
Abstract
Vascular endothelial growth factor (VEGF) plays an important role in maintaining the regular homeostasis of vascular walls. VEGF binds its receptor (VEGFR) promoting the regular survival and function of endothelial cells. Anti-VEGF and anti-VEGFR drugs inhibit the action of VEGF and VEGFR. These drugs can cause cardiovascular toxic effects such as arterial hypertension, thromboembolism, myocardial ischemia and heart failure. The monoclonal antibody bevacizumab and tyrosine kinase inhibitors (sorafenib, sunitinib, pazopanib, regorafenib, axitinib, cabozantinib, ponatinib) are the main inhibitors of VEGF, VEGFR and other tyrosine kinases. In this chapter we will illustrate the cardiovascular toxic effects of these drugs, their mechanism of action, strategy to early diagnose and treat these complications. We will also illustrate strategy to prevent cardiovascular toxicity. It is important to know cardiovascular toxic effect of these drugs widely used in oncological field, to avoid the development of severe future complications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Vascular endothelial growth factor and its receptor (VEGF and VEGFR) play an important role in maintaining the regular homeostasis of vascular walls. Endothelium is not a simple cellular monolayer that separates the blood from the vascular walls, but it plays a key role in the regulation of vascular function, by producing vasoconstrictor and vasodilator substances, such as endothelin-1 (ET-1), angiotensin II (Ang II), thromboxane A2, reactive oxygen species, nitrogen monoxide (NO), and prostacyclin [1]. Mature endothelial cells (ECs), endothelial progenitor cells, and circulating ECs participate in the physiological maintenance of cardiovascular tissue homeostasis, including vascular tone, permeability and intima thickness, vessel remodeling and angiogenesis, coagulation, and fibrinolysis. In contrast, endothelial dysfunction is involved in the pathophysiology of several diseases, including atherothrombosis, diabetes, sepsis, pulmonary hypertension, microangiopathy associated with neurodegenerative diseases, hepatic steatosis, and cancer metastasis [2, 3]. VEGF and VEGFR transduce signaling that promote survival and function of endothelial cells. Thus inhibition of VEGF and VEGFR can cause cardiovascular toxic effects [4]. Bevacizumab is a monoclonal antibody that inhibits VEGF; it causes predominantly arterial hypertension and thromboembolic events. Tyrosine kinase inhibitors (sorafenib, sunitinib, pazopanib, regorafenib, axitinib, ponatinib) cause vascular toxicity directly through inhibition of VEGFR or indirectly by interfering with other tyrosine kinases. Heart failure caused by inhibition of VEGF and VEGFR has been rarely documented. Arterial hypertension and thromboembolic events are more frequent cardiovascular side effects. In this chapter we will illustrate the mechanism of action of anti-VEGF/anti-VEGFR drugs, their toxic effects, and the strategy to prevent, to diagnose, and to treat these cardiovascular complications.
VEGF and VEGFR
Vascular endothelial growth factor (VEGF) is the main member of a family of structurally and functionally related cytokines, which plays a critical role in angiogenesis and promotes cell survival and growth and proliferation of endothelial cells by binding to specific receptors (VEGFR-1, VEGFR-2, neuropilin) [5, 6]. VEGF includes a family of seven members such as VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, and PIGF. They all have a common homologous domain. VEGF-A is the most representative compound. VEGF-A mRNA is expressed in several tissues, including the lung, kidney, heart, and adrenal glands. VEGF-A is a glycoprotein that exists in at least seven isoforms, from 34 to 42 kDa of molecular weight, which are derived by alternative splicing of the eight exons of the VEGF human gene. To date, researchers have identified three different receptors that bind VEGF, such as VEGFR-1 (Flt-1), VEGFR-2 (KDR or Flk-1), and VEGFR-3. The binding of VEGF to its receptors activates signaling pathways that promote the growth of vascular endothelial cells derived from arteries, veins, and lymphatic vessels. Each receptor has seven immunoglobulin-like domains in the extracellular portion, a single transmembrane portion, and an intracellular tyrosine kinase domain. The different receptors differ in activity and affinity for ligand:
-
A.
VEGFR-1 (Flt-1): it is the first VEGF receptor discovered, although its function is not yet clear. The binding of VEGF-A with this receptor seems to modulate the division of endothelial cells during the early stages of vascular development, although with a weak activity [7].
-
B.
VEGFR-2 (KDR or Flk-1) appears to be the most important receptor in the regulation of mitogenesis and permeability by VEGF. The effects of VEGF binding to VEGFR-2 during angiogenesis include the production of platelet-activating factor by endothelial cells, stimulation of mitosis, and migration of these cells, as well as an increase in vascular permeability. It has been shown that Flk-1 null mice are characterized by the absence of vasculogenesis. This evidence highlights the importance of VEGF binding to VEGFR-2. VEGF binding to this receptor leads to activation of the inositol 3 phosphate kinase pathway, which results in an increase in intracellular inositol triphosphate. This event leads to activation of protein kinase B (Akt/PKB) and endothelial nitric oxide synthase. The first enzyme inhibits caspase-9, promoting cell survival, while the second enzyme leads to NO formation which, in turn, promotes vasodilation and increases permeability and cell migration [8].
