
Abstract
Neurofibromatosis type 1 is a genetically determined multisystemic disease that affects up to 1:2500–3500 people throughout the world. Several organs and systems can be affected by the disorder, including the eye and orbit. Some ocular manifestations, such as Lisch nodules in the iris, choroidal nodules, or retinal vascular abnormalities are completely innocuous and require no treatment. Other manifestations such as optic nerve gliomas and plexiform neurofibromas can carry significant morbidity, including vision loss and facial disfigurement. Therefore, the role of the ophthalmologist in managing patients with ophthalmic manifestations of NF1 is significant and varied.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a genetic disease caused by a defect in a single gene encoding for neurofibromin, a cytoplasmic protein involved in control of the cell cycle [1,2,3]. More specifically, neurofibromin acts as a negative regulator of the Ras protoncogene, which represents a key molecule in cell growth [4, 5].
NF1 has a classic Mendelian inheritance pattern, autosomal dominant with complete penetrance but variable expression [2]. It affects 1:2500–3500 people throughout the world, with no gender or ethnicity predilection [1,2,3]. Around 50% of cases are caused by sporadic mutations, given the high rate of spontaneous mutation of the NF1 gene (about 1:10,000) [1]. Less frequently, somatic mosaicism may also occur leading to segmental NF1 in which characteristic features of the disease are displayed only in certain body segments, including the eye [6]. Genetic testing is currently available to identify specific mutations in the NF1 gene and confirm the diagnosis. However, diagnosis remains a clinical decision due to the complexity of genetic testing considering the large size of the gene, the variety and lack of clustering of possible defects and the existence of pseudogenes [1].
In order to guide clinicians in the recognition of the disease, in 1998 the National Institutes of Health (NIH) established the following clinical criteria for diagnosis of this disorder [7]:
-
1.
Six or more café-au-lait macules >5 mm in greatest diameter in children and >15 mm in greatest diameter in adults
-
2.
Two or more neurofibromas or one plexiform neurofibroma
-
3.
Axillary or inguinal freckling
-
4.
Optic pathway glioma
-
5.
Two or more Lisch nodules
-
6.
Characteristic skeletal dysplasia (sphenoid wing, long bones)
-
7.
First-degree relative with NF1 diagnosed by the above criteria
The presence of at least 2 of the above-mentioned criteria is sufficient for the diagnosis. Most of these clinical features are age dependent, so the vast majority of patients (around 95%) meet diagnostic criteria by age 8 and virtually all of them do so by their 20s [8]. The most common ocular manifestations are Lisch nodules (50–90% of patients), [9] followed by optic pathway gliomas (around 15% of patients) [10] and plexiform neurofibromas (less than 10% of patients) [11]. These entities represent hallmark lesions of NF1, indicating the pivotal role ophthalmologists may have in the diagnosis of this disorder.
6.2 Ocular Adnexa
The most common ocular adnexal finding in NF1 is plexiform neurofibromas (PNs), even though their overall incidence is fairly low as previously mentioned (less than 10% of children affected by NF1) [11]. PNs are complex nerve sheath tumors that follow multiple nerve branches and are at risk for malignant transformation. In contrast, discrete neurofibromas (dermal or subcutaneous) arise from small nerves or nerve endings, appear later in life and have no risk for malignant transformation [11].
Most PNs are identified in early childhood (usually before age 5 years) and may grow rapidly during this period, as well as during puberty and pregnancy. Clinically they commonly present with ptosis (“S-shaped ptosis” in cases of predominantly lateral infiltration of upper eyelid), proptosis, eyelid swelling, orbital dystopia, and strabismus [1, 11, 12]. PNs can involve the upper eyelid, brow, orbit, and temple and may grow to the point that they become disfiguring for patients [1, 11]. In addition to affecting children’s aesthetic appearance, decreased visual acuity and deprivational, refractive or strabismic (less frequently) amblyopia may occur in up to 50% of cases [13].
One of the rare secondary complications of PNs is sphenoid wing dysplasia, defined as absence or marked thinning of the sphenoid bone that comprises the posterolateral wall of the orbit. It has been found in 1–6% of children with NF1 and is usually found ipsilateral to the plexiform neurofibroma [14]. This dysplasia allows protrusion of the anterior temporal lobe into the orbit, causing proptosis, pulsatile exophthalmos, strabismus, and optic nerve compression [11].
Biopsy for tissue diagnosis is usually unnecessary in patients with NF1. However, all children with newly diagnosed PNs should undergo magnetic resonance imaging (MRI) of the brain and orbits regardless of whether a diagnosis of NF1 has been confirmed [11].
