology Questions A and B). However, an alternative mechanism to stabilize reactive organic species involves their adsorption onto the surfaces of minerals. This allows stabilization of considerably less material in the formation of a bulk organic mineral. However, as one proceeds toward oligomeric RNA, stabilization of even small amounts of material is likely useful.
Precisely because of their low solubility, phosphate minerals, especially apatite, were early candidates for those seeking minerals as to concentrate and catalyze the assembly of scarce organic building blocks to make biological polymers, including RNA, proteins, and even DNA. The more daring researchers considered the apatite group of minerals, which added arsenates and vanadates to phosphates. The resulting mineral collection has a hexagonal or pseudohexagonal monoclinic structure. The general formula is A5(BO4)3 (OH, F, Cl), where the A cations can be metal ions such as calcium, barium, sodium, lead, strontium, lanthanum, and/or cerium. The B cations can be phosphorus, vanadium, or arsenic. Carbonate and silicate anions can substitute to a limited extent for the BO4 groups. This group includes apatite itself, mimetite (a lead arsenate), pyromorphite (a lead phosphate), and vanadinite (a lead vanadate).
The need to concentrate originated in the thermodynamic impossibility of dehydrating building blocks in aqueous environment from dilute solution (“Paradox” 4). Fox had used high temperature for this purpose (Fox 1965). However, the surfaces of soluble minerals may give access to dehydrated products at lower temperatures.
For apatite, much of this was aided by a detailed understanding of the surface of apatite that was developed by Taves (Taves 1963; Taves and Reedy 1969). Thus, Neuman measured small amounts of adsorption of adenosine monophosphate on the surface of apatite. Adenine and adenosine did not adsorb. Therefore, Newman argued that apatite surfaces might allow the concentration of diluted nucleoside monophosphate from aqueous solution. Neuman carried these studies further, heating apatite-adsorbed monophosphates and observing formation of RNA like biopolymers. As the analytical methods were insufficiently advanced to characterize the products to modern standards, these results must be taken as tentative.
3.4.4.2 Adsorption onto Carbonate and Sulfate Minerals
It is easy enough to observe the adsorption of dissolved species onto solid surfaces. It is much more difficult to quantitate that adsorption. When two species interact in aqueous solution, their interaction energy can be determined by the concentration dependence of the two, an interaction that involves two rate processes, a biomolecular association process, and a unimolecular dissociation process.
However, when one of the two interacting components is a mineral, its concentration cannot be determined. Thus, one can easily measure the fraction of a dissolved species that becomes mineral-bound, but this has different meanings in different concentration regimes. Therefore, if the number of dissolved molecules in solution in total is larger than the number of binding sites on the surface of the mineral, then the fraction of solute bound must be less than unity, simply because the surface of the mineral is saturated. That fraction will increase by adding more mineral.
However, in the concentration regime where the amount of solute is less than the number of binding sites, the fraction bound can still be unity, if the affinity of the solute is quite high. Here, the binding rate can be assumed to be limited by the diffusion-controlled encounter of the solute with the surface. The affinity then can be usefully quantitated by measuring the rate of dissociation of the adsorbed solute into a solvent having infinite volume.
However, different solids have different levels of porosity, their surfaces have different levels of smoothness, steps on the surface can greatly alter the number of sites available, and these together make the actual number of sites unknowable. Attempts to determine these parameters by using, for example, noble gas adsorption on powdered minerals are often off point. The number of sites or crevices available to, for example, argon has little relevance to the number of sites on the surface that are available to, for example, an RNA molecule.
Adding to this challenge is the fact that no two natural mineral samples are identical. Natural minerals vary in chemical composition away from their canonical composition from specimen to specimen and certainly from locale to locale. For example, the name of apatite comes from the Greek word meaning “to deceive,” which reflects the fact that it comes in many different colors. These are due to different inclusions of atomic species that are not in the canonical formula of the mineral. This, of course, includes substitution of elements within the apatite crystal structures that are discussed above.
In our laboratory, Biondi et al. (2016) recently introduced a general strategy to manage these problems. In this work, we compared the adsorbance of radiolabeled RNA on to binary mineral species obtained in various ways.
In one, the species was precipitated as a synthetic mineral via a double decomposition reaction between the two chemical components. This allowed for an exact (and exacting) level of purity which is the kind of “controlled experiment” that chemists like. However, it may be criticized as being “artificial.” Moreover, solid phases with high surface areas, precipitates in particular, are general adsorbents for many molecules, especially macromolecules. Therefore, it is difficult to know, if RNA is seen to adsorb onto a surface, whether the adsorption is in any sense specific or whether it is just another example of a general feature that big molecules adsorb on big surfaces.
Thus, in a second approach, the mineral itself was obtained from a natural source, and the fraction of RNA bound to it was measured.
In a third approach, two precipitated minerals were combined, and the partition of radiolabeled RNA between the two was measured in a competition experiment.
This strategy then asked whether the trend in RNA adsorption was consistent across the same minerals in their various forms and presentations, especially within a set of minerals having a common anion (e.g., all carbonates) but differing in their cationic components (e.g., magnesium carbonate, calcium carbonate, strontium carbonate, and barium carbonate). Binary carbonate minerals within Row II (alkaline earth) cations were especially interesting, not only because carbon dioxide is likely to have been an abundant component of an early Earth atmosphere, but also because alkaline earths form a well-known set of binary carbonates that include insoluble magnesium, calcium, strontium, and barium carbonates (magnesite, calcite, strontianite, and witherite, respectively).
In the natural mineral experiments, we observed a periodic table trend. For example, while only a quarter of the radioactivity remained bound to the surface of the specimen of magnesite (with magnesium), ~94% of the reactivity was bound to the surface of the specimen of witherite (with barium). The fraction bound to calcite (calcium) and strontianite (strontium) were intermediate, 47% and 83%. Thus, a periodic table trend was observed with the binding of RNA to the carbonates relatively Ba > Sr > Ca > Mg. We then asked whether the same results could be qualitatively observed with precipitated synthetic minerals, and they could. Here, the percentage adsorption ranged from 95% to 77%, again with the ranking Ba > Sr > Ca > Mg.
To complete the analysis, various pairs of synthetic carbonates were then co-precipitated by mixing aqueous solutions of the alkali metal chlorides with an aqueous solution of sodium carbonate in a 1:1 ratio. These were then separated gravitationally, as the different carbonates have different densities (CaCO3 2.71 g/cm3, MgCO3 2.96 g/cm3, SrCO3 3.5 g/cm3, BaCO3 4.29 g/cm3). The partition of RNA between each pair was then observed by pre-equilibration of the radiolabeled RNA in a column with the two minerals, followed by freezing and dissection of the column, counting various slices within it. The labeled RNA partitioned as before; Ba > Sr > Ca > Mg.
As the rationale, if the same trends are observed both in precipitated synthetic minerals and in natural minerals, and if RNA is partitioned consistently between two mineral species precipitated together, the effects cannot easily be nonspecific general adsorption of big biopolymers on to big surfaces. Rather they must be intrinsic statements about the affinity of RNA for the different mineral species.
A series of sulfate minerals was more difficult to obtain as binary alkaline earth minerals, since the first member of the series (magnesium sulfate, epsomite) is quite soluble in water. However, with precipitated synthetic minerals, the same trend was observed, with barium sulfate binding RNA more than strontium sulfate, which bound more RNA than calcium sulfate. The corresponding trend was also observed in the specimens of the natural minerals, with barite > celestine > gypsum (59% > 49% > 20%).
