
Abstract
Gold nanoparticles have properties useful in biomedical imaging and cancer therapy. This biocompatible metal has been used for centuries in medicine. In the last 20 years, the rapid developments in nanotechnology have revealed several applications of nanosized gold, which are now being evaluated for clinical procedures. For instance, gold nanoparticles can be used to develop sensors due to their optical properties; they also make possible the development of new hyperthermia and drug delivery treatments. However, gold nanoparticles could find more immediate and direct applications in medical physics procedures, such as X-ray imaging and radiotherapy. First, this chapter provides an overview of the different synthesis routes for the production of biomedical gold nanoparticles. Then, an overview of the physical principles of photon–matter interactions, that are fundamental to the concept of X-ray attenuation in biological tissues, is presented. The properties of gold nanoparticles as contrast agents for X-ray and computed tomography (CT) imaging are reviewed, along with the principles of the radiosensitization effect useful in medical physics and oncology. The main mechanisms leading to dose enhancement, to cell damage and to cell death, are described in the light of the specific interactions taking place between ionizing photons and high-Z materials such as gold (Au) when these are distributed in biological tissues such as tumours. Finally, the performance of gold nanoparticles as CT contrast agents and radiosensitizers in oncology is discussed, in the perspective of their consideration for clinical applications.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Gold nanoparticles
- Radiotherapy
- Brachytherapy
- Computed tomography
- Radiosensization
- Cancer treatment
- Theranostics
1 Introduction
Over the last decades, gold nanoparticles (Au NPs) have emerged as a new promising material for a large variety of medical applications [13, 41, 51, 52, 55, 78, 95, 97, 100, 120, 131, 142, 176, 178, 190, 197, 199, 200]. Au NPs bear several physico-chemical properties useful for the development of diagnosis tools as well as for imaging contrast agents. They can also be used as radiosensitizers: these are products that, when they are injected in biological tissues, have the capacity to increase the impact of radiotherapy [25, 39, 45, 76, 111, 204, 208]. Finally, nanosized gold is resistant to oxidation in biological media and overall, it is accepted by the cells at relatively high concentrations [64, 95, 113, 173].
A very large number of studies and reviews have been written about the properties of Au NPs used as ‘plasmonic’ materials [57, 186, 200]. Plasmons are oscillations of free electrons taking place at the surface of conductor nanomaterials (such as gold). The surface electrons of Au NPs can couple with electromagnetic radiation of certain wavelengths that are far larger than the particle. Plasmonic nanoparticles exhibit interesting scattering, absorbance and coupling properties, and several proof-of-concept studies useful to the biomedical field have been made by exploiting the resonant properties of Au NPs irradiated by visible and near-infrared light. For instance, new sensor technologies have been developed relying on the principle that molecules reacting with a specific molecular function present at the surface of Au NPs, induce slight changes to specific resonance peaks. These can be sensitively detected with appropriate spectroscopic tools. The resonant properties of Au NPs under visible and infrared light can also be exploited to increase the temperature of cells, a phenomenon referred to as ‘hyperthermia’ [53, 182].
Although the plasmonic characteristics of Au NPs appear very useful for in vitro applications, their potential for in vivo procedures has always been somewhat limited to skin diseases and to superficial cancer (e.g. skin cancer). In fact, visible and near-infrared photons diffuse strongly in the biological tissues. Diffusion limits the depth of penetration of such low-energy photons in biological tissues to a few millimetres only. Therefore, to be useful for a large variety of applications in vivo, Au NPs must preferably be irradiated with mid-to-high energy electromagnetic radiation—mainly photons—that have the capacity to penetrate deep enough in the body.
In fact, biomedical imaging and cancer treatment by radiotherapy are two areas of medicine where the injection of Au NPs in vivo could be recommended for clinical diagnoses and therapies [45, 70, 71, 76]. High-energy photons (from 1 keV and above) are already exploited in medicine. This is precisely the range of energies useful to medical physics. Irradiated with photons in the ionizing energy range, Au NPs can be used as effective contrast agents for X-ray computed tomography (CT), as well as for ‘boosting’ the radiation dose delivered in radiotherapy treatments [25, 84, 131].
Therefore, in vivo applications of Au NPs based on their interactions with higher energy photons, have emerged in modern medicine and in particular for the treatment of cancer. Au NPs can be used to enhance the differences of density between biological tissues (e.g. using X-ray and CT imaging). They can also be used in radiosensitizing procedures to induce more damages to cells irradiated with radiotherapy beams or with internal radioactive sources. In fact, Au NPs are a very dense material; they can attenuate high-energy photons in the biological tissues even if they are distributed at relatively moderate concentrations. Also, photons interacting with high-Z materials such as gold generate many by-products such as low-energy photons and electrons. These ‘secondary products’ have the capacity to increase the therapeutic impact of the primary photons beams, as well as the radioactive sources used in radiotherapy. In medical physics, Au NPs are often referred to as ‘radiosensitizers’, i.e. products that have the capacity to enhance radiotherapy treatments.
This chapter begins with an introduction to the different synthesis routes leading to the production of Au NPs, as well as to their surface functionalization. Then, an overview about the physical mechanisms by which photons (~10–500 keV) interact with the atoms present in biological tissues is given. Photons of energy higher than 10 keV can penetrate deep enough in the biological tissues (e.g. sub-visible light wavelength range), and therefore can be exploited either for imaging or therapeutic applications. Finally, the applications of Au NPs as contrast agents for CT (X-ray imaging), and as radiosensitizers in radiotherapy (medical physics in oncology), are presented in the context of their ongoing evaluation towards clinical applications.
2 Synthesis of Gold Nanoparticles for Biomedical Applications
Colour change is the most evident signature of colloidal gold. Depending on the size of the nanoparticles, gold presents either an intense red colour for small to medium-sized particles, purple for larger particles and blue for aggregates [52, 78]. Because the properties and the applications of Au NPs so closely depend on their size and shape, the colour of aqueous suspensions of Au NPs provides very efficient indications to the chemist at every step of the nanoparticle preparation.
The first experiments for the synthesis of gold sols under controlled conditions were reported by Michael Faraday in the 1850s [58, 152]. The scientist accidentally generated a ruby red solution while mounting pieces of gold leaves onto microscope slides. He referred to this solution as ‘activated gold’. Then, he used phosphorus to reduce a solution of gold chloride. Already interested in the properties of light and matter, Faraday further investigated the optical properties of these colloidal gold solutions. For a long time, the composition of ‘ruby’ or ‘activated’ gold was unclear, but the colour-particle size relationship was already acknowledged. The studies on Au NPs did not make significant advances until the end of the twentieth century, with the exponential development of advanced analytical technologies, such as atomic force microscopy and electron microscopy. Due to their relatively easy synthesis routes, high stability and high density per particle, colloidal gold has also been used in the development of physical separation techniques such as ultracentrifugation, as well as electron microscopy dyes.
Gold nanoparticles produced for biomedical applications are usually generated from precursor solutions containing chloroauric acid (H[AuCl4]). After H[AuCl4] is dissolved in water, the solution is rapidly stirred while a reducing agent is added. To prevent the particles from aggregating, a stabilizing agent is usually added for coating the nanoparticle surface. A comprehensive review of the different classes of Au NPs synthesis was written by Daniel et al. [48]. A selection of the most significant ones is reviewed here.
2.1 Direct Reduction: The Turkevitch Method
The chemical reduction of AuCl4 by citric acid in a hot aqueous solution was investigated in 1951 [185]. In this synthesis, citrate acts as the reducing agent, then as the capping agent that stabilizes the Au NPs through electrostatic interactions. The Turkevitch method allows the growth of NPs with many different shapes and diameters usually ranging from 10 to 50 nm (Fig. 1). A significant increase in the particle diameters is observed at lower citrate concentration, leading to the generally accepted conclusion that the total particle surface area is determined by the number of citrate ions available to cover it [65].

Source The authors, following procedures adapted from [185] including the use of PEG-thiol as a surfactant
Gold nanoparticles synthesized by the Turkevitch method in citrate: a, b as-synthesized; c, d grafted with polyethylene glycol PEG 1000 mw; and e, f grafted with polyethylene glycol PEG 5000 mw. The longer the chains, the larger the spacing between the particles.
More recently, an alternative approach showed that a high concentration of Cl− ions also leads to larger sizes for Au NPs reduced by citric acid, since it decreases their surface charge and thereby promotes coarser particle size and even aggregation [205]. A similar result was obtained by increasing the pH of the NP solution just after initiating the reduction step. An increase of the pH limits the nucleation process and decreases the final number of grown Au NPs [167]. Although simple, the direct chemical reduction synthesis route usually leads to the production of NPs of relatively large sizes, showing anisotropy and a certain degree of size polydispersity.
2.2 Seed-Mediated Growth for Smaller and Narrower Particle Size Distributions
The constantly increasing demand for Au NPs in photonics, biology and medicine, has led to the development of new synthesis routes for achieving better control over size distributions. One of the most promising approaches in this goal is the seed-mediated growth technique. In this concept, small nanoparticle seeds of a typical size smaller than 15 nm, act as catalytic nucleation centres for the growth of larger NPs [86]. The nucleation of very small NP seeds is usually made through the reduction of gold ions by sodium borohydride (NaBH4), a relatively toxic reducing agent that is tolerated as an initiator of Au NPs seeds. However, it is not indicated in the final synthesis steps of biomedical Au NPs. Shape uniformity in the seed suspension is of paramount importance to achieve both shape and size uniformity of the final NPs [141].
Several articles have been published on the topic of seed-mediated growth of Au NPs. One of the most comprehensive ones is a three-step process that was used to produce uniform Au NPs with a diameter up to 300 nm (Fig. 2) [209]. The main limitation of this method is that an important population of smaller NPs often remains in solution additionally to the grown seeds, requiring further purification to achieve very narrow particle size distributions. Perrault et al. overcame this problem by using hydroquinone as a reducing agent [147]. Hydroquinone reduces selectively the gold ions that are located in the immediate vicinity of Au0 seed nanoclusters, leaving isolated gold ions unreduced. This chemical route allows a selective growth of the seeds while avoiding further nucleation of Au clusters in the reaction fluid.

Reprinted with permission from [209]. Copyright 2011 American Chemical Society
TEM images of a 15 ± 2 nm seeds, b 31 ± 3 nm, c 69 ± 3 nm, d 121 ± 10 nm, and e 151 ± 8 nm and SEM image of f 294 ± 17 nm gold nanoparticles. Scale bars are 200 nm for parts a–c and 500 nm for parts d–f.
2.3 Syntheses Taking Place in Organic Media
Nanoparticle synthesis routes performed in aqueous conditions are easy to implement, and as a major advantage, they avoid the introduction of potentially toxic solvents into the chain of NP production. Unfortunately, the size distribution of Au NPs synthesized in aqueous media is relatively difficult to keep to narrow and small values. It is generally well established and acknowledged in the field of nanotechnology, that syntheses performed in organic media often result in much smaller and narrower particles size distributions. The Brust–Schiffrin method is one of the first reported to synthesize Au NPs in organic media, and it is until now one of the most widely performed to synthesize small particles of narrow distributions (Fig. 3) [22]. Typically, it consists of using tetraoctylammonium bromide (TOAB) to transfer the gold salts into an organic phase (e.g. toluene, ethanol and ethylene glycol). Then, NPs are nucleated through a reduction step through thiol ligands and NaBH4. This approach produces very small and stable NPs ranging from 1.5 to 5 nm that can be easily functionalized with other ligands. Recent developments related to the Brust–Schiffrin method include mostly fundamental studies of mechanisms such as charge transfer, nucleation and growth during the synthesis process [54, 146].

Source The authors, following procedures described in [22]
Gold nanoparticles synthesized by the Brust–Schiffrin method. Among the different colloidal synthesis techniques, this one usually leads to very small particle size distributions.
Another class of synthesis procedures taking place in an organic solvent is the so-called polyol process. It allows the synthesis of monodisperse Au NPs, or alternatively to particles of various well-controlled geometries [60]. In this procedure, gold salts are dispersed in high boiling point alcohols (e.g. ethylene glycol), which also act as a reducing and as a capping agent. By using a polymeric stabilizer, highly symmetrical polyhedron-shaped Au NPs with a narrow size distribution can be obtained in a wide variety of sizes. Xia et al. have used a polyol process with polyvinylpyrrolidone (PVP) as the stabilizer to synthesize very uniform silver nanocubes with 50–115 nm of edge length, and used them as sacrificial templates in a solution of HAuCl4 for the production of gold nanoboxes with perfectly smooth surfaces [179]. Song et al. dissolved HAuCl4 and PVP in 1,5-pentanediol, instead of ethylene glycol for its higher boiling point, and added small concentrations of AgNO3 [165]. Depending on the concentration of silver ions, octahedral (ratio Ag/Au = 1/600) or cubic (Ag/Au = 1/200) shape nanostructures were obtained, among others. This chemical behaviour was attributed to the selective deposition of silver species on the seed surface during the reaction. Overall, the polyol process is a convenient and low-cost technology for the large-scale production of highly symmetrical Au NPs of homogeneous sizes. However, the complete elimination of the solvent residues, as well as the presence of potentially toxic chemicals, necessitates the introduction of tedious filtration, dialysis or chromatography procedures that are not necessarily easy to upscale.
2.4 Au NPs Purification Prior to Coatings and Functionalization
The purification and functionalization steps to remove potentially toxic reagents and to cover the NPs surface with biocompatible ligands are very important in the development of stable as well as functional Au NP formulations for biomedical applications. The purification is usually performed by dialysis, chromatography or centrifugation. Those methods are generally sufficient to remove the majority of the contaminants. As mentioned above, the majority of Au NPs used for biomedical applications are synthesized either by the Turkevitch [56] method, by the seed-mediated growth approach [150] or by the Brust–Schiffrin [42] technique. In the first case, citrate molecules cover the surface of the Au NP. In the second one, hexadecyltrimethylammonium bromide (CTAB) is usual, whereas alkanethiols are employed in the Brust–Schiffrin method. For biomedical applications, the Au NPs must be dispersed in aqueous solutions, and they must be free of even low traces of contaminants (e.g. organic solvents, reduction agents and excess of surfactants). They must be stable in physiological media, which are rich in diverse ions, proteins and many other molecules. Thus, in order to preserve the colloidal stability and assure biocompatibility, the Au NPs obtained by the previously described methods must be functionalized with biocompatible molecules.
2.5 Ligand-Free Au NP Suspensions
The conventional Au NP synthesis approaches do have several limitations in terms of toxicity risks induced by surfactants and chemical residues. For instance, sodium borohydride (NaBH4) is a harsh reagent that must be entirely removed from the solutions [146]. Then, the gold ions used in most Au NP synthesis techniques usually come from chloroauric acid, a potent acid which must be entirely cleared from the biomedical solutions [5]. In addition, ligand exchange procedures involve several manipulation steps that lead to material loss, as well as to agglomeration. Several alternatives to the more conventional chemical synthesis approaches have emerged recently to synthesize purer Au NPs, generally free from ligands or reducing agents, and therefore ready to be efficiently functionalized. These synthesis techniques could represent a major advantage in the quest for compounds that limit the toxicity risks related to the presence of residues in Au NPs compounds.
2.5.1 Pulsed Laser Ablation in Liquid (PLAL)
The synthesis technique that possibly enables the production of the smallest and the purest Au NPs solutions, is pulsed laser ablation in liquid (PLAL). This synthesis method was introduced by Cotton and Henglein in 1993 [63, 138]. It is performed by immersing a gold metal target in a fluid, followed by irradiation of the target surface for a certain time with a pulsed laser [153]. The target absorbs the laser pulse energy, resulting in heating and photoionization of the irradiated area. Solid, liquid and vaporized materials are emitted in the form of a plasma plume, which expands outwards in a confining liquid [8, 9, 81]. Then, the plasma plume starts to cool down and a cavitation bubble is formed [8]. At this moment, the supersaturation point is achieved due to the concentration of metal ions, to the high pressure and to the temperature attained because of liquid confinement. This causes NPs to nucleate: metal atoms condense and coalesce in the form of NP nuclei. The newly formed NPs start to diffuse into the expanding cavitation bubble, where further growth, coalescence or aggregation happen. At last, when the cavitation bubble collapses, all the NPs are released into the surrounding liquid [8]. A schematic representation of PLAL for the synthesis of Au NPs is shown in Fig. 4. The ultra-small Au NPs colloids synthesized by this technique can be stabilized by PEG grafting to enhance their stability in biomedical media [169].