Surgery is the mainstay of treatment for PNs, but a detailed discussion of surgical techniques is beyond the scope of this chapter. However, it is important to highlight that the non-encapsulated and highly vascular tumor has an infiltrative nature, which increases the potential for recurrence and complications such as bleeding, in response to surgical intervention [1, 15]. Some authors suggest that conservative management via close observation with serial MRIs might be an acceptable alternative in selected cases [11]. The main indications for surgery include clinical progression causing anatomical and functional damage (amblyopia, optic neuropathy, corneal exposure) or facial disfigurement [15].
Given the variety of opinions on timing and selection of treatment, a multidisciplinary task force of experts from tertiary care centers proposed a consensus statement for ophthalmic monitoring and management of PNs as outlined below [11]:
-
1.
Adoption of the uniform terminology “Orbital-Periorbital Plexiform Neurofibroma” or OPPN for plexiform neurofibromas involving the eyelid, orbit, periorbital, and facial structures.
-
2.
Children with OPPN are at highest risk for rapid growth of OPPN before the age of 8. Comprehensive ophthalmic evaluation is recommended every 6 months until visual maturity. After that, frequency of examination should be guided by the clinical course.
-
3.
Patients with OPPN confined to the upper eyelid may not need to undergo neuroimaging. For patients with orbital, periorbital, or facial involvement, high resolution MRI scanning with and without contrast of the orbit, face, and cavernous sinus should be performed.
-
4.
Treatment for related ophthalmic issues, such as ptosis, lacrimal involvement, or amblyopia is supportive. Early intervention is recommended with the exception of strabismus surgery. Strabismus caused by orbital or periorbital tumor involvement while the tumor is in its rapid growth phase carries a high risk for recurrence after strabismus surgery. Associated problems such as amblyopia and refractive error should be managed aggressively and surgery deferred until the tumor growth has stabilized, if clinically appropriate to do so.
-
5.
Debulking surgery may be indicated for the following:
-
(a).
Visual decline.
-
(b).
Progressive tumor growth involving a vital structure.
-
(c).
Progressive disfigurement or functional decline.
Debulking is more successful in older patients and adults. Younger patients have a high risk of recurrent progression and need for more surgery.
-
(a).
-
6.
Clinical trials using biologic agents (i.e., MEK inhibitors) are underway but no definitive recommendations can be made at this time.
6.3 Anterior Segment
Lisch nodules (Fig. 6.1) represent the most common finding of NF1 in the anterior segment and they are specific to the disease, thus qualifying as one of its hallmark manifestations [1]. These lesions are melanocytic hamartomas of the iris, consisting of a condensation of spindle cells on the anterior surface of the iris [16]. They may be visualized without the aid of a microscope occasionally, but slit-lamp examination is frequently required to assess the exact number and location of the nodules. In adults, Lisch nodules lack intrinsic vasculature and present as multiple, bilateral elevated nodules located in the inferior half of the iris, ranging from white to yellow or brown in color [8, 9, 17].

NIR (near infrared) anterior segment picture of a 53-year-old man (a) and a 64-year-old woman (c) affected by NF1 showing multiple Lisch nodules. AS-OCT (anterior segment optical coherence tomography) scans demonstrate the iris lesions casting a shadow posteriorly (b, d)
Lisch nodules might not be evident in early years, but they become apparent later in life, as their prevalence is known to correlate with age, but not with the severity of the disease or the number of café-au-lait macules or neurofibromas [1, 17]. Several studies have investigated the prevalence of Lisch nodules by age groups. According to Beauchamp, they are found in virtually all adults with a confirmed diagnosis of NF1, while their prevalence in children is lower (around 53% in patients under the age of 10 years) [9]. In another work by Lubs and colleagues, the prevalence of Lisch nodules was studied by age group and was found to be only 5% in children under 3 years of age, 42% in children between 3 and 4 years of age (the largest incremental increase among age subdivisions), 55% in children between 5 and 6 years of age, and 100% among adults over 21 years of age [17]. Furthermore, the prevalence of Lisch nodules was greater than that of neurofibromas in all but the youngest age group (under 3 years of age) [17]. These findings indicate that the lack of these nodules in young children does not rule out NF1, as they may present at a later age.
