
Abstract
Two concepts have gained a growing audience these last years: the pressure amplification between central and peripheral arteries in response to arterial stiffening and pressure wave reflection and the left ventricle (LV)-arterial system coupling in heart failure (notably with preserved ejection fraction also referred to as diastolic heart failure) as arterial stiffening can result in impaired active ventricular relaxation and passive ventricular compliance. The aims of this chapter are (1) to detail the haemodynamic characteristics of the arterial circulation in order to explain why it is important to measure arterial stiffness and central BP in hypertensive patients, (2) to describe the various non-invasive methods currently available to measure arterial stiffness and central BP and (3) to discuss how arterial stiffness and central BP measurements can help understanding the relationship between hypertension and heart failure.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
The relationship between brachial blood pressure (BP), hypertension and heart failure is well established. However, two concepts have gained a growing audience these last years: the pressure amplification between central and peripheral arteries in response to arterial stiffening and pressure wave reflection and the left ventricle (LV)-arterial system coupling in heart failure (notably with preserved ejection fraction also referred to as diastolic heart failure) as arterial stiffening can result in impaired active ventricular relaxation and passive ventricular compliance. An increasing number of physiological studies, as well as pathophysiological, epidemiological and pharmacological studies, have underlined the importance of measuring not only brachial systolic and pulse pressures but also central systolic and pulse (i.e. systolic minus diastolic) pressures and arterial stiffness.
The aims of this chapter are (1) to detail the haemodynamic characteristics of the arterial circulation in order to explain why it is important to measure arterial stiffness and central BP in hypertensive patients, (2) to describe the various non-invasive methods currently available to measure arterial stiffness and central BP and (3) to discuss how arterial stiffness and central BP measurements can help understanding the relationship between hypertension and heart failure.
1 Pathophysiology of Central Blood Pressure and Wave Reflection in Hypertension
1.1 Haemodynamic and Reflection of Pressure Waves
During ventricular contraction, a part of the stroke volume is forwarded directly to the peripheral tissues, while the remainder is momentarily stored in the aorta and central arteries, thereby stretching the arterial walls and raising local blood pressure (Fig. 9.1). Part of the energy produced by the heart is thus used for the distension of arteries and is transferred to the vessel walls as potential energy by passive loading of elastic elements in the wall. During diastole, the aorta recoils, and the “stored” energy is restored to the arterial system, squeezing the accumulated blood forwards into the peripheral tissues, ensuring quasi-continuous flow, especially during diastole (Fig. 9.1). Cardiac work has two components: potential energy (pressure generation-dP/dT) and kinetic energy (volumic pump, related to SEV and blood velocity). The systolic work is delivered during 1/3 of the time, i.e. any increase in pulse pressure will lead to an increase in dP/dT. In order to optimize the cardiac work during ventricular ejection, the energy spent to increase pressure (i.e. to distend arteries) should be retrieved as kinetic energy during diastole. Thus the optimum is obtained for a slow and small increase in pulse pressure, inducing large distension. The relation between increase in pulse pressure and increase in arterial volume defines arterial compliance (or its inverse arterial stiffness). Thus the efficiency of heart vessel coupling depends on the stiffness and geometry of the arteries, mainly the proximal aorta [2]. When arterial stiffness is low, arterial wall opposes low resistance to distension, and dP/dT is minimized, while high compliance is retrieved during diastole. On the contrary, when the arterial system becomes rigid and distension limited, most of the stroke volume will flow through the arterial system and peripheral tissues only during systole with two consequences: intermittent high pulsatility flow and short capillary transit time with reduced metabolic exchanges.


Schematic representation of the role of arterial stiffness on assuring continuous blood flow through the peripheral circulation and how the aortic stiffening leads to increased SBP and PP. Adapted from Briet et al. [1] with permission from Elsevier
The ejection of blood into the aorta generates a pressure wave that propagates along the aorta towards the peripheral arterial tree. The velocity of wave propagation along the aorta, i.e. pulse wave velocity (PWV), is a direct measure of arterial stiffness. The pressure waveform differs whether measured centrally close to the heart or more distally. The central arterial pressure waveform is a composite of the forward pressure wave created by ventricular contraction and a reflected wave [1] (Fig. 9.2). Indeed, the arterial tree is not a simple tube but a complex structure that can be seen as a branched tube with a reflection site at its distal end. From the heart towards the periphery, arteries continuously decrease in diameter (i.e. geometric taper) and increase in stiffness (i.e. elastic taper, also named “arterial stiffness gradient”) while also continuously branching [3]. In fact, the notion of reflection site is statistic; the sum of multiple reflection sites (bifurcations, tapering, diameter mismatch, peripheral resistances) acts as a unique site, which grossly correspond to the renal arteries (see below). The stiffness gradient, together with the geometric taper, local arterial branching and lumen narrowing, creates an impedance mismatch causing partial reflections of forward pressure waves travelling back to the central aorta (reflected wave) [3, 4] (Fig. 9.3).