Reproduced from [169] with permission from the Royal Society of Chemistry
Pulsed laser ablation in liquid for the nucleation of Au NPs in ultrapure water (a), produces Au colloids of chemically pure surfaces that are stabilized by electrostatic forces (b). To increase their colloidal stability in biological fluids, the Au NPs are stabilized by polyethylene glycol. Additionally, the Au NP@PEG was functionalized with DMSA-DTPA-Mn2+ to be applied as a contrast agent for MRI (c). PLAL synthesis followed by PEG and DMSA-DTPA-Mn2+ grafting enables the production of very small particle size distributions (d).
The laser wavelength, the laser fluence, the repetition rate, the ablation time and the composition of the liquid solution can be tuned to achieve a specific concentration of NPs and size distribution [8, 153]. The most suitable wavelengths to synthesize Au NPs are in the NIR region (e.g. 1064 nm) [184]. Subsequently, post-irradiation with wavelengths coinciding with plasmon absorption or interband transition of Au NPs can be used to tune the size and to reduce polydispersity [124].
PLAL allows the production of Au NPs in water or organic solvents [10, 180]. However, organic solvents are prone to pyrolysis during laser ablation and the degraded molecules can adsorb on the surface of the NPs, which may raise biocompatibility issues. In pure water, PLAL-synthesized Au NPs show a size range of 10–40 nm; they are electrostatically stabilized by negative charges that result from the formation of Au–O− species [8, 153, 180]. To improve the stability and to control the Au NPs size in water, it is possible to use low salt concentration. The anions of the salts adsorb on the surface of the Au NPs and increase the negative charge density, thus enhancing the repulsive forces between the NPs. This effect prevents both the growth of the NPs by avoiding nuclei coalescence and improves the colloidal stability prior to further grafting of the nanoparticles with biocompatible molecules (e.g. to create steric hindrance) [130, 153].
In a biomedical context, the main advantage of PLAL over conventional Au NP synthesis colloidal chemistry routes is the production of NPs directly in water without the use of reducing or stabilizing chemicals (e.g. NaBH4) [153]. As a result, the surface of the NPs is free of ligands and it is thereby readily available for the conjugation with other molecules [153]. Moreover, many reducing chemicals used in colloidal Au NP synthesis routes can be toxic, and this hazard is totally eliminated in laser ablation. Thus, the ligand-free surface allows to avoid extensive purification processes and aggregation associated with ligand exchange procedures [194]. The main limitations of PLAL for Au NP synthesis are the necessity to use very expensive lasers as well as low concentrations of solutions synthesized, which require several concentration steps in order to reach biomedical applicability.
2.5.2 Atmospheric Plasma Electrochemistry
Atmospheric plasma synthesis, or plasma electrochemistry, allows the rapid nucleation and growth of Au NPs directly in water, without the need of any chemical reducing agent. Compared to pulsed laser ablation, it has the advantage of producing more concentrated solutions of Au NPs while requiring less expensive equipment. Plasma is a charged gas containing free electrons, positive and negative ions, neutral species in the ground or excited state, and photons. This mixture of charged species gives plasmas unique physico-chemical properties. The most common and easily attained method for forming plasma is to apply an electrical voltage between two electrodes, solid or liquid, separated by a gas. With aqueous solution, plasma electrochemistry must be operated near atmospheric pressure and at low temperature to avoid solvent evaporation.
A great variety of chemical reactions take place at the plasma–liquid interface. In the presence of nitrogen as the main plasma component or as a contaminant from the presence of air, the formation and dissolution of nitric acid can cause a significant decrease in pH [108]. The electrolysis of water \(\left( {2{\text{H}}_{2} {\text{O}} + 2{\text{e}}^{ - } \to 2{\text{OH}}^{ - } + {\text{H}}_{2} } \right)\) at the plasma cathode and the formation of chlorine gas \((2{\text{Cl}}^{ - } \to {\text{Cl}}_{2} + 2{\text{e}}^{ - } )\) at the anode were also reported [158]. The formation of reactive ions H+, H−, O−, OH−, radicals H·, O· and OH· and H2O2 is also a very important factor in the cascade of chemistry events taking place in the nucleation and growth of nanoparticles [66, 125].
The exact mechanism for the nucleation of Au NPs by plasma–liquid electrochemistry is still not entirely understood. The electrons generated in the plasma are accelerated to the surface of the liquid by the electric field between the electrodes and pass into solution in the form of solvated electrons [1, 125]. Several reactive oxygen and nitrogen species generated by the plasma treatment act as metal ion reducers. These ‘fugitive’ or metastable reducing agents make possible the nucleation of Au NPs while avoiding the use of external toxic chemicals (e.g. NaBH4) for the synthesis of water-dispersed Au NPs. Gold ions in solution can reach the plasma–liquid interface by the combined effects of the electric field, the convection forces and the concentration gradient [2]. When an ion reaches the interface, it is reduced into atomic gold directly by the electrons: \(\left[ {{\text{AuCl}}_{4} } \right]^{ - } { + }\,3{\text{e}}^{ - } \to {\text{Au}}^{0} { + }\,4{\text{Cl}}^{ - }\). Once reduced, the gold atoms collide and form clusters, which diffuse away from the plasma–liquid interface. These clusters act as nucleation sites of NPs [2]. Size control is usually performed by adjusting only the concentrations of metal salt and surfactant in solution. Increasing the current can also lead to the production of smaller NPs since it allows a higher nucleation rate [79].
In the last 10 years, new ‘cold plasma’ reactors operating at atmospheric pressure brought the possibility to synthesize NPs from metal salts directly in water. The first type of atmospheric plasma reactor that was used to nucleate Au NPs was the microplasma [104, 160], and the most commonly used geometry of microplasma is the microhollow cathode [20, 104, 155]. It consists of a gas stream flowing in a cylindrical hollow cathode placed a few millimetres above a liquid solution containing metal ions. A DC voltage, typically several thousand V/cm, is applied between the cathode and the solution. The plasma discharge generated at the output of the microhollow cathode thus extends to the surface of the liquid, where the reduction of the metal ions and the nucleation of NPs take place.
Richmonds and Sankaran demonstrated the possibility to synthesize gold and silver NPs from the dissolution of a metal sheet in a slightly acidic aqueous solution [155]. Using fructose as a stabilizing agent, the reduction by argon plasma has formed 10 nm diameter NPs for both metals. This device shows the conventional concept of an electrochemical cell, where the metal film acts as a sacrificial anode, and the microplasma as a cathode [160]. This concept was adapted several times for the synthesis of gold and silver NPs of different sizes and morphologies, with and without stabilizer [127, 188, 189]. Unfortunately, the microplasma technologies have a rather limited surface coverage, and other types of plasma must be considered to allow the development of technologies enabling the synthesis of large volumes of solutions.
A promising alternative to the microplasma is the dielectric barrier discharge (DBD). Through the DBD technology, plasma–liquid interface area of several cm2 can be generated [16]. This technology was recently employed to synthesize radioactive Au NPs for applications in oncology [19]. The main limitation of Au NPs synthesized by plasma electrochemistry remains the relatively high polydispersity of particles, requiring further separation steps. Further work aimed at understanding the exact electrochemical mechanisms behind the reduction of Au ions should help reaching a better control over size.
2.6 Surface Treatment of Au NPs
The size, shape and surface properties of the Au NPs are determinant factors for their successful biomedical applications [4, 35, 40, 55, 59, 95, 134]. To be used in vivo, the particles should be dispersible in water; they should form a stable colloid in biological media, which are aqueous solutions with significant ionic strengths and rich in proteins; they should not induce cytotoxicity; they should not adsorb too many proteins on their surfaces to avoid uptake by the mononuclear phagocytic system (MPS) cells; and they should be easily conjugated with specific ligands for targeting studies [35, 59, 172]. Therefore, the Au NPs produced by the previous techniques should undergo ligand exchange processes for their surface to be coated with different types of biocompatible, antifouling and/or biologically active molecules [119, 159].
The surface of Au NPs can be functionalized using molecules containing thiols, amines, carboxylic acids and phosphines [140]. The most resistant coatings are formed by making use of ligands that have the highest affinity to Au. Therefore, thiol modification is usually preferred, as the sulphur atoms form a coordinate covalent bond with the metal surface [119]. The surface of Au NPs stabilized with citrate or CTAB can be easily functionalized by exchanging these molecules with thiol containing ligands [159]. The initial stabilizing agent is quickly replaced during the adsorption of sulphur atoms, followed by a slower reorganization and packing of the incoming molecules [52, 119]. The surface of Au NPs stabilized by alkanethiols can be modified by thiol–thiol exchange [159]. This process requires higher molar excess of the incoming ligands [119]. In addition, this method can also be used to produce mixed organic monolayers containing different types of ligands at specific ratios [159]. Finally, polyethylene glycol (PEG) is a commonly used antibiofouling ligand for Au NPs [52]. When this highly hydrophobic polymer is attached at the surface of Au NPs, the long chains prevent the aggregation of the nanoparticles by steric repulsion. PEG also prevents the adhesion of proteins at the surface of the NPs.
2.7 Cell Toxicity of Au NPs: In Vitro Studies
Several studies and reviews have reported on the viability of a wide selection of cell types incubated with Au NPs [64, 90, 95, 113, 173]. Several sizes of Au NPs were reported, as well as different surface charges and molecular coatings. The concentration thresholds for which cytotoxicity effects are detected with the main types of Au NPs used in biomedicine can be found in this literature.
3 Principles of Physical Interactions Between Photons and High-Z Elements
High-energy photons (X or γ rays) in the range typically used for computed tomography and medical physics, can penetrate long distances in the biological tissues. By interacting with the different chemical elements present in vivo, they undergo specific interactions which makes them useful for whole body medical imaging and therapy. Photons used in medical physics and imaging usually have an energy included between 10 keV and 1 MeV. In that range, photons can be attenuated by four major types of interactions upon colliding with materials: (1) the photoelectric effect, which dominates at the lower energies; (2) the Compton effect, which increases at higher energies and for higher atomic numbers; (3) Rayleigh scattering; and finally (4) pair production, which occurs only at very high energies. A schematic representation of each one of these phenomena is represented in Fig. 5.

Source The authors (Review, in press at Advanced Healthcare Materials, with permission from Wiley-Verlag)
Illustration of interactions of photons with the atoms that constitute materials and tissues.
Attenuation coefficients calculated for each element, at different energies, and for each one of the interaction mechanisms, allow a comprehensive quantitation of the photon–matter interaction process. The cross section is a measure of the probability for an interaction to occur, and it depends on the atomic number (Z). For high-Z elements, the probability of interaction is generally higher due to their larger cross section. The total mass attenuation coefficient (μ/ρ) corresponds to the sum of the individual attenuation coefficients (Eq. 1). It represents the probability of interaction per mass unit of a given material (in cm2/g):
where ρ is the density of the material, and τ, σ, κ and σR are the cross sections for the photoelectric effect, the Compton effect, pair production as well as Rayleigh scattering, respectively.
The photoelectric effect occurs when a photon collides with an electron from one of the inner orbitals of the impacted atom (Fig. 5). The photon is absorbed and the electron is ejected (i.e. the ‘photoelectron’) with a kinetic energy (Ek) corresponding to the difference between the energy of the incident photon (hυ) and the binding energy of the electron (EB). When the photoelectron is ejected from the inner-shell, an electron of an upper shell fills the vacant place, which leads to the emission of either characteristic X-rays or Auger electrons. Characteristics X-rays are emitted from electronic transition rearrangement and their energy is specific to each element. The Auger effect occurs when the energy involved in the transition serves to eject an electron (i.e. the ‘Auger electron’) present on a higher orbital.
Figure 6a, b illustrates the mass attenuation coefficients (μ/ρ) for each one of the processes occurring in gold and as well as in water. For gold, the photoelectric effect is very strong at energies of 200 keV and below, whereas similar photoelectric interaction rates are reached in water at photon energies of 30 keV and below. The probability of the photoelectric effect to occur is very high for materials made of high-Z elements: in fact, it is proportional to ~Z4 [14, 89]. Figure 6c illustrates the relative importance of each mechanism for different atomic numbers and photon energies. For conventional external beam therapy with 100–250 keV photons, the Compton effect dominates in the biological elements (C, O, N), whereas the photoelectric effect is preponderant in the interactions with gold. At lower energies, however, the photoelectric effect dominates.

a, b Mass attenuation coefficients for gold and water according to the photon energy (data from the NIST) [136]. c Diagram of the different photon–matter interactions taking place depending on the atomic number (Z) and the energy of photons. Reproduced from [14] with permission from Wiley-VCH Verlag. d Comparison of the photon mass energy absorption coefficients for gold and soft tissue. The ratio of the mass energy absorption coefficients is shown as a function of energy. Reproduced from [25], based on data originally from [80], with permission from the Royal Chemical Society
The Compton effect occurs when a photon collides with an electron of the outer shell and ejects it from the atom (Fig. 5). The photon loses part of its energy and is scattered at an angle ϕ. Either it continues its course, or it undergoes further interactions such as Compton, photoelectric or Rayleigh scattering. The ejected electron is referred to as ‘Compton electron’, and travels in the surrounding material where it may cause subsequent excitation and ionization of atoms. The incidence of the Compton effect is significant at photon energies ranging from 100 keV to 10 MeV (Fig. 6a, b). It is almost independent of Z, and it predominates at intermediate therapeutic energies [89].
In Rayleigh scattering or coherent scattering, the incoming photon interacts with the whole atom. The electrons oscillate in phase and release an extra quantity of energy in the form of a secondary photon having the same energy as the incident photon, however emitted in a slightly different direction (Fig. 5) [14, 89]. Overall, this interaction does not contribute to the dose enhancement in radiotherapy, since no energy is transferred to orbital electrons, and therefore, ionization does not occur. Usually negligible at high energies, Rayleigh scattering is more probable with high-Z materials such as gold, as well as for low and very low-energy photons (Fig. 6a, b) [89].
Finally, pair production arises from the trajectory of a high-energy photon (>1.02 MeV) passing near the nucleus of the atom. In this specific situation, the energy of the photon is converted into mass and an electron–positron pair is created (Fig. 5) [14, 89]. The energy in excess (E = 1.02 MeV) is equally distributed in the form of kinetic energy (e.g. 511 keV) between the positron and the electron, and this energy is lost via ionization as the particles travel through matter. When the positron comes to rest, it interacts with an electron of the material, resulting in the emission of two annihilation photons of 511 keV, travelling in opposite directions. Pair production interactions are dominant at high energies (>10 MeV, e.g. external beam radiotherapy) and are more likely to happen for high-Z elements, since the cross section for this kind of interaction varies according to Z2 (Fig. 6a, b) [14, 89].
Figure 6d shows a comparison of the photon mass energy absorption coefficients for gold and soft tissue. The ratio of the mass energy absorption coefficients is shown as a function of energy. The difference of mass absorption energy coefficients is maximal in the 10–200 keV energy range, the same as used for CT imaging and for the vast majority of radiotherapeutics procedures used in oncology. The coupling of these modalities with Au NPs is discussed later in this chapter: in Sect. 5 for imaging, and in Sect. 6 radiosensitization and radiooncology.
4 Impact of Radiation and Au-Mediated Radiosensitization Products on Cells and Tissues
Ionizing radiation impacts on the biological systems by generating several defects in organelles and at the cell membranes. In particular, the impairment to the DNA molecules is by far the impact that has the strongest and the most lasting influence on the fate of the cells. The interaction of ionizing radiation with biological systems can generally be divided into three phases: the physical, the chemical and the biological [76]. Each phase and related mechanism is illustrated in Fig. 7.