In terms of management, Lisch nodules are not correlated with ocular complications, visual impairment or ocular morbidity of any sort; therefore, they do not require any treatment [1, 17]. However, careful clinical examination is essential to differentiate these lesions from other conditions with similar presentations, such as iris nevi, which usually appear as flat or variably elevated darkly pigmented lesions with ill-defined margins and iris mammillations, a condition associated with ocular melanocytosis that presents as uniformly distributed nipple-like protuberances in the anterior part of a deeply pigmented iris [1, 17].
Juvenile xanthogranuloma (JXG) is a benign histiocytic proliferation that occurs in young children most frequently in the skin but can manifest in the eye also. Ocular involvement typically occurs with adnexal involvement, as a circumscribed iris nodule that can occasionally lead to hyphema and elevated intraocular pressure and rarely as a diffuse iris infiltration [18]. Several reports have suggested an association between NF1 and JXG, with the latter manifesting as the initial presenting feature of NF1. In a series of 288 patients, 17 of 77 patients under 3 years of age with NF1 had JXG [19]. Furthermore, an association between JXG with NF1 and a particular kind of leukemia called juvenile myelomonocytic leukemia (JMML) has been described. More specifically, the presence of both NF1 and JXG seems to create a 20–30 times higher risk for developing JMML compared to NF1 alone [20]. However, the existence of this relationship has been disputed in a recent study [21]. Therefore, it is questionable whether or not children diagnosed with NF1 and clinical evidence of JXG benefit from a hematological evaluation to rule out JMML.
Other very rare anterior segment findings reported in the literature include conjunctival neurofibromas and diffuse hypertrophy of corneal stromal nerves (“lignes grise”) [1, 8].
6.4 Retina
Retinal abnormalities associated with NF1 are not very common and range from retinal vascular abnormalities (RVAs) to several benign retinal tumors.
RVAs were first described in 2002 in a cohort of 12 patients out of 32 subjects affected by NF1 (37.5%) [22]. The anomalies described in this paper ranged from a single affected vessel (“forme fruste”) to the full-blown manifestation (“complete form”). The most common finding consisted of minuscule second or third order tortuous venules called “corkscrew vessels,” usually one to two disc diameters in dimensions, isolated and most often located temporally. Fluorescein angiography (FA) better characterized these lesions but showed no leakage. Less common vascular abnormalities included a venous-venous anastomosis that was found in the nasal retina in one patient and an extensive arteriovenous malformation associated with an epiretinal membrane that was detected in another patient [22].
More recently, a series of 17 NF1 patients was reported in which 6 patients (35%) presented with distinctive microvascular abnormalities, consisting of small tortuous “spiral” or “corkscrew” vessels, often located overlying choroidal alterations that were visible with near infrared reflectance (NIR) [23].
Interestingly, in a much bigger series of 294 patients affected by NF1 only 18 patients (6.1%) presented with RVAs, defined as small, tortuous retinal vessels with a “spiral/corkscrew” appearance originating from small tributaries of retinal veins [24]. RVAs were mostly unilateral (94%) and single (83%), located along the temporal vascular arcades in two third of cases and at the posterior pole in the remaining one thirds of cases. On FA, RVAs were first visible during the arteriovenous phase and did not develop late leakage. On optical coherence tomography angiography (OCTA), RVAs were located in the superficial vascular plexus in all cases, with associated localized abnormal congested capillary networks in the deep vascular plexus [24]. Also of interest, the presence of RVAs did not correlate with the presence of other specific ocular or systemic NF1 features [24].
In terms of benign neoplastic lesions associated with NF1, several entities have been described [25]. Astrocytic hamartomas in neurofibromatosis present as small whitish to yellowish masses with a “mulberry-like” appearance usually involving the optic nerve, similar to lesions most commonly seen in tuberous sclerosis. Combined hamartomas of the retina and retinal pigment epithelium and retinal capillary hemangiomas have also been associated with NF1. Occasionally, these lesions might cause vision-threatening complications such as massive exudation, retinal detachment, neovascular glaucoma, and vitreous hemorrhage [25]. Finally, in a more recent series of 275 patients with retinal vasoproliferative tumors, 6 (2.2%) were found to have NF1 [26]. The tumors were located between the equator and the ora serrata in all cases and were variably associated with subretinal fluid and exudation, epiretinal membrane, retinal and vitreous hemorrhage, retinal neovascularization, and cystoid macular edema [26].
6.5 Choroid
Choroidal neurofibromatosis was once considered a rare variant of NF1, but is now known to be a common feature of the disease. Improved imaging modalities such as indocyanine-green fundus angiography and confocal microscopy using infrared light can penetrate the retinal pigment epithelium (RPE), which allows imaging of the choroid [1, 27, 28]. Histopathologic examination of enucleated eyes with choroidal neurofibromatosis revealed choroidal thickening with ovoid bodies and proliferation of connective tissue with pigment-containing cells and ganglion-like cells, features consistent with choroidal ganglioneuroma [29].