Representation of forward and reflected pressure wave travelling and the influence of their timing and overlap on recorded aortic and peripheral pressure waves. Tsh time to shoulder. Adapted from Briet et al. [1] with permission from Elsevier


Upper panel: in the presence of arterial stiffness gradient (aortic PWV < peripheral PWV), partial pressure wave reflection occurs distant from microcirculation and returns at low PWV to the aorta in diastole maintaining central-to-peripheral amplification. Partial pressure wave reflections limit the transmission of pulsatile pressure energy to the periphery and protect the microcirculation. Lower panel: When the stiffness gradient disappears or is inverted (aortic PWV > peripheral PWV), pulsatile pressure is not sufficiently dampened and is transmitted, thus damaging the microcirculation. In parallel, the central-to-peripheral pressure amplification is attenuated. Adapted from Briet et al. [1] with permission from Elsevier
The wave reflections will considerably change the pressure wave amplitude and shape along the arterial tree. Forward and reflected pressure waves overlap, and the final amplitude and shape of the pulse pressure wave are determined by the phase relationship (the timing) between these component waves. The overlap between the two waves depends on the site of pressure recording along the arterial tree. Peripheral arteries are close to reflection sites, and the reflected wave occurs at the impact of forward wave, i.e. the waves are in phase producing an additive effect. The ascending aorta and central arteries are distant from reflecting sites, and the return of the reflected wave is variably delayed depending on PWV and travelling distances (Figs. 9.2 and 9.3) [1, 2]. In the aorta or central arteries, the forward and reflected waves are not in phase. In subjects with low PWV, reflected waves impact on central arteries during end systole or early diastole, increasing the aortic pressure in early diastole and not during systole. This is physiologically advantageous, since the increased diastolic pressure boosts the coronary perfusion without increasing the LV pressure load. When PWV is high, the reflected wave comes back early during systole, increases central SBP and PP and increases LV work.
Pressure waves are reflected from the periphery, mainly at branch points or sites of impedance mismatch. The reflection pattern is thus complex, but may be seen as a “net” or “effective” reflection pattern, where all the forward and backward running waves seem to add up to one single forward and backward wave, representing the global effect of all reflections present [2]. The phenomenon of wave reflection can be quantified through the augmentation index (AIx)—defined as the difference between the second (P2) and first (P1) systolic peaks (P2 – P1 = AP, i.e. augmentation pressure) expressed as a percentage of pulse pressure (AIx = AP/PP) (Fig. 9.4).


The phenomenon of wave reflection can be quantified through the augmentation index (AIx)—defined as the difference between the second (P2) and first (P1) systolic peaks (P2 − P1 = AP, i.e. augmentation pressure) expressed as a percentage of PP: AIx = AP/PP. Adapted from Laurent et al. [5] with permission from Oxford University Press
Thus, apart from a high PWV, also changes in reflection sites can influence central SBP, PP and AIx. The major determinant of central SBP and PP is, by definition, the forward pressure wave, since the reflected wave cannot physically carry more energy than the forward wave, but can be of high amplitude if reflections are coming from a major site [2]. In clinical investigation, not only DBP and height, which are related to total peripheral resistance and reflection sites, but also age and aortic PWV are the main determinants of AIx.
1.2 Central-to-Peripheral Amplification Phenomenon
When SBP is recorded invasively and simultaneously in the aortic arch and at multiple peripheral sites, it is possible to detect an “amplification phenomenon”, i.e. under resting conditions in healthy men, brachial SBP is about 10% higher than aortic SBP [6] (Fig. 9.3). Indeed, in the presence of the physiological arterial stiffness gradient (aortic PWV < peripheral PWV), partial pressure wave reflection occurs distant from microcirculation and return at low PWV to the aorta in diastole; thus reflected wave arrives back at the aortic root during late systole, whereas at the site of peripheral artery (i.e. brachial artery), the pressure wave travels rapidly, and the reflected wave (from peripheral branching sites and small arteries) arrives at the recording site very close to the forward wave, i.e. in early systole, thus rising local SBP. In average, central SBP is lower than distal SBP, leading to the so-called central-to-peripheral amplification. By contrast, when the stiffness gradient disappears or is inverted (aortic PWV > peripheral PWV), pulsatile pressure is not sufficiently dampened at the central level, and the central-to-peripheral pressure amplification is attenuated [2, 6] (Fig. 9.3).