4.1 The Physical Phase
The physical phase takes place upon collision of the photon with the atoms present in the cells and more globally in the tissues (e.g. water, proteins, lipids and DNA). Through these interactions, energy from the photons is deposited in the tissues, causing thereby several direct damages to the molecules. Low-energy electrons and free radicals can also be generated through the interaction of the primary photons with the tissues [74, 76]. The interaction of photons with high-Z elements such as gold atoms results in the emission of further secondary species: photoelectrons, Auger electrons and characteristic X-rays [76]. Thus, in addition to the damage created by the energy transferred from the incident photon, the DNA, proteins and lipids of the irradiated cells, can be further injured by the secondary emissions coming from the Au NPs. The low-energy electrons can also ionize the water molecules located in their vicinity, thus causing the formation of reactive oxygen species (ROS). They can also interact with other Au NPs and lead to the emission of further Auger electrons [26, 74, 102, 143]. The contribution of these secondary species has a strong impact on the overall dose deposition.
4.2 The Chemical Phase
The free radicals and other reactive and metastable species, diffuse and react with atoms and molecules present in the biological systems. This is the ‘chemical phase’ mentioned above, which causes further damage and cell stress [76, 157]. Several studies concluded that, at the chemical phase, Au NPs contribute to cell damage essentially by increasing the production of ROS. These highly reactive oxygen-containing chemical species (e.g. •OH, H2O2 and •O2) have the capacity to oxidize biomolecules in the cell and thereby inducing a chain reaction of potentially toxic free radicals.
In fact, it has been demonstrated that Au NPs alone, and in particular the smaller ones (<5 nm) not entirely covered with surfactants, can induce the formation of ROS in water or in biological medium. This occurs spontaneously because Au NPs have an electronically active surface. This is a consequence of the small size and high surface curvature that modifies the energy levels of the electrons in the surface atoms compared with bulk structures [139]. Therefore, depending on the ‘bareness’ of their surfaces, Au NPs can catalyze the production of ROS through surface-mediated electron transfer to oxygen molecules [76, 139].
Misawa and Takahashi suggested that the production of ROS by Au NPs is potentiated after irradiation, because secondary emissions cause more ionization of water molecules [132]. In addition to this, Cheng et al. observed that the Au surface atoms can also be activated by radiation-induced ROS [37]. Porcel et al. proposed that following electron depletion after secondary emissions, positively charged Au NP surfaces can destabilize the nearby water molecules, facilitating their dissociation and increasing the production of ROS [151]. Furthermore, Zheng et al. observed that small (e.g. 5 nm) and positively charged Au NPs can bind to the DNA backbone through electrostatic interactions, and as a result weaken the DNA structure. This type of interactions can make the DNA more susceptible to permanent damage, in particular when it is impacted by low-energy electrons produced through the interaction of photons with Au NPs [76, 208].
4.3 Measurement of Physical and Chemical Impact of Au NPs on DNA
It is difficult to clearly dissociate the impact of physical damages, from the impact of purely chemical damages caused by impacting photons to the biological tissues. The mechanisms leading to increased DNA damage and to cell death, after irradiation in the presence of Au NPs, are not completely understood. However, Au NPs have a critical effect in each phase of radiation-induced cell damage. DNA impairment is the most important parameter to take into account when evaluating the efficacy of radiotherapy [74, 76, 157]. Overviews of the mechanisms of radiobiology can be found in comprehensive books on the topic [73]. Au NPs used as radiosensitizers increase dose deposition and localize their impact more precisely into targeted tissues such as in tumours [74]. Several experimental studies indicated that dose enhancement in the vicinity of Au NPs is much higher than initially predicted by theoretical simulations [88, 111, 129]. Such observations led to a number of investigations aiming to understand the mechanisms behind the radiosensitizing effect induced by Au NPs and its influence on the radiobiological mechanisms.
Traditionally, the most direct means for quantifying the impact of ionizing radiation on cells was by measuring the frequency of chromosomal aberrations in function of the absorbed dose [73]. Nowadays, the damage caused by ionizing radiation to cells in presence of Au NPs is generally quantified by clonogenic assays [27, 39, 47, 88, 123, 129, 166, 181, 207]. This analysis provides information about cell survival after irradiation, and it allows a quantification of the sensitizing effect generated by Au NPs [27, 123, 181].
However, more analytical tests are necessary to fully quantify the impact of the radiosensitizing effect on the cells. Double-strand breaks to the DNA can be quantified by using the γ-H2AX or the 53BP1 foci formation assays [27, 39, 47, 129, 166, 181, 207]. The presence of these proteins is an indicator that irreparable damage was caused to the DNA. The quantification of ROS production is usually performed using the dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay [27, 123]. By combining the complementary information provided by different assays, Butterworth et al. demonstrated that irradiation of PC3 cells containing Au@DTDTPA NPs induced a 1.7-fold increase in residual DNA double-strand breaks (e.g. 24 h after irradiation) compared with cells treated with radiation only. In addition, the authors observed that incubation of PC3 cells with Au@DTDTPA NPs did not cause a statistically significant increase in the production of ROS, compared with non-treated cells [27]. Overall, these observations indicate that the natural DNA repair mechanisms can be impeded when increasingly complex damages are caused to the helixes. However, this effect seems to be cell-dependent [27]. On the other hand, after X-ray irradiation, Ma et al. observed a 1.6-, 1.2- and 1.1-fold increase of ROS concentration in KB cells incubated with Au NPs, Au nanospikes and nanorods, compared with the control group. These studies suggested that increased ROS production is one of the possible mechanisms behind Au NPs radiosensitization in the chemical phase [123].
4.4 The Biological Phase
The biological phase begins when repair mechanisms are triggered in response to the physical and to the chemical damages caused to vital structures (DNA, mitochondria and other organelles) [76, 157]. In particular, the increased ROS production and the indirect DNA damage initiated during the chemical phase can (1) induce oxidative stress, (2) disrupt the cell cycle and (3) delay or inhibit DNA repair [76, 123, 157]. Oxidative stress can induce damage to almost all organelles, and therefore lead to cell death in a variety of ways. Although the exact impact of Au NPs on oxidative stress induction is not entirely well elucidated at the moment, Au NPs can lead to an increased production of ROS through catalytic reactions. High intracellular concentrations of ROS impair mitochondrial function, and the presence of Au NPs in cells can inhibit the mechanisms of proteins implied in cellular oxidation homeostasis [76, 116, 157, 187].
The cell cycle also plays an important role in cell sensitivity and survival to radiation. It has been demonstrated that cells are more sensitive to radiation-induced damage when they are at the end of cycle step G2 and during mitosis. On the other hand, cells in late S phase are more resistant to radiation [76, 157]. The analysis of cell cycle is often performed by flow cytometry, after DNA staining with propidium iodide and 5-bromo-2′-deoxyuridine [47, 123, 207]. This method quantifies the DNA content per cell, and with this information, one can identify the phase of the cell cycle where the impact of radioactivity is the highest. Roa et al. observed that Au NPs (e.g. glucose-coated Au NPs) can synchronize and stop cells in the G2/mitosis phase. This mechanism, in turn, increases cell sensitivity to radiation damage [193]. After exposing cells to different types of Au NPs, Ma et al. observed a slight increase on the percentage of cells in the G2/M phase for Au NP-treated cells unexposed to radiation; after X-ray treatment, the percentage of cells in the G2/M phase appeared to be significantly increased [123]. However, the Au NP-mediated ‘cell cycle synchronization effect’ reported in the previously cited papers is being debated, since other research groups have not detected any cell cycle change after treatment with Au NPs [47, 207]. One can argue that the variation in the results between the studies can be attributed to different experimental conditions, such as the physico-chemical properties of Au NPs and the cell lines [76, 157]. A systematic and more comprehensive methodology should clearly help to identify and to quantify this effect in the near future.
When DNA is affected by physically or chemically induced defects, repair mechanisms are activated that, in principle, help the cell to recover its vital functions. However, as seen in the previous sections, the presence of Au NPs in the cells can multiply the impacts on the DNA, potentially leading to a delay or to the inhibition of the DNA repair mechanisms. Usually, counting of double-stranded DNA damage takes place immediately after (e.g. about 1 h) and at 24 h post-irradiation. If the damage persists or even increases at 24 h, the residual damage reflects an inhibition or a delay of the repair process. Chithrani et al. observed higher concentrations of γ-H2AX and 53BP1 foci at 4 and 24 h after irradiation of HeLa cells incubated with Au NPs [39], whereas Cui et al. and Butterworth et al. did not observe increase in the double-strand DNA damage immediately after irradiation (e.g. 30 min to 1 h). Instead, they detected an increase in the residual DNA damage at 24 h post-irradiation [27, 47]. Chen et al. detected a 2.02 and 1.95-fold increase in the amount of γ-H2AX foci at 2 and 4 h after irradiation of cells incubated with BSA-coated Au NPs [36]. The concentration of double-stranded DNA damage was similar at 24 h after irradiation, suggesting that Au NPs did not interfere with the repair mechanism [36]. Jain et al. also concluded that Au NPs neither increase radiation-induced double-stranded DNA damage formation nor inhibit DNA repair after irradiation of cells at clinically relevant MV X-ray energies [83]. Thus, further studies are necessary to clarify several aspects of the influence of Au NPs on the DNA repair mechanisms [76, 157].
5 Performance of Au NPs as X-Ray Computed Tomography Contrast Agents
X-ray computed tomography (CT) is a common tool in modern medicine. It is essential for the diagnosis of bone pathologies and trauma, to identify the calcification of tissues, as well as for visualizing several lung diseases [191]. In addition, the relatively short acquisition times typical of CT imaging enables a variety of cardiac and vascular diagnostic procedures [92, 101]. In the preclinical field, microCT acquires images of higher spatial resolutions than clinical CT (<50 μm), making the imaging modality an essential tool in the development of small-animal models of various pathologies [96, 191].
CT imaging is based on the attenuation of high-energy photons (typically in the range 25–140 keV) by biological tissues. In this energy range, the main photon–matter interactions taking place are the photoelectric and the Compton effects. Photons of a given energy irradiating a material, are attenuated a certain linear rate (per depth of penetration in this substance). This rate is the linear attenuation coefficient (μ), and it is proper to each compound [24]. This characteristic is intrinsic to each tissue, depending on its elemental composition. During CT acquisition, X-rays are projected at different angles across the body, and a detector collects the transmitted photons. Then, the numerical analysis allows the 3D reconstruction of images representing the attenuation of X-rays by the biological tissues, which are largely guided by their respective attenuation coefficients (μ). The differential X-ray attenuation maps are presented in Hounsfield units; these are obtained by normalizing the total μ measured in each voxel to the μ of a known reference (e.g. water) using Eq. (2) [69, 92].
The resulting map is a 3D representation of the various volumes presenting different densities in the biological system (e.g. bone, muscle, fat, etc.).
5.1 Attenuation of X-Rays by High-Z Elements
The attenuation of X-rays by the different biological tissues in the body, or by exogenous materials such as gold, is dictated by the probability of interaction of each element in function of the energy of X-rays. Attenuation of X-rays is quantified by the mass attenuation coefficient (ρ) which is often normalized to the density of the material (μ/ρ) and expressed in units of cm2/g. Figure 8 illustrates the mass attenuation coefficients of several elements, in function of the energy of X-ray photons. Compared with carbon, calcium has a higher mass attenuation coefficient at photon energies common for CT imaging (e.g. 40–100 keV). This is the main reason why bones and calcified tissues, appear strongly contrasted in CT imaging.

(data from [136])
X-ray mass attenuation coefficients for gold, iodine, calcium and carbon within the X-ray energy range used for CT imaging
In order to provide strong contrast enhancement in the blood, or for targeting certain types of diseases that are not characterized by strong differences of tissue density, it is necessary to rely on contrast agents. These are made with elements that have the capacity to strongly attenuate X-ray radiation. Any element with a high atomic number (Z) is a good X-ray attenuator and can be used as a contrast agent for CT, given that it is not too toxic for biological applications [121]. In the energy range of CT imaging, attenuation by high-Z elements mainly results from the photoelectric absorption effect. Moreover, the probability of an X-ray photon to interact with a given element varies as (Z/E)3, where Z is the atomic number and E is the photon energy [24]. Just above the binding energy of the core electrons, the absorption of photons by the atoms is significantly higher [24, 69]. These ‘jumps’ in the mass attenuation coefficient graphs (Fig. 8) are referred to as the ‘k-edges’. The presence of such discontinuities must be taken into account in the selection of a photon energy that maximizes the contrast effect. For instance, the k-edge of gold being 80.7 keV; performing a CT-imaging scan at an energy of 81 keV multiplies by at least a factor of 4, the mass attenuation coefficient compared to a scan performed at an energy of 80 keV. At 81 keV, the mass attenuation coefficient of Au is much higher than that of iodine, which is the most widely used element for the fabrication of clinical CT contrast agents nowadays.
5.2 Conventional Iodinated Contrast Agents
CT contrast agents are usually made of iodinated hydrosoluble molecules [114]. Iodine is an element that is endogenous to the body, and it strongly attenuates the photons (Fig. 8). Among most common formulations approved by the health authorities, figure monomeric or dimeric water-soluble tri-iodinated derivates of benzoic acid [106, 175]. After intravenous injection, these small molecules diffuse to all vascularized tissues through the fenestrations of blood vessels. Then, they are removed by the blood and they are eliminated mostly by the kidneys [106]. As these contrast agents have a short circulation time (e.g. 15–40 s), the images must be acquired quickly after the injection. This is a significant limitation in preclinical studies, because the small animals eliminate these products faster and the acquisition times in microCT usually take longer than with clinical CT scanners [96]. The main strategy to overcome this limitation is through the administration of multiple or long, continuous injections of the contrast agents in the small animals [96, 174]. This unfortunately raises toxicity concerns. To maintain the image quality as the contrast effect fades away, the X-ray exposure could also be increased. However, the dose of a single anatomical image is around 0.1 Gy. Higher imaging doses of 1.5 Gy have been reported for cardiac gated imaging in mice but should never exceed 6 Gy even in multiple sequential scans procedures. This threshold is considered lethal for small rodents [156].
5.3 Blood-Pool Contrast Agents
In order to prolong the contrast-enhancement effect, blood-pool contrast agents were developed [114]. Blood-pool contrast agents cannot go across the gaps between the endothelial cells of normal vessels, and therefore they remain for longer times in blood circulation, thus providing an improved time window for visualization of the vascular system. By increasing the size of contrast agents up to the 50–100 nm, renal elimination through glomerular filtration is reduced. The extended blood half-life of these molecules is also a result of a delayed recognition by the cells of the mononuclear phagocyte system (MPS), due to the use of well-selected surfactants [148]. Different methods and materials have been used to produce blood-pool contrast agents, and many of them are commercially available for preclinical research [96, 106, 175]. They are usually made of macromolecules, or of oil emulsions containing iodine-modified lipids. They can also be made of iodine-encapsulating micelles and liposomes [183]. Blood-pool CT contrast agents can also be based on inorganic NPs: bismuth-sulphide, barium, alkaline-earth metals or gold [96, 121]. Among these, Au-based nanoparticles have provided the most promising proofs-of-concepts for the development of a new generation of blood-pool contrast agents for CT imaging.