-
C.
VEGFR-3 differs from the other two receptors because it moves toward proteolytic cleavage of the extracellular portion. Only VEGF-C and VEGF-D bind to this receptor, and its presence is limited to the endothelial cells of lymphatic vessels [9].
Anti-VEGF and Anti-VEGFR Drugs
The inhibition of VEGF and its receptors represents the main (but not sole) mechanism by which antiangiogenic drugs can cause vascular toxicity (Table 5.1) [10].
Bevacizumab is a monoclonal antibody that targets VEGF-A, thus preventing its interaction with VEGFR and leading to inhibition of tumor angiogenesis. It can cause high blood pressure, left ventricular dysfunction (LV), heart failure (HF), myocardial ischemia, and atherothrombotic events (ATEs). The incidence of severe ATEs in patients treated with bevacizumab was reported to be around 1.8%, with an incidence of AMI equal to 0.6% [11,12,13]. Left ventricular dysfunction was reported in 1.7–3% of cases [14].
Sunitinib is a multi-target tyrosine kinase inhibitor (TKI). It targets the VEGF receptor (VEGFR) 1–3, PDGFR, c-Kit, FMS-like tyrosine kinase-3 (FLT3), colony-stimulating factor-1 receptor (CSF-1R), and the product of the RET human gene (RET, mutated in medullary thyroid carcinomas/multiple endocrine neoplasia). It can cause high blood pressure and HF in 4–11% [14, 15].
Sorafenib is a multi-target TKI that, at clinically relevant concentrations in vitro kinase assay, inhibits at least 15 kinases, including VEGFR, PDGFR, Raf-1, B-Raf, c-Kit, and FLT3 [16]. It can cause high blood pressure, myocardial ischemia, and, rarely, heart failure [17].
Pazopanib is a small molecule, multi-target inhibitor of PDGFR, VEGFR, and c-KIT. It can cause high blood pressure and congestive heart failure [18, 19].
Axitinib is a potent second-generation inhibitor of VEGFR. It can cause high blood pressure but also myocardial infarction and arrhythmias [20].
Regorafenib is a multi-target TKI. It targets VEGFR-2–3, RET, KIT, PDGFR, and RAF. It can cause high blood pressure and myocardial ischemia [21].
Cabozantinib is a potent inhibitor of receptor tyrosine kinases, including VEGF, MET, RET, KIT, Flt-3, AXL, and Tie-2. It can cause venous and, more rarely, arterial thrombosis (myocardial infarction and stroke) [22].
Mechanisms of Cardiovascular Toxicity
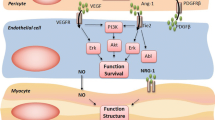
VEGF cascade induces proliferation of endothelial cells and promotes vascular integrity. Hence, inhibition of VEGF/VEGFR signaling pathway seems to be the main cause of vascular injury, endothelial dysfunction, and atherothrombotic events [23]. In fact, VEGF/VEGFR inhibition can lead to endothelial dysfunction and exposure of subendothelial collagen. This can facilitate the activation of the coagulation cascade by tissue factor binding and occurrence of thrombotic events. VEGF binding with the VEGFR-2 activates several intracellular signaling pathways, including the phosphatidylinositol-3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK) pathways. When VEGF interacts with its receptors VEGFR-1, VEGFR-2, and VEGFR-3, PI3K and phospholipase C (PLC) are triggered. On the one hand, PI3K induces the conversion of PIP2 into PIP3, which stimulates Akt supported by the action of PD1K. Akt determines the stimulation of eNOS (endothelial nitric oxide synthase) , thus causing the production of NO. In addition, Akt inhibits caspase-9, promoting cell survival. On the other hand, PLC determines the cleavage of PIP2 to inositol trisphosphate (IP3) and diacylglycerol (DAG). The second messenger IP3 facilitates the entry of Ca2+ ions in the cell. This can lead to eNOS induction and increase of NO production [24]. NO can increase cGMP production through induction of guanylyl cyclase activity. This produces vasodilation with a reduction of platelet aggregation and smooth muscle cell growth. Thus, anti-VEGF therapies promote an unbalance between vasodilation and vasoconstriction through the reduction of NO and prostacyclin, leading to arterial hypertension and to an increase of blood viscosity through the overproduction of erythropoietin [25]. Vasoconstriction is accompanied by endothelial dysfunction. Hence, increased blood viscosity related to cancer, inhibition of the VEGF/VEGFR, and endothelial dysfunction can contribute to increase the risk of arterial thrombosis (stroke and myocardial infarction) in cancer patients (Fig. 5.1) [23].