While a periodic table trend is easy to observe in these cases, there is no reason a priori why such a trend should exist. For example, one might speculate that RNA would adsorb better onto a surface if the pattern of anion and cation sites on the surface matched more closely the distances of the anionic sites (phosphates) on the RNA molecule. While one might expect different cations in a mineral would change the spacing of those sites, there is no reason why the heaviest cation would have sites that match RNA the best. Indeed, if this were the mechanism for different surfaces having different affinities for RNA, one might expect a trend to have a mineral that maximally adsorbs somewhere in the middle of the series, rather the end of the series.
This may have been observed. For example, among the precipitated phosphates, the calcium species bound RNA (93%) more than the magnesium phosphate (64%), the strontium phosphate (84%), and barium phosphate (32%). Since “strontium apatite” and “barium apatite” are very seldom found in nature, natural minerals were not available for this study.
Another layer of complexity comes from the fact that same atoms can form different crystal forms. For example, calcium carbonate can precipitate as calcite, aragonite, or vaterite. Calcite crystallizes a trigonal space group; aragonite and vaterite are orthorhombic (Dal Negro and Ungaretti 1971; Maslen et al. 1993). Calcite is the more stable and consequently most common phase, while aragonite is less common, although it does occur in nature as a metastable phase (Fyfe and Bischoff 1965). Vaterite, also known as μ-CaCO3, is a third metastable phase of CaCO3. It occurs much less commonly in nature because it is the least thermodynamically stable. It generally and rapidly transforms itself into one of the other two forms (Grasby 2003). Vaterite is mostly seen when biological systems intervened to precipitate calcium carbonate.
To complete the analysis of the CaCO3 system, natural specimens of aragonite and calcite were examined. Aragonite consistently adsorbed more radiolabeled RNA than calcite. To obtain a synthetic mineral by precipitation, we reasoned that if RNA prefers to bind to aragonite over calcite, then perhaps RNA would nucleate the formation of an aragonite precipitate over a calcite precipitate.
In forming the minerals synthetically, calcite dominates CaCO3 that precipitates upon mixing CaCl2 and Na2CO3 in water at near-neutral pH and room temperature and pressure; absent contaminant (McCauley and Roy 1974), aragonite is not formed. We easily reproduced this general result, establishing the structure of the precipitated phases that we obtained by both staining with Feigl stain (silver sulfate and manganese sulfate) (Feigl 1937) and by powder X-ray diffraction.
Initial results were auspicious. Feigl stain suggested that CaCO3 precipitated preferentially as aragonite in the presence of RNA.
We then did powder X-ray diffraction to confirm the crystalline form of the precipitated calcium carbonate. Here, results were variable, but the precipitate formed in the presence of RNA was often identified as being primarily vaterite. We do not have a molecular interpretation of these observations, but this might be a first example of RNA-induced mineral polymorphism.
Interestingly, we also found that RNA bound to aragonite was more thermally stable than the same RNA in aqueous solution. Indeed, ~70% of the RNA bound to aragonite remained full-length after incubation at 95 °C for 2 h, while RNA treated the same way but in aqueous solution showed high levels of degradation.
Thus, we showed that, where it is possible, a comparison of natural minerals, synthetically precipitated minerals, and co-precipitated mineral combinations can be used to drive the conclusion that the adsorbance data collected are relevant to the mineral species themselves and do not merely reflect the adherence of large macromolecules to large surfaces.
The most obvious limitation of this comparative approach comes from nature herself. The rarity of minerals having different elemental compositions determines their availability for these experiments. Some elemental compositions are simply not found in nature at all.
The most striking outcome of these results is the periodic table relationship in the adsorbance of RNA to alkaline earth minerals, both the carbonates and the sulfates. All of these minerals are likely to have been present on early Earth. They are also known on Mars (Ehlmann et al. 2008). Today, most calcium carbonate is the result of biological activity. Where that activity is not present today (perhaps, but perhaps not, on Mars), we might expect to find stabilized RNA formed abiologically.
3.4.4.3 Adsorption onto Opals
Oxygen and silicon are the first and second most abundant elements in Earth’s crust (Cox 1989). Therefore, it is not surprising that Earth’s near-surface geology is dominated by silicate minerals. Indeed, species containing only silicon and oxygen (“silica”) come in multiple forms, including quartz (Agricola 1530), perhaps best known as large crystals forming from supercritical water at high pressures deep in the crust. However, at lower pressures near the Earth’s surface, alkaline water dissolves silica in small amounts, from which silica is reprecipitated. In the first phases of reprecipitating silica, the result is “opal.”
Thus, it is not surprising that silica has attracted the attention of those interested in the interactions between minerals and organic species during the emergence of the first life on Earth (Kim and Benner 2010; Lambert et al. 2010a, b).
Opal has a very high surface area, raising the possibility that silica might adsorb RNA (Nishiyama et al. 2013). Further, it has long been known that nucleic acids are adsorbed on glass substrates in prep kits that recover small amounts of DNA/RNA from complex biological mixtures (Decher et al. 1994). These, of course, involve chaotropic salts unlikely to have been present on early Earth in abundance.
In our laboratory, we asked, much like we did for carbonates above, whether silica adsorption might have played a role in the stabilization of RNA (Biondi et al. 2016).
Here we showed that synthetic opal does indeed adsorb RNA, surprisingly, much better than to amorphous silica prior to aqueous reworking (ASPAR). Further, RNA adsorbed to opal remains intact, as shown by its ability to give full-length products using reverse transcriptase. Full-length product could also be recovered from ASPAR, but RT-PCR was required to obtain the product in detectable amounts.
For these experiments, opal and amorphous silica were created using a modified Stöber process (Stöber et al. 1968) by acid-catalyzed hydrolysis of tetraethoxysilane. This initially forms fully amorphous SiO2. Incubation with water over periods of months allows the reworking of the silica to give natural-like opal. These two types of silica are ~ 30% porous, with pore sizes of ~ 20 Å for amorphous silica and ~ 90 Å for opal. For comparison, we note that RNA nucleotide averages ~12 Å in dimension, measured from the Watson-Crick edge of the nucleobase to the phosphate.
Adsorption on SiO2 surfaces was measured by using a 32P-5′-labeled 83 nt long RNA molecule. For opal, the fraction of RNA tightly bound to the mineral was calculated to be 1.355 pmoles/cm2 of surface (a calculation that ignored possible movement of RNA from the surface into the bulk solid material). Even a wash treatment with NaF pH 9.5 released only ~5.4% total of the bound radiation after 2 days incubation. As the label is still associated with macromolecular RNA, this was consistent with the protection of RNA by silica against alkaline degradation. For amorphous SiO2 on the other hand, adsorption was only 0.32 pmoles/cm2, about a quarter of what was seen with the synthetic opal. This lower adsorption might be ascribed to the smaller pore size of this mineral compared to the opal version of SiO2. This would also indicate either that the (main) RNA-mineral interaction is based on occupation of mineral pores, or that the total surface was inaccessible by way of the small pores (20 Å) to large molecules such as RNA, compared to the pore size in structured opal of ~ 90 Å. Considering that the 83 nt long RNA molecule used in this study might range from 228 Å in size for the fully relaxed (but mostly double-stranded) form to 48 Å in diameter in a fully folded form [calculated from the Flory law of gyration radius: RG = 5.5x(number of nts)1/3 (Hyeon et al. 2006)], the RNA could only partially enter the 20 Å pores of SiO2 rod through its axial single- or double-stranded edges (10–20 Å), while it could gain access to the pores of opal in various degrees of folding.