5.4 Au-Based Blood-Pool Agents
Among all elements considered for CT contrast media applications, gold shows good biocompatibility and has higher X-ray attenuation coefficients than iodine at the energy range of both CT and microCT [67, 82, 85]. As mentioned in the previous section, the photoelectric effect is inversely related to the energy of the photons (1/E3), except for discontinuities (i.e. absorption edges) close to the binding energies of the inner-shell electrons, at which the probability of interaction increases significantly [82]. For example, Au has a k-edge absorption at 80.7 keV [67, 82], thus when peak voltages (kVp) from 100 to 140 are used, the mass attenuation of Au (μ/ρ) increases up to 8.9 cm2 g−1. In comparison, the mass attenuations of C, Ca and I in the same energy range are 0.161, 0.366 and 3.51 cm2 g−1, respectively [80]. In that range, gold attenuates X-rays approximately 55 times more than C (present in soft tissues), 24 more than calcium (present in bones) and at least 2.5 times more than iodine (Fig. 8). As a consequence, in this range of energies, Au NPs can be administered at lower elemental concentrations than the iodine-based contrast agents, for a similar level of contrast enhancement [67, 137]. Au NPs also attenuate X-ray photons more efficiently by Compton scattering than soft tissues (e.g. carbon), bone (e.g. calcium and phosphorus) and iodine-based contrast agents, because of higher electron density.
In 2006, Hainfeld and colleagues were the first to report the use of Au NPs as contrast agents for vascular imaging [71]. In this study, 1.9 nm Au NPs were injected in a mouse model, and provided evidence of longer retention times and superior contrast to iodine with resolution of vessels as small as 100 μm [71] Despite high initial blood concentrations (10 mg/ml blood), no haematological nor biochemical abnormalities were detected at 11 and 30 days post-injection. Au NPs were found to be excreted by the kidneys.
Since then, several Au NPs-based formulations have been developed and used in preclinical studies [93, 98]. A variety of synthesis techniques have been employed: Turkevich [56], Brust–Schiffrin [42] and seed-mediated growth [150]. Galper and colleagues demonstrated that the maximum contrast enhancement for Au NPs dispersed in water (core diameter: 3.6 ± 1.1 nm), was achieved at 120 kVp (Fig. 9) and it was 1.9 times higher than with a commercial iodine contrast agent for an equivalent elemental concentration [67]. Jackson et al. showed that Au NPs provide better contrast over iodine at peak voltages lower than 50 kVp. However, no significant improvement was found between 70 and 90 kVp [82].

From [67], reproduced with permission from Lippincott, Williams and Wilkins
a Graph (in Hounsfield units: HU) of attenuation versus concentration of Au NP and iodine-CA scanned in air at 100 mA and 140 kV. Error bars are standard deviations; where not seen, the error bars are hidden by the data point. Attenuation of b iodine-CA and c Au NP scanned in water with a calcium phosphate matrix at various tube voltages. CT images of d 157 mM of iodine-CA and e 119 mM of Au NP (windowing, 800Y3000 HU). Data from phantom 2. Error bars omitted for clarity.
The development of fine Au NPs of well-controlled size and targeting ligands opened the possibility to improve image contrast by increasing the concentration of Au NPs in disease tissues [56]. NPs made of high-density lipoprotein (HDL) [42, 43] and low-density lipoprotein (LDL) [6] containing Au NPs also showed promising results. More specifically, Au-HDL NPs were used to specifically target and image macrophage content in atherosclerotic plaque. In this system, the targeting was mediated by the apolipoprotein A–I [42, 43]. On the other hand, Au-LDL NPs were used to target tumour associated macrophages. In this case, the targeting properties were attributed to the apolipoprotein B100 and to the EPR effect [6].
Dendrimer-entrapped Au NPs (2–4 nm diameter cores) have also been synthesized and tested in vivo [103, 145, 196]. Ultra-small Au NPs (~2.4 nm diameter core) coated with dithiolated polyaminocarboxylate (DTDTPA), and injected intravascularly in the mouse model, were found massively excreted by the urinary ways (>60% of injected initial dose after 24 h) [7]. A significant fraction of the product was also found in the faeces after 72 h (2–3% cumulative, on initial dose injected).
One of the strategies that has been suggested to increase the sensitivity of Au NP-based contrast agents is the integration of a large number of ultra-small Au NP cores (e.g. 1.9 nm in diameter such as Aurovist™) into polymeric micelles (‘gold-loaded polymeric micelles’, or GPMs) [3]. It was demonstrated that GPMs in the form of micelles containing a large number of ultra-small Au NPs (1.9 nm diam.), remain in the blood for several hours (Fig. 10a, b) and accumulate at high concentrations in tumours (Fig. 10c, d). They also show much higher liver and spleen retention compared with their ultra-small 1.9 nm diameter individual counterparts (Fig. 10d). This could be a strong limitation to their approval for clinical applications by the health authorities.

Reprinted from [3] with permission. Copyright 2014, the American Chemical Society
Blood clearance profile of gold-loaded polymeric micelles (GMPs), measured by elemental analysis (n = 3). b Serial CT coronal views of a mouse following retro-orbital injection of 200 μL of GPM solution (650 mg/kg). Coronal views of heart and liver (top) and inferior vena cava and kidneys (bottom) are shown. A very strong contrast is seen at t = 30 min. c In vivo CT images of nu/nu nude mice with HT1080 flank tumours, injected with GMPs, as well as the ultra-small Au NPs Aurovist (1.9 nm diameter NPs, also enclosed in the GMPs compound). Representative CT images in the axial plane prior to injection (precontrast) and 30 min, 24 and 48 h post-injection of GPMs (n = 3) or AuroVist (n = 3). Tumour boundaries are indicated by white arrows. d Elemental analysis in excised and dissolved organs of gold distribution at 48 h following the administration of GPMs or AuroVist. The asterisk indicates statistical significance (p < 0.05).
Overall, Au NPs used as CT contrast agents, and in particular, very small ones (e.g. ~2 nm) provide longer long blood half-lives than the small iodinated molecules conventionally used in CT imaging. Few evidences of toxic reactions in the mouse model have been found in preclinical studies until now [28, 71, 137]. The smallest Au NPs are eliminated by glomerular filtration in the kidneys within minutes. By increasing the size of the contrast agents, either by using a polymer backbone, dendrimers or a core of inorganic material, the blood half-life of the contrast agent can be longer. However, the largest NPs cannot be filtered by the kidneys and therefore represent a potential toxicity risk. Indeed, their hydrodynamic diameter is usually larger than the pore size of the glomerular wall [35, 134]. This is one of the main reasons why the majority of blood-pool contrast agents, and in particular, the ones based on particles larger than 10 nm in diameter, have not been approved for clinical applications until now.
6 Performance of Gold Nanoparticles as Radiosensitizers in Oncology
As described in the previous sections, gold has a great potential for X-ray imaging, at least in preclinical applications and thanks to the fact that is chemically inert in biological media [182]. In addition, the size and shape of Au NPs are relatively easy to control, and ligands can be readily attached to their surface [52]. In addition to imaging, Au NPs can also be used as radiosensitizing agents for the radiotherapy of different tumours [25, 44, 70, 102]. The strong attenuation of photons by high-Z materials generates secondary products (e.g. Auger electrons, characteristic X-rays and photoelectrons) that contribute to enhancing the dose deposited by the primary photons.
In radiooncology, the treatments are performed by irradiating tumours with a beam of photons at a specific energy. The photons interact with the atoms of the tissues through the particle–matter interaction mechanisms described in Sect. 3. In particular, secondary emissions of various natures (electrons and photons) are ejected from the atoms of the irradiated materials, and these account for the most important fraction of the deposited dose in the tissues [14, 89]. The secondary emissions subsequently interact with atoms present in the surrounding environment, producing cascades of ionizing events [14, 89]. Generally speaking, radiotherapy treatments that could accommodate the use of Au NPs are mainly divided in two variants: (1) external beam radiotherapy, where the radiation source is external to the patient, and which exploits a photon energy range between ~0.1 and 8 MeV; and (2) low-dose brachytherapy, in which radioactive seeds are inserted directly in the cancer tissues, for a photon energy range between 20 and 28 keV [161].
Over the recent years, several strategies have been investigated to maximize the dose enhancement generated by Au NPs distributed in tissues; simulation studies and experimental approaches have been tested (e.g. external beam radiation therapy combined to Au NPs, brachytherapy combined to Au NPs and radioactive NPs). In vivo experiments have been performed to demonstrate the therapeutic effect caused by the combination of Au NPs with various radiation sources on xenograft tumours. Here, we review the comprehensive aspect and the main outcomes related to the use of Au NPs as radiosensitizers in radiation therapy.
6.1 Main Mechanisms of Radiosensitization
By using a radiosensitizer, the sought effect is a maximization of the fraction of attenuated photons and consequently, an increase in the overall quantity of electrons cascading in the tissues. The mechanisms leading to the radiosensitization effect generated by gold atoms irradiated by photons in a biological tissue can be highlighted by a careful examination of Fig. 6a, b (mass attenuation coefficients of Au and water). First, at an energy of 20 keV, the total mass attenuation coefficients of gold and water differ by a factor of 97. This means that, for low-energy photons such as those used in low dose-rate brachytherapy, gold attenuates substantially more energy per gram than water. At 20 keV, the mass attenuation coefficient of gold is 78.83 cm2/g; it drops to 2.19 and 5.16 cm2/g at 80 and 100 keV, respectively. Therefore, the probability of interaction of photons with gold is much higher at 20 keV, where the photoelectric effect is the predominant interaction mechanism. As the photoelectric effect generates a large number of secondary electrons and photons, Au NPs radiosensitization is maximized with low-energy photons. Although these secondary emissions (photoelectrons, Auger electrons and characteristics X-rays) have low energies, they significantly contribute to dose (energy) deposited in the tissue. In terms of dose enhancement, the lower energies of the secondary products are compensated by their relative abundance in comparison to the flux of primary photons.
Energy deposition occurs when secondary electrons gradually lose their kinetic energy through many ‘collisions’ with orbital electrons present in the biological matter (Fig. 11). The collisional behaviour causes ionizations and excitations along the path of the electron, and these events can be defined as points in space where energy is transferred to the medium (in the biological tissues: mainly water). The amount of energy transferred per unit volume is defined as the energy lost by the electrons following each interaction (ΔE = E1 − E2). The electron continues to travel in the medium where it interacts with other atoms. At each collision, it transfers a small amount of energy until it comes entirely to rest (Ekin = 0).

Source The authors (Review, in press at Advanced Healthcare Materials, with permission from Wiley-Verlag)
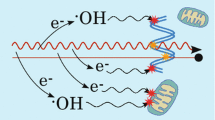
Schematic representation of the mechanisms leading to energy deposition in biological tissues. First, an incident photon (1) interacts with the Au NPs and cause the emission of a secondary electron (1). This electron travels until it collides with an orbital electron of an atom in the medium (2). At this point, a fraction of the kinetic energy (Ekin) of the electron is transferred to an orbital electron of this atom (energy deposition). This results in electron ejection (ionization) or migration to a higher shell (excitation) (3).
6.2 Au NPs as Radiosensitizers in External Beam Therapy
The use of Au NPs as radiosensitizers during radiotherapy treatments has emerged as one of the most promising applications of gold colloids. By directly injecting Au NPs in cancer tissues, followed by adequate irradiation by mid-to-high energy photons, it is possible to generate a large range of low-energy products (electrons and photons) in the immediate vicinity of the Au NPs [25]. In turn, these low-energy electrons and photons lead to dose enhancement in the surrounding tissues, to the production of reactive oxygen species inside or in the close vicinity of the cells. Overall, these products cause significant further damages to the tissues [25, 37, 76, 128, 132, 139, 151, 208]. It is generally assumed that the internalization of Au NPs brings them closer to the cell nucleuses, with potentially stronger impact on the DNA.
The first experimental evidences of the radiosensitization effect of Au NPs on tumoural tissues have been exhibited by Hainfeld et al. [72]. The experiment was conducted with external beam radiation (EBRT), where mice bearing xenograft tumour (breast) were injected intravenously with Au NPs. A delay in the tumour growth was observed for a group of animals treated with a combination of Au NPs and external radiation. Since then, many studies using external beam source have been reported in the literature.
Among the main parameters and conditions investigated, the Au NPs size and administration routes showed interesting results. Au NPs of 15–30 nm seems to be the most effective for tumour growth inhibition [204, 198]. Lately, Shi et al. showed significant differences between the concentrations of Au NPs in tumours following an intravenous or an intratumoral injection of Au NPs (i.t.: 496 ± 106 μg Au/g; i.v.: 5.27 ± 1.17 μg Au/g), 8 h after the injection [166]. Those discrepancies were reflected in the tumour growth response to radiotherapy, since a 25% difference in the inhibition was observed between the two groups.
Different combinations of treatments have also been investigated through the years to enhance the therapeutic effect of Au NPs. Radiation and hyperthermia therapies, as well as radiation and drug delivery, have showed interesting results [46, 75, 87, 99, 105, 122, 126, 144]. Targeting molecules present at the surface of Au NPs also contributed to a greater accumulation within the tumour, which helped in achieving higher therapeutic efficacy [33, 87, 149, 177, 195, 198, 202, 206]. These alternatives showed benefit gains in terms of tumour growth inhibition around 25% as compared to the ones using untargeted Au NPs.
However, due to the various conditions used in each one of these studies, it is still difficult to have definitive conclusions about the optimal conditions allowing the highest dose enhancement. Among the drawbacks related to the combination of EBRT and Au NPs, there is the energy range of the EBRT that is not suitable to benefit the radiosensitization effect, as well as the potential toxicity issues related to the Au NPs concentration that need to be injected to observe therapeutic effect.
6.3 Dose Simulations Have Confirmed a Good Match Between Au NPs and Brachytherapy Sources
One of the most important aspects related to clinical radiation therapy is treatment planning. This medical physics procedure is performed before radiotherapy treatments, and they consist in dose simulations prior to irradiation of the tissues. Several codes and programmes based on Monte Carlo calculation have been developed through the years to simulate radiation passage through biological tissues (e.g. Geant4, EGS, MCNP and PENELOPE: [38]). Early in the development of Au NPs as radiosensitizers, simulation studies have been conducted to identify the most critical parameters enabling dose enhancement. Quickly after the inception of the concept of Au NPs as radiosensitizers, it became necessary to evaluate the respective impact of size, concentration and distribution of Au NPs in vivo, when they are irradiated by fluxes of photons. Dosimetric studies were critical in identifying the more interesting combinations of parameters to be tested in experimental radiotherapy studies involving Au NPs.
The first simulation studies were based on calculating in a relatively conventional manner, the ratio between the mass energy attenuation coefficients of gold and tissue at the macroscopic scale (e.g. tumour scale). Dose enhancement factors were calculated as a quantitative value that exhibits the effect of Au NPs presence in tissue. Dose enhancement factors correspond to the ratio of the dose deposited in a tumour volume when Au NPs are present over the dose when no Au NPs are in the tissue. It was highlighted that this macroscopic approach had some drawbacks and did not represent accurately the dose enhancement phenomenon observed thorough experimental in vitro studies. A new dosimetric approach called microdosimetry was developed to bridge the experimental results and the dose enhancement deposition created by the generation of secondary electrons at the nanoscale (cellular scale). Through these studies, it was confirmed that low-energy photon sources such as brachytherapy seeds (e.g. 103Pd and 125I) offered a higher dose enhancement, as well as the benefit to use smaller Au NPs to reduce the amount of auto absorption of secondary electrons directly in the Au NPs [91, 111, 112]. These simulations offered new insights about dose deposition at the nanometric scale and confirmed the strong potential for radiooncology, of irradiating Au NPs with low-energy photons such as low dose-rate brachytherapy seeds.
6.4 Radioactive Au NPs
Historically, radioactive Au NPs have been first introduced to oncology procedures in the mid-1950s in the form of colloidal Au NPs [23, 61, 62]. However, it was highlighted that heterogeneous distribution of the solution inside the tumour influenced the efficacy of the treatment, as well as their quick excretion from the body. The size variability and the poor uniformity of Au NPs forced to abandon this strategy until recent advances in nanotechnology and materials characterization techniques (e.g. transmission electron microscopy), which helped to develop new synthesis routes that offer very controlled sizes and shapes. These advances allowed to revisit the concept of radioactive Au NPs for potential interstitial treatments.