The amplification phenomenon is attenuated by ageing and hypertension [6] because of arterial stiffening. Indeed, by favouring early wave reflections, arterial stiffening increases peak- and end-systolic pressures in the ascending aorta, increasing myocardial pressure load (left ventricular hypertrophy) and oxygen consumption and decreasing the diastolic blood pressure and subendocardial blood flow. Thus central SBP is higher in elderly subjects and hypertensive patients than in young normotensive subjects and closer to the brachial SBP value, reducing the difference. Indeed, at the site of brachial artery, arterial stiffness is not influenced by age and little by hypertension, and the timing of forward and reflected waves is similar to those in young normotensive subjects.
Central-to-peripheral amplification can be expressed either as absolute value (peripheral SBP minus central SBP; peripheral PP minus central PP) or relative value (peripheral SBP/central SBP; peripheral PP/central PP).
1.3 Reflection Magnitude
The time-domain analysis of the pulse waveform, called pulse waveform analysis (PWA), allows quantifying the effects of pressure wave reflection on the central arterial waveform. The determination of forward and backward waveforms requires a pressure-flow analysis in the time domain (Fig. 9.5). In early systole, prominent wave reflections are reduced; thus early systolic pressure and flow can be interpreted according to a simple model originally proposed by Westerhof et al. [8], referred as “the standard” Windkessel model for the systemic circulation: Zc = Δ Pressure/Δ Flow, where Zc is characteristic impedance and Δ Pressure and Δ Flow are calculated at the time point at which flow and pressure reach 95% of their peak value (Fig. 9.5). Because of the superimposition of reflected (backward) and incident (forward) waves in the early systole, measured pressure equals the sum of forward and backward pressures, and measured flow equals the sum of forward and backward flows (backward flow having a negative sign). Pressure and flow waves can be quantitatively related to each other, through Zc. A procedure, commonly called wave separation analysis (WSA) (Fig. 9.5), can be used to decompose the pressure signal into its forward (Pf) and reflected (backward, Pb) components:


Wave separation analysis. Once Zc is known (see text), pressure and flow waves can be quantitatively “scaled” in the vertical axis (a), allowing for identification of the difference in their waveforms (grey area), which is assumed to be the result of wave reflections. Pressure can be separated into forward and backward pressure (Pf and Pb), and the amplitude ratio of Pb/Pf (reflection magnitude) can be computed (b). Adapted from Chirinos et al. [7] with permission from Wolters Kluwer Health
In practice, this way of calculating Pf and Pb is not very different from the one resulting from frequency analysis where each individual harmonic is studied and then adding all harmonics. The ratio of their amplitudes defines the reflection magnitude:
Interestingly, this computation of RM does not depend too much on the calibration of the flow waveform, and some authors [4] have proposed an approximated approach using pressure information only, assuming a triangular or a physiologic flow waveform. Although very appealing from a theoretical point of view, time domain or frequency domain wave magnitude determination has produced little breakthrough advances in the comprehension of heart vessel coupling.
2 Methods for Determining Arterial Stiffness and Central Blood Pressure
A large number of reviews have made recommendations for adequate measurements of arterial stiffness and central BP [5, 9, 10]. We will shortly review here the main methods.
2.1 Arterial Stiffness Measurements
Arterial stiffness can be evaluated at the systemic, regional and local levels. In contrast to systemic arterial stiffness, which can only be estimated from models of the circulation, regional and local arterial stiffness can be measured directly, and non-invasively, at various sites along the arterial tree. A major advantage of the regional and local evaluations of arterial stiffness is that they are based on direct measurements of parameters strongly linked to wall stiffness. Table 9.1 details the various methods currently used for determining arterial stiffness.
2.1.1 Regional Measurements of Arterial Stiffness
The aorta is a major vessel of interest when determining regional arterial stiffness for at least two reasons: the thoracic and abdominal aorta makes the largest contribution to the arterial buffering function, and aortic PWV is an independent predictor of outcome in a variety of populations.