The hypothesis that RNA in SiO2 rod is adsorbed via the small pores of the mineral was also supported by results of reverse transcription of the adsorbed RNA. This was used to demonstrate that full-length material was adsorbed. Reverse transcripts were in good yields, even after 2 days incubation in NaF pH 9.5, indicating that the adsorption mechanism did not compromise the integrity of the molecule, but rather stabilized the RNA against alkaline degradation.
On the other hand, with amorphous SiO2, full-length reverse transcripts could not be obtained in detectable amounts, even though shorter amplified molecular fragments could be observed. Full-length cDNA could only be detected after 25 cycles of PCR amplification. This indicated that very little intact RNA survived.
These results suggested that as silica progresses from its completely amorphous state (the state presumably created the instant that a tetraalkoxysilane derivative hydrolyzes) to more organized (but still bulk amorphous) states, RNA oligonucleotides are better able to be adsorbed and stabilized. These experiments do not allow us to clearly distinguish, as an explanation of these results, between the possibility that the greater crystalline order after months of aqueous reworking is directly responsible for the improved adsorption/stabilization, or whether it is indirectly responsible as a result of greater pore size, greater accessible surface area, or both.
Interestingly, very little RNA adsorbed on the surface of a fully grown natural quartz crystal (here, a natural “Herkimer diamond”) or a natural agate. Natural obsidian (a volcanic glass) adsorbed some RNA better. However, among the natural silica species, natural opals adsorbed the best (Biondi et al. 2017).
In the “discontinuous synthesis model” for the origin of RNA (Neveu et al. 2013), RNA emerged in dry valleys fed intermittently by aqueous runoff from basaltic high lands, such as valleys currently found on Mars or Death Valley in California. It is in exactly these types of dry valleys that silica minerals would be reworked to give opals. Indeed, much of the gem opals found widely in jewelry are created in these environments. Through results reported here that show the stabilization of RNA in the environments on these minerals, additional steps might be added to this model, providing stabilized oligoribonucleotides that might be useful as starting points for molecular Darwinism. Indeed, the length of the 83 nt RNA molecule reported in this study, which is stabilized on opal, is comparable to the length of the RNA that Holliger, Joyce, and others suggest might be able to catalyze its own self-replication.
3.5 The Big Picture: The Discontinuous Synthesis Model for the Prebiotic Formation of RNA
We are now prepared to put this together in a “big picture,” which we have called the “discontinuous synthesis model” (DSM) for the prebiotic formation of RNA in a mineral environment (Fig. 3.8) (Neveu et al. 2013). Because it incorporates ideas and data from many labs, the DSM has no particular ownership. It requires these elements:

The Discontinuous Synthesis Model (DSM) (Neveu et al. 2013) for RNA formation, based on results from labs worldwide, with atmospheric steps (green), steps in water environments (blue), and steps in nonaqueous “exotic” solvents such as formamide and urea. Each step has experimental support, with key papers indicated. First, HCHO, glycolaldehyde, NCNH2 (cyanamide), and HCN are formed in a CO2-CH4-N2-H2O post-veneer atmosphere by UV, electrical discharge, silent discharge (Löb 1913), and cosmic energy (Pinto et al. 1980; Holland 1984). These are rained into aquifers in contact with serpentinizing basalts containing peridotite and igneous tourmalines, which create aqueous alkali that convert HCN to formamide and ammonium formate, cyanamide to urea, and formaldehyde to carbohydrates, the last being stabilized as their borate complexes. The effluent is then delivered to an arid evaporite at −20 to 60 °C, where atmospheric CO2 lowers the pH to ~ 6.5 (Sleep et al. 2011), allowing formation of polyphosphates and phosphate-formate anhydrides and, with Mo6+, in turn allowing Bilik reactions for carbohydrate processing (Kim et al. 2011; Petrus et al. 2001). Desert dehydration enriches exotic solvents such as formamide and urea, also precursors for nucleobases in local geothermal environments (Anumukonda et al. 2011; Yuasa et al. 1984). Nucleosides arise by Becker et al. (2016), Powner et al. (2009), or Kim and Benner (2017) routes. Next, borophosphates (e.g., lüneburgite) form 5′-nucleoside phosphates. Later, mixed anhydrides (formate, phosphate, thiophosphate) generate oligomeric RNA, adsorbing in homochiral form on mineral surfaces and/or stabilized by adsorption by opals (Biondi et al. 2016). Flooding of evaporite basin returns water, leading to the hydrolysis of formate esters and N-formylated nucleobases (half-life ∼1 month at 80 °C, pH 6). Subsequent steps involving catalysis by oligomeric RNA rely on work from Lehman, Szostak, and Joyce (Lehman 2003; Lincoln and Joyce 2009)
-
1.
A basaltic watershed region containing serpentinizing olivines, igneous apatites, and igneous tourmalines that are eroded by rain falling from a post-veneer atmosphere containing HCHO, HNCNH, HCN, HCCCN, and trace glycolaldehyde (HOCH2CHO). The liquid is alkaline (pH 10–11) and contains borate at 35–100 mM concentrations; it supports the aqueous processes in the DSM (blue in Fig. 3.8), including the borate-moderated formose process (Kim et al. 2011) that creates borate-carbohydrate complexes. These conditions also support hydrolysis of HCN and NCNH2, generating formamide, urea, ammonium formate, and carbonate.
-
2.
The effluent emerges into an arid evaporite environment with intermittent water. When water evaporates, formamide, ammonium formate, and urea remain behind. Formamide boils with decomposition at over 200 °C at 1 atm; it is conceivably a liquid on the surface of Mars today.
-
3.
Temperatures in the arid environment range from −20 to 60 °C. However, when encountering local geothermal environments, higher temperatures allow formamide, ammonium formate, and other products derived from HCN (e.g., diaminomaleonitrile, DAMN) to form nucleobases (Saladino et al. 2003). Borate minerals assist this (Saladino et al. 2011). These environments also allow chemistry of Becker et al. (2016) and Kim and Benner (2017) to form nucleosides and nucleoside phosphates.
-
4.
The pH in the arid environment is buffered by atmospheric CO2 (Sleep et al. 2011) but is intermittently raised as alkaline solutions are reintroduced. At pH ~6, molybdate can complete the formation of ribose, which is stabilized as an organic mineral.
-
5.
The arid environment is also exposed to volcanic gasses, including sulfur dioxide. These may stabilize simple carbohydrates like glyceraldehyde and glycolaldehyde, preserving them in reservoirs in sufficient amounts to rescue the Powner et al. (2009) model for the formation of pyrimidine nucleoside phosphates.
-
6.
Dehydration followed by rehydration is frequent. This gives polyphosphates, thiophosphates (if volcanic H2S is available, or with a combination of phosphite and S8), and phosphate-formate anhydrides. These condense to form linear RNA, which is stabilized and concentrated by adsorption on mineral surfaces.
We titled DSM as “discontinuous” to illustrate why it is not yet (and should not yet be) accepted as a solution to the “origins of Darwinism” problem. Too many steps are disconnected, requiring a chemist to clean up the output of the first step to prepare it for input into the second step. However, we know when we are succeeding in this framework when this cleanup becomes less severe or goes away entirely.