Radioactive gold (198Au; t1/2 = 2.7 days) is a high-energy beta-emitter (Emoy = 312 keV) that also emits high-energy photons (Emoy = 412 keV). It has been revisited recently in several preclinical studies involving tumour injections in animal models (Fig. 12b, c) [17, 94, 110, 168]. Beta-emitters have been investigated in internal therapy for many years, since they can create dense damages to the DNA. However, the high-energy photons emissions of 198Au are not perfectly well suited for clinical applications. More recently, low-energy beta-emitters have been used to radiolabel Au NPs (e.g. 177Lu) [29, 30, 201]. This radiolabeling is performed via chelate of linker molecules attached at the surface of the Au NPs. Au NPs radiolabeled through chelates can lead to the cleavage of the radioisotope in vivo. Recently, a new approach based on the low-energy photon emitter 103Pd commonly used in brachytherapy has emerged in the form of core–shell Pd:Au NPs. Core–shell Au NPs incorporating radioisotope 103Pd (20–23 keV photons) have demonstrated a strong potential in delaying tumour growth (Fig. 12) [50, 110, 170]. Moreover, the combination of low-energy photon and gold offers the best conditions to benefit dose enhancement via the radiosensitization effect [111].

a Intratumoural (i.t.) administration of Au NPs in xenograft tumours (murine model; source The authors). b Tumour growth curves in a prostate cancer model injected with radioactive 198Au NPs: these results figure among the first in the recent years to have revealed the potential of radioactive Au NPs in oncology (reprinted from [168], with permission from the National Academy of Sciences of the United States of America). c Schematic representation of 103Pd:Pd@Au-PEG NPs (single radioisotope) and 103Pd:Pd@198Au:Au-PEG NPs (dual radioisotopes, represented by the enhanced corona) synthesized by a direct reduction route forming core–shell particles (reprinted from [110], with permission from Wiley). d Tumour volume follow-up of prostate cancer, after injection of ~1 mCi of 103Pd:Pd@Au-PEG NPs (reprinted from [110] with permission from Wiley)
6.5 Current Status of Au NPs as Radiosensitizers for Clinical Applications
Each of the experimental studies mentioned above showed interesting outcomes in terms of tumour growth delay. However, it is still very difficult to compare the efficacy of each one due to the various conditions employed. Every strategy has advantages and drawbacks that need to be balanced in the perspective of clinical implementation. The impact of key parameters such as the radiation source, the physico-chemical properties of Au NPs (e.g. size, concentration and functionalization), the dose or the administration route must be evaluated systematically to enable comparisons between the efficacy of each one of the Au NPs formulations.
For Au NPs to be translated into clinical procedures, a certain number of challenges must be overcome. First, the long-term retention of Au NPs into the tumours, and their excretion pathways, must be comprehensively investigated. Long-term biocompatibility issues, as well as the biodistribution and clearance routes, must be thoroughly studied. In addition to this, treatment planning and dose measurements tools currently used in medical physics must be adapted to the specific reality of Au NPs. Recent studies have clearly highlighted the importance of understanding Au NPs diffusion in tissues and the impact of this diffusion on treatment efficacy and on the dose given to the surrounding organs [21, 68, 109, 171]. It is also important to develop simulation tools for the diffusion of Au NPs that could be added to existing dose planification platforms. In conclusion, it is evident that more experiments are needed before health agencies approve Au NPs procedures in radiation therapy. Continued interdisciplinary research efforts are key to develop optimal radiosensitization, ‘nanobrachytherapy’ products and dose calculation tools that could make a wide impact in the future of oncology practice.
7 Biodistribution, Clearance and in Vivo Toxicity
7.1 Biodistribution and Clearance Routes of Gold Nanoparticles
The size, shape, surface chemistry, charge and concentration of Au NPs are determinant factors for their successful application in biomedical imaging and in radiotherapy [74, 76, 162]. These parameters influence the biodistribution, the accumulation, the diffusion and the uptake of the NPs by cells and tumours [55, 74, 77, 95, 162]. Therefore, the physico-chemical properties must be optimized according to the final application, so that the highest image quality or therapeutic efficiency could be achieved, while using the minimum concentration of Au NPs possible. Comprehensive reviews on the toxicity and biodistribution of Au NPs have been written by Dykman and Khlebstov [55, 95].
7.2 The Intravascular Injection Route
NPs can be administered through different routes and the intravenous (i.v.) has been the most common procedure reported in preclinical studies. It is not invasive, it provides a quick distribution of the NPs through the body [90], and it also opens possibilities for targeted CT imaging [33, 56, 114, 149, 150, 154, 195]. Upon their i.v. administration, Au NPs diffuse in the blood pool [77]. PEG is commonly used as a coating for Au NPs. This polymer efficiently stabilizes the particles by providing steric hindrance and surface charge modulation. The adsorption of opsonins at the surface of NPs, usually occurring very fast as soon as the nanoparticles are injected into the blood, is thereby delayed. As a consequence, it takes longer time for the mononuclear phagocytic system (MPS) cells to recognize the NPs [28]. After some hours in circulation, they end up accumulating in the liver and in the spleen [28, 71, 77, 148, 164]. Depending on surface properties and size, Au NPs can also significantly accumulate in the lungs [18] and in the kidneys [28, 71, 164]. To a lower extent, Au NPs can also be detected in the lymph nodes, in the small and large intestines, in the heart, in the skeletal muscle and in the brain [28, 164].
The effect of size on the blood clearance of NPs is clearly revealed by comparing the works performed by Hainfeld et al. [71] and Cai et al. [28]. In the first study, the authors used Au NPs with an estimated HD of 5.8 nm, while on the second work, NPs with an HD of 38 nm were used. As a consequence, 5 h after the injection, Hainfeld et al. observed that 77.5% of the injected dose (i.d.) had been eliminated by the kidneys, while Cai et al. reported that 72 h after the injection, only 6% of Au i.d. had been eliminated in the faeces. Also, Hirn et al. studied the biodistribution of negatively charged Au NPs with different core diameters (e.g. 1.4, 5, 18, 80 and 200 nm). The authors observed that the NPs of 5, 18, 80 and 200 nm were quickly eliminated from the bloodstream and accumulated mainly in the liver (91.9–96.9% of the i.d.). The 1.4 nm diameter NPs were also cleared relatively fast, and there was lower retention in the liver (e.g. 51.3%). The spleen was the second organ with the highest accumulation of Au NPs; however, the percentage of retained NPs, about 2%, was similar for all the particle sizes studied [77].
As mentioned in Sect. 5, the long blood half-life of Au NPs systems, and in particular the larger PEGylated ones, is a consequence of the inability of kidneys to excrete them. It is also due to the delayed uptake of the NPs by the macrophages residing in the organs of the MPS, such as the liver and the spleen [77, 148]. NPs with an HD smaller than 6 nm are easily excreted by the kidneys because the size is under the renal filtration threshold [148]. NPs with sizes ranging from 6 to 10 nm can still be eliminated by the kidneys, but usually, a certain retention in other organs such as liver and spleen also occurs. It is generally assumed that particles with positively charged surfaces are eliminated faster than neutral or negatively charged surfaces [117]. Finally, if the Au NPs are not eliminated relatively quickly by the kidneys, they accumulate in different organs and are assumed to be slowly eliminated by the biliary system [148].
When the nanoparticles are designed to remain in the blood for several minutes or even hours, they can reach and accumulate in tumours by the enhanced, permeability and retention (EPR) effect. In fact, tumours that grow fast present an irregular and leaky vasculature [4, 117]. For the NPs to accumulate in the tumour through the EPR effect, their size (i.e. their hydrodynamic diameter—HD), their surface charge, their hindrance (e.g. by PEGylation) and their shape must be carefully tuned to maximize the blood circulation times. The heterogeneity of blood vessels around and inside the tumours must be taken into account. Usually, the periphery of tumours is well vascularized, and therefore, the NPs can more easily accumulate in this area. However, the core of the tumour is generally less vascularized, and this effect must be taken into account when evaluating the potential of nanoparticles injected i.v. for oncology procedures [192].
Lately, there has been much debate on the efficiency, and therefore on the relevance of developing new therapeutics based on the EPR effect. In fact, an analysis of 232 studies reporting on the i.v. injection of NPs concluded that the median of NPs percentage accumulating in the tumours was 0.7% of the injected dose. Active targeting, in which biomolecules are attached at the surface of NPs to enable their binding to surface antigens, only raised the efficiency of delivery to 0.9% i.d. [192]. These observations suggest that approximately 99% of the overall doses of NPs described in the scientific literature where i.v. injections were performed in cancer xenograft animal models, accumulate non-specifically in organs, where they can possibly raise negative side effects by long-term exposition [77, 192]. Finally, the inefficient delivery of NPs to tumours would necessarily be translated into higher manufacturing costs per therapeutic dose [192].
7.3 The Intratumoral Injection Route
The percentage of Au NPs reaching tumours after i.v. injections in small-animal models have been reportedly low [192], and for this reason, alternative administration routes have been investigated. A few studies have reported on the intraperitoneal injection [32], whereas direct intratumoral injections (i.t.) are increasingly performed in the context of brachytherapy (see Fig. 12a). This appears as a promising way to improve the NP delivery efficiency in the tumour while minimizing the accumulation of Au NPs in non-specific organs [29, 33, 94, 110, 166].
The i.t. injection of Au NPs as radiosensitizers for the treatment of prostate cancer tumours, results in much lower accumulations of the product in non-specific organs (liver, spleen), compared with the i.v. route [94, 110, 166]. Chattopadhyay et al. clearly demonstrated the advantages of this approach by comparing the biodistribution of trastuzumab-111In-Au NPs injected by either the i.v. or the i.t. route in mice bearing HER-2 positive breast tumours. Quantification of organ uptake at 48 h after the injections demonstrated that i.t. administration yielded 24 times higher concentration of trastuzumab-111In-Au NPs in the tumour (29.6% i.d./g vs. 1.2% i.d./g for i.v. injection). In addition, this administration route lead to 10 times lower accumulation of the NPs in the spleen (1.8% i.d./g vs. 19.2% i.d./g for i.v. injection), and about 1.7 of the concentration in the liver (1.6% i.d./g vs. 2.7% i.d./g for i.v. injection) [34].
Shi et al. also compared the biodistribution of Au NPs injected either i.t. or i.v. In this work, tiopronin-coated Au NPs (core diameter = 2.77 ± 0.69 nm) were administered to mice bearing an HCT116 tumour (e.g. colorectal tumour) [166]. The results revealed that 8 h after i.t. or i.v. injection, tumour uptake was 94 times higher in the animals injected i.t. (496 ± 106 μg Au/g) compared with i.v.-injected ones (5.27 ± 1.17 μg Au/g).
Another work demonstrating the advantages of i.t. injection was performed by Laprise-Pelletier et al. [110]. In this study, 4 µL of radioactive Pd@Au NPs (e.g. 103Pd:Pd@Au-PEG, HDnumber-w. = 36.4 nm) suspended in alginate gel was injected directly into prostate tumours (PC3). The alginate gel was used to maintain the particles in the tumour, because this gel polymerizes in the presence of Ca2+ ions, which are naturally available in the tissues. Biodistribution data was collected after harvesting the tumour (e.g. 27–33 or 88–90 d after i.t. injection), the results showed that most of the injected NPs (e.g. >75% of the total activity) were trapped in the tumour. Amounts of NPs were found in the liver (e.g. approximately 16% of the total activity) and in the spleen (e.g. ≈3% of the total activity). Weekly inspections performed after i.t. injections, using a Geiger counter, indicated that NPs reach the liver and spleen within the first 2 weeks after the injection, and this suggests that improvements to the binding of Au NPs in the tumours must be made at the injection step. TEM observations demonstrated that ultra-small NPs (<5 nm) were the most likely to escape the tumour matrix in the first hours following i.t. injection [110].
7.4 In Vivo Toxicity Studies
To transfer Au NPs in clinical applications, either as contrast or as radiosensitizing agents, it is necessary to undertake comprehensive studies on their potential side effects should they remain in the body for several months [168]. The impact of long-term exposure of organs to various amounts of Au NPs, is studied through in vivo toxicity assays. Each new compound has unique physico-chemical properties, and these have a specific impact on the interaction with the biologic system.
Several research groups developing Au NP-based radiosensitizers have conducted in vivo toxicity assays (Table 1). Among the most commonly conducted tests figure (1) body weight monitoring; (2) organ histology (e.g. heart, liver, spleen, lung, kidney and reproductive organs); (3) complete blood count (CBC); and (4) liver and kidney function tests. A CBC examination often includes the quantification of white blood cells (WBC), red blood cell (RBC), hematocrit (HCT), mean corpuscular volume (MCV), haemoglobin (HGB), platelets (PLT), mean corpuscular haemoglobin (MCH) and mean corpuscular haemoglobin concentration (MCHC). On the other hand, liver and kidney function tests investigate the concentration of alanine aminotransferase (ALT), aspartate aminotransferase (AST), total protein (TP), albumin (ALB), alkaline phosphatase (ALP), gamma-glutamyl transferase (GGT), blood urea nitrogen (BUN), creatinine (CREA), globulin (GLOB) and total bilirubin (TB) [33, 203, 204].
Most of the studies summarized in Table 1 concluded that Au NPs do not induce significant systemic toxicity. Only two studies performed with Au NPs with an HD larger than the renal filtration threshold and which were injected intraperitoneally, detected liver damage [203, 204]. This was concluded after histologic observation of liver samples and due to alteration of the AST and RBC values for BSA-coated Au NPs [203], and because of AST and ALT abnormal results for PEG-coated Au NPs [204]. Data from recent systemic in vivo toxicity studies conducted in the context of radiosensitizing with Au NPs are listed in Table 1. These studies point to the low systemic effects of Au NPs at the concentration required for radiosensitization, and in particular if the NPs are administered intratumorally.
However, longer and more standardized studies must be performed to identify the potential impacts in the long term, of long retentions of Au NPs in the liver and in other organs. In addition, assays to detect the effects of oxidative stress in the liver (e.g. lipid peroxidation, protein carbonylation and inflammatory markers such as TNF-α, IL-1ß, IL-6 and IL-10) [118] and abnormal gene expression should also be performed to clarify the interaction between the retained NPs and the organs [203]. Effects of long-term exposure to nanoparticles must be collected in the critical organs, in order to clearly and precisely identify the toxicity limits for guaranteeing a safe use of Au NPs in the human body.
8 Perspectives: Current Status of Au NPs in Clinical Trials
The clinical use of NPs is still scarce, and most of the approved systems are made of protein–drug conjugates or liposomes [49, 163]. Regarding inorganic nanoparticles, only iron-based ones have received approval so far [12]. The approved compounds are used for the treatment of cancer, for fungal infections, for macular degeneration, or as imaging agents, vaccines and anaesthetics [12].
Several Au NP systems have been investigated in clinical studies for cancer treatment. A list of current and past trials can be found on the U.S. National Library of Medicine website (www.clinicaltrials.gov), as well as in several recent reviews in [11]. Examples include tumour necrosis factor-bound colloidal gold (CYT-6091, NCT00436410 and NCT00356980), which has been evaluated in Phase 0 and I studies for treatment of primary, advanced and metastatic solid tumours [107, 115, 135]. PEGylated silica-gold nanoshells (AuroLase®) have also been tested for photothermal therapy in head and neck tumours (NCT00848042). Technical and biological challenges associated with these Au NPs systems hindered their clinical approval. In particular, relatively weak tumour targeting values were found following systemic administration [11]. To date, no clinical study has evaluated Au NPs as radiosensitization agents for cancer treatment; however, the fact that other nanoparticle systems (e.g. gadolinium and hafnium oxide-based) are being tested as radiosensitizers in Phase I, II and III oncology studies [45], opens the door to more clinical investigations in the field, including with Au NPs.
To reach clinical trials, more comprehensive studies must be performed at the preclinical stage on a per-compound basis. These studies must include (1) a thorough physico-chemical characterization of Au NPs before and after exposure to biological media; (2) a reproducible demonstration of the therapeutic efficiency of Au NPs in vivo and of an improved contrast enhancement; (3) a detailed evaluation of systemic toxicity; (4) a comprehensive assessment of pro-inflammatory and immunity response for each one of these compounds; and (5) a clear demonstration that these new compounds can be systematically produced with the utmost level of precision under good manufacturing practices.
9 Conclusion
As demonstrated in various preclinical studies, gold nanoparticles (Au NPs) have emerged as a promising platform for imaging and therapy. In this chapter, we introduced the main concepts underlying the potential of Au NPs as imaging agents for CT, as well as radiosensitizers for cancer therapy. The main synthesis routes for the production and functionalization of Au NPs were presented, followed by a detailed description of the mechanisms of interaction occurring between high-energy photons and Au NPs. The main developments in the field of Au NPs as contrasts agents for CT (X-ray imaging), as well as Au NPs radiosensitizers for radiotherapy (medical physics in oncology), were also described. More clinical trials will be necessary in the near future in order to circumscribe precisely the types of cancer as well as their grades that could be best treated with such technologies. Prior to this, the biodistribution and clearance data, the toxicity thresholds, the pro-inflammatory and the immune response characteristics of each new compound will have to be revealed through comprehensive in vitro and preclinical studies. In resume, Au NPs could provide more efficient imaging agents, as well as radiosensitizing products making possible the use of much lower doses of ionizing radiation in radiation oncology treatments.
References
Abel B, Buck U, Sobolewski AL, Domcke W (2011) On the nature and signatures of the solvated electron in water. Phys Chem Chem Phys 14:22–34. https://doi.org/10.1039/C1CP21803D
Akolkar R, Sankaran RM (2013) Charge transfer processes at the interface between plasmas and liquids. J Vac Sci Technol, A 31:050811. https://doi.org/10.1116/1.4810786
Al Zaki A, Joh D, Cheng Z, De Barros ALB, Kao G, Dorsey J, Tsourkas A (2014) Gold-loaded polymeric micelles for computed tomography-guided radiation therapy treatment and radiosensitization. ACS Nano 8:104–112. https://doi.org/10.1021/nn405701q
Albanese A, Tang PS, Chan WCW (2012) The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng 14:1–16. https://doi.org/10.1146/annurev-bioeng-071811-150124
Alkilany AM, Nagaria PK, Hexel CR, Shaw TJ, Murphy CJ, Wyatt MD (2009) Cellular uptake and cytotoxicity of gold nanorods: molecular origin of cytotoxicity and surface effects. Small 5:701–708. https://doi.org/10.1002/smll.200801546
Allijn IE et al (2013) Gold nanocrystal labeling allows low-density lipoprotein imaging from the subcellular to macroscopic level. ACS Nano 7:9761–9770. https://doi.org/10.1021/nn403258w
Alric C et al (2013) The biodistribution of gold nanoparticles designed for renal clearance. Nanoscale 5:5930–5939. https://doi.org/10.1039/c3nr00012e
Amendola V, Meneghetti M (2009) Laser ablation synthesis in solution and size manipulation of noble metal nanoparticles. Phys Chem Chem Phys 11:3805. https://doi.org/10.1039/b900654k
Amendola V, Meneghetti M (2013) What controls the composition and the structure of nanomaterials generated by laser ablation in liquid solution? Phys Chem Chem Phys 15:3027. https://doi.org/10.1039/c2cp42895d
Amendola V, Polizzi S, Meneghetti M (2006) Laser ablation synthesis of gold nanoparticles in organic solvents. J Phys Chem B 110:7232–7237. https://doi.org/10.1021/jp0605092
Anselmo A, Mitragotri S (2015) A review of clinical translation of inorganic nanoparticles. AAPS J 17:1041–1054. https://doi.org/10.1208/s12248-015-9780-2
Anselmo AC, Mitragotri S (2016) Nanoparticles in the clinic. Bioeng Transl Med 1:10–29. https://doi.org/10.1038/520609a
Arvizo RR, Bhattacharyya S, Kudgus RA, Giri K, Bhattacharya R, Mukherjee P (2012) Intrinsic therapeutic applications of noble metal nanoparticles: past, present and future. Chem Soc Rev 41:2943–2970. https://doi.org/10.1039/c2cs15355f
Attix FH (1986) Introduction to radiological physics and radiation dosimetry. Wiley, New York
Axiak-Bechtel SM et al (2014) Gum arabic-coated radioactive gold nanoparticles cause no short-term local or systemic toxicity in the clinically relevant canine model of prostate cancer. IJN 9:5001–5011. https://doi.org/10.2147/ijn.s67333
Bednar N, Matović J, Stojanović G (2013) Properties of surface dielectric barrier discharge plasma generator for fabrication of nanomaterials. J Electrostat 71:1068–1075. https://doi.org/10.1016/j.elstat.2013.10.010
Black KCL et al (2014) Radioactive 198Au-doped nanostructures with different shapes for in vivo analyses of their biodistribution tumor uptake, and intratumoral distribution. ACS Nano 8:4385–4394
Boote E et al (2010) Gold nanoparticle contrast in a phantom and juvenile swine. Acad Radiol 17:410–417. https://doi.org/10.1016/j.acra.2010.01.006
Bouchard M et al (2015) Rapid nucleation of iron oxide nanoclusters in aqueous solution by plasma electrochemistry. Langmuir 31:7633–7643. https://doi.org/10.1021/acs.langmuir.5b01235
Bouchard M, Laprise-Pelletier M, Turgeon S, Fortin M-A (2017) Efficient and rapid synthesis of radioactive gold nanoparticles by dielectric barrier discharge. Part Part Syst Charact 34:1600231. https://doi.org/10.1002/ppsc.201600231
Brivio D, Nguyen PL, Sajo E, Ngwa W, Zygmanski P (2017) A Monte Carlo study of I-125 prostate brachytherapy with gold nanoparticles: dose enhancement with simultaneous rectal dose sparing via radiation shielding. Phys Med Biol 62:1935–1948
Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R (1994) Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid–liquid system. J Chem Soc, Chem Commun:801–802. https://doi.org/10.1039/c39940000801
Bulkley GJ, O’Conor VJ (1959) Treatment of carcinoma of the prostate by interstitial irradiation with radioactive gold. Experimental and clinical studies. Trans Am Assoc Genito-Urin Surg 51:126–133
Bushberg JT, Seibert JA, Leidholdt EM, Boone JM (2011) The essential physics of medical imaging. Wolters Kluwer Health
Butterworth KT, McMahon SJ, Currell FJ, Prise KM (2012) Physical basis and biological mechanisms of gold nanoparticle radiosensitization. Nanoscale 4:4830–4838. https://doi.org/10.1039/c2nr31227a
Butterworth KT, McMahon SJ, Taggart LE, Prise KM (2013) Radiosensitization by gold nanoparticles: effective at megavoltage energies and potential role of oxidative stress. Transl Cancer Res 2
Butterworth KT et al (2016) Preclinical evaluation of gold-DTDTPA nanoparticles as theranostic agents in prostate cancer radiotherapy. Nanomedicine (Lond) 11:2035–2047. https://doi.org/10.2217/nnm-2016-0062
Cai QY et al (2007) Colloidal gold nanoparticles as a blood-pool contrast agent for X-ray computed tomography in mice. Invest Radiol 42:797–806. https://doi.org/10.1097/RLI.0b013e31811ecdcd
Cai Z, Chattopadhyay N, Yang K, Kwon YL, Yook S, Pignol J-P, Reilly RM (2016) 111In-labeled trastuzumab-modified gold nanoparticles are cytotoxic in vitro to HER2-positive breast cancer cells and arrest tumor growth in vivo in athymic mice after intratumoral injection. Nucl Med Biol 43:818–826. https://doi.org/10.1016/j.nucmedbio.2016.08.009
Cai Z, Yook S, Lu Y, Bergstrom D, Winnik MA, Pignol J-P, Reilly RM (2017) Local radiation treatment of HER2-positive breast cancer using trastuzumab-modified gold nanoparticles labeled with 177Lu. Pharm Res 34:579–590. https://doi.org/10.1007/s11095-016-2082-2
Chanda N et al (2010a) Radioactive gold nanoparticles in cancer therapy: therapeutic efficacy studies of GA-198AuNP nanoconstruct in prostate tumor-bearing mice. Nanomedicine 6:201–209. https://doi.org/10.1016/j.nano.2009.11.001
Chanda N et al (2010b) Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. Proc Natl Acad Sci 107:8760–8765. https://doi.org/10.1073/pnas.1002143107
Chattopadhyay N et al (2012) Role of antibody-mediated tumor targeting and route of administration in nanoparticle tumor accumulation in vivo. Mol Pharm 9:2168–2179. https://doi.org/10.1021/mp300016p
Chattopadhyay N, Cai Z, Kwon YL, Lechtman E, Pignol J-P, Reilly RM (2013) Molecularly targeted gold nanoparticles enhance the radiation response of breast cancer cells and tumor xenografts to X-radiation. Breast Cancer Res Treat 137:81–91. https://doi.org/10.1007/s10549-012-2338-4
Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK (2011) Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng 2:281–298. https://doi.org/10.1146/annurev-chembioeng-061010-114300
Chen N, Yang W, Bao Y, Xu H, Qin S, Tu Y (2015) BSA capped Au nanoparticle as an efficient sensitizer for glioblastoma tumor radiation therapy. RSC Adv 5:40514–40520. https://doi.org/10.1039/c5ra04013b
Cheng NN, Starkewolf Z, Davidson RA, Sharmah A, Lee C, Lien J, Guo T (2012) Chemical enhancement by nanomaterials under X-ray irradiation. J Am Chem Soc 134:1950–1953. https://doi.org/10.1021/ja210239k
Chetty I et al (2007) Report of the AAPM Task Group No. 105: issues associated with clinical implementation of Monte Carlo-based photon and electron external beam treatment planning. Med Phys 34:4818–4853
Chithrani DB et al (2010) Gold nanoparticles as radiation sensitizers in cancer therapy. Radiat Res 173:719–728. https://doi.org/10.1667/RR1984.1
Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG, Frangioni JV (2010) Design considerations for tumour-targeted nanoparticles. Nat Nanotechnol 5:42–47. https://doi.org/10.1038/nnano.2009.314
Cobley CM, Chen JY, Cho EC, Wang LV, Xia YN (2011) Gold nanostructures: a class of multifunctional materials for biomedical applications. Chem Soc Rev 40:44–56. https://doi.org/10.1039/b821763g
Cormode DP et al (2008) Nanocrystal core high-density lipoproteins: a multimodality contrast agent platform. Nano Lett 8:3715–3723. https://doi.org/10.1021/nl801958b
Cormode DP et al (2010) Atherosclerotic plaque composition: analysis with multicolor CT and targeted gold nanoparticles. Radiology 256:774–782. https://doi.org/10.1148/radiol.10092473
Coulter JA, Hyland WB, Nicol J, Currell FJ (2013) Radiosensitising nanoparticles as novel cancer therapeutics—pipe dream or realistic prospect? Clin Oncol 25:593–603
Cui L, Her S, Borst GR, Bristow RG, Jaffray DA, Allen C (2017) Radiosensitization by gold nanoparticles: will they ever make it to the clinic? Radiother Oncol 124:344–356. https://doi.org/10.1016/j.radonc.2017.07.007
Cui L et al (2014) Hypoxia and cellular localization influence the radiosensitizing effect of gold nanoparticles (AuNPs) in breast cancer cells. Radiat Res 182:475–488. https://doi.org/10.1667/RR13642.1
Cui L et al (2017) Significant radiation enhancement effects by gold nanoparticles in combination with cisplatin in triple negative breast cancer cells and tumor xenografts. Radiat Res 187:147–160
Daniel MC, Astruc D (2004) Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem Rev 104:293–346. https://doi.org/10.1021/cr030698+
Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG, Searson PC (2014) State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Controlled Release 187:133–144. https://doi.org/10.1016/j.jconrel.2014.05.036
Djoumessi D, Laprise-Pelletier M, Chevallier P, Lagueux J, Cote MF, Fortin MA (2015) Rapid, one-pot procedure to synthesise 103Pd:Pd@Au nanoparticles en route for radiosensitisation and radiotherapeutic applications. J Mater Chem B 3:2192–2205. https://doi.org/10.1039/c4tb01663g
Doane TL, Burda C (2012) The unique role of nanoparticles in nanomedicine: imaging, drug delivery and therapy. Chem Soc Rev 41:2885–2911. https://doi.org/10.1039/c2cs15260f
Dreaden EC, Alkilany AM, Huang X, Murphy CJ, El-Sayed MA (2012) The golden age: gold nanoparticles for biomedicine. Chem Soc Rev 41:2740–2779. https://doi.org/10.1039/c1cs15237h
Dreaden EC, Mackey MA, Huang X, Kang B, El-Sayed MA (2011) Beating cancer in multiple ways using nanogold. Chem Soc Rev 40:3391. https://doi.org/10.1039/c0cs00180e
Duong Q, Tan Y, Corey J, Anz S, Sun P (2015) Mechanism of the transfer of AuCl4− and TOA+ ions across the liquid/liquid interface. J Phys Chem C 119:10365–10369. https://doi.org/10.1021/jp512909t
Dykman L, Khlebtsov N (2012) Gold nanoparticles in biomedical applications: recent advances and perspectives. Chem Soc Rev 41:2256–2282. https://doi.org/10.1039/c1cs15166e
Eck W, Nicholson AI, Zentgraf H, Semmler W, Bartling S (2010) Anti-CD4-targeted gold nanoparticles induce specific contrast enhancement of peripheral lymph nodes in X-ray computed tomography of live mice. Nano Lett 10:2318–2322. https://doi.org/10.1021/nl101019s
Eustis S, El-Sayed MA (2006) Why gold nanoparticles are more precious than pretty gold: noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem Soc Rev 35:209–217. https://doi.org/10.1039/b514191e
Faraday M (1857) X. The Bakerian Lecture.—Experimental relations of gold (and other metals) to light. Philos Trans R Soc Lond 147:145–181
Ferrari M (2005) Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer 5:161–171. https://doi.org/10.1038/nrc1566
Fievet F, Lagier JP, Blin B, Beaudoin B, Figlarz M (1989) Homogeneous and heterogeneous nucleations in the polyol process for the preparation of micron and submicron size metal particles. Solid State Ionics 32–33(Part 1):198–205. https://doi.org/10.1016/0167-2738(89)90222-1
Flocks RH, Kerr HD, Elkins HB, Culp D (1952) Treatment of carcinoma of the prostate by interstitial radiation with radio-active gold (Au 198): a preliminary report. J Urol 68:510–522
Flocks RH, Kerr HD, Elkins HB, Culp DA (1954) The treatment of carcinoma of the prostate by interstitial radiation with radioactive gold (Au198): a follow-up report. J Urol 71:628–633
Fojtik A, Henglein A (1993) Laser ablation of films and suspended particles in a solvent: formation of cluster and colloid solutions. Ber Bunsenges Phys Chem:252–254
Fratoddi I, Venditti I, Cametti C, Russo MV (2015) How toxic are gold nanoparticles? The state-of-the-art. Nano Res 8:1771–1799. https://doi.org/10.1007/s12274-014-0697-3
Frens G (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature 241:20–22. https://doi.org/10.1038/10.1038/physci241020a0
Furuya K, Hirowatari Y, Ishioka T, Harata A (2007) Protective agent-free preparation of gold nanoplates and nanorods in aqueous HAuCl4 solutions using gas–liquid interface discharge. Chem Lett 36:1088–1089. https://doi.org/10.1246/cl.2007.1088