Consequences of VEGF pathway inhibition. Reproduced from: Bronte G et al. Conquests and perspectives of cardio-oncology in the field of tumor angiogenesis-targeting tyrosine kinase inhibitor-based therapy. Expert Opin Drug Saf. 2015 Feb;14(2):253–67
Arterial Hypertension
Several mechanisms of arterial hypertension have been postulated. These mechanisms include both functional (inactivation of eNOS and production of vasoconstrictors such as endothelin-1) and structural (capillary rarefaction) modifications [26].
VEGF inhibition decreases the production of NO leading to vasoconstriction, elevated peripheral vascular resistance, and hypertension. NO deficiency also leads to increased proliferation of vascular medial cells, creating a more resistant hypertensive state [27]. Additionally, impaired NO production affects renal sodium homeostasis, leading to sodium retention and further elevations in blood pressure [28]. Capillary rarefaction is another postulated mechanism through which VEGF inhibition can lead to hypertension. This process involves a decrease in capillary density at the peripheral level leading to increased vascular resistance. Rarefaction can be functional (vasoconstriction of arterioles) or structural (true capillary loss), although both processes are often interrelated [29]. In a study evaluating 20 patients treated with the VEGF inhibitor bevacizumab, microvascular rarefaction and hypertension were observed in all subjects [30]. This phenomenon is thought to be reversible after discontinuation of the VEGF inhibitor [31]. Whether rarefaction is the cause of hypertension or a consequence of elevated pressures remains uncertain [32]. The loss of pericytes due to inhibition of PDGFR, along with inhibition of angiogenesis, due to the VEGFR inhibition, is supposed to be the main mechanisms for capillary rarefaction [33].
VEGF inhibition may also lead to increased production of other vasoactive substances which can contribute to the development of hypertension. For example, VEGF inhibition causes endothelial dysfunction and increased endogenous sFlt-1 and endothelin-1 (ET-1) production, which leads to a phenotype resembling preeclampsia with significant hypertension and proteinuria [34, 35]. Finally, hypertension due to anti-VEGF may be exacerbated by their effects on other organ systems. Renal dysfunction due to angiogenesis inhibition may also play a role in the development and maintenance of hypertension. Thrombotic microangiopathy has been observed and can lead to hypertension, proteinuria, and hemolysis. These agents can also lead to the deprivation of functional VEGF in the glomeruli resulting in deterioration of kidney function, reduced glomerular filtration rate (GFR), volume retention, and ultimately the hypertensive response [36, 37]. VEGF-mediated suppression of nephrin, which is important for the maintenance of glomerular function, can contribute to the development of arterial hypertension [38]. In addition, vascular injury can be “direct,” i.e., caused directly by the target therapies such as VEGF/VEGFR, or “indirect,” i.e., caused by arterial hypertension secondary to target treatment (Fig. 5.2).

Mechanisms leading to arterial hypertension induced by anti-VEGF treatments
Arterial hypertension was reported in patients treated with bevacizumab, axitinib, sorafenib, sunitinib, axitinib, pazopanib, and regorafenib. It is the most common cardiovascular adverse effect. In a meta-analysis including 13 clinical trials and a total of 4999 patients, the incidence of all-grade hypertension was 21.6%. The incidence of hypertension warranting the addition or adjustment to dosing of more than one medication (grade 3 or grade 4) was 6.8%. The RR of grade 3 or grade 4 hypertensions using sunitinib compared to placebo was 23 [39, 40].
Qi et al. in a meta-analysis showed that the average incidence of all-grade hypertension among patients receiving pazopanib was 35.9%. High-grade (grade 3 or grade 4) hypertension was associated with significant morbidity and subsequent dose reduction or discontinuation of pazopanib treatment. The trials reported an average incidence of high-grade hypertension among patients receiving pazopanib of 6.5% [41].
Arterial hypertension is also the most frequent cardiovascular adverse event associated with axitinib. Indeed, all-grade hypertension showed a frequency of 40%, while grade 3 or higher had a frequency of 13% [42].