References
Abramov O, Mojzsis SJ (2009) Microbial habitability of the Hadean Earth during the late heavy bombardment. Nature 459:419–422
Agricola G (1530) Quarzum. In: Bermannus, Sive De Re Metallica, in Aedibus Frobenianis. Basileae, p 129
Allègre CJ, Manhès G, Göpel G (2008) The major differentiation of the Earth at ∼4.45 Ga. Earth Planet Sci Lett 267:386–398
Andreani M, Munoz M, Marcaillou C et al (2013) μXANES study of iron redox state in serpentine during oceanic serpentinization. Lithos 178:70–83
Anet FA (2004) The place of metabolism in the origin of life. Curr Opin Chem Biol 8:654–659
Anumukonda LN, Young A, Lynn DG et al (2011) Adenine synthesis in a model prebiotic reaction: connecting origin of life chemistry with biology. J Chem Educ 88:1698–1701
Appayee C, Breslow R (2014) Deuterium studies reveal a new mechanism for the formose reaction involving hydride shifts. J Am Chem Soc 136:3720–3723
Bach W, Paulick H, Garrido CJ et al (2006) Unravelling the sequence of serpentinization reactions: petrography, mineral chemistry, and petrophysics of serpentinites from MAR 15_N (ODP Leg 209, Site 1274). Geophys Res Lett 33:L13306
Bada JL, Chalmers JH, Cleaves HJ (2016) Is formamide a geochemically plausible prebiotic solvent? Phys Chem Chem Phys 18:20085–20090
Becker H (2006) Highly siderophile element composition of the Earth’s primitive upper mantle: constraints from new data on peridotite massifs and xenoliths. Geochim Cosmochim Acta (17):4528–4550
Becker S, Thoma I, Deutsch A et al (2016) A high-yielding, strictly regioselective prebiotic purine nucleoside formation pathway. Science 352(6287):833–836
Bell EA, Boehnke P, Harrison TM et al (2015) Potentially biogenic carbon preserved in a 4.1 billion-year-old zircon. Proc Natl Acad Sci U S A 112(47):14518–14521
Benner SA (2009) The life, the universe and the scientific method. FfAME Press, .Gainesville, 312 pp.
Benner SA (2013) Synthesis as a route to knowledge. Biol Theory 8:357–367
Benner SA (2017a) Detecting Darwinism from molecules in the Enceladus plumes, Jupiter’s moons, and other planetary water lagoons. Astrobiology 17(9):840–851
Benner SA (2017b) Uniting natural history with the molecular sciences. The ultimate multidisciplinarity. Acc Chem Res 50:498–502
Benner SA, Kim HJ (2015) The case for a Martian origin for Earth life. In: Hoover RB, Levin GV, Rozanov, AY, Wickramasinghe NC (eds) Instruments, methods, and missions for astrobiology XVII, SPIE Optical Engineering+ Applications, 9606, 96060C
Benner SA, Ellington AD, Tauer A (1989) Modern metabolism as a palimpsest of the RNA world. Proc Natl Acad Sci U S A 86:7054–7058
Benner SA, Devine KG, Matveeva LN et al (2000) The missing organic molecules on Mars. Proc Natl Acad Sci U S A 97:2425–2430
Benner SA, Caraco MD, Thomson JM et al (2002) Planetary biology. Paleontological, geological, and molecular histories of life. Science 293:864–868
Benner SA, Bains W, Seager S (2013) Models and standards of proof in cross-disciplinary science: the case of arsenic DNA. Astrobiology 13:510–513
Benner SA, Karalkar NB, Hoshika S et al (2016) Alternative Watson-Crick synthetic genetic systems. Synthetic Biology. Cold Spring Harb Perspect Biol 8(11). doi: https://doi.org/10.1101/cshperspect.a023770
Bernal JD (1951) The physical basis of life. Routledge and Kegan Paul, London
Berndt ME, Allen DE, Seyfried WE (1996) Reduction of CO2 during serpentinization of olivine at 300 °C and 500 bar. Geology 24(4):351–354
Biondi E, Branciamore S, Maurel MC et al (2007a) Montmorillonite protection of an UV-irradiated hairpin ribozyme. Evolution of the RNA world in a mineral environment. BMC Evol Biol 7(Suppl 2):S2
Biondi E, Branciamore S, Fusi L et al (2007b) Catalytic activity of a hammerhead ribozyme in a clay mineral environment: implications for the RNA World. Gene 389:10–18
Biondi E, Howell L, Benner SA (2016) Opal adsorbs and stabilizes RNA – a hierarchy of prebiotic silica minerals. Syn Lett 27:A–E
Biondi E, Furukawa Y, Kwai J et al (2017) Adsorption of RNA on mineral surfaces and mineral precipitates. Beilstein J Org Chem 13:393–404
Blöchl E, Keller M, Wächtershäuser G et al (1992) Reactions depending on iron sulfide and linking geochemistry with biochemistry. Proc Natl Acad Sci U S A 89(17):8117–8120
Boehnken P, Harrison TM (2016) Illusory late heavy bombardments. Proc Natl Acad Sci U S A 113(39):10802–10806
Boily JF, Persson P, Sjöberg S (2000a) Benzenecarboxylate complexation at the goethite-water interface: I. A mechanistic description of pyromellitate surface complexes from the combined evidence of infrared spectroscopy, potentiometry, adsorption data and surface complexation modeling. Langmuir 16:5719–5729
Boily JF, Persson P, Sjöberg S (2000b) Benzenecarboxylate surface complexation at the goethite (α-FeOOH)/water interface: II. Linking IR spectroscopic observations to mechanistic surface complexation models for phthalate, trimellitate, and pyromellitate. Geochim Cosmochim Acta 64(20):3453–3470
Bonner WA, Kavasmaneck PR, Martin FS et al (1974) Asymmetric adsorption of alanine by quartz. Science 186(4159):143–144
Bonner WA, Kavasmaneck PR, Martin FS et al (1975) Asymmetric adsorption by quartz: a model for the prebiotic origin of optical activity. Orig Life 6(3):367–376
Borisov AA (2016) Mutual interaction of redox pairs in silicate melts: equilibria involving metallic phases. Petrology 24(2):117
Brandes JA, Boctor NZ, Cody GD et al (1998) Abiotic nitrogen reduction on the early Earth. Nature 395(6700):365–367
Brasser R, Mojzsis SJ (2017) A colossal impact enriched Mars’ mantle with noble metals. Geophys Res Lett. https://doi.org/10.1002/(2017GL074002
Brasser R, Mojzsis SJ, Werner SC et al (2016) Late veneer and late accretion to the terrestrial planets. Earth Planet Sci Lett 455:85–93
Bregestovski PD (2015) “RNA World”, a highly improbable scenario of the origin and early evolution of life on Earth. J Evol Biochem Physiol 51:72–84
Burcar BT, Barge LM, Trail D et al (2015) RNA oligomerization in laboratory analogues of alkaline hydrothermal vent systems. Astrobiology 15:509–522
Burcar BT, Pasek M, Gull M et al (2016) Darwin’s warm little pond: a one-pot reaction for prebiotic phosphorylation and the mobilization of phosphate from minerals in a urea-based solvent. Angew Chem Int Ed 55(42):13249–13253
Burton FG, Neuman MW, Neuman WF (1969) On the possible role of crystals in the origins of life. I. The adsorption of nucleosides, nucleotides and pyrophosphate by apatite crystals. Biosystems 3(1):20–26