Galper MW et al (2012) Effect of computed tomography scanning parameters on gold nanoparticle and iodine contrast. Invest Radiol 47:475–481. https://doi.org/10.1097/RLI.0b013e3182562ab9
Ghorbani M, Bakhshabadi M, Golshan A, Knaup C (2013) Dose enhancement by various nanoparticles in prostate brachytherapy. Australas Phys Eng Sci Med 36:431–440
Gore JC, Yankeelov TE, Peterson TE, Avison MJ (2009) Molecular imaging without radiopharmaceuticals? J Nucl Med 50:999–1007. https://doi.org/10.2967/jnumed.108.059576
Hainfeld J, Smilowitz H, O’Connor M, Dilmanian F, Slatkin D, Oconnor M (2013) Gold nanoparticle imaging and radiotherapy of brain tumors in mice. Nanomed: Nanotechnol Biol Med 8:1601–1609
Hainfeld JF, Slatkin DN, Focella TM, Smilowitz HM (2006) Gold nanoparticles: a new X-ray contrast agent. Br J Radiol 79:248–253. https://doi.org/10.1259/bjr/13169882
Hainfeld JF, Slatkin DN, Smilowitz HM (2004) The use of gold nanoparticles to enhance radiotherapy in mice. Phys Med Biol 49:N309–N315. https://doi.org/10.1088/0031-9155/49/18/n03
Hall EJ, Giaccia AJ (2006) Radiobiology for the radiologist. Lippincott Williams & Wilkins
Haume K et al (2016) Gold nanoparticles for cancer radiotherapy: a review. Cancer Nanotechnol 7:8. https://doi.org/10.1186/s12645-016-0021-x
Hazkani I et al (2017) Can molecular profiling enhance radiotherapy? Impact of personalized targeted gold nanoparticles on radiosensitivity and imaging of adenoid cystic carcinoma. Theranostics 7:3962–3971. https://doi.org/10.7150/thno.19615
Her S, Jaffray DA, Allen C (2017) Gold nanoparticles for applications in cancer radiotherapy: mechanisms and recent advancements. Adv Drug Deliv Rev 109:84–101. https://doi.org/10.1016/j.addr.2015.12.012
Hirn S et al (2011) Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur J Pharm Biopharm 77:407–416. https://doi.org/10.1016/j.ejpb.2010.12.029
Hu M et al (2006) Gold nanostructures: engineering their plasmonic properties for biomedical applications. Chem Soc Rev 35:1084. https://doi.org/10.1039/b517615h
Huang X, Li Y, Zhong X (2014) Effect of experimental conditions on size control of Au nanoparticles synthesized by atmospheric microplasma electrochemistry. Nanoscale Res Lett 9:572. https://doi.org/10.1186/1556-276X-9-572
Hubbell JH, Seltzer SM. Tables of X-Ray mass attenuation coefficients and mass energy-absorption coefficients from 1 keV to 20 MeV for elements Z = 1 to 92 and 48 additional substances of dosimetric interest, May 1996 edn
Itina TE (2011) On nanoparticle formation by laser ablation in liquids. J Phys Chem C 115:5044–5048. https://doi.org/10.1021/jp1090944
Jackson PA, Rahman WNWA, Wong CJ, Ackerly T, Geso M (2010) Potential dependent superiority of gold nanoparticles in comparison to iodinated contrast agents. Eur J Radiol 75:104–109. https://doi.org/10.1016/j.ejrad.2009.03.057
Jain S et al (2011) Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int J Radiat Oncol Biol Phys 79:531–539. https://doi.org/10.1016/j.ijrobp.2010.08.044
Jain S, Hirst DG, O’Sullivan JM (2012) Gold nanoparticles as novel agents for cancer therapy. Br J Radiol 85:101–113. https://doi.org/10.1259/bjr/59448833
Jakhmola A (2012) Inorganic nanoparticles based contrast agents for X-ray computed tomography. Adv Healthc Mater 1:413–431
Jana NR, Gearheart L, Murphy CJ (2001) Evidence for seed-mediated nucleation in the chemical reduction of gold salts to gold nanoparticles. Chem Mater 13:2313–2322. https://doi.org/10.1021/cm000662n
Jeong S-Y, Park S-J, Yoon SM, Jung J, Woo HN, Yi SL (2009) Systemic delivery and preclinical evaluation of Au nanoparticle containing beta-lapachone for radiosensitization. J Controlled Release 139. https://doi.org/10.1016/j.jconrel.2009.07.007
Jeynes JCG, Merchant MJ, Spindler A, Wera AC, Kirkby KJ (2014) Investigation of gold nanoparticle radiosensitization mechanisms using a free radical scavenger and protons of different energies. Phys Med Biol 59:6431
Johns HE, Cunningham JR (1983) The physics of radiology. Charles C. Thomas
Johnston HJ, Hutchison G, Christensen FM, Peters S, Hankin S, Stone V (2010) A review of the in vivo and in vitro toxicity of silver and gold particulates: particle attributes and biological mechanisms responsible for the observed toxicity. Crit Rev Toxicol 40:328–346. https://doi.org/10.3109/10408440903453074
Jones BL, Krishnan S, Cho SH (2010) Estimation of microscopic dose enhancement factor around gold nanoparticles by Monte Carlo calculations. Med Phys 37:3809–3816. https://doi.org/10.1118/1.3455703
Kachelriess M (2006) Clinical X-ray computed tomography. In: Schlegel W, Bortfeld T, Grosu A-L (eds) New technologies in radiation oncology. Medical radiology. Springer, Berlin, pp 41–80. https://doi.org/10.1007/3-540-29999-8_7
Kattumuri V (2007) Gum arabic as a phytochemical construct for the stabilization of gold nanoparticles: in vivo pharmacokinetics and X-ray-contrast-imaging studies. Small 3:333–341
Khan MK et al (2008) Fabrication of {198Au0} radioactive composite nanodevices and their use for nanobrachytherapy. Nanomed Nanotechnol Biol Med 4:57–69. https://doi.org/10.1016/j.nano.2007.11.005
Khlebtsov N, Dykman L (2011) Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem Soc Rev 40:1647–1671
Kiessling F, Pichler BJ (2011) Small animal imaging: basics and practical guide. Springer, Berlin
Kim CK, Ghosh P, Rotello VM (2009) Multimodal drug delivery using gold nanoparticles. Nanoscale 1:61–67. https://doi.org/10.1039/b9nr00112c
Kim D, Park S, Lee JH, Jeong YY, Jon S (2007) Antibiofouling polymer-coated gold nanoparticles as a contrast agent for in vivo X-ray computed tomography imaging. J Am Chem Soc 129:7661–7665. https://doi.org/10.1021/ja071471p
Kim K et al (2016) Doxorubicin/gold-loaded core/shell nanoparticles for combination therapy to treat cancer through the enhanced tumor targeting. J Controlled Release 228:141–149. https://doi.org/10.1016/j.jconrel.2016.03.009
Kim ST, Chompoosor A, Yeh YC, Agasti SS, Solfiell DJ, Rotello VM (2012) Dendronized gold nanoparticles for siRNA delivery. Small 8:3253–3256. https://doi.org/10.1002/smll.201201141
Kircher MF, Willmann JK (2012) Molecular body imaging: MR imaging, CT, and US. Part I. Principles. Radiology 263:633–643. https://doi.org/10.1148/radiol.12102394
Kobayashi K, Usami N, Porcel E, Lacombe S, Le Sech C (2010) Enhancement of radiation effect by heavy elements. Mutat Res 704:123–131. https://doi.org/10.1016/j.mrrev.2010.01.002
Kojima C, Umeda Y, Ogawa M, Harada A, Magata Y, Kono K (2010) X-ray computed tomography contrast agents prepared by seeded growth of gold nanoparticles in PEGylated dendrimer. Nanotechnology 21:245104. https://doi.org/10.1088/0957-4484/21/24/245104
Koo IG, Lee MS, Shim JH, Ahn JH, Lee WM (2005) Platinum nanoparticles prepared by a plasma-chemical reduction method. J Mater Chem 15:4125–4128. https://doi.org/10.1039/B508420B
Koonce NA et al (2015) Combination of gold nanoparticle-conjugated tumor necrosis factor-α and radiation therapy results in a synergistic antitumor response in murine carcinoma models. Int J Radiat Oncol Biol Phys 93:588–596. https://doi.org/10.1016/j.ijrobp.2015.07.2275
Krause W (1999) Delivery of diagnostic agents in computed tomography. Adv Drug Deliv Rev 37:159–173. https://doi.org/10.1016/S0169-409X(98)00105-7
Kumar A, Zhang X, Liang X-J (2013) Gold nanoparticles: emerging paradigm for targeted drug delivery system. Biotechnol Adv 31:593–606. https://doi.org/10.1016/j.biotechadv.2012.10.002
Kuwahata H, Mikami I (2014) Generation of nitric acid and nitrous acid in distilled water irradiated with atmospheric-pressure plasma jet. e-J Surf Sci Nanotechnol 12:410–413. https://doi.org/10.1380/ejssnt.2014.410
Lai P, Lechtman E, Mashouf S, Pignol J-P, Reilly RM (2016) Depot system for controlled release of gold nanoparticles with precise intratumoral placement by permanent brachytherapy seed implantation (PSI) techniques. Int J Pharm 515:729–739. https://doi.org/10.1016/j.ijpharm.2016.11.001
Laprise-Pelletier M, Lagueux J, Côté M-F, Lagrange T, Fortin M-A (2017) Low-dose prostate cancer brachytherapy with radioactive palladium-gold nanoparticles. Adv Healthc Mater 6:1601120. https://doi.org/10.1002/adhm.201601120
Lechtman E, Chattopadhyay N, Cai Z, Mashouf S, Reilly R, Pignol JP (2011) Implications on clinical scenario of gold nanoparticle radiosensitization in regards to photon energy, nanoparticle size, concentration and location. Phys Med Biol 56:4631–4647. https://doi.org/10.1088/0031-9155/56/15/001
Leung MK, Chow JC, Chithrani BD, Lee MJ, Oms B, Jaffray DA (2011) Irradiation of gold nanoparticles by x-rays: Monte Carlo simulation of dose enhancements and the spatial properties of the secondary electrons production. Med Phys 38:624–631
Lewinski N (2008) Cytotoxicity of nanoparticles. Small 4:26–49
Li X, Anton N, Zuber G, Vandamme T (2014) Contrast agents for preclinical targeted X-ray imaging. Adv Drug Deliv Rev 76:116–133. https://doi.org/10.1016/j.addr.2014.07.013
Libutti SK et al (2010) Phase I and pharmacokinetic studies of CYT-6091, a novel PEGylated colloidal gold-rhTNF nanomedicine. Clin Cancer Res: Off J Am Assoc Can Res 16:6139–6149. https://doi.org/10.1158/1078-0432.CCR-10-0978
Liu R, Wang Y, Yuan Q, An D, Li J, Gao X (2014) The Au clusters induce tumor cell apoptosis via specifically targeting thioredoxin reductase 1 (TrxR1) and suppressing its activity. Chem Commun (Camb) 50:10687–10690. https://doi.org/10.1039/c4cc03320e
Longmire M, Choyke PL, Kobayashi H (2008) Clearance properties of nano-sized particles and molecules as imaging agents: consideration and caveats. Nanomedicine 3. https://doi.org/10.2217/17435889.3.5.703
Lopez-Chaves C, Soto-Alvaredo J, Montes-Bayon M, Bettmer J, Llopis J, Sanchez-Gonzalez C (2017) Gold nanoparticles: distribution, bioaccumulation and toxicity. In vitro and in vivo studies. Nanomedicine. https://doi.org/10.1016/j.nano.2017.08.011
Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM (2005) Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem Rev 105:1103–1170. https://doi.org/10.1021/cr0300789
Lu F, Doane TL, Zhu JJ, Burda C (2012) Gold nanoparticles for diagnostic sensing and therapy. Inorg Chim Acta 393:142–153. https://doi.org/10.1016/j.ica.2012.05.038
Lusic H, Grinstaff MW (2013) X-ray-computed tomography contrast agents. Chem Rev 113:1641–1666. https://doi.org/10.1021/cr200358s
Ma N et al (2016) Enhanced radiosensitization of gold nanospikes via hyperthermia in combined cancer radiation and photothermal therapy. ACS Appl Mater Interfaces 8:28480–28494. https://doi.org/10.1021/acsami.6b10132
Ma N et al (2017) Shape-dependent radiosensitization effect of gold nanostructures in cancer radiotherapy: comparison of gold nanoparticles, nanospikes, and nanorods. ACS Appl Mater Interfaces 9:13037–13048. https://doi.org/10.1021/acsami.7b01112
Mafune F, Kohno J, Takeda Y, Kondow T, Sawabe H (2001) Formation of gold nanoparticles by laser ablation in aqueous solution of surfactant. J Phys Chem B 105:5114–5120. https://doi.org/10.1021/jp0037091
Mariotti D, Patel J, Švrček V, Maguire P (2012) Plasma-liquid interactions at atmospheric pressure for nanomaterials synthesis and surface engineering. Plasma Processes Polym 9:1074–1085. https://doi.org/10.1002/ppap.201200007
Masood R et al (2012) Gold nanorod–sphingosine kinase siRNA nanocomplexes: a novel therapeutic tool for potent radiosensitization of head and neck cancer. Integr Biol 4:132–141
McKenna J, Patel J, Mitra S, Soin N, Švrček V, Maguire P, Mariotti D (2011) Synthesis and surface engineering of nanomaterials by atmospheric-pressure microplasmas. Eur Phys J Appl Phys 56. https://doi.org/10.1051/epjap/2011110203
McMahon SJ, Mendenhall MH, Jain S, Currell F (2008) Radiotherapy in the presence of contrast agents: a general figure of merit and its application to gold nanoparticles. Phys Med Biol 53:5635–5651. https://doi.org/10.1088/0031-9155/53/20/005
McQuaid HN et al (2016) Imaging and radiation effects of gold nanoparticles in tumour cells. Sci Rep 6:19442. https://doi.org/10.1038/srep19442
Merk V, Rehbock C, Becker F, Hagemann U, Nienhaus H, Barcikowski S (2014) In situ non-DLVO stabilization of surfactant-free, plasmonic gold nanoparticles: effect of Hofmeister’s anions. Langmuir 30:4213–4222. https://doi.org/10.1021/la404556a
Mieszawska AJ, Mulder WJM, Fayad ZA, Cormode DP (2013) Multifunctional gold nanoparticles for diagnosis and therapy of disease. Mol Pharm 10:831–847. https://doi.org/10.1021/mp3005885
Misawa M, Takahashi J (2011) Generation of reactive oxygen species induced by gold nanoparticles under X-ray and UV Irradiations. Nanomed: Nanotechnol Biol Med 7:604–614. https://doi.org/10.1016/j.nano.2011.01.014
Moeendarbari S et al (2016) Theranostic nanoseeds for efficacious internal radiation therapy of unresectable solid tumors. Sci Rep 6:20614. https://doi.org/10.1038/srep20614
Moghimi SM, Hunter AC, Andresen TL (2012) Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu Rev Pharmacol Toxicol 52:481–503. https://doi.org/10.1146/annurev-pharmtox-010611-134623
Movia D et al (2014) A safe-by-design approach to the development of gold nanoboxes as carriers for internalization into cancer cells. Biomaterials 35:2543–2557. https://doi.org/10.1016/j.biomaterials.2013.12.057
National Institute of Standards and Technology. http://www.nist.gov/pml/data/xraycoef. Accessed 17 Sept 2017
Nebuloni L, Kuhn GA, Muller R (2013) A comparative analysis of water-soluble and blood-pool contrast agents for in vivo vascular imaging with micro-CT. Acad Radiol 20:1247–1255. https://doi.org/10.1016/j.acra.2013.06.003
Neddersen J, Chumanov G, Cotton TM (1993) Laser ablation of metals: a new method for preparing sers active colloids. Appl Spectrosc 47:1959–1964. https://doi.org/10.1366/0003702934066460
Nel A, Xia T, Mädler L, Li N (2006) Toxic potential of materials at the nanolevel. Science 311:622
Neouze M-A, Schubert U (2008) Surface modification and functionalization of metal and metal oxide nanoparticles by organic ligands. Monatsh Chem 139:183–195. https://doi.org/10.1007/s00706-007-0775-2