In 1998, Rescaldani and colleagues were the first to use indocyanine-green angiography (ICGA) to investigate the choroidal features of 2 cases of NF1 [30]. In both cases, early phases of the examination showed multiple extensive areas of hypofluorescence, that became smaller in the late phases. The authors speculated that the early hypofluorescence could be due to slow choroidal filling caused by alterations to the walls of the choroidal arterioles induced by the disease. Late hypofluorescent areas were presumed to be either persistent nonperfused lobules of choriocapillaris or choroidal nodules [30].
Later, Yasunari and colleagues investigated the use of infrared monochromatic light examination by confocal scanning laser ophthalmoscope (cSLO) in 33 eyes of 17 patients with NF1 [27]. They detected multiple bright patchy regions at and around the posterior pole of all 33 eyes examined, which corresponded to the hypofluorescent areas on ICGA. No abnormalities were noted at corresponding areas under conventional ophthalmoscopic examination or FA [27]. The authors pointed out that bright patchy regions under infrared light may indicate the presence of refractile tissue or material in the choroid, corresponding to choroidal neurofibromas, which are thought to be refractile in nature [27].
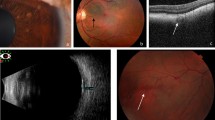
Another study from 2012 used cSLO to examine 95 consecutive adult and pediatric patients (190 eyes) [28]. Bright patchy choroidal nodules (Fig. 6.2) were detected by NIR in 79 (82%) patients, including 15 children (71%), while conventional fundus ophthalmoscopy, fundus autofluorescence, and red-free imaging did not disclose any abnormalities in the corresponding areas. Lesions were more frequently located in the posterior pole, but occurred diffusely throughout the fundus. Optical coherence tomography (OCT) showed irregular hyperreflective foci located under the RPE, corresponding to the alterations detected by NIR imaging [28]. Interestingly, the authors found no significant correlation between Lisch nodules and choroidal nodules, but a correlation was found between increased patient age and more diffused involvement of the fundus [28]. The prevalence of choroidal nodules detected by NIR in the overall NF1 population (82%) was similar to the average prevalence of the 4 most common NIH diagnostic criteria (cafè-au-lait, freckling, Lisch nodules, and neurofibromas). In the pediatric population, choroidal nodules were present in a much higher frequency (71%) than that of the NIH diagnostic criteria of for iris Lisch nodules (43%) [28]. Therefore, the authors suggested the presence of choroidal abnormalities detected by NIR imaging should be used as an additional diagnostic criterion for NF1.

NIR (near infrared) fundus picture of the left eye (OS) of a 16-year-old boy with NF1 discloses multiple bright patches at the posterior pole and along the arcades (a). OCT (optical coherence tomography) scans through 2 of these lesions show that the bright areas correspond to choroidal nodules (b–e, arrowheads)
Following this observation, Parrozzani and colleagues evaluated the diagnostic performance of NF1-related choroidal abnormalities detected by NIR as a diagnostic criterion in the pediatric population [31]. They evaluated 140 consecutive pediatric patients aged 0–16 years old and found choroidal abnormalities in 72 affected (61%) and 1 suspected (2%) children. Compared with standard NIH criteria, the presence of choroidal abnormalities detected by NIR was the third parameter for positive predictive value and the fourth for sensitivity, specificity, and negative predictive value. Compared with Lisch nodules, this criterion had higher specificity and positive predictive value [31].
A similar study by Vagge and colleagues investigated the presence of choroidal abnormalities detected by NIR in pediatric patients with NF1 and documented similar findings [32]. Choroidal abnormalities were found in 54 patients (69%), were most frequently located at the posterior pole and the number of involved areas correlated with patient age [32].
Finally, NIR and OCT imaging modalities have been useful in detecting other interesting choroidal findings. In a series of 34 eyes of 17 patients with NF1 and typical choroidal nodules, bilateral anomalous choroidal vessels were observed with NIR in 4 patients. Enhanced depth imaging (EDI)-OCT revealed unusually dilated choroidal vessels and an absence of the choriocapillaris or Sattler’s layer above the dilated vessels [33]. In another study EDI-OCT was able to identify 2 distinctive morphologies of choroidal nodules detected by NIR imaging, “dome-shaped” or “placoid” [34]. In the same study, authors reported a reduction in the mean choroidal thickness in patients with NF1, as well as in the neuroepithelium, photoreceptor-retinal pigment epithelium, and outer nuclear layer thickness [34].
6.6 Optic Nerve
Optic pathway gliomas (OPGs) occur in 15–20% of patients, and they represent the most common orbital and intracranial manifestation of NF1, as well as the most common central nervous system tumor in children with NF1 [35]. They can affect any part of the visual pathway from the pre-chiasmatic tracts of the optic nerves to the chiasmatic-hypothalamic region to the posterior optic pathway (Fig. 6.3) [1, 36]. Rarely, gliomas in the brainstem may also occur in children with NF1 [1]. Histologically, they are classified as grade I astrocytomas or juvenile pilocytic astrocytomas according to the World Health Organization [37].

Axial scans (a, b) and coronal scans (c, d) of MRI of the brain and orbits disclose large bilateral fusiform optic nerve masses with extension into the suprasellar region and extending posteriorly to involve the chiasm and hypothalamus. The lesions are compatible with optic pathway gliomas found in NF1
OPGs usually develop during the first decade of life (with children aged 6 years or younger at greatest risk), even though later onset has been described in the literature [35]. They are usually unilateral, but bilateral presentation has been described in about one third of cases [10]. Clinical symptoms at time of diagnosis have been reported in about 60% of cases, with clinical presentation varying according to tumor location [38]. Clinical behavior of OPGs is highly variable [35]. For tumors located in the optic nerve, typical presentation is with gradual onset of painless unilateral loss of vision, with visual acuity ranging from 20/20 to no light perception. Clinical examination may disclose a combination of dyschromatopsia, centrocecal scotoma on visual field (VF) testing, and/or a relative afferent pupillary defect. The optic nerve may appear normal, swollen or atrophic. Proptosis, nystagmus or strabismus can also occur. Other rare findings include headaches and papilledema secondary to high intracranial pressure in larger chiasmal-hypothalamic gliomas that are more likely to occur in sporadic cases [1, 2, 39,40,41]. If the location of the tumor is in the chiasm, bilateral visual loss, bilateral swelling or atrophy of the optic nerves, and bitemporal hemianopsia on VF might occur [42]. Finally, if the hypothalamus is invaded, precocious puberty can be seen in up to 40% of patients older than 6 years of age. This finding can represent the presenting symptom or the first sign of progression of the disease [35].
In terms of natural history, OPGs are usually benign and show a more indolent course compared to sporadic cases. However, hazardous evolution with significant morbidity, vision loss, and endocrine abnormalities has been reported in one third of cases [36, 38, 43]. In a recent study on 414 consecutive patients with NF1 referred before age 6, 52 (13%) developed OPGs during follow-up, with females more commonly affected [36]. OPGs were more frequently detected in patients with suggestive symptoms compared to patients who underwent an MRI for screening purposes. Clinical management was conservative in most patients, with only 8 (2%) requiring therapy due to visual deterioration [36]. This study confirmed findings from previous studies, in which the presence of symptoms at the time of diagnosis was shown to be the best predictor of the need for treatment, with asymptomatic children rarely requiring treatment [2, 38].
The preferred screening protocol for asymptomatic children affected by NF1 is controversial. Some centers prefer performing more frequent examinations in the first year of life and then gradually increasing the intervals between visits [35, 40, 41]. However, the recommendations from the American Academy of Pediatrics (AAP) indicate annual ophthalmic examinations from ages 1 to 7 years and then every 2 years from ages 8 to 18 years [44]. Several papers indicate that visual deterioration is considered the best indicator of presence or progression of OPGs, therefore visual acuity should be quantitatively assessed at every visit [35, 39]. When visual deterioration (defined as a 2 line decrease in visual acuity) not attributable to other causes is detected, prompt MRI of the brain and orbits should be performed [35].
The role of systematic MRI screening for OPGs in children with NF1 has been widely discussed in the literature. In 1997, the OPG Task Force determined that there was no conclusive evidence that early detection of asymptomatic gliomas led to reduced vision loss [40]. These findings, combined with the potential neurotoxic effects of repeated sedation in children, led the AAP not to recommend routine MRI screening for asymptomatic NF1 patients. However, in a more recent study on 826 individuals with NF1, Prada and colleagues found that chiasmatic and postchiasmatic OPGs carried the highest risk for progression and vision loss and that early identification with MRI screening in asymptomatic cases may lead to improved visual outcomes [45]. Nevertheless, current indications identify MRI of the brain and orbits as the preferred imaging modality to confirm the diagnosis of OPG once an abnormal ophthalmological evaluation has been documented [35]. Neuroradiological appearance is fairly typical: OPGs appear as fusiform masses in the optic nerve and chiasm region, hypointense or isointense on T1 images, and hyperintense in T2 images [46].
Other screening modalities for OPGs in older and more cooperative children include VF testing and OCT. Kinetic (Goldmann) VF testing can be easier for younger children, however, VF testing can be difficult to perform in a repeatable and reliable way [35]. It has been found that subjects with NF1 and OPGs have a thinner retinal nerve fiber layer (RNFL) on OCT [47]. Interestingly, Parrozzani and colleagues found that RNFL assessment using spectral domain OCT can be superior to visual function assessment and optic nerve evaluation as a screening tool for OPGs [48].
As previously mentioned, the clinical course of OPGs can be varied and unpredictable, making the management quite challenging. In a large study from 2003, no single specific epidemiological factor that could serve as a predictor of the need for future treatment was identified [41]. Also, there is still debate about the optimal choice and timing of treatment. Generally speaking, most authors agree that documented clinical decline and/or radiographic progression represent the main indications for treatment [35, 38, 40, 42]. Clinical progression includes decreased visual acuity or color vision, VF defect progression, progressive proptosis or endocrine, and neurological dysfunctions [38, 42]. Radiographic progression is defined as tumor enlargement, change in enhancement or progressive involvement of the posterior visual pathway [35]. It is important to keep in mind that initial management often involves careful observation, as about one half of OPGs do not cause clinical symptoms.
A detailed discussion about different treatment protocols for OPGs in NF1 patients is beyond the scope of this chapter. However, it is important to mention that chemotherapy currently represents the first-line treatment for all age groups [35, 42]. Standard combination includes vincristine and carboplatin. This regimen is overall well tolerated, does not carry significant long-term toxicity and is effective in the control of both newly diagnosed and recurrent OPGs [49]. However, there is a risk of acute toxicity (myelosuppression). Radiotherapy was once considered the first-line treatment in children older than 6 years of age, but a risk for secondary tumors and unacceptable complications have been identified [35, 42]. This is particularly dangerous in patients with NF1, that are already predisposed to both benign and malignant lesions. Finally, surgery has very limited role in OPGs from NF1. It is reserved for biopsies in atypical cases or when the mass effect causes painful and disfiguring proptosis associated with corneal exposure of a blind eye [35, 39, 42].
6.7 Glaucoma
Glaucoma rarely occurs in patients with NF1 (about 1–2%), but these patients are at increased risk of developing elevated intraocular pressure for multiple reasons [1].
Patients with NF1 and orbitofacial involvement (mainly plexiform neurofibroma of the upper lid) have a higher incidence of glaucoma, found in up to 23% of these patients [50]. In this subset of NF1 patients, glaucoma seems to be associated with poor visual outcomes and surgical intervention is often required [50]. More specifically, unilateral glaucoma associated with buphthalmos can be found in patients with NF1 and ipsilateral plexiform neurofibroma of the upper eyelid [51]. The association of unilateral buphthalmos, ipsilateral plexiform neurofibroma, and ipsilateral facial hemihypertrophy is referred to as François syndrome [1].
Potential mechanisms of increased intraocular pressure include angle abnormalities such as abundant iris processes, anteriorization of angle insertion, pigmentary disturbances, secondary angle closure from synechiae formation, and angle infiltration by Lisch nodules or neurofibromas [52].
Assessment for glaucoma, including gonioscopy, intraocular pressure evaluation, visual field testing, and optic nerve assessment is, therefore, advised in patients with NF1 [8].
References
Kinori M, Hodgson N, Zeid JL. Ophthalmic manifestations in neurofibromatosis type 1. Surv Ophthalmol. 2018;63:518.
Savar A, Cestari DM. Neurofibromatosis type I: genetics and clinical manifestations. Semin Ophthalmol. 2008;23(1):45–51.
Friedman JM. Neurofibromatosis : phenotype, natural history, and pathogenesis. Baltimore: Johns Hopkins University Press; 1999.
DeClue JE, Cohen BD, Lowy DR. Identification and characterization of the neurofibromatosis type 1 protein product. Proc Natl Acad Sci U S A. 1991;88(22):9914–8.
Gutmann DH, Collins FS. The neurofibromatosis type 1 gene and its protein product, neurofibromin. Neuron. 1993;10(3):335–43.
Tinschert S, Naumann I, Stegmann E, Buske A, Kaufmann D, Thiel G, et al. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet. 2000;8(6):455–9.
National Institutes of Health Consensus Development Conference Statement: neurofibromatosis. Bethesda, Md., USA, July 13–15, 1987. Neurofibromatosis. 1988;1(3):172–8.
Abdolrahimzadeh B, Piraino DC, Albanese G, Cruciani F, Rahimi S. Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography. Clin Ophthalmol. 2016;10:851–60.
Beauchamp GR. Neurofibromatosis type 1 in children. Trans Am Ophthalmol Soc. 1995;93:445–72.
Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. 1989;114(5):788–92.
Avery RA, Katowitz JA, Fisher MJ, Heidary G, Dombi E, Packer RJ, et al. Orbital/periorbital plexiform neurofibromas in children with neurofibromatosis type 1: multidisciplinary recommendations for care. Ophthalmology. 2017;124(1):123–32.
Chaudhry IA, Morales J, Shamsi FA, Al-Rashed W, Elzaridi E, Arat YO, et al. Orbitofacial neurofibromatosis: clinical characteristics and treatment outcome. Eye. 2012;26(4):583–92.
Oystreck DT, Morales J, Chaudhry I, Alorainy IA, Elkhamary SM, Pasha TM, et al. Visual loss in orbitofacial neurofibromatosis type 1. Ophthalmology. 2012;119(10):2168–73.
Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–43.
Keren S, Dotan G, Ben-Cnaan R, Leibovitch L, Leibovitch I. A combined one-stage surgical approach of orbital tumor debulking, lid reconstruction, and ptosis repair in children with orbitotemporal neurofibromatosis. J Plast Reconstr Aesthet Surg. 2017;70(3):336–40.
Williamson TH, Garner A, Moore AT. Structure of Lisch nodules in neurofibromatosis type 1. Ophthalmic Paediatr Genet. 1991;12(1):11–7.
Lubs ML, Bauer MS, Formas ME, Djokic B. Lisch nodules in neurofibromatosis type 1. N Engl J Med. 1991;324(18):1264–6.
Sanders TE. Intraocular juvenile xanthogranuloma (nevoxanthogranuloma): a survey of 20 cases. Trans Am Ophthalmol Soc. 1960;58:59–74.
Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated with neurofibromatosis 1: 14 patients without evidence of hematologic malignancies. Pediatr Dermatol. 2004;21(2):97–101.
Zvulunov A, Barak Y, Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol. 1995;131(8):904–8.
Liy-Wong C, Mohammed J, Carleton A, Pope E, Parkin P, Lara-Corrales I. The relationship between neurofibromatosis type 1, juvenile xanthogranuloma, and malignancy: a retrospective case-control study. J Am Acad Dermatol. 2017;76(6):1084–7.
Muci-Mendoza R, Ramella M, Fuenmayor-Rivera D. Corkscrew retinal vessels in neurofibromatosis type 1: report of 12 cases. Br J Ophthalmol. 2002;86(3):282–4.
Abdolrahimzadeh S, Felli L, Piraino DC, Mollo R, Calvieri S, Recupero SM. Retinal microvascular abnormalities overlying choroidal nodules in neurofibromatosis type 1. BMC Ophthalmol. 2014;14:146.
Parrozzani R, Pilotto E, Clementi M, Frizziero L, Leonardi F, Convento E, et al. Retinal vascular abnormalities in a large cohort of patients affected by neurofibromatosis type 1: a study using optical coherence tomography angiography. Retina. 2018;38(3):585.
Destro M, D'Amico DJ, Gragoudas ES, Brockhurst RJ, Pinnolis MK, Albert DM, et al. Retinal manifestations of neurofibromatosis. Diagnosis and management. Arch Ophthalmol. 1991;109(5):662–6.
Shields JA, Pellegrini M, Kaliki S, Mashayekhi A, Shields CL. Retinal vasoproliferative tumors in 6 patients with neurofibromatosis type 1. JAMA Ophthalmol. 2014;132(2):190–6.
Yasunari T, Shiraki K, Hattori H, Miki T. Frequency of choroidal abnormalities in neurofibromatosis type 1. Lancet. 2000;356(9234):988–92.
Viola F, Villani E, Natacci F, Selicorni A, Melloni G, Vezzola D, et al. Choroidal abnormalities detected by near-infrared reflectance imaging as a new diagnostic criterion for neurofibromatosis 1. Ophthalmology. 2012;119(2):369–75.
Woog JJ, Albert DM, Craft J, Silberman N, Horns D. Choroidal ganglioneuroma in neurofibromatosis. Graefes Arch Clin Exp Ophthalmol. 1983;220(1):25–31.
Rescaldani C, Nicolini P, Fatigati G, Bottoni FG. Clinical application of digital indocyanine green angiography in choroidal neurofibromatosis. Ophthalmologica. 1998;212(2):99–104.
Parrozzani R, Clementi M, Frizziero L, Miglionico G, Perrini P, Cavarzeran F, et al. In vivo detection of choroidal abnormalities related to NF1: feasibility and comparison with standard NIH diagnostic criteria in pediatric patients. Invest Ophthalmol Vis Sci. 2015;56(10):6036–42.
Vagge A, Camicione P, Capris C, Sburlati C, Panarello S, Calevo MG, et al. Choroidal abnormalities in neurofibromatosis type 1 detected by near-infrared reflectance imaging in paediatric population. Acta Ophthalmol. 2015;93(8):e667–71.
Abdolrahimzadeh S, Parisi F, Abdolrahimzadeh B, Cruciani F. Unusual choroidal vessels in neurofibromatosis type 1 observed with near-infrared reflectance and spectral domain optical coherence tomography. Acta Ophthalmol. 2016;94(8):e815–e6.
Abdolrahimzadeh S, Felli L, Plateroti R, Plateroti AM, Giustini S, Calvieri S, et al. Morphologic and vasculature features of the choroid and associated choroid-retinal thickness alterations in neurofibromatosis type 1. Br J Ophthalmol. 2015;99(6):789–93.
Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61(3):189–98.
Trevisson E, Cassina M, Opocher E, Vicenzi V, Lucchetta M, Parrozzani R, et al. Natural history of optic pathway gliomas in a cohort of unselected patients affected by Neurofibromatosis 1. J Neuro-Oncol. 2017;134(2):279–87.
Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215–25; discussion 26-9.
Thiagalingam S, Flaherty M, Billson F, North K. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology. 2004;111(3):568–77.
Avery RA, Fisher MJ, Liu GT. Optic pathway gliomas. J Neuroophthalmol. 2011;31(3):269–78.
Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143–9.
King A, Listernick R, Charrow J, Piersall L, Gutmann DH. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet A. 2003;122A(2):95–9.
Liu GT. Optic gliomas of the anterior visual pathway. Curr Opin Ophthalmol. 2006;17(5):427–31.
Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718–22.
Hersh JH. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121(3):633–42.
Prada CE, Hufnagel RB, Hummel TR, Lovell AM, Hopkin RJ, Saal HM, et al. The use of magnetic resonance imaging screening for optic pathway gliomas in children with neurofibromatosis type 1. J Pediatr. 2015;167(4):851–6.e1.
Mentzel HJ, Seidel J, Fitzek C, Eichhorn A, Vogt S, Reichenbach JR, et al. Pediatric brain MRI in neurofibromatosis type I. Eur Radiol. 2005;15(4):814–22.
Chang L, El-Dairi MA, Frempong TA, Burner EL, Bhatti MT, Young TL, et al. Optical coherence tomography in the evaluation of neurofibromatosis type-1 subjects with optic pathway gliomas. J AAPOS. 2010;14(6):511–7.
Parrozzani R, Clementi M, Kotsafti O, Miglionico G, Trevisson E, Orlando G, et al. Optical coherence tomography in the diagnosis of optic pathway gliomas. Invest Ophthalmol Vis Sci. 2013;54(13):8112–8.
Packer RJ, Lange B, Ater J, Nicholson HS, Allen J, Walker R, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11(5):850–6.
Morales J, Chaudhry IA, Bosley TM. Glaucoma and globe enlargement associated with neurofibromatosis type 1. Ophthalmology. 2009;116(9):1725–30.
Greenwald MJ, Paller AS. Ocular and dermatologic manifestation of neurocutaneous syndromes. Dermatol Clin. 1992;10(3):623–39.
Grant WM, Walton DS. Distinctive gonioscopic findings in glaucoma due to neurofibromatosis. Arch Ophthalmol. 1968;79(2):127–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Di Nicola, M., Viola, F. (2020). Ocular Manifestations in Neurofibromatosis Type 1. In: Tadini, G., Legius, E., Brems, H. (eds) Multidisciplinary Approach to Neurofibromatosis Type 1. Springer, Cham. https://doi.org/10.1007/978-3-319-92450-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-92450-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-92449-6
Online ISBN: 978-3-319-92450-2
eBook Packages: MedicineMedicine (R0)