
Abstract
Ever increasing environmental cadmium presence consequent of industrial activities is considered a health hazard and is closely linked to deteriorating global health status as general animal cadmium exposure expands from cigarette smoke and ingestion of foodstuffs sourced from heavily polluted hotspots to widespread contaminated air and water, including cadmium-containing microplastics found in household water. Cadmium exerts myriads of cellular perturbances based on its abilities to directly interact with macromolecules and to mimic or displace essential physiological ions. Cell organelles are membrane-bound structures that form complex tightly regulated compartmentalized networks with specialized functions which are fundamental to life. Interorganellar communication is mediated either by release of signaling molecules, mechanical force through change in organelle shape or direct membrane contacts and is crucial to orchestrate correct cell behavior and adaptive stress responses. In this chapter, cadmium effects on organellar structure and function will be reviewed with particular consideration to disruption of organelle physiology in vertebrates. Mitochondrial dysfunction (electron transport chain, mitochondrial membrane potential, permeability transition), mitochondrial dynamics, intralumenal homeostasis and stress response in the endoplasmic reticulum, altered nuclear architecture and chromatin organization, lysosomal expansion, instability and membrane permeabilization, autophagic flux, and disruption of vesicle trafficking will be discussed in the context of cadmium.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

4.1 Introduction
Cell organelles are characterized by a lipid membrane boundary creating distinct structures and a microenvironment optimal for specialized organellar function, such as storage, e.g., lipids in lysosomes, protein synthesis in endoplasmic reticulum (ER), ATP synthesis in mitochondria, internalization of extracellular signals and signal transduction through the endo-/lysosomal pathway, and cargo trafficking and degradation in autophago(lyso)somes [1]. Interorganellar communication is essential for cell function, behavior, and adaptation to extracellular cues therefore organelles cannot and do not exist as single entities but rather as an extended interactive network [2]. In emphasis, the ER is an extension of the nuclear membrane, the ER has intimate contact with mitochondria and plasma membrane (PM), trafficking vesicles fuse with multiple organelles as well as maturing into different vesicles, and existing organelle membranes are used to generate new membrane-bound structures.
Lipid bilayers are essential to organelle function providing structural separation, appropriate membrane curvature, optimal lipid microenvironment to functionalize membrane proteins, maintaining intraorganellar homeostasis, and dictating fusogenicity. Lipid composition varies between organelles albeit differing ratios of phospholipids, cholesterol and sphingolipids are found across all organelles. Cadmium uptake and transport mechanisms into the intracellular space have been well elucidated in comparison to cadmium effects on the lipid bilayer. Using liposomes to mimic erythrocyte outer leaflet membranes, cadmium interacts preferentially with phosphatidylethanolamine, but not with cholesterol [3], causing tighter lipid packing and increased membrane rigidity without changes in lateral organization (reviewed in [4]). In conjunction with cadmium-induced altered sphingolipid metabolism [5, 6], membrane-associated cellular processes, such as transport or second messenger signaling, as well as organelle structure could be modulated by cadmium interactions with membrane lipids.
4.2 Cadmium and Mitochondria (Fig. 4.1)
Mitochondria form the central hub of bioenergetic metabolism through ATP synthesis. Comprising of a relatively permeable outer mitochondrial membrane (OMM), a highly selective inner mitochondrial membrane (IMM), the intermembrane space (IMS) and the matrix core, electron donors are formed through the citrate/Krebs cycle and fed into the IMM-located electron transport chain (ETC) to drive ATP production. Energy released from electron shuttling generates a matrix-directed electrochemical H+ gradient necessary to fuel F1-F0 ATP synthase-mediated conversion of kinetic energy to chemical energy, which is stored into phosphoanhydride bonds in the ATP molecule during oxidative phosphorylation (OXPHOS). Energy is released in exergonic hydrolysis reactions by ATPases to power energy-dependent cellular processes. Damaging reactive oxygen species (ROS), usually superoxide anion (O2•−) or hydrogen peroxide (H2O2), are generated as byproducts of single electron escape from the ETC, OXPHOS, and matrix biochemical reactions but are detoxified by antioxidants, such as glutathione or ROS-metabolizing enzymes.