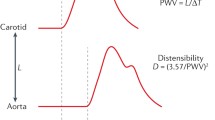
Two-site pulse wave velocity measurements are recommended. The gold standard is the measurement of pulse wave velocity (PWV), which is considered as the most simple, non-invasive, robust and reproducible method. Carotid-femoral PWV is a direct measurement and is usually performed using the foot-to-foot velocity method from various waveforms. These are usually obtained, transcutaneously at the right common carotid artery and the right femoral artery (i.e. “carotid-femoral” PWV), and the time delay (Δt, or transit time) measured between the feet of the two waveforms (Fig. 9.6). The “foot” of the wave is defined at the end of diastole, when the steep rise of the wave front begins. The transit time is the time of travel of the “foot” of the wave over a known distance. A variety of different waveforms can be used including pressure, distension and flow (Doppler), with similar values. The distance (D) covered by the waves is usually assimilated to the surface distance between the two recording sites, i.e. the common carotid artery (CCA ) and the common femoral artery (CFA ). PWV is calculated as PWV = D (m)/Δt (s). However, the pressure wave splits at the origin of the brachiocephalic trunk, the one travelling forwards to the carotid bifurcation, the other travelling down the aorta to the femoral artery. Thus the descending thoracic aorta is reached by the pressure wave after another pressure wave, originating from the same cardiac contraction, arrives at the carotid site. For correcting for this uneven pathway, it has been recommended in a recent consensus paper to measure the direct distance and to apply a 0.8 coefficient, to take into account the shorter pathway of the pressure [11]. Reference values for carotid-femoral PWV have been established in 1455 healthy subjects and a larger population of 11,092 subjects with CV risk factors [12]. Multiple devices using pressure waveforms recorded simultaneously are validated to provide automated measurement of PWV. They are detailed in Table 9.1. They include the brachial-ankle PWV (baPWV), the cardiac-ankle PWV, and the finger-toe PWV.


Measurement of carotid-femoral pulse wave velocity with the foot-to-foot method. The waveforms are usually obtained transcutaneously at the right common carotid artery and the right femoral artery. The time delay (Δt, or transit time) is measured between the feet of the two waveforms. The distance (ΔL) covered by the waves is usually assimilated to the surface distance between the two recording sites, i.e. the common carotid artery and the common femoral artery. PWV is calculated as PWV = 0.8 × ΔL (m)/Δt (s). From Laurent et al. [5] with permission from Oxford University Press
Single-site pulse wave velocity measurements would simplify measurement. Several methods have challenged the reference methods described above. An increasing number of methods indeed calculate PWV over a given arterial pathway from the analysis of the brachial pressure wave. Brachial pressure wave is determined with a brachial cuff. PWV is thus referred as “single-site”- or “brachial cuff”-derived PWV and apparatus as “brachial cuff”-based devices. Importantly, PWV is estimated from various parameters, but not directly measured. These methods include the determination of the time difference between Q wave at ECG and Korotkov sounds at the brachial level (QKD) with ambulatory blood pressure measurement, the Arteriograph® system that estimates PWV from a single-site brachial-cuff oscillometric determination of the suprasystolic waveform at the brachial artery site and the Mobil-O-Graph® system that takes advantage of oscillometric recording of brachial artery pressure waveform to synthesize the central pulse wave by applying a transfer function. This latter method has phenomenological adjustments since age and blood pressure are used to refine PWV estimation.
2.1.2 Local Determination of Arterial Stiffness
Local arterial stiffness of superficial arteries can be directly determined using high-resolution echotracking devices. Carotid stiffness may be of particular interest, since in that artery atherosclerosis is frequent. The advantage of high-resolution echotracking devices is their high precision for determining diameter at diastole and stroke changes in diameter, compared with classical video-image analysis. Thoracic magnetic resonance imaging (MRI) is increasingly popular since it allows combined determination of cardiac and aortic structure and function with undisputed anatomical precision, but at the cost of lower spatial and temporal resolution. However, most of pathophysiological and pharmacological studies have used echotracking techniques.
2.1.3 Systemic Arterial Stiffness
A methodology based on an electrical circuit using a modified Windkessel model has been developed to determine a proximal capacitive compliance and a distal oscillatory compliance. Systemic arterial compliance can also be determined using the “area method” which requires measurement of aortic blood flow (velocimeter at the suprasternal notch) and associated driving pressure by applanation tonometry over the proximal right common carotid artery. A number of theoretical, technical and practical limitations impair their widespread application in the clinical setting.
2.2 Central Blood Pressure Measurements
Arterial pressure waveform should be analysed at the central level, i.e. the ascending aorta, since it represents the true load imposed to the heart, the brain, the kidney and more generally the central large artery walls. Table 9.2 details the various methods currently used for determining central blood pressure. The pressure waveform can be recorded non-invasively with a pencil-type probe incorporating a high-fidelity Millar strain gauge transducer (SPT-301, Millar Instruments). The most widely used approach is to perform radial artery tonometry and then apply a transfer function (SphygmoCor, AtCor, Sydney Australia) to calculate the aortic pressure waveform from the radial waveform (Table 9.2). Indeed, the radial artery is well supported by bony tissue, making optimal applanation easier to achieve.
Aortic pressure waveform can also be estimated from the common carotid waveforms (Table 9.2). Carotid tonometry requires a higher degree of technical expertise, but a transfer function is not necessary since the arterial sites are very close and waveforms are similar. Alternatively, distension waveforms from carotid echotracking can be rescaled and used. A large number of pathophysiological and pharmacological studies have been published using these methods. Apart from methods determining the pressure waveform at the central site, novel methods have been developed, which aim at determining the discrete value of central SBP using the second systolic peak (SBP2) on the radial or brachial pressure waves (Table 9.2). Whatever the method used, an external calibration is necessary because neither tonometry nor echotracking can give access to absolute values of blood pressure. This is usually done by using brachial SBP and DBP to calibrate radial artery tonometry (at the cost of some imprecision due to brachial radial amplification) and then using the radial MBP and DBP to calibrate either aortic or carotid waveforms [5, 6].
Reference values for central BP [6] have been established in 18,183 healthy subjects and a larger population of 27,253 subjects with CV risk factors.
3 Arterial Stiffness, Central Blood Pressure and Systolic Dysfunction
Although the role of arterial stiffness/central BP in the pathophysiology of heart failure with reduced ejection fraction (HFrEF) is known for decades, there is less evidence from longitudinal studies in humans reporting incident HFrEF in patients with high arterial stiffness and central BP.
3.1 Pathophysiological Mechanisms
The mechanistic relationships between HFrEF and arterial stiffness/central BP have been analysed in several reviews and a large number of studies [2, 13,14,15]. We have shown that arterial stiffness acted as “pure” pressure overload, being powerfully associated with LV concentric remodelling, whereas arterial geometry played a “pure” volume overload, being associated with LV dilatation [13]. Arterial stiffness can aggravate myocardial ischaemia, which is a major determinant of HFrEF. Indeed, arterial stiffness, through an early return of wave reflection, increases peak systolic BP and thus myocardial load, triggering left ventricular hypertrophic (LVH) remodelling and reducing diastolic coronary perfusion. The mismatch in myocardial oxygen supply-demand that is conferred by both LVH and lower central diastolic blood pressure can reduce coronary perfusion and increase subendocardial ischaemia [2, 16,17,18]. Aortic stiffness is higher in patients with coronary heart disease (CHD) than in non-CHD patients, as well as in HFrEF patients [14, 15], and is associated with CHD events independently of classical CV risk factors [19]. Aortic stiffness is associated with greater atherosclerotic burden [13, 15, 20], and coronary atherosclerosis may be aggravated by elevated aortic stiffness through intimal damage.
Neurohumoral activation in response to the decreased cardiac output can lead to a vicious circle. Indeed, neurohumoral-induced vasoconstriction increases resistance vessel tone to maintain mean arterial pressure but also increase vascular smooth muscle mass, tone and fibrosis, resulting in increased stiffness and central pulse pressure. A direct relationship between neurohumoral activation (activation of the sympathetic nervous system and renin-angiotensin system) and increased carotid stiffness has been seen in HFrEF [14, 15].
3.2 Longitudinal Studies
Several longitudinal studies have reported incident HF, but did not discriminate between HFrEF and HFpEF. In the 5960 participants in the Multiethnic Study of Atherosclerosis (MESA), free of apparent cardiovascular disease and benefiting from a 7.61-year follow-up, Chirinos et al. [7] demonstrated that reflection magnitude—RM—an index of pressure wave reflection, was strongly and independently predictive of new-onset congestive heart failure (CHF), either systolic or diastolic. In the 2602 participants to the Chronic Renal Insufficiency Cohort (CRIC), a multiethnic, multicentre prospective observational study of patients with chronic kidney disease, who were free of HF at baseline and had 3.5 years of follow-up, Chirinos et al. [21] reported that patients with cf-PWV in the middle and top tertiles had a two- to threefold higher chance of developing new-onset hospitalized HF than patients in the first tertile of cf-PWV. Brachial systolic and pulse pressure were also independently associated with incident hospitalized HF, whereas central pressures were less consistently associated with this end point. The association between cf-PWV and incident HF persisted after adjustment for systolic blood pressure.
More recent studies, which discriminated between HFrEF and HFpEF, reported variable results. Tsao et al. [22] studied 2539 participants in the Framingham Study without clinical HF at baseline and who had 10.1-year follow-up. In multivariable-adjusted analyses, cf-PWV was associated with incident HF in a continuous and graded fashion. HFrEF and HFpEF were not defined using current guidelines but as left ventricular ejection fraction <45% and ≥45%, respectively, as defined by echocardiography or radionuclide angiography within the year of incident HF diagnosis. Cf-PWV was associated with HFrEF in age- and sex-adjusted models, which was attenuated in multivariable-adjusted models. Pandey et al. [23] studied 2290 participants to the Health ABC study (Health, Aging and Body Composition) without prevalent HF who had arterial stiffness measured as cf-PWV at baseline and a 11.4-year follow-up. In adjusted analysis, higher cf-PWV was associated with greater risk of HF after adjustment for age, sex, ethnicity, mean arterial pressure, and heart rate. However, cf-PWV was not associated with risk of HFrEF after adjustment for potential confounders. Thus, more studies are needed, with a precise determination of combined interactions between arterial stiffness and incident HF according to its category.
4 Arterial Stiffness, Central Blood Pressure and Diastolic Dysfunction
A common causal mechanistic pathway may be present, whereby increased aortic stiffness and central BP promote the ventricular remodelling and the development of diastolic dysfunction through delayed diastolic relaxation. Indeed, carotid-femoral PWV [12] and central BP increase with ageing [6], and their prognostic value for cardiovascular events has been demonstrated independent of traditional risk factors. In addition, the prevalence of left ventricular (LV) diastolic dysfunction increases with ageing, and LV diastolic dysfunction is a predictor of all-cause mortality [24]. Diastolic dysfunction and arterial stiffness share not only predominance in elderly subjects and hypertensives, predictive value for cardiovascular morbidity and mortality but also remodelling that includes tissue fibrosis, stiffening of cardiac myocytes and vascular smooth muscle cells and advanced glycation end-products (AGEs) deposition in type 2 diabetes.
Paulus et al. recently suggested [25] a new paradigm for HFpEF development, which identified a systemic pro-inflammatory state induced by comorbidities as the cause of myocardial structural and functional alterations, but overlooked the influence of arterial stiffness. Indeed, the systemic pro-inflammatory state in response to comorbidities such as overweight, obesity, diabetes mellitus, chronic obstructive pulmonary disease and salt-sensitive hypertension may lead not only to coronary microvascular inflammation but also to large artery inflammation. Thus, it is likely that besides coronary microvascular inflammation leading to stiff cardiomyocytes and interstitial fibrosis, and contributing to high diastolic LV stiffness and heart failure, large artery inflammation leading to increased stiffness may exaggerate diastolic dysfunction through delayed diastolic relaxation.
In addition, the role of vascular stiffness in the mechanical behaviour of the LV has not been studied properly. The large- and medium-size coronary arteries represent a vascular scaffold which is stretched during diastole. Any increase in coronary artery stiffness might lead to improper filling of the LV because of reduced LV compliance of vascular origin. This phenomenon might explain HFpEF in diabetics and hypertensives [26].
4.1 Pathophysiology of the LV Loading Sequence
A pathophysiological link between arterial stiffness and LV diastolic dysfunction may involve an increased afterload (estimated by characteristic impedance—Zc) and wave reflection (estimated by reflection magnitude, late systolic peak or aortic pulse pressure) in response to arterial stiffening, both impairing diastolic relaxation through temporal changes in myocardial wall stress, i.e. the loading sequence of the LV (Fig. 9.7). Indeed, myocardial and arterial load are time-varying phenomena, and distinct arterial phenomena determine early versus late systolic load on the heart. Specifically, arterial wave reflections generally arrive at the central aorta in mid-to-late systole, selectively increasing late systolic LV afterload and pressure. Evidence in favour of this pathophysiological link (Fig. 9.7) is summarized below.


Arterial stiffness and LV diastolic dysfunction: an increased afterload (estimated by characteristic impedance—Zc) and wave reflection (estimated by reflection magnitude, late systolic peak or aortic pulse pressure) in response to arterial stiffening can impair diastolic relaxation through temporal changes in myocardial wall stress, i.e. delaying the loading sequence of the LV
In a case-control study including 233 subjects with normal diastolic function, mild diastolic dysfunction, and moderate or severe diastolic dysfunction, Abhayaratna et al. [27] showed that brachial pulse pressure, central pulse pressure, and PWV progressively increased according to the severity of diastolic dysfunction, independent of age and sex. Most importantly, the overall performance of PWV was superior to central pulse pressure for the detection of any diastolic dysfunction, suggesting that aortic pressure measurement may not reflect major haemodynamic determinants of diastolic dysfunction. Borlaug et al. [28] studied the impact of loading sequence on left ventricular tissue velocities in 48 subjects, using carotid characteristic impedance (Zc) for assessing early systolic load, carotid augmentation index (cAIx) for assessing late systolic load, and tissue Doppler echocardiography (TDE) for determining LV tissue velocities. In multivariate analysis [28], Zc, arterial compliance and cAIx were independent determinants of early diastolic mitral annular velocity (E′), suggesting an incremental influence of proximal aortic stiffness, aortic lumen, and wave reflection on early diastolic relaxation.
In the 1214 healthy volunteers of the Asklepios study who had measurements with applanation tonometry and speckle-tracking echocardiography, Chirinos et al. [29] demonstrated that proximal aortic Zc but not reflection magnitude—RM—was a multivariate independent determinant of peak myocardial stress, whereas this was the reverse for end-systolic myocardial stress, i.e. RM but not Zc was a multivariate independent determinant of end-systolic myocardial stress. In addition, they showed that early-ejection phase myocardial wall stress was positively associated, whereas late-ejection phase myocardial wall stress was negatively associated, with early diastolic mitral annular velocity (E′) [29]. Altogether, these data indicate that reflected waves (i.e. backward travelling waves), which arrive in late systole, have little effect on peak myocardial stress, but rather on end-systolic stress [29]. Thus, by contrast to a LV early systolic load which is associated with faster relaxation, the shift in the LV loading sequence towards late systole impairs diastolic relaxation (Fig. 9.7).
These findings can be translated into the diagnosis level. Indeed, in patients who had dyspnoea as a major symptom, and in whom the diagnosis was based on invasively derived filling pressures and natriuretic peptide levels, Weber et al. [18] demonstrated that aortic PWV, aortic PP and backward wave amplitude (Pb) were effective in correctly classifying patients as HFpEF or no HFpEF and as effective as tissue Doppler echocardiography.
4.2 Evidence from Longitudinal Studies
By contrast to the large number of cross-sectional studies relating HFpEF to arterial stiffness, only a small number of longitudinal studies have been performed. As a proof of concept, these studies attempted to show that an increased arterial stiffness and/or a hyperpulsatile haemodynamics were associated with increased incidence of HFpEF. Unfortunately, results were mainly negative. In the Framingham Study described above, Tsao et al. [22] did not find any significant association between aortic stiffness and incident HFpEF. In the Health ABC study described above, Pandey et al. [23] showed no association between cf-PWV and risk of HFpEF after adjustment for potential confounders. The lack of significant association could have been attributable to low event rates and the relative health of the samples. More studies are needed in larger cohort, with more appropriate comorbidities, higher CV risk and longer follow-up.
5 Conclusion
This chapter highlighted the importance of arterial stiffness, central BP and wave reflection, for a better understanding of the relationships between hypertension and heart failure, either systolic or diastolic. We also detailed the haemodynamic characteristics of the arterial circulation and explained why it is important to measure arterial stiffness and central BP in hypertensive patients. Finally, we described the various non-invasive methods currently available to measure arterial stiffness and central BP.
6 Future Directions
Although a large number of cross-sectional studies are already available to decipher the various mechanisms by which increased arterial stiffness and central BP are associated with heart failure, more studies are needed, on a longitudinal approach, to determine, as a proof of concept, whether increased arterial stiffness and central BP favour incident heart failure or congestive decompensation.
References
Briet M, Boutouyrie P, Laurent S, London G. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int. 2012;82:388–400.
Nichols WW, O’Rourke MF, Vlachopoulos C. Cardiac failure: clinical implications. In: Arnold H, editor. McDonald’s blood flow in arteries. Theoretical, experimental and clinical principles. 6th ed. London: CRC Press; 2011. p. 344–56.
Laurent S, Boutouyrie P. The structural factor in hypertension: large and small artery alterations. Circ Res. 2015;116:1007–21.
Westerhof BE, Guelen I, Westerhof N, Karemaker JM, Avolio A. Quantification of wave reflection in the human aorta from pressure alone: a proof of principle. Hypertension. 2006;48:595–601.
Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H. Expert consensus document on arterial stiffness: methodological aspects and clinical applications. Eur Heart J. 2006;27:2588–605.
Herbert A, Cruickshank K, Laurent S, Boutouyrie P, on behalf of The Reference Values for Arterial Measurements Collaboration. Establishing reference values for central blood pressure and its amplification in a general healthy population and according to cardiovascular risk-factors. Eur Heart J. 2014;35:3122–33.
Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 1: pressure and flow measurements and basic principles of wave conduction and reflection. Hypertension. 2010;56:555–62.
Westerhof N, Sipkema P, van den Bos CG, Elzinga G. Forward and backward waves in the arterial system. Cardiovasc Res. 1972;6:648–56.
Laurent S, Boutouyrie P. What is the best method to evaluate central blood pressure? Dialog Cardiovasc Med. 2015;20(3):3–14.
Laurent S, Marais L, Boutouyrie P. The non-invasive assessment of vascular aging. Can J Cardiol. 2016;32:669–79.
Van Bortel LM, Laurent S, Boutouyrie P, Chowienczyk P, Cruickshank JK, De Backer T, Filipovsky J, Huybrechts S, Mattace-Raso FU, Protogerou AD, Schillaci G, Segers P, Vermeersch S, Weber T, on behalf of the Artery Society, the European Society of Hypertension Working Group on Vascular Structure and Function and the European Network for Noninvasive Investigation of Large Arteries. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J Hypertens. 2012;30:445–8.