O’Brien MN, Jones MR, Brown KA, Mirkin CA (2014) Universal noble metal nanoparticle seeds realized through iterative reductive growth and oxidative dissolution reactions. J Am Chem Soc 136:7603–7606. https://doi.org/10.1021/ja503509k
Otsuka H, Nagasaki Y, Kataoka K (2003) PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev 55:403–419. https://doi.org/10.1016/s0169-409x(02)00226-0
Pan X, Cloutier P, Hunting D, Sanche L (2003) Dissociative electron attachment to DNA. Phys Rev Lett 90:208102. https://doi.org/10.1103/PhysRevLett.90.208102
Park J et al (2015) Multifunctional hollow gold nanoparticles designed for triple combination therapy and CT imaging. J Controlled Release 207:77–85. https://doi.org/10.1016/j.jconrel.2015.04.007
Peng C et al (2012) PEGylated dendrimer-entrapped gold nanoparticles for in vivo blood pool and tumor imaging by computed tomography. Biomaterials 33:1107–1119. https://doi.org/10.1016/j.biomaterials.2011.10.052
Perala SRK, Kumar S (2013) On the mechanism of metal nanoparticle synthesis in the Brust-Schiffrin method. Langmuir 29:9863–9873. https://doi.org/10.1021/la401604q
Perrault SD, Chan WCW (2009) Synthesis and surface modification of highly monodispersed, spherical gold nanoparticles of 50–200 nm. J Am Chem Soc 131:17042–17043. https://doi.org/10.1021/ja907069u
Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW (2009) Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett 9:1909–1915. https://doi.org/10.1021/nl900031y
Popovtzer A et al (2016) Actively targeted gold nanoparticles as novel radiosensitizer agents: an in vivo head and neck cancer model. Nanoscale 8:2678–2685. https://doi.org/10.1039/C5NR07496G
Popovtzer R, Agrawal A, Kotov NA, Popovtzer A, Balter J, Carey TE, Kopelman R (2008) Targeted gold nanoparticles enable molecular CT imaging of cancer. Nano Lett 8:4593–4596. https://doi.org/10.1021/nl8029114
Porcel E et al (2010) Platinum nanoparticles: a promising material for future cancer therapy? Nanotechnology 21:85103. https://doi.org/10.1088/0957-4484/21/8/085103
Reddy VR (2006) Gold nanoparticles: synthesis and applications. Synlett 2006:1791–1792. https://doi.org/10.1055/s-2006-944219
Rehbock C, Merk V, Gamrad L, Streubel R, Barcikowski S (2013) Size control of laser-fabricated surfactant-free gold nanoparticles with highly diluted electrolytes and their subsequent bioconjugation. Phys Chem Chem Phys 15:3057. https://doi.org/10.1039/c2cp42641b
Reuveni T, Motiei M, Romman Z, Popovtzer A, Popovtzer R (2011) Targeted gold nanoparticles enable molecular CT imaging of cancer: an in vivo study. Int J Nanomed 6:2859–2864. https://doi.org/10.2147/IJN.S25446
Richmonds C, Sankaran RM (2008) Plasma-liquid electrochemistry: rapid synthesis of colloidal metal nanoparticles by microplasma reduction of aqueous cations. Appl Phys Lett 93:131501–131503. https://doi.org/10.1063/1.2988283
Ritman EL (2007) Small-animal CT: its difference from, and impact on, clinical CT. Nucl Instrum Methods Phys Res, A 580:968–970. https://doi.org/10.1016/j.nima.2007.06.040
Rosa S, Connolly C, Schettino G, Butterworth KT, Prise KM (2017) Biological mechanisms of gold nanoparticle radiosensitization. Cancer Nanotechnol 8:2. https://doi.org/10.1186/s12645-017-0026-0
Rumbach P, Witzke M, Sankaran RM, Go DB (2013) Plasma-liquid interactions: separating electrolytic reactions from plasma/gas phase reactions. Proceedings of ESA Annual Meeting on Electrostatics
Saha K, Agasti SS, Kim C, Li X, Rotello VM (2012) Gold nanoparticles in chemical and biological sensing. Chem Rev 112:2739–2779. https://doi.org/10.1021/cr2001178
Sankaran RM (2011) Electrochemical cell including a plasma source and method of operating the electrochemical cell. US20110048960 A1, 3 Mar 2011
Scarboro SB, Followill DS, Howell RM, Kry SF (2011) Variations in photon energy spectra of a 6 MV beam and their impact on TLD response. Med Phys 38:2619–2628. https://doi.org/10.1118/1.3575419
Schuemann J et al (2016) Roadmap to clinical use of gold nanoparticles for radiation sensitization. Int J Radiat Oncol Biol Phys 94:189–205. https://doi.org/10.1016/j.ijrobp.2015.09.032
Searson PC (2014) Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments:1–13. https://doi.org/10.3389/fchem.2014.00069/abstract
Semmler-Behnke M et al (2008) Biodistribution of 1.4- and 18-nm gold particles in rats. Small 4:2108–2111. https://doi.org/10.1002/smll.200800922
Seo D, Park JC, Song H (2006) Polyhedral gold nanocrystals with Oh symmetry: from octahedra to cubes. J Am Chem Soc 128:14863–14870. https://doi.org/10.1021/ja062892u
Shi M, Paquette B, Thippayamontri T, Gendron L, Guerin B, Sanche L (2016) Increased radiosensitivity of colorectal tumors with intra-tumoral injection of low dose of gold nanoparticles. Int J Nanomed 11:5323–5333. https://doi.org/10.2147/IJN.S97541
Shiba F (2013) Size control of monodisperse Au nanoparticles synthesized via a citrate reduction process associated with a pH-shifting procedure. CrystEngComm 15:8412–8415. https://doi.org/10.1039/C3CE41516C
Shukla R et al (2012) Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. Proc Natl Acad Sci U S A 109:12426–12431. https://doi.org/10.1073/pnas.1121174109
Simao T et al (2016) Laser-synthesized ligand-free Au nanoparticles for contrast agent applications in computed tomography and magnetic resonance imaging. J Mater Chem B 4:6413–6427. https://doi.org/10.1039/C6TB01162D
Sina M et al (2016) Theranostic nanoseeds for efficacious internal radiation therapy of unresectable solid tumors. Sci Rep 6:1–9. https://doi.org/10.1038/srep20614
Sinha N et al (2015) Brachytherapy application with in situ dose painting administered by gold nanoparticle eluters. Int J Radiat Oncol Biol Phys 91:385–392. https://doi.org/10.1016/j.ijrobp.2014.10.001
Skotland T, Iversen TG, Sandvig K (2010) New metal-based nanoparticles for intravenous use: requirements for clinical success with focus on medical imaging. Nanomed: Nanotechnol Biol Med 6:730–737. https://doi.org/10.1016/j.nano.2010.05.002
Soenen SJ, Rivera-Gil P, Montenegro J-M, Parak WJ, De Smedt SC, Braeckmans K (2011) Cellular toxicity of inorganic nanoparticles: common aspects and guidelines for improved nanotoxicity evaluation. Nano Today 6:446–465. https://doi.org/10.1016/j.nantod.2011.08.001
Solomon SB, Silverman SG (2010) Imaging in interventional oncology. Radiology 257:624–640. https://doi.org/10.1148/radiol.10081490
Speck U (2008) Contrast agents: X-ray contrast agents and molecular imaging—a contradiction? In: Semmler W, Schwaiger M (eds) Molecular imaging I, vol 185/1. Handbook of experimental pharmacology. Springer, Berlin, pp 167–175. https://doi.org/10.1007/978-3-540-72718-7_8
Sperling RA, Rivera gil P, Zhang F, Zanella M, Parak WJ (2008) Biological applications of gold nanoparticles. Chem Soc Rev 37:1896–1908. https://doi.org/10.1039/b712170a
Su N, Dang Y, Liang G, Liu G (2015) Iodine-125-labeled cRGD-gold nanoparticles as tumor-targeted radiosensitizer and imaging agent. Nanoscale Res Lett 10:160. https://doi.org/10.1186/s11671-015-0864-9
Su S, Zuo XL, Pan D, Pei H, Wang LH, Fan CH, Huang W (2013) Design and applications of gold nanoparticle conjugates by exploiting biomolecule-gold nanoparticle interactions. Nanoscale 5:2589–2599. https://doi.org/10.1039/c3nr33870c
Sun Y, Xia Y (2002) Shape-controlled synthesis of gold and silver nanoparticles. Science 298:2176–2179. https://doi.org/10.1126/science.1077229
Sylvestre J-P, Poulin S, Kabashin AV, Sacher E, Meunier M, Luong JHT (2004) Surface chemistry of gold nanoparticles produced by laser ablation in aqueous media. J Phys Chem B 108:16864–16869. https://doi.org/10.1021/jp047134
Taggart LE, McMahon SJ, Currell FJ, Prise KM, Butterworth KT (2014) The role of mitochondrial function in gold nanoparticle mediated radiosensitisation. Cancer Nanotechnol 5:5. https://doi.org/10.1186/s12645-014-0005-7
Thakor AS, Jokerst J, Zavaleta C, Massoud TF, Gambhir SS (2011) Gold nanoparticles: a revival in precious metal administration to patients. Nano Lett 11:4029–4036. https://doi.org/10.1021/nl202559p
Torchilin VP, Frank-Kamenetsky MD, Wolf GL (1999) CT visualization of blood pool in rats by using long-circulating, iodine-containing micelles. Acad Radiol 6:61–65. https://doi.org/10.1016/s1076-6332(99)80063-4
Tsuji T, Iryo K, Nishimura Y, Tsuji M (2001) Preparation of metal colloids by a laser ablation technique in solution: influence of laser wavelength on the ablation efficiency (II). J Photochem Photobiol, A 145:201–207. https://doi.org/10.1016/s1010-6030(01)00583-4
Turkevich J, Stevenson PC, Hillier J (1951) A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss Faraday Soc 11:55–75. https://doi.org/10.1039/DF9511100055
Vincenzo A, Roberto P, Marco F, Onofrio MM, Maria Antonia I (2017) Surface plasmon resonance in gold nanoparticles: a review. J Phys: Condens Matter 29:203002
Wahab R et al (2014) Statistical analysis of gold nanoparticle-induced oxidative stress and apoptosis in myoblast (C2C12) cells. Colloids Surf, B Biointerfaces 123:664–672. https://doi.org/10.1016/j.colsurfb.2014.10.012
Wang R, Zuo S, Wu D, Zhang J, Zhu W, Becker KH, Fang J (2014) Microplasma-assisted synthesis of colloidal gold nanoparticles and their use in the detection of cardiac Troponin I (cTn-I). Plasma Processes Polym. https://doi.org/10.1002/ppap.201400127
Wang R, Zuo S, Zhu W, Wu S, Nian W, Zhang J, Fang J (2014) Microplasma-assisted growth of colloidal silver nanoparticles for enhanced antibacterial activity. Plasma Processes Polym 11:44–51. https://doi.org/10.1002/ppap.201300038
Webb JA, Bardhan R (2014) Emerging advances in nanomedicine with engineered gold nanostructures. Nanoscale 6:2502–2530. https://doi.org/10.1039/c3nr05112a
Weissleder R (2002) Scaling down imaging: molecular mapping of cancer in mice. Nat Rev Cancer 2:11–18. https://doi.org/10.1038/nrc701
Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW (2016) Analysis of nanoparticle delivery to tumours. Nat Rev Mater 1:16014. https://doi.org/10.1038/natrevmats.2016.14. https://www.nature.com/articles/natrevmats201614#supplementary-information
Wilson R et al (2009) Gold nanoparticle sensitize radiotherapy of prostate cancer cells by regulation of the cell cycle. Nanotechnology 20:375101
Woehrle GH, Brown LO, Hutchison JE (2005) Thiol-functionalized, 1.5-nm gold nanoparticles through ligand exchange reactions: scope and mechanism of ligand exchange. J Am Chem Soc 127:2172–2183. https://doi.org/10.1021/ja0457718
Wolfe T et al (2015) Targeted gold nanoparticles enhance sensitization of prostate tumors to megavoltage radiation therapy in vivo. Nanomed: Nanotechnol Biol Med 11:1277–1283. https://doi.org/10.1016/j.nano.2014.12.016
Xiao T et al (2013) Facile synthesis of acetylated dendrimer-entrapped gold nanoparticles with enhanced gold loading for CT imaging applications. J Mater Chem B 1:2773–2780. https://doi.org/10.1039/c3tb20399a
Yah CS (2013) The toxicity of Gold Nanoparticles in relation to their physiochemical properties. Biomed Res-India 24:400–413
Yang Y et al (2016) Tumor angiogenesis targeted radiosensitization therapy using gold nanoprobes guided by MRI/SPECT imaging. ACS Appl Mater Interfaces 8:1718–1732. https://doi.org/10.1021/acsami.5b09274
Yeh YC, Creran B, Rotello VM (2012) Gold nanoparticles: preparation, properties, and applications in bionanotechnology. Nanoscale 4:1871–1880. https://doi.org/10.1039/c1nr11188d
Yong KT, Swihart MT, Ding H, Prasad PN (2009) Preparation of gold nanoparticles and their applications in anisotropic nanoparticle synthesis and bioimaging. Plasmonics 4:79–93. https://doi.org/10.1007/s11468-009-9078-2
Yook S, Cai Z, Lu Y, Winnik MA, Pignol J-P, Reilly RM (2016) Intratumorally injected 177Lu-labeled gold nanoparticles: gold nanoseed brachytherapy with application for neoadjuvant treatment of locally advanced breast cancer. J Nucl Med 57:936–942. https://doi.org/10.2967/jnumed.115.168906
Zhang X-D et al (2015) Use of epidermal growth factor receptor antibody–gold cluster conjugates with good renal excretion in targeted cancer radiation treatment. J Mater Chem B: Mater Biol Med 3:4735–4741
Zhang X-D et al (2014) Enhanced tumor accumulation of sub-2 nm gold nanoclusters for cancer radiation therapy. Adv Healthc Mater 3:133–141. https://doi.org/10.1002/adhm.201300189
Zhang XD, Wu D, Shen X, Chen J, Sun YM, Liu PX, Liang XJ (2012) Size-dependent radiosensitization of PEG-coated gold nanoparticles for cancer radiation therapy. Biomaterials 33:6408–6419. https://doi.org/10.1016/j.biomaterials.2012.05.047
Zhao L, Jiang D, Cai Y, Ji X, Xie R, Yang W (2012) Tuning the size of gold nanoparticles in the citrate reduction by chloride ions. Nanoscale 4:5071–5076. https://doi.org/10.1039/C2NR30957B
Zhao N, Yang Z, Li B, Meng J, Shi Z, Li P, Fu S (2016) RGD-conjugated mesoporous silica-encapsulated gold nanorods enhance the sensitization of triple-negative breast cancer to megavoltage radiation therapy. Int J Nanomed 11:5595–5610. https://doi.org/10.2147/IJN.S104034
Zheng Q, Yang H, Wei J, Tong JL, Shu YQ (2013) The role and mechanisms of nanoparticles to enhance radiosensitivity in hepatocellular cell. Biomed Pharmacother 67:569–575. https://doi.org/10.1016/j.biopha.2013.04.003
Zheng Y, Cloutier P, Hunting DJ, Sanche L (2008) Radiosensitization by gold nanoparticles: comparison of DNA damage induced by low and high-energy electrons. J Biomed Nanotechnol 4:469–473. https://doi.org/10.1166/jbn.2008.012
Ziegler C, Eychmüller A (2011) Seeded growth synthesis of uniform gold nanoparticles with diameters of 15–300 nm. J Phys Chem C 115:4502–4506. https://doi.org/10.1021/jp1106982
Acknowledgements
Mr. Mahmoud Omar and Mrs. Beatriz Ribeiro Nogueira are gratefully acknowledged for their work on the syntheses related to Fig. 1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Fortin, MA., Simão, T., Laprise-Pelletier, M. (2018). Gold Nanoparticles for Imaging and Cancer Therapy. In: Gonçalves, G., Tobias, G. (eds) Nanooncology. Nanomedicine and Nanotoxicology. Springer, Cham. https://doi.org/10.1007/978-3-319-89878-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-89878-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89877-3
Online ISBN: 978-3-319-89878-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)