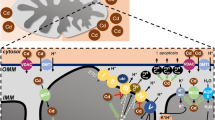
Cadmium effects on mitochondria. Low cadmium-induced mitochondrial fusion, contraction, and biogenesis are elicited by mild oxidative stress and engage adaptive responses. High cadmium leads to mitochondrial damage, such as cristae loss, fragmentation, swelling, and high ROS. Cadmium putatively permeates the outer mitochondrial membrane (OMM) via VDAC or DMT1 and the inner mitochondrial membrane (IMM) through the mitochondrial calcium uniporter (MCU). From within the matrix, cadmium blocks complex III, increasing superoxide anion which oxidizes cytochrome c (cytC to cytCox) facilitating its release. Cadmium also activates aquaporin-8 (AQP8) for swelling and K+-cycling for contraction as well as dissipating mitochondrial membrane potential (Δψm). See Sect. 4.2 for further details
4.2.1 Mitochondrial Membranes
The IMM and OMM function together to maintain intramitochondrial spaces with defined ionic and proteinaceous composition that are highly regulated by a multitude of channels and transporters mainly expressed in the selective IMM.
The lipid microenvironment functionalizes resident proteins. Mitochondrial membranes contain relatively low amounts of cholesterol compared to the PM. CdAc (2 mg/kg/day, i.p. 7–30 days) increases cholesterol at the expense of certain phospholipids (phosphatidylethanolamine, phosphatidic acid) in mitochondrial membranes derived from rat liver [7] and brain [8] albeit with no change in membrane fluidity. Intriguingly, sphingomyelin is also increased [8]. Sphingomyelin and cholesterol are key components of lipid rafts, specialized membrane domains, wherein recognition and transporting proteins are activated, and both lipids are well documented to be augmented in tumor cells [9]. In fact, increased cholesterol has been suggested to underlie the metabolic switch from OXPHOS to anaerobic glycolysis in cancerous cells [10] and could form part of cell alterations initiated during the process of cadmium carcinogenesis [11, 12].
Cardiolipin, a mitochondrial phospholipid, is localized exclusively in the IMM, whereupon the ETC shuttling and proapoptotic protein, cytochrome c (cytC), is tightly bound by electrostatic interactions. Oxidized cardiolipin serves two purposes: release of cytC from the tightly bound pool into the IMS and translocation from the IMM to OMM to facilitate cytC leakage into the cytosol. Cadmium weakly interacts with cardiolipin, increasing membrane rigidity and liposome aggregation [13], and could possibly promote cytC release in apoptotic signaling [14] (Fig. 4.1) through interference with electrostatic interactions and membrane biophysical properties.
Invaginations of the IMM called cristae serve to increase surface area for permeability and ETC proteins and bring ETC complexes into closer proximity for efficient electron transfer and ultimately, greater capacity for ATP synthesis. Numerous reports using transmission electron microscopy (TEM) evidence deleterious effects of cadmium on mitochondrial cristae, such as their reduction in number and their shortening, in various animal systems [15,16,17,18,19] and has been correlated with reduced expression of cytochrome c oxidases (COX), essential components of ETC complexes, indicating compromised mitochondrial function [20].
4.2.2 Mitochondrial Dynamics
As cellular energy demands fluctuate, mitochondria adapt through redistribution within the cell, trafficked to the sites of high metabolic demand, and undergo fusion and fission/fragmentation. Fusion events occur during high energy demands as well as during stress conditions to mitigate cell stress responses and limit the number of damaged mitochondria by mixing them with healthy mitochondria. Conversely, fission quality controls mitochondrial health, helping to sort out dysfunctional/damaged mitochondria, and usually precedes cell death execution [21]. Mitochondrial fragmentation is induced by high cadmium in the brain [22] but also by low cadmium in cultured liver cells [23]. Unexpectedly, yeast mitochondrial fusion mutants were more resistant to cadmium [24] suggesting that fusion could a preliminary step to mitochondrial fission and elimination.
To cope with demand, chronic stress and high metabolic demand can induce mitochondrial biogenesis, which is partially regulated by the transcription factors peroxisome proliferator-activated receptor (PPAR) and PPAR coactivator 1 (PGC-1), members of the nuclear co-regulator family [25].