Cardiac Dysfunction
Mechanisms of cardiac dysfunction induced by anti-VEGF drugs are not completely understood and some hypothesis has been formulated. For example, sunitinib may cause systolic dysfunction by inhibiting the mitogen-activated protein kinase (MAPK), a regulator of myocyte stress response. This inhibition leads to the reduction of energy production and mitochondrial and consequently ventricular dysfunction [43]. In animal models treated with sunitinib, an increase in the expression of genes involved in the response to hypoxia was observed, including the prolyl hydroxylase domain-containing protein, which is important in the regulation of the hypoxia-inducible factor 1α (HIF-1α). Hypotheses have suggested that a chronic unregulated activation of genes involved in the response to hypoxia, especially HIF-1α, may result in cardiac dysfunction. Further confirmations are required [44]. Also PDGFR inhibition induced by sorafenib and sunitinib could contribute to cardiac dysfunction. PDGFR plays an important role in promoting cell survival and cardioprotection in conditions of pathological stress [45]. Inhibition of stem cell growth factor (c-Kit or CD 117) that is expressed by hematopoietic stem cell precursors and endothelial progenitor cells may also contribute to cardiac dysfunction [46]. VEGF inhibition, in mice subjected to pressure overload, resulted in reduced capillary density, reduced compensatory hypertrophy, left ventricular dilatation, and contractile dysfunction [47]. In animal models of nonischemic cardiomyopathy, overexpression of VEGF resulted in a reduction of apoptosis and proapoptotic signals and delayed progression versus heart failure after tachy-pacing [48]. Data suggest that inhibition of VEGF may worsen myocardial function, especially in the course of pathological stresses such as increased post-loading and arterial hypertension [46]. Moreover inhibition of the ERK factor favoring cell survival through inhibition of the BRAF proto-oncogene and other molecules was hypothesized [49, 50].
Thrombotic Events
Arterial thrombotic events (ATEs) have been reported with bevacizumab through VEGF inhibition [51]. Increased blood viscosity related to cancer and the presence of cardiovascular risk factors, in association to VEGF inhibition (endothelial dysfunction, production of vasoactive substances, increased inflammation, and plaque instability), further increase the risk of arterial thrombosis in oncological patients [25].
Bevacizumab through VEGF inhibition may increase inflammation and plaque instability causing thrombus formation; it increases the release of inflammatory cytokines which activate the coagulation system [52, 53]. The incidence of thrombotic events is greater in patients treated with bevacizumab plus chemotherapy compared to patients treated with chemotherapy alone [54]. Particularly Economopoulou et al. reported ATEs in 5.5% of patients treated with bevacizumab plus chemotherapy compared to 3.1% of patients treated with chemotherapy alone. The incidence of myocardial infarction was 1.5% in patients treated with bevacizumab compared to 1% in the control group. Older age (>65 years) and a previous thrombotic episode can increase thrombotic risk [55].
In a recent meta-analysis of 12,617 patients, treatment with bevacizumab was associated with a significant increase of ATEs and particularly of myocardial ischemia but not stroke [56]. The incidence of ATEs (stroke and IMA) was 3.8% in patients with metastatic colonic carcinoma, non-small cell lung cancer, and breast cancer [57].
ATEs can also be caused by sorafenib, sunitinib, pazopanib, and axitinib. In a meta-analysis that included over 10,000 patients, the incidence of arterial thromboembolic events was analyzed. The relative risk (RR) of ATEs for TKI in comparison with controls was of 3.03. RR for sorafenib was 3.1, and it was 2.39 for sunitinib [58, 59].
In a trial by Sternberg et al., the authors showed that arterial thrombotic events occurred in 3% of pazopanib-treated patients, among these MI/ischemia 2%, cerebrovascular accident <1%, and transient ischemic attack <1% compared with the placebo arm, in which there were none [60].
Bevacizumab should be discontinued in patients who develop severe ATE during therapy; there are no guidelines regulating restart of the drug in these patients. Treatment of ATEs during anti-VEGF and anti-VEGFR treatment is the same as that used in the absence of treatment with anticancer drugs.
Pharmacological prophylaxis for thrombotic events using cardioaspirin could be considered in oncological patients at high cardiovascular risk, before starting treatment with anti-VEGF-VEGFR if there are no contraindications. Cardioaspirin has improved survival in patients with cancer and myocardial ischemia, regardless of thrombocytopenia [61].
Venous thromboembolic events can also occur in patients treated with bevacizumab although the incidence of this event is conflicting in the different studies. From a meta-analysis, the risk of venous thromboembolic events was high in patients treated with bevacizumab; from a second one, no significant increase in the venous events was found [62].
Venous thromboembolic events are reported also in the course of treatment with TKI especially if used in combination with chemotheraphy [63]. Venous thrombotic events are currently managed using low-molecular-weight heparin; however recent evidences suggest that direct oral anticoagulant is safe and effective (see chapter on venous thromboembolism) [64].