Butlerov A (1861) Bildung einer zuckerartigen Substanz durch Synthese. Ann Chem 120:295–298
Cairns-Smith AG (1977) Takeover mechanisms and early biochemical evolution. Biosystems 9(2–3):105–109
Cairns-Smith AG (1982) Genetic takeover and the mineral origins of life. Cambridge University Press, Cambridge
Cairns-Smith AG (2005) Sketches for a mineral genetic material. Elements 1:157–161
Calvin M, Calvin GI (1964) Atom to Adam. Am Sci 52:163
Cech TR (2000) The ribosome is a ribozyme. Science 289(5481):878–879
Cech TR, Bass BL (1986) Biological catalysis by RNA. Annu Rev Biochem 55:599–629
Chapman CR, Cohen BA, Grinspoon DH (2007) What are the real constraints on the existence and magnitude of the late heavy bombardment? Icarus 189(1):233–245
Cherniak DJ, Hanchar JM, Watson EB (1997) Rare-earth diffusion in zircon. Chem Geol 134:289–301
Cleaves HJ (2008) The prebiotic geochemistry of formaldehyde. Precambrian Res 164:111–118
Cody GD (2004) Transition metal sulfides and the origins of metabolism. Geophysical Laboratory, Carnegie Institution of Washington, Washington, DC, pp 569–599
Cody GD (2005) Geochemical connections to primitive metabolism. Elements 1:139–143
Cody GD, Boctor NZ, Filley T et al (2000) The primordial synthesis of carbonylated iron-sulfur clusters and the synthesis of pyruvate. Science 289:1339–1339
Cody GD, Boctor NZ, Hazen RM et al (2001) Geochemical roots of autotrophic carbon fixation: Hydrothermal experiments in the system citric acid, H2O-(±FeS)-(±NiS). Geochim Cosmochim Acta 65(20):3557–3576
Cody GD, Boctor NZ, Brandes JA et al (2004) Assaying the catalytic potential of transition metal sulfides for abiotic carbon fixation. Geochim Cosmochim Acta 68(10):2185–2196
Cohen BA, Swindle TD, Kring DA (2000) Support for the lunar cataclysm hypothesis from lunar meteorite impact melt ages. Science 290:1754–1756
Condie KC (2018) A planet in transition: the onset of plate tectonics on Earth between 3 and 2 Ga. Geosci Front 9(1):51–60
Cox PA (1989) The elements: their origin, abundance, and distribution. Oxford University Press, Oxford
da Silva JAL, Holm NG (2014) Borophosphates and silicophosphates as plausible contributors to the emergence of life. J Colloid Interface Sci 431:250–254
Dal Negro A, Ungaretti L (1971) Refinement of the crystal structure of aragonite. Am Mineral 56:768–772
Danielson LR, Righter K, Newville M et al (2011) Molybdenum valence in basaltic silicate melts: effects of temperature and pressure. In: 42nd Lunar and planetary science conference, 2609
Dauphas N, Schauble EA (2016) Mass fractionation laws, mass-independent effects, and isotopic anomalies. Annu Rev Earth Planet Sci 44:709–783
Dauphas N, Chen J, Papanastassiou D (2015) Testing Earth–Moon isotopic homogenization with calcium-48. In: Lunar and planetary science conference XXXXVI, 2436
Day JM, Walker RJ (2015) Highly siderophile element depletion in the Moon. Earth Planet Sci Lett 423:114–124
Decher G, Lehr B, Lowack K et al (1994) New nanocomposite films for biosensors – layer-by-layer adsorbed films of polyelectrolytes, proteins or DNA. Biosens Bioelectron 9:677–684
Decker P, Schweer H, Pohlamnn R (1982) Bioids: X. Identification of formose sugars, presumable prebiotic metabolites, using capillary gas chromatography/gas chromatography—mas spectrometry of n-butoxime trifluoroacetates on OV-225. J Chromatogr A 244:281–291
Dyson F (1985) Origin of life. Cambridge University Press, Cambridge
Ehlmann BL, Mustard JF, Murchie SL et al (2008) Orbital identification of carbonate-bearing rocks on Mars. Science 322:1828–1832
Ertem G, Ferris JP (1996) Synthesis of RNA oligomers on heterogeneous templates. Nature 379(6562):238–240
Ertem G, Ferris JP (1997) Template-directed synthesis using the heterogeneous templates produced by montmorillonite catalysis. A possible bridge between the prebiotic and RNA worlds. J Am Chem Soc 119(31):7197–7201
Ertem G, Ertem MC, McKay CP et al (2017) Shielding biomolecules from effects of radiation by Mars analogue minerals and soils. Int J Astrobiol 16(03):280–285
Eschenmoser A (1997) Towards a chemical etiology of nucleic acid structure. Orig Life Evol Biosph 27(5–6):535–553
Evgenii K, Wolfram T (2000) The role of quartz in the origin of optical activity on earth. Orig Life Evol Biosph 30(5):431–434
Feely RA, Trefry JH, Lebon GT et al (1998) The relationship between P/Fe and V/Fe ratios in hydrothermal precipitates and dissolved phosphate in seawater. Geophys Res Lett 25:2253–2256
Feigl F (1937) Qualitative analysis by spot tests. Nordemann, New York, p 400
Ferris JP (2005) Mineral catalysis and prebiotic synthesis: montmorillonite-catalyzed formation of RNA. Elements 1(3):145–149
Ferris JP, Ertem G (1992) Oligomerization of ribonucleotides on montmorillonite: reaction of the 5′-phosphorimidazolide of adenosine. Science 257(5075):1387–1389
Ferris JP, Ertem G (1993) Montmorillonite catalysis of RNA oligomer formation in aqueous solution. A model for the prebiotic formation of RNA. J Am Chem Soc 115(26):12270–12275
Ferris JP, Sanchez RA, Orgel LE (1968) Studies in prebiotic synthesis: III. Synthesis of pyrimidines from cyanoacetylene and cyanate. J Mol Biol 33(3):693–704
Feynman R (1974) Cargo cult science. Caltech commencement address. Reproduced in “Surely You’re Joking, Mr. Feynman”. Norton, New York
Fiore M, Strazewski P (2016) Prebiotic lipidic amphiphiles and condensing agents on the early Earth. Life 6. https://doi.org/10.3390/life6020017
Fox SW (1965) A theory of macromolecular and cellular origins. Nature 205:328
Fuchs LH (1969) The phosphate mineralogy of meteorites. In: Meteorite research. Springer, Dordrecht, pp 683–695
Fyfe WS, Bischoff JL (1965) The calcite-aragonite problem. In Pray LC, Murray RC (eds), Dolomitization and limestone diagenesis: a symposium. Society of Economic Paleontologists and Mineralogists, Special Publication, 13, pp 3–13
Galison PL (1987) How experiments end. University of Chicago Press, Chicago
Genda H, Fujita T, Kobayashi H et al (2017) Impact erosion model for gravity-dominated planetesimals. Icarus 294:234–246
Goldschmidt VM (1952) Geochemical aspects of the origin of complex organic molecules on the earth, as precursors to organic life. New Biol 12:97–105
Grasby SE (2003) Naturally precipitating vaterite (μ-CaCO3) spheres: Unusual carbonates formed in an extreme environment. Geochim Cosmochim Acta 67:1659–1666
Grosjean M, Geyh MA, Messerli B et al (1995) Late-glacial and early Holocenelake sediments, groundwater formation and climate in the Atacama altiplano 22–241S. J Paleolimnol 14:241–252
Guerrier-Takada C, Gardiner K, Marsh T et al (1983) The RNA moiety of ribonuclease-P is the catalytic subunit of the enzyme. Cell 35:849–857
Gull M, Mojica MA, Fernández FM et al (2015) Nucleoside phosphorylation by the mineral schreibersite. Sci Rep 5:17198. https://doi.org/10.1038/srep17198
Hamano K, Abe Y, Genda H (2013) Emergence of two types of terrestrial planet on solidification of magma ocean. Nature 497:607–610
Hanczyc MM, Fujikawa SM, Szostak JW (2003) Experimental models of primitive cellular compartments: encapsulation, growth, and division. Science 302(5645):618–622
Harman CE, Kasting JF, Wolf ET (2013) Atmospheric production of glycolaldehyde under hazy prebiotic conditions. Orig Life Evol Biosph 43:77–98
Hartmann WK (1975) Lunar “cataclysm”: a misconception? Icarus 24(2):181–187
Hazen RM (2005) Genesis. The scientific quest for life’s origins. Joseph Henry Press, Washington, DC
Hazen RM, Papineau D, Bleeker W et al (2008) Mineral evolution. Am Mineral 93(11–12):1693–1720
Hedenquist JW, Arribas A, Gonzalez-Urien E (2000) Exploration for epithermal gold deposits. Rev Econ Geol 13:45–77
Herdewijn P (2001) TNA as a potential alternative to natural nucleic acids. Angew Chem Int Ed 40(12):2249–2251
Holland H (1984) The chemical evolution of the atmosphere and oceans. Princeton University Press, Princeton, 598 p
Holm NG, Ertem G, Ferris JP (1993) The binding and reactions of nucleotides and polynucleotides on iron oxide hydroxide polymorphs. Orig Life Evol Biosph 23(3):195–215
Huber C, Wächtershäuser G (1997) Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 276(5310):245–247
Huber C, Wächtershäuser G (1998) Peptides by activation of amino acids with CO on (Ni,Fe)S surfaces: implications for the origin of life. Science 281(5377):670–671
Huber C, Eisenreich W, Hecht S et al (2003) A possible primordial peptide cycle. Science 301(5635):938–940
Huber C, Kraus F, Hanzlik M et al (2012) Elements of metabolic evolution. Chem Eur J 18(7):2063–2080
Hud NV, Jain SS, Li X et al (2007) Addressing the problems of base pairing and strand cyclization in template-directed synthesis. Chem Biodivers 4(4):768–783
Hyeon C, Dima RI, Thirumalai D (2006) Size, shape, and flexibility of RNA structures. J Chem Phys 125(19):194905
Ichida JK, Zou K, Horhota A et al (2005) An in vitro selection system for TNA. J Am Chem Soc 127(9):2802–2803
Inoue T, Orgel LE (1982) Oligomerization of (guanosine 5′-phosphor)-2-methylimidazolide on poly (C): an RNA polymerase model. J Mol Biol 162(1):201–217
Islam S, Bučar DK, Powner MW (2017) Prebiotic selection and assembly of proteinogenic amino acids and natural nucleotides from complex mixtures. Nat Chem 9:584–589
Izgu EC, Oh SS, Szostak JW (2016) Synthesis of activated 3′-amino-3′-deoxy-2-thio-thymidine, a superior substrate for the nonenzymatic copying of nucleic acid templates. Chem Commun 52(18):3684–3686
Javoy M, Kaminski E, Guyot F et al (2010) The chemical composition of the Earth: enstatite chondrite models. Earth Planet Sci Lett 293:259–268
Jermann TM, Opitz JG, Stackhouse J et al (1995) Reconstructing the evolutionary history of the artiodactyl ribonuclease superfamily. Nature 374:57–59
Jones LC, Rosenbauer R, Goldsmith JI et al (2010) Carbonate control of H2 and CH4 production in serpentinization systems at elevated P-Ts. Geophys Res Lett 37:L14306
Joyce GF, Orgel LE (1999) Prospects for understanding the origin of the RNA world. In: Gestland RF, Cech RTR, Atkins JF (eds) The RNA World, 2nd edn. Cold Spring Harbor Press, Cold Spring Harbor, NY, pp 49–78
Keefe AD, Miller SL (1996) Was ferrocyanide a prebiotic reagent? Orig Life Evol Biosph 26(2):111–129
Kim HJ, Benner SA (2010) Comment on “The silicate-mediated formose reaction: bottom-up synthesis of sugar silicates”. Science 329(5994):902-a
Kim HJ, Benner, SA (2017) Prebiotic stereoselective synthesis of purine and noncanonical pyrimidine nucleotide from nucleobases and phosphorylated carbohydrates. Proc Natl Acad Sci U S A (on line)
Kim HJ, Ricardo A, Illangkoon HI et al (2011) Synthesis of carbohydrates in mineral-guided prebiotic cycles. J Am Chem Soc 133:9457–9468
Kim HJ, Furukawa Y, Kakegawa T et al (2016) Evaporite borate-containing mineral ensembles make phosphate available and regiospecifically phosphorylate ribonucleosides: borate as a multifaceted problem solver in prebiotic chemistry. Angew Chem 55:15816–15820
Kring DA, Cohen BA (2002) Cataclysmic bombardment throughout the inner solar system 3.9–4.0Ga. J Geophys Res 107(E2):4-1–4-6
Kruijer TS, Kleine T, Fischer-Gödde M et al (2015) Lunar tungsten isotopic evidence for the late veneer. Nature 5:534–537
Lahav N (1994) Minerals and the origin of life – hypotheses and experiments in heterogeneous chemistry. Heterog Chem Rev 1:159–179
Lahav N, White D, Chang S (1978) Peptide formation in the prebiotic era: thermal condensation of glycine in fluctuating clay environments. Science 201(435 O):67–69
Lambert JB, Gurusamy-Thangavelu SA, Ma KBA (2010a) The silicate-mediated formose reaction: bottom-up synthesis of sugar silicates. Science 327:984–986
Lambert JB, Gurusamy-Thangavelu SA, Ma KBA (2010b) Response to comment on “The silicate-mediated formose reaction: bottom-up synthesis of sugar silicates”. Science 329(5994):902-b
Lapen TJ, Righter M, Brandon AD et al (2010) A younger age for ALH84001 and its geochemical link to shergottite sources in Mars. Science 328:347–351
Larralde R, Robertson MP, Miller SL (1995) Rates of decomposition of ribose and other sugars. Implications for chemical evolution. Proc Natl Acad Sci U S A 92:8158–8160
Lehman N (2003) A case for the extreme antiquity of recombination. J Mol Evol 56:770–777
Leman LJ, Orgel LE, Ghadiri MR (2006) Amino acid dependent formation of phosphate anhydrides in water mediated by carbonyl sulfide. J Am Chem Soc 128(1):20–21
Levy M, Miller SL (1998) The stability of the RNA bases: implications for the origin of life. Proc Natl Acad Sci U S A 95:7933–7938
Levy M, Miller SL, Oró J (1999) Production of guanine from NH4CN polymerizations. J Mol Evol 49:165–168
Li L, Prywes N, Tam CP et al (2017) Enhanced nonenzymatic RNA copying with 2-aminoimidazole activated nucleotides. J Am Chem Soc 139(5):1810–1813
Lincoln TA, Joyce GF (2009) Self-sustained replication of an RNA enzyme. Science 323:1229–1232
Löb W (1913) Uber das Verhalten des Formamids unter der Wirkung der stillen Entladung Ein Beitrag zur Frage der Stickstoff-Assimilation. Ber Dtsch Chem Ges 46:684–697
Lohrmann R (1972) Formation of urea and guanidine by irradiation of ammonium cyanide. J Mol Evol 1:263–269
Maher KA, Stevenson DJ (1988) Impact frustration of the origin of life. Nature 331:612–614
Malaterre C (2013) Synthetic biology and synthetic knowledge. Biol Theory 8:346–356
Mann U, Frost DJ, Rubie DC et al (2012) Partitioning of Ru Rh Pd Re Ir and Pt between liquid metal and silicate at high pressures and high temperatures – Implications for the origin of highly siderophile element concentrations in the Earth’s mantle. Geochim Cosmochim Acta 84:593–613
Marchi S, Bottke WF, Cohen BE et al (2013) High-velocity collisions from the lunar cataclysm recorded in asteroidal meteorites. Nat Geosci 6:303–307
Markovitch O, Lancet D (2014) Multispecies population dynamics of prebiotic compositional assemblies. J Theor Biol 357:26–34
Maslen EN, Streltsov VA, Streltsova NR (1993) X-ray study of the electron density in calcite, CaCO3. Acta Cryst B49:636–641
McCauley JW, Roy R (1974) Controlled nucleation and crystal growth of various CaCO3 phases by the silica gel technique. Am Mineral 59:947–963
McCollom TM (2013) Miller-Urey and beyond: What have we learned about prebiotic organic synthesis reactions in the past 60 years? Annu Rev Earth Planet Sci 41:207–229
McCollom TM, Seewald JS (2001) A reassessment of the potential for reduction of dissolved CO2 to hydrocarbons during serpentinization of olivine. Geochim Cosmochim Acta 65(21):3769–3778
Miller SL (1953) A production of amino acids under possible primitive earth conditions. Science 117:528–529
Miyakawa S, Cleaves HJ, Miller SL (2002) The cold origin of life: B. Implications based on pyrimidines and purines produced from frozen ammonium cyanide solutions. Orig Life Evol Biosph 32:209–218
Mojzsis SJ, Arrhenius G, McKeegan KD et al (1996) Evidence for life on Earth before 3,800 million years ago. Nature 384(6604):55–59
Molster FJ, Lim TL, Sylvester RJ et al (2001) The complete ISO spectrum of NGC 6302. Astron Astrophys 372:165–172
Morbidelli A, Marchi S, Bottke WF et al (2012) A sawtooth-like timeline for the first billion years of lunar bombardment. Earth Planet Sci Lett 355-356:144–151
Müller J, Fabricius F (1978) Lüneburgite [Mg3(PO4)2B2O(OH)4 × 6 H2O] in Upper Miocene sediments of the Eastern Mediterranean Sea. Init Rep DSDP 42:661–664. https://doi.org/10.2973/dsdp.proc.42-1.127.1978
Mutschler H, Wochner A, Holliger P (2015) Freeze-thaw cycles as drivers of complex ribozyme assembly. Nat Chem 7:502–508
Neveu M, Kim HJ, Benner SA (2013) The “Strong” RNA World hypothesis. Fifty years old. Astrobiology 13:391–403
Nishiyama T, Kagami Y, Yamauchi T et al (2013) Assembly of stimulus-sensitive gel particles with DNA-dye complexes. Polym J 45:659–664
Okafor CD, Lanier KA, Petrov AS et al (2017) Iron mediates catalysis of nucleic acid processing enzymes: support for Fe (II) as a cofactor before the great oxidation event. Nucleic Acids Res 45:3634–3642
Orgel LE (2000a) A simpler nucleic acid. Science 290(5495):1306–1307
Orgel LE (2000b) Self-organizing biochemical reactions. Proc Natl Acad Sci U S A 97:12503–12507
Oró JJ (1965) Investigation of organo-chemical evolution. In: Mamikunian G, Briggs MH (eds) Current aspects of exobiology. Pergamon Press, Oxford, pp 13–39
Parsons I, Lee MR, Smith JV (1998) Biochemical evolution II: origin of life in tubular microstructures on weathered feldspar surfaces. Proc Natl Acad Sci 95(26):15173–15176
Pasek MA (2016) Schreibersite on the early earth: scenarios for prebiotic phosphorylation. Geosci Front 8(2):329–335
Pearce BKD, Pudritz RE (2015) Seeding the pregenetic Earth: meteoritic abundances of nucleobases and potential reaction pathways. Astrophys J 807:85–94
Pearce BKD, Pudritz RE, Semenov DA et al (2017) Origin of the RNA world: the fate of nucleobases in warm little ponds. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1710339114
Petrus L, Petrusová M, Hricovíniová Z (2001) The Bilik reaction. In: Stutz AE (ed) Topics in current chemistry: glycoscience, epimerisation, isomerisation and rearrangment reactions of carbohydrates, vol 215. Springer, Berlin, pp 15–41
Piccoli PM, Candela P (2002) Apatite in igneous systems. Rev Miner Geochem 48:255–292
Pinto J, Gladstone G, Yung Y (1980) Photochemical production of formaldehyde in Earth’s primitive atmosphere. Science 210:183–185
Powner MW, Gerland B, Sutherland JD (2009) Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459:239–242
Puchtel IS, Walker RJ, Touboul M et al (2014) Insights into early Earth from the Pt–Re–Os isotope and highly siderophile element abundance systematics of Barberton komatiites. Geochim Cosmochim Acta 125:394–413
Qin L, Alexander CMD, Carlson RW (2010) Contributors to chromium isotope variation of meteorites. Geochim Cosmochim Acta 74:1122–1145
Ricardo A, Carrigan MA, Olcott AN et al (2004) Borate minerals stabilize ribose. Science 303:196
Rich A (1962) On the problems of evolution and biochemical information transfer. In: Kasha M, Pullmann B (eds) Horizons in biochemistry. Academic Press, NY, pp 103–126
Richter Y, Fischer B (2003) Characterization and elucidation of coordination requirements of adenine nucleotides complexes with Fe (II) ions. Nucleosides Nucleotides Nucleic Acids 22(9):1757–1780
Righter K, Yang H, Costin G et al (2008) Oxygen fugacity in the Martian mantle controlled by carbon: New constraints from the nakhlite MIL 03346. Meteorit Planet Sci 43:1709–1723
Righter K, Danielson LR, Pando KM et al (2015) Highly siderophile element (HSE) abundances in the mantle of Mars are due to core formation at high pressure and temperature. Meteorit Planet Sci 50:604–631
Rosing MT (1999) 13C-Depleted carbon microparticles in >3700-Ma sea-floor sedimentary rocks from West Greenland. Science 283:674–676
Rubie DC, Jacobson SA, Morbidelli A et al (2015a) Accretion and differentiation of the terrestrial planets with implications for the compositions of early-formed Solar System bodies and accretion of water. Icarus 248:89–108
Rubie DC, Nimmo F, Melosh HJ (2015b) Formation of the Earth’s core. In: Schubert G (ed) Treatise on geophysics, vol 9: Evolution of the Earth, 2nd edn. Elsevier, Oxford, pp 43–79
Russell MJ, Hall AJ (1997) The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J Geol Soc 154(3):377–402
Russell MJ, Martin W (2004) The rocky roots of the acetyl-CoA pathway. Trends Biochem Sci 29(7):358–363
Russell MJ, Daniel RM, Hall AJ et al (1994) A hydrothermally precipitated catalytic iron sulphide membrane as a first step toward life. J Mol Evol 39(3):231–243
Sahai N, Kaddour H, Dalai P (2016) The transition from geochemistry to biogeochemistry. Elements 12(6):389–394
Saladino R, Ciambecchini U, Crestini C et al (2003) One-pot TiO2-catalyzed synthesis of nucleic bases and acyclonucleosides from formamide: implications for the origin of life. Chembiochem 4:514–521
Saladino R, Barontini M, Cossetti C et al (2011) The effects of borate minerals on the synthesis of nucleic acid bases, amino acids and biogenic carboxylic acids from formamide. Orig Life Evol Biosph 41:317–330
Sanchez RA, Orgel LE (1970) Studies in prebiotic synthesis: V. Synthesis and photoanomerization of pyrimidine nucleosides. J Mol Biol 47:531–543
Sanchez RA, Ferris JP, Orgel LE (1966) Cyanoacetylene in prebiotic synthesis. Science 154:784–785
Santos AR, Agee CB, McCubbin FM et al (2013) Apatite and merrillite from Martian meteorite NWA 7034. In: Lunar and planetary science conference 44, 2601
Schoonen M, Smirnov A (2016) Staging life in an early warm ‘seltzer’ ocean. Elements 12(6):395–400
Schoonen M, Smirnov A, Cohn C (2004) A perspective on the role of minerals in prebiotic synthesis. Ambio 33(8):539–551
Sekimoto K, Takayama M (2012) Formation of hydrogen cyanide HCN under limited discharge conditions in non-reduced ambient air. ESCAMPIG XXI, Viana do Castelo, Portugal, 10–14 July
Shapiro R (1995) The prebiotic role of adenine: a critical analysis. Orig Life Evol Biosph 25:83–98
Shapiro R (1999) Prebiotic cytosine synthesis: a critical analysis and implications for the origin of life. Proc Natl Acad Sci U S A 96:4396–4401
Shapiro R (2007) A simpler origin for life. Sci Am 296:46–53
Sleep NH, Meibom A, Fridriksson T et al (2004) H2-rich fluids from serpentinization: geochemical and biotic implications. Proc Natl Acad Sci U S A 101:12818–12823
Sleep NH, Bird DK, Pope EC (2011) Serpentinite and the dawn of life. Philos Trans R Soc B 366:2857–2869
Smith JV (1998) Biochemical evolution. I. Polymerization on internal, organophilic silica surfaces of dealuminated zeolites and feldspars. Proc Natl Acad Sci U S A 95(7):3370–3375
Smith JV, Arnold FP, Parsons I et al (1999) Biochemical evolution III: polymerization on oganophilic silica-rich surfaces, crystal-chemical modeling, formation of first cells, and geological clues. Proc Natl Acad Sci U S A 96(7):3479–3485
Stephenson JD, Hallis LJ, Nagashima K et al (2013) Boron enrichment in Martian clay. PLoS One 8(6):e64624
Stephenson JD, Popović M, Bristow TF et al (2016) Evolution of ribozymes in the presence of a mineral surface. RNA 22:1893–1901
Stober W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in micron size range. J Colloid Interface Sci 26:62–69
Summers DP, Chang S (1993) Prebiotic ammonia from reduction of nitrite by iron (II) on the early Earth. Nature 365(6447):630–633
Sutherland JD (2016) The origin of life. Out of the blue. Angew Chem Int Ed 55(1):104–121
Taves DR (1963) Similarity of octacalcium phosphate and hydroxyapatite structures. Nature 200(4913):1312–1313
Taves DR, Reedy RC (1969) A structural basis for the transphosphorylation of nucleotides with hydroxyapatite. Calcif Tissue Int 3(1):284–292
Tera F, Papanastassiou DA, Wasserburg GJ (1974) Isotopic evidence for a terminal lunar cataclysm. Earth Planet Sci Lett 22(1):1–21
Touboul M, Puchtel IS, Walker RJ (2012) 182W evidence for long term preservation of early mantle differentiation products. Science 335:1065–1069
Touboul M, Liu J, O’Neil J et al (2014) New insights into the Hadean mantle revealed by 182 W and highly siderophile element abundances of supracrustal rocks from the Nuvvuagittuq greenstone belt, Quebec Canada. Chem Geol 383:63–75
Trail D, Watson EB, Tailby ND (2011) The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 480(7375):79–82
Van Vleck JH (1928) The correspondence principle in the statistical interpretation of quantum mechanics. Proc Natl Acad Sci U S A 14(2):178–188
Wächtershäuser G (1988a) Before enzymes and templates: theory of surface metabolism. Microbiol Rev 52(4):452–484
Wächtershäuser G (1988b) Pyrite formation, the first energy source for life: a hypothesis. Syst Appl Microbiol 10(3):207–210
Wächtershäuser G (1990a) Evolution of the first metabolic cycles. Proc Natl Acad Sci U S A 87(1):200–204
Wächtershäuser G (1990b) The case for the chemoautotrophic origin of life in an iron-sulfur world. Orig Life Evol Biosph 20(2):173–176
Wächtershäuser G (1993) The cradle chemistry of life: on the origin of natural products in a pyrite-pulled chemoautotrophic origin of life. Pure Appl Chem 65(6):1343–1348
Warren PH (2011) Stable-isotopic anomalies and the accretionary assemblage of the Earth and Mars: a subordinate role for carbonaceous chondrites. Earth Planet Sci Lett 311:93–100
Weber AL (1995) Prebiotic polymerization: oxidative polymerization of 2,3-dimercapto-l-propanol on the surface of iron(III) hydroxide oxide. Orig Life Evol Biosph 25(1–3):53–60
Willbold M, Elliot T, Moorbath S (2011) The tungsten isotopic composition of the Earth’s mantle before the terminal bombardment. Nature 477:195–198
Willbold M, Mojzsis SJ, Chen HW et al (2015) Tungsten isotope composition of the Acasta Gneiss Complex. Earth Planet Sci Lett 419:168–177
Young ED, Kohl IE, Warren PH et al (2016) Oxygen isotopic evidence for vigorous mixing during the Moon-forming giant impact. Science 351:493–496
Yuasa S, Flory D, Basile B et al (1984) Abiotic synthesis of purines and other heterocyclic compounds by the action of electrical discharges. J Mol Evol 21:76–80
Zhang J, Dauphas N, Davis AM et al (2012) The proto-Earth as a significant source of lunar material. Nat Geosci 5:251–255
Zhang S, Blain JC, Zielinska D et al (2013) Fast and accurate nonenzymatic copying of an RNA-like synthetic genetic polymer. Proc Natl Acad Sci U S A 110(44):17732–17737
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Benner, S.A., Kim, HJ., Biondi, E. (2018). Mineral-Organic Interactions in Prebiotic Synthesis. In: Menor-Salván , C. (eds) Prebiotic Chemistry and Chemical Evolution of Nucleic Acids. Nucleic Acids and Molecular Biology, vol 35. Springer, Cham. https://doi.org/10.1007/978-3-319-93584-3_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-93584-3_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93583-6
Online ISBN: 978-3-319-93584-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)