Reference values for carotid-femoral pulse wave velocity in the reference values for arterial stiffness’ collaboration database. Eur Heart J. 2010;31:2338–50.
Boutouyrie P, Laurent S, Girerd X, Benetos A, Lacolley P, Abergel E, Safar M. Common carotid artery stiffness and patterns of left ventricular hypertrophy in hypertensive patients. Hypertension. 1995;25(4 Pt 1):651–9.
Giannattasio C, Achilli F, Failla M, Capra A, Vincenzi A, Valagussa F, Mancia G. Radial, carotid and aortic distensibility in congestive heart failure: effects of high-dose angiotensin-converting enzyme inhibitor or low-dose association with angiotensin type 1 receptor blockade. J Am Coll Cardiol. 2002;39:1275–82.
Weber T, Auer J, O’Rourke MF, Kvas E, Lassnig E, Berent R, Eber B. Arterial stiffness, wave reflections, and the risk of coronary artery disease. Circulation. 2004;109:184–9.
Hoffman J, Buckberg GD. The myocardial supply: demand ratio—a critical review. Am J Cardiol. 1978;41:327–32.
Safar ME. Pulse pressure in essential hypertension: clinical and therapeutical implications. J Hypertens. 1989;7:769–76.
Watanabe H, Ohtsuka S, Kakihana M, Sugishita Y. Coronary circulation in dogs with an experimental decrease in aortic compliance. J Am Coll Cardiol. 1993;21:1497–506.
Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, Laurent S. Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension. 2002;39:10–5.
van Popele NM, Mattace-Raso FU, Vliegenthart R, Grobbee DE, Asmar R, van der Kuip DA, Hofman A, de Feijter PJ, Oudkerk M, Witteman JC. Aortic stiffness is associated with atherosclerosis of the coronary arteries. Circulation. 2005;112:2254–62.
Chirinos JA, Khan A, Bansal N, et al. CRIC study investigators. Arterial stiffness, central pressures, and incident hospitalized heart failure in the chronic renal insufficiency cohort study. Circ Heart Fail. 2014;7:709–16.
Tsao CW, Lyass A, Larson MG, Levy D, Hamburg NM, Vita JA, Benjamin EJ, Mitchell GF, Vasan RS. Relation of central arterial stiffness to incident heart failure in the community. J Am Heart Assoc. 2015;4(11):e002189. https://doi.org/10.1161/JAHA.115.002189.
Pandey A, Khan H, Newman AB, Lakatta EG, Forman D, Butler J, Berry JD. Arterial stiffness and risk of overall heart failure, heart failure with preserved ejection fraction, and heart failure with reduced ejection fraction. The Health ABC Study (Health, Aging, and Body Composition). Hypertension. 2017;69:267–74.
Redfield MM, Jacobsen SJ, Burnett JC Jr, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289:194–202.
Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–71.
Stassen FR, Fazzi GE, Leenders PJ, Smits JF, De Mey JG. Coronary arterial hyperreactivity and mesenteric arterial hyporeactivity after myocardial infarction in the rat. J Cardiovasc Pharmacol. 1997;29:780–8.
Abhayaratna WP, Srikusalanukul W, Budge MM. Aortic stiffness for the detection of preclinical left ventricular diastolic dysfunction: pulse wave velocity versus pulse pressure. J Hypertens. 2008;26:758764.
Borlaug BA, Melenovsky V, Redfield MM, Kessler K, Chang HJ, Abraham TP, Kass DA. Impact of arterial load and loading sequence on left ventricular tissue velocities in humans. J Am Coll Cardiol. 2007;50:1570–7.
Chirinos JA, Segers P, Gillebert TC, Gupta AK, De Buyzere ML, De Bacquer D, St John-Sutton M, Rietzschel ER, Asklepios Investigators. Arterial properties as determinants of time-varying myocardial stress in humans. Hypertension. 2012;60:64–7.
Acknowledgements
This work was supported by INSERM, Assistance Publique—Hopitaux de Paris, and Paris Descartes University (SL).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter
Cite this chapter
Laurent, S., Hulot, JS., Boutouyrie, P. (2019). Role of Central Blood Pressure and Arterial Stiffening. In: Dorobantu, M., Mancia, G., Grassi, G., Voicu, V. (eds) Hypertension and Heart Failure. Updates in Hypertension and Cardiovascular Protection. Springer, Cham. https://doi.org/10.1007/978-3-319-93320-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-93320-7_9
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93319-1
Online ISBN: 978-3-319-93320-7
eBook Packages: MedicineMedicine (R0)