Early Diagnosis of Cardiovascular Toxicity
In order to prevent the occurrence of cardiovascular events in patients treated with anti-VEGF-VEGFR drugs and avoid the need of treatment discontinuation, it is important that patients undergo a comprehensive cardiovascular evaluation before starting the treatment and during its course. Particularly it is mandatory to identify and manage cardiovascular risk factors and to optimize treatment of current cardiac disease [65, 66]. Cardiovascular evaluation should include objective examination including peripheral pulses evaluation, blood pressure measurement, ECG, and echocardiography. Antihypertensive treatment should be started if high blood pressure is found or therapy implemented if at control it is found to be nonoptimal. During anticancer therapy, according to the National Cancer Institute, blood pressure should be measured every week during the first cycle, subsequently every 2 to 3 weeks [67]. Antihypertensive treatment should be individualized on the basis of the risk profile of the patient; only in same cases of resistant arterial hypertension, temporary suspension of anti-VEGF-VEGFR drugs should be considered.
Echocardiography beyond measurement of ejection fraction with biplane Simpson method should possibly include 3D evaluation and left ventricle global longitudinal strain analysis to identify early signs of left ventricular dysfunction.
Carotid ultrasound should be performed in patients with risk factors to refine risk stratification. Recent studies reported the usefulness of carotid-femoral pulse wave velocity (cf-PWV) and augmentation index to early detect vascular damage in patients treated with anti-VEGFR drugs. Specifically, blood pressure (BP) and cf-PWV and systolic (global longitudinal strain) and diastolic function have been shown to change in patients after the initiation of the anti-VEGFR treatment [68]. Interestingly, changes in BP and stiffness seemed to be reversible upon discontinuation of treatment, while LV systolic and diastolic functions were persistently abnormal [69].
Treatment of Cardiovascular Toxicity
There are no specific guidelines or expert consensus regarding the optimal treatment of anti-VEGF-induced arterial hypertension and other cardiovascular complications in oncological patients. Treatment of cardiovascular complication induced by anti-VEGF-VEGFR follows the current recommendations about heart failure, myocardial infarction, and arterial hypertension [70, 71]. Standard recommendations on arterial hypertension can also be applied in this patient population (restriction of sodium intake, exercise, diet, lifestyle changes). ACEI/sartans, calcium dihydropyridine antagonists and beta-blockers are the preferred drugs in this setting [65]. Some studies showed that ACEIs and sartans have antitumor properties; they can reduce angiogenesis and microvascular rarefaction, although recent studies have suggested that these drugs can develop a pro-tumor microenvironment [72, 73].
Diltiazem and verapamil that inhibit the cytochrome P450 should be avoided for their possible interference with anti-VEGF inhibitors, and diuretics are not drugs of choice for possible induction of electrolyte disturbances that may favor QT interval prolongation.
Considering mechanism of action of VEGF-VEGFR, drugs that increase the release of nitric oxide should be considered in resistant arterial hypertension [74]. In fact, some cases reported the efficacy of long-release nitrates in patients with antiangiogenic drugs induced hypertension despite optimized medical therapy [75].
Also sildenafil (phosphodiesterase inhibitor) and nebivolol can potentiate the vasodilatory properties of NO, and they can be effective in such patients [76]. Nebivolol, B1 adrenergic antagonist, may enhance the nitric oxide signal; it may induce vasodilatation increasing the bioavailability of NO. The exact mechanism is not known, but it may be secondary to nitric oxide synthase activation through the stimulation of β3-adrenergic receptors expressed in embryonic cells and the reduction of dimethyl arginine and free oxygen radicals [77]. Also antagonists of endothelin receptors may play a role in resistant hypertension even if their use has been validated in the treatment of pulmonary hypertension (Fig. 5.3).

Treatment of arterial hypertension induced by anti-VEGF treatments
In cases of resistant arterial hypertension, despite optimal medical therapy, dose reduction or temporary suspension of anti-VEGF treatment should be considered [78].
In patients at high cardiovascular risk, treatment with aspirin could be warranted before starting anti-VEGF treatment [61]. In case of documented myocardial ischemia during angiogenic inhibitor therapy, treatment should be suspended and eventually therapy resumed, after healing if benefits outweigh the risks. In case of myocardial infarction, the permanent discontinuation of the drug should be considered.
If asymptomatic LV dysfunction occurs, angiogenic inhibitors should be continued in cases of mild (ejection fraction reduction >15%, with ejection fraction >50%) or moderate decrease (ejection fraction 50–40%). Only in case of severe LV dysfunction (ejection fraction <40%) and in symptomatic patients, it is recommended to stop anticancer therapy [74].
LV dysfunction should be treated according to current recommendation. Resumption of the drug can be considered upon ejection fraction improvement and the normalization of symptoms, but the evidences on this regard are still unclear [79].
Conclusions
Anti-VEGF-VEGFR drugs cause cardiac and vascular toxicity, especially arterial hypertension and arterial thrombotic events. Given the high incidence of arterial hypertension induced by VEGF/VEGFR inhibitors, it is very important to control blood pressure before starting any treatment and accurately monitor its variations during the course of therapy. It is also mandatory for the control of cardiovascular risk factors such as diabetes mellitus and dyslipidemia, which can make patients more prone to vascular injury. Moreover, optimization of treatment of concomitant cardiovascular disease is advisable. Echocardiographic monitoring of cardiac function is also reasonable. There are no standardized guidelines regarding the timing of monitoring patients undergoing this treatment; however baseline evaluation and routine periodical surveillance during treatment (i.e., every 3 to 6 months in the absence of symptoms) is reasonable.
More and wider prospective studies are needed to clarify pathophysiological mechanism of cardiovascular damage induced by anti-VEGF inhibitors and to better delineate how to manage patients.
References
Schiffrin EL. A critical review of the role of endothelial factors in the pathogenesis of hypertension. J Cardiovasc Pharmacol. 2001;38(Suppl 2):S3–6.
Green D, Jones H, Thijssen D, Cable NT, Atkinson G. Flow-mediated dilation and cardiovascular event prediction does nitric oxide matter? Hypertension. 2011;57:363–9.
Park KA, Park WJ. Endothelial dysfunction: clinical complications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. 2015;30:1213–25.
Di Lisi D, Madonna R, Zito C, Bronte E, Badalamenti G, Parrella P, et al. Anticancer therapy-induced vascular toxicity: VEGF inhibition and beyond. Int J Cardiol. 2017;227:11–7.
Kurzyk A. Angiogenesis-possibilities, problems and perspectives. Postepy Biochem. 2015;61:25–34.
Tocchetti CG, Gallucci G, Coppola C, Piscopo G, Cipresso C, Maurea C, et al. The emerging issue of cardiac dysfunction induced by antineoplastic angiogenesis inhibitors. Eur J Heart Fail. 2013;15:482–9.
Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86.
Anisimov A, Alitalo A, Korpisalo P, Soronen J, Kaijalainen S, Leppänen VM, et al. Activated forms of VEGF-C and VEGF-D provide improved vascular function in skeletal muscle. Circ Res. 2009;104:1302–12.
Madonna R, De Caterina R. VEGF receptor switching in heart development and disease. Cardiovasc Res. 2009;84:4–6.
Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A, et al. Vascular toxicities of cancer therapies: the old and the new – an evolving avenue. Circulation. 2016;133:1272–89.
Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. 2007;49:186–93.
Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a re-view. Eur J Cancer. 2006;42:3127–39.
Sugrue MM, Yi J, Purdie D, et al. Serious arterial thromboembolic events (sATE) in patients (pts) with metastatic colorectal cancer (mCRC) treated with bevacizumab (BV): results from the BRiTE registry. J Clin Oncol. 2007;25(18):4136.
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–9.
Mena AC, Pulido EG, Guillen-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anti-Cancer Drugs. 2010;21(Suppl. 1):S3–11.
Lyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: a clinical and pharmacologic review. Expert Opin Pharmacother. 2010;11:1943–55.
Youn JY, Wang T, Cai H. An ezrin/calpain/PI3K/AMPK/eNOSs1179 signaling cascade mediating VEGF-dependent endothelial nitric oxide production. Circ Res. 2009;104:50–9.
Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, Hong BF, et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2010;28:475–80.
Van Marcke C, Ledoux B, Petit B, Seront E. Rapid and fatal acute heart failure induced by pazopanib. BMJ Case Rep. 2015;2015 pii: bcr2015211522.
Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–62.
Bronte E, Bronte G, Novo G, Bronte F, Bavetta MG, Lo Re G, et al. What links BRAF to the heart function? New insights from the cardiotoxicity of BRAF inhibitors in cancer treatment. Oncotarget. 2015;6(34):35589–601.
Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol. 2013;31:3639–46.
Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96:1788–95.
Facemire CS, Nixon AB, Griffiths R, , Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension 2009;54:652–658.
Kilickap S, Abali H, Celik I. Bevacizumab, bleeding, thrombosis, and warfarin. J Clin Oncol. 2003;21:3542.
Izzedine H, Massard C, Spano JP, Goldwasser F, Khayat D, Soria JC. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur J Cancer. 2010;46:439–48.
Brinda BJ, Viganego F, Vo T, Dolan D, Fradley MG. Anti-VEGF-induced hypertension: a review of pathophysiology and treatment options. Curr Treat Options Cardiovasc Med. 2016;18:33.
Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–52.
Steeghs N, Gelderblom H, Roodt JO, Christensen O, Rajagopalan P, Hovens M, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. 2008;14:3470–6.
Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19:927–34.
Steeghs N, Rabelink TJ, op’t Roodt J, Batman E, Cluitmans FH, Weijl NI, et al. Reversibility of capillary density after discontinuation of bevacizumab treatment. Ann Oncol. 2010;21:1100–5.
Hansen-Smith FM, Morris LW, Greene AS, Lombard JH. Rapid microvessel rarefaction with elevated salt intake and reduced renal mass hypertension in rats. Circ Res. 1996;79:324–30.
Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5.
Vigneau C, Lorcy N, Dolley-Hitze T, Jouan F, Arlot-Bonnemains Y, Laguerre B, et al. All anti-vascular endothelial growth factor drugs can induce ‘pre-eclampsia-like syndrome’: a RARe study. Nephrol Dial Transplant. 2014;29:325–32.
Amraoui F, Spijkers L, Hassani Lahsinoui H, Vogt L, van der Post J, Peters S, et al. SFlt-1 elevates blood pressure by augmenting endothelin-1-mediated vasoconstriction in mice. PLoS One. 2014;9(3):45.e91897.
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic micro-angiopathy. N Engl J Med. 2008;358:1129–36.
Izzedine H. Anti-VEGF cancer therapy in nephrology practice. Int J Nephrol Renov Dis. 2014;2014:143426.
DeJesus GN, Robinson E, Moslehi J, Humphreys BD. Management of antiangiogenic therapy-induced hypertension. Hypertension. 2012;60(3):607–15.
Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103(9):763–73.
Chen ZI, Ai DI. Cardiotoxicity associated with targeted cancer therapies. Mol Clin Oncol. 2016;4(5):675–81.
Qi WX, Lin F, Sun YJ, Tang LN, He AN, Yao Y, et al. Incidence and risk of hypertension with pazopanib in patients with cancer: a meta-analysis. Cancer Chemother Pharmacol. 2013;71(2):431–9.
Qi WX, He AN, Shen Z, Yao Y. Incidence and risk of hypertension with a novel multi-targeted kinase inhibitor axitinib in cancer patients: a systematic review and meta-analysis. Br J Clin Pharmacol. 2013;76:348–57.
Cheng H, Force T. Molecular mechanisms of cardiovascular toxicity of targeted cancer therapeutics. Circ Res. 2010;106:21–34.
Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, et al. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation. 2010;122:1004–16.
Chintalgattu V, Ai D, Langley RR, Zhang J, Bankson JA, Shih TL, et al. Cardiomyocyte PDGFR-beta signaling is an essential component of the mouse cardiac response to load-induced stress. J Clin Invest. 2010;120:472–84.
Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, et al. Cardioprotective c-kit± cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–77.
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–18.
Pepe M, Mamdani M, Zentilin L, Csiszar A, Qanud K, Zacchigna S, et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ Res. 2010;106:1893–903.
Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. 2009;2(1):15–25.
Khakoo AY, Kassiotis CM, Tannir N, Plana JC, Halushka M, Bickford C, et al. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer. 2008;112:2500–8.
Totzeck M, Mincu RI, Rassaf T. Cardiovascular adverse events in patients with cancer treated with bevacizumab: a meta-analysis of more than 20,000 patients. J Am Heart Assoc. 2017;6(8):e006278.
Hesser BA, Liang XH, Camenisch G, Yang S, Lewin DA, Scheller R, et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104(1):149–58.
Kuenen BC, Levi M, Meijers JC, Kakkar AK, van Hinsbergh VW, Kostense PJ, et al. Analysis of coagulation cascade and endothelial cell activation during inhibition of vascular endothelial growth factor/vascular endothelial growth factor receptor pathway in cancer patients. Arterioscler Thromb Vasc Biol. 2002;22:1500–5.
Hall PS, Harshman LC, Srinivas S, Witteles RM. The frequency and severity of cardiovascular toxicity from targeted therapy in advanced renal cell carcinoma patients. JACC Heart Fail. 2013;1:72–8.
Economopoulou P, Kotsakis A, Kapiris I, Kentepozidis N. Cancer therapy and cardiovascular risk: focus on bevacizumab. Cancer Manag Res. 2015;7:133–43.
Ranpura V, Hapani S, Chuang J, Wu S. Risk of cardiac ischemia and arterial thromboembolic events with the angiogenesis inhibitor bevacizumab in cancer patients: a meta-analysis of randomized controlled trials. Acta Oncol. 2010;49:287–97.
Scappaticci FA, Skillings JR, Holden SN, Gerber HP, Miller K, Kabbinavar F, et al. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. 2007;99:1232–9.
Choueiri TK, Schutz FA, Je Y, Rosenberg JE, Bellmunt J. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010;28(13):2280–5.
Bronte G, Bronte E, Novo G, Pernice G, Lo Vullo F, Musso E, et al. Conquests and perspectives of cardio-oncology in the field of tumor angiogenesis-targeting tyrosine kinase inhibitor-based therapy. Expert Opin Drug Saf. 2015;14(2):253–67.
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28(6):1061–8.
Sarkiss MG, Yusuf SW, Warneke CL, Botz G, Lakkis N, Hirch-Ginsburg C, et al. Impact of aspirin therapy in cancer patients with thrombocytopenia and acute coronary syndromes. Cancer. 2007;109:621–7.
Hurwitz HI, Saltz LB, Van Cutsem E, Cassidy J, Wiedemann J, Sirzén F, et al. Venous thromboembolic events with chemotherapy plus bevacizumab: a pooled analysis of patients in randomized phase II and III studies. J Clin Oncol. 2011;29:1757–64.
Zamorano JL. Specific risk of atrial fibrillation and stroke in oncology patients. Eur Heart J. 2016;37(36):2747–8.
Mandalà M, Falanga A, Roila F. ESMO Guidelines Working Group. Management of venous thromboembolism (VTE) in cancer patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2011;22(Suppl 6):vi85–92.
Zamorano J. An ESC position paper on cardio-oncology. Eur Heart J. 2016;37(36):2739–40.
Tocchetti CG, Cadeddu C, Di Lisi D, Femminò S, Madonna R, Mele D, et al. From molecular mechanisms to clinical management of antineoplastic drug-induced cardiovascular toxicity: a translational overview. Antioxid Redox Signal. 2017; https://doi.org/10.1089/ars.2016.6930.
Maitland ML, Bakris GL, Black HR, Chen HX, Durand JB, Elliott WJ, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst. 2010;102:596–604.
Vallerio P, Stucchi M, Moreo A, Ricotta R, Pozzi M, Giupponi L, et al. Possible role of arterial function in cancer treatment targeting vascular endothelial growth factor receptor oncologic response. J Hypertens. 2015;33(Suppl 1):e111.
Moreo A, Vallerio P, Ricotta R, Stucchi M, Pozzi M, Musca F, et al. Effects of cancer therapy targeting vascular endothelial growth factor receptor on central blood pressure and cardiovascular system. Am J Hypertens. 2016;29:158–62.
López-Fernández T, Martín García A, Santaballa Beltrán A, Montero Luis Á, García Sanz R, Mazón Ramos P, et al. Cardio-onco-hematology in clinical practice. Position paper and recommendations. Rev Esp Cardiol (Engl Ed). 2017;70(6):474–86.
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37(36):2768–801.
Siddiqui AJ, Mansson-Broberg A, Gustafsson T, Grinnemo KH, Dellgren G, Hao X, et al. Antagonism of the renin-angiotensin system can counteract cardiac angiogenic vascular endothelial growth factor gene therapy and myocardial angiogenesis in the normal heart. Am J Hypertens. 2005;18:1347–52.
Okwan-Duodu D, Landry J, Shen XZ, Diaz R. Angiotensin-converting enzyme and the tumor microenvironment: mechanisms beyond angiogenesis. Am J Physiol Regul Integr Comp Physiol. 2013;305:R205–15.
Curigliano G, Cardinale D, Suter T, Plataniotis G, de Azambuja E, Sandri MT, et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann Oncol. 2012;23(Suppl 7):vii155–66.
Kruzliak P, Kovacova G, Pechanova O. Therapeutic potential of nitric oxide donors in the prevention and treatment of angiogenesis-inhibitor-induced hypertension. Angiogenesis. 2013;16:289–95.
Okamoto LE, Gamboa A, Shibao CA, Arnold AC, Choi L, Black BK, et al. Nebivolol, but not metoprolol, lowers blood pressure in nitric oxide-sensitive human hypertension. Hypertension. 2014;64:1241–7.
Dessy C, Saliez J, Ghisdal P, Daneau G, Lobysheva II, Frérart F, et al. Endothelial beta3-adrenoreceptors mediate nitric oxide-dependent vasorelaxation of coronary microvessels in response to the third-generation beta-blocker nebivolol. Circulation. 2005;112:1198–205.
Dirix LY, Maes H, Sweldens C. Treatment of arterial hypertension (AHT) associated with angiogenesis inhibitors. Ann Oncol. 2007;18:1121–2.
Maurea N, Spallarossa P, Cadeddu C, Madonna R, Mele D, Monte I, et al. A recommended practical approach to the management of target therapy and angiogenesis inhibitors cardiotoxicity: an opinion paper of the working group on drug cardiotoxicity and cardioprotection, Italian Society of Cardiology. J Cardiovasc Med (Hagerstown). 2016;17 Suppl 1 Special issue on Cardiotoxicity from Antiblastic Drugs and Cardioprotection:e93–104. Review.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Novo, G., Di Lisi, D., Bronte, E., Fiuza, M., Pinto, F.J. (2019). Cardiovascular Damage Induced by Anti-VEGF Therapy. In: Russo, A., Novo, G., Lancellotti, P., Giordano, A., Pinto, F. (eds) Cardiovascular Complications in Cancer Therapy. Current Clinical Pathology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-93402-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-93402-0_5
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-93401-3
Online ISBN: 978-3-319-93402-0
eBook Packages: MedicineMedicine (R0)