In cultured renal proximal tubule cells (RPTCs), PPARγ and mitochondrial DNA (mtDNA) were augmented by 1–10 µM CdCl2 for 24 h and correlated with glutathione loss and low rates of apoptosis [26]. In contrast, 30 µM CdCl2 attenuated PPARα, PGC-1β, and mtDNA, despite sustained PPARγ increase, and was associated with apoptotic markers. Similarly in subtoxic subchronic CdCl2-treated Fischer rats (1 mg/kg/day, s.c., 2 weeks), PPARα, and mtDNA significantly increased whereas glutathione was unchanged compared to saline-treated controls [26], further confirming mitochondrial biogenesis as part of an adaptive mechanism to chronic oxidative stress by cadmium. Conversely, PGC-1α activity was turned off (assessed by its acetylation status), mtDNA content and mitochondrial mass were reduced in hepatocellular carcinoma cells exposed to <10 µM CdCl2 for 12 h [27]. Despite increased ROS, and even at nontoxic CdCl2, no adaptive responses involving mitochondria were recorded. This discrepancy can only be explained by the different cell models used, that is, noncancerous versus cancerous cells, which likely harbor divergent antioxidant status and could result in cadmium-induced generation of ROS subspecies to varying degrees.
4.2.3 Permeation into Mitochondria
Mitochondrial transport of cadmium is reviewed in depth in the chapter “Membrane Transport Proteins and Receptors for Cadmium and Cadmium Complexes”.
The most abundant OMM protein called porin or voltage-dependent anion channel (VDAC), which permits molecules of up to 5 kDa, is thought to mediate passage of molecules into mitochondria, however, recent evidence revealed the presence of the divalent metal transporter (DMT1), which transports cadmium, in the OMM where it is thought to regulate mitochondrial iron levels [28]. Conversely, the IMM is equipped with an array of transport proteins that tightly regulate movements across the membrane. Ion movement requires uniporters, symports, and antiports, such as the mitochondrial calcium uniporter (MCU) and K+/H+ exchanger, whereas small nucleotides and nascent proteins require the adenine nucleotide transporter (ANT) and TIM/TOM complex, respectively. Due to their similar ionic radii, calcium and cadmium can imitate each other at recognition sites [29]. To this end, cadmium permeation of the IMM via the MCU has been demonstrated in isolated kidney or liver mitochondria using MCU inhibitors ruthenium red, Ru360 or La3+ [30,31,32,33].
4.2.4 Electron Transport Chain
The ETC comprises five multimeric complexes (CI–CV) localized in the IMM wherein electrons are shuttled from the multivalent metal core of one complex to the next, helped by ubiquinone and cytC on either side of CIII, generating energy for shunting of protons from the matrix to the IMS. Consequently, a proton-motive force and a mitochondrial membrane potential (Δψm) is created across the IMM. CV, the F1-F0 ATP synthase, uses the energy stored in the H+ gradient to drive its turbine to form ATP.
Mitochondria are the major site of ROS production as a consequence of electron shuttling, in particular, CI and CIII produce highly reactive superoxide anions (O2•−). Cadmium is well evidenced to increase ROS levels either by affecting mitochondrial function, ROS-producing/metabolizing enzymes, or negatively targeting antioxidants, such as glutathione. In an elegant study in isolated mitochondria, cadmium inhibited ETC complex activities (CIII > CII ≫ CIV > CI) where CIII was maximally inhibited by ~75% at 20 µM CdCl2 that could be reversed by EDTA [34], corroborating an earlier study wherein electron transfer from ubisemiquinone to cytochrome bT, a component of CIII, is blocked by 30 µM CdCl2 [35]. Cadmium targets CIII through competitive binding at the zinc binding site, preventing electron transfer and resulting in increased superoxide [34]. Since the oxidative status of cytC appears to be prerequisite for its transfer to a loosely bound pool [36], which is then ready for liberation in apoptosis signaling, ROS production by cadmium in the immediate vicinity of cytC makes for a favorable mechanism by enhancing cytC apoptogenicity (Fig. 4.1).
4.2.5 Mitochondrial Membrane Potential (Δψm)
Consequent to the ETC, Δψm is generated where the matrix is negatively charged compared to the IMS. Dissipation of Δψm indicates mitochondrial dysfunction, has been well documented for cadmium [11, 14, 27, 31] and could be linked to ETC block [34, 35].
4.2.6 Mitochondrial Permeability Transition (mPT) and Permeability Transition Pore (PTP)
Apoptotic stimuli, such as calcium or ROS, can induce the IMM to undergo permeability transition such that water and solutes can freely pass into the matrix, concluding with an increase in matrix volume, consequent of osmotic pressure, and swelling of mitochondria. With sufficient matrix expansion, the IMM, with its larger surface area, causes the OMM to disrupt culminating in the release of proapoptotic factors, such as cytC, from the IMS and mitochondrial dysfunction. PTP formation at IMM-OMM contact sites, permitting solutes up to 1500 Da across the IMM, is thought to underlie mPT. Once thought to be comprised of OMM VDAC, IMM ANT, and matrix cyclophilin D, apoptosis execution was observed despite genetic deletion of VDAC [37] or ANT [38], interrogating the molecular composition of PTP and interpretation of studies using ANT modulators bongkrekic acid or atractyloside.
Does cadmium induce PTP opening? Numerous light scattering studies have reported PTP participation in cadmium cell death signaling using isolated mitochondria from rodent liver or kidney and monitoring mitochondrial volume/swelling [30, 32, 33, 39]. Considering the current literature, wherein VDAC and ANT as PTP components has been challenged [37, 38], it seems even more important to draw conclusions only from modulation of cyclophilin D, which has escaped molecular scrutiny. In fact, ablation of calcium-induced mitochondrial swelling and cytC release in cyclophilin D-null mice leaves no doubt to its contribution to PTP [40]. The ineffectiveness of cyclosporine A (CsA), a cyclophilin D inhibitor, on cadmium-induced mitochondrial swelling [30, 32, 39] indicates that the PTP is not a ubiquitous mitochondrial swelling mechanism elicited by cadmium. Rather, the opening of aquaporin-8 by cadmium permits water influx into the matrix to cause swelling [30].
4.2.7 Mitochondrial Volume Dynamics
In response to cellular energetic demands, mitochondria do not only undergo fusion and fission but can also swell and contract through monovalent cation cycling to regulate chemical reactions [41]. Swollen mitochondria exhibit decreased β-oxidation, Krebs cycle activity, and respiration and can be made to contract by ATP, ADP, Mg2+, or potassium cyanide, depending on the swelling stimulus. Furthermore, it has been proposed that mitochondrial shape changes through swelling serve as mechanical signals to communicate with other cell organelles [42].
Isolated rat kidney cortex mitochondria, suspended in KCl buffer and energized with rotenone/succinate, undergo swelling followed by contraction completed within 2 min after 5–20 µM CdCl2 addition [31]. Nonenergized mitochondria swell but do not contract. Using pharmacological inhibitors, swelling was MCU- and K+-influx-dependent but PTP-independent while contraction was Δψm-independent and mediated by action of the K+/H+-exchanger, which depended on the ETC-generated pH gradient across the IMM [31] (Fig. 4.1). Transient limited mitochondrial swelling by nontoxic cadmium doses may represent a mechanical signal to neighboring organelles as part of an adaptive stress response and may precede mitochondrial fusion/fission, damaged mitochondria removal by mitophagy, temporary switches in energy metabolism, and altered expression of mitochondrial proteins.
4.3 Cadmium and ER
The ER is an expansive and highly dynamic network, maintaining contacts with all other organelles and could be regarded as the delegator, by sensing signals and giving instruction in cellular responses. Through mitochondria-associated membranes (MAMs), the ER directs calcium flux to and from mitochondria in addition to dictating and aiding mitochondrial fission [43]. Furthermore, ER membranes supply autophagophore formation [44]. ER-PM contacts mediate store-operated calcium entry through STIM1/Orai and maintain lipid homeostasis at the PM, for instance, during second messenger signaling (reviewed in [45]).
Primary ER functions are mRNA translation, protein folding, some protein modifications, and lipid synthesis that demand an oxidizing and calcium-rich environment. Protein folding is prone to errors therefore several quality control and damage-limiting mechanisms protect from potential stress elicited by large amounts of misfolded proteins. ER-resident chaperones (GRP78/BiP, GRP94) occupy ER stress sensor proteins, maintaining them in an inactive state, and bind polypeptides to aid protein folding as well as offering refolding opportunities, if mistakes are made. Should these refolding endeavors prove unsuccessful, unfolded proteins are directed to ER-associated degradation (ERAD) machinery that results in cytosolic proteasome-driven destruction. Increased unfolded protein load shifts ER chaperone distribution, such that ER stress sensor proteins become unoccupied, and initiates the unfolded protein response (UPR), which initially delays cell damage by reducing mRNA translation but subsequently engages cell death-promoting pathways, culminating in upregulation of proapoptotic GADD153/CHOP and caspase-12 activation [46].
4.3.1 Intralumenal Homeostasis
Folding a polypeptide into its tertiary conformation requires formation of disulfide bridges. The intralumenal oxidizing environment of the ER is optimal for this process; but too oxidizing or too reducing results in aberrant disulfide bridge formation and thus, malformed protein structure. It is not yet clear as to exactly how the ER maintains correct redox balance though glutathione could be involved [47] and targeting by cadmium affects ER lumen redox status, increases misfolded proteins, and initiates the UPR.
High ER lumenal calcium is maintained by a pump-leak system, whereby the thapsigargin-sensitive sarco-/endoplasmic reticulum calcium ATPase (SERCA) actively transports calcium back into the ER lumen following passive leakage into the cytosol. Using aequorin-based probes, cadmium diminishes SERCA activity without effect on leakage that results in ER calcium depletion and evokes the ER stress response in parallel with the mitochondrial apoptosis pathway suggesting interorganellar communication through calcium [48].
4.3.2 ER Stress
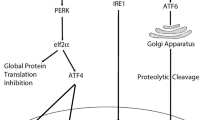
ER stress is initiated by decreased intralumenal calcium and oxidative stress that contribute to the accumulation of unfolded proteins, activating UPR and ERAD. When ER stress sensor proteins PERK, IRE1, and ATF6 are non-chaperone bound, they become active. The PERK-eIF2α-ATF4 pathway blocks further mRNA translation to prevent ER overloading so the cell has time and capacity to attempt correction of misfolded proteins. In acute and prolonged ER stress, IRE1 activation of XBP1 mRNA splicing and ATF6 truncation in the Golgi culminate in upregulation of proapoptotic genes [46].
Amongst heavy metals, cadmium is particularly effective in inducing ER stress [49], which has been well documented in various models (reviewed in [11, 14]), wherein ER chaperones (GRP78, GRP94, and BiP), all UPR arms, and CHOP are upregulated. In mammalian cells, it appears that ROS/reactive nitrogen species (RNS), specifically superoxide anion (O2•−) or peroxynitrite (ONOO−) [50], or calcium precede ER stress and UPR, however, caution should be taken when potential cadmium chelators, such as BAPTA, N-acetylcysteine, and Fura-2, are used. In contrast, ER accumulation of cadmium in yeast elicits UPR but not through inhibition of disulfide bond formation [51]. Analogous to autophagy, mild ER stress by cadmium triggers the PERK-eIF2α-ATF4 pathway and precedes signs of damage [49], acting in a protective manner, whereas major ER stress recruits the prodeath UPR arms culminating in CHOP.
4.3.3 ER Restructuring
Swollen, disorganized and damaged rough ER is caused by cadmium in various models, including insect cells (66 µM CdCl2, 24 h) [17] and rat liver (0.84 mg/kg, i.p. CdAc, 48–96 h) [16]. The effect of cadmium on distribution of physical ER contacts has not yet been investigated.
4.4 Cadmium and the Nucleus
Genetic material in the nucleus is partitioned from the cytosol by the nuclear membrane, consisting of outer and inner membranes and an underlying nuclear lamina that surrounds the nuclear matrix, which comprises cytoskeletal proteins and nuclear sap, acting as a support framework for intranuclear macromolecules. Nuclear pores span both nuclear membranes and permit communication and translocation of gene regulatory molecules between the cytosol and nucleus albeit in a regulated manner.
The central subcellular localization of the nucleus poses a spatial hurdle for cadmium as it must cross the PM followed by the cytosolic minefield to encounter the nucleus. Atomic absorption spectroscopy evidenced cadmium uptake into nuclei, plateauing at ~1 nM [52], and radioactive cadmium (109Cd or 115Cd) data evidence nuclear cadmium, driven by diffusion, within and peaking at 1 h of exposure [53, 54], weakly bound, and subsiding over time. Upregulation of cytosolic cadmium-binding sites shifts equilibrium towards the cytosolic compartment wherein cadmium is complexed [54], strongly suggesting that cadmium-induced effects from within the nucleus are short-lived and dependent on the level of cadmium-sequestering proteins in the extranuclear compartment. Thus, primary cadmium exposures probably trigger genetic and epigenetic changes from within the nucleus as its journey encounters less hurdles whereas secondary and repeated cadmium exposures will have less impact because augmented intracellular cadmium-binding sites sequester cadmium before it can reach the nucleus [55].
Genomic instability and DNA mutational effects by cadmium will be discussed in the chapter “Cadmium and its Impact on Genomic Stability”.
4.4.1 Nuclear Architecture
Ultrastructural transmission electron microscopy (TEM) studies of cadmium-exposed animal tissues and cell lines generally indicate dilated nuclei, dilated or ringed nucleoli, nuclear indentation and aberrant chromatin condensation [19, 56, 57], accumulation of RNA storage perichromatin granules (PG) at the nucleolar edge [19, 58], and micronuclei formation [19, 59, 60], wherein damaged chromosomes reside and indicates chromosome instability. Isolated nuclei evidenced millimolar cadmium-induced redistribution of lamin A, but not lamin B, from nuclear periphery to across the nuclear matrix [61] and could represent adaptive stiffening of the nuclear lamina in response to mechanical signals [62], such as mitochondrial swelling. Incidentally, large holes in the nuclear membrane after cadmium [58] could be a result of increased membrane rigidity [4] causing the nuclear membrane to become fragile and brittle, and therefore prone to breakages.
4.4.2 Chromatin Organization and Epigenetics
Chromatin comprises repeating units called nucleosomes and describes DNA wound around and condensed by histone proteins, influencing the accessibility of a gene. Histone proteins are susceptible to modifications, for example, acetylation or methylation, and can affect gene transcription through changes in DNA winding/unwinding and masking or exposing sites for transcription. Low cadmium (1 µM CdCl2) treatment causes changes in chromatin structure [58] and appearance of perichromatin granules (PGs) [58,59,60], which are storage sites for newly synthesized heterogeneous nuclear RNA, including pre-mRNA, indicating its maturation and export are impaired by cadmium. How can cadmium affect gene transcription when mRNA processing is hindered? A possible explanation is biphasic responses concerning RNA: low/acute cadmium augments whereas high/chronic cadmium attenuates RNA synthesis and mRNA activity, probably due to interactions with zinc-dependent enzymes, such as RNA polymerase [51]. Further, topoisomerase IIα, which alters the topological state of nucleic acids, is redistributed in cadmium-exposed K562 cells [63] and could affect gene transcription.
Epigenetics describes inheritable changes in gene expression without modifications to the DNA sequence and is strongly influenced by environmental factors [64]. Histone modification and DNA methylation have been implicated in protection against cadmium toxicity as well as in malignant transformation (summarized in [11]), whereas cadmium effects on noncoding RNAs, namely microRNAs, are currently emerging (reviewed in [65]). Recently, monomethylation of histone H3 at K27 (H3K27me1) was decreased by IC25 CdCl2 in mouse embryonic stem cells and was associated with prolonged mitosis, decreased population doublings, and compensatory increased total histone protein production. Importantly, daughter cells inherited these alterations [66]. In cultured lung cells, carcinogenesis-associated histone methylation marks H3K4me3 and H3K9me2 were elevated by CdCl2 (≤2.5 µM, 6–48 h), which inhibited histone demethylases [67]. In transformation studies with 2 µM CdCl2, H3K4me3 and H3K9me2 were elevated at 4 weeks but no longer at 20 weeks suggesting transient changes in histone methylation is sufficient to bring about persistent genetic changes associated with transformation [67].
4.4.3 Nuclear Matrix Proteins
The nuclear matrix and perichromatin space contains a plethora of regulatory proteins that maintain the composition of the nuclear sap, execute signaling cues, govern turnover of proteins, and modulate gene transcription.
DNA fragmentation is the penultimate step in the apoptotic signaling cascade, prior to apoptotic body formation and engulfment by macrophages, and is executed by a Ca2+/Mg2+-dependent endonuclease that cleaves DNA at intranucleosomal linker regions [68]. In isolated bovine liver nuclei, cadmium activates the endonuclease and induces DNA laddering only at low concentrations (<10 µM free Cd2+), corroborating apoptosis is associated with low cadmium, but also potently inhibited DNA laddering induced by calcium [69]. The same group reported cadmium blockade of nuclear calcium ATPase, resulting in lower nuclear calcium [52], therefore, it is unclear whether the cadmium effects are consequent of nuclear calcium uptake inhibition or direct endonuclease blockade.
Activation of nuclear protein kinase C (PKC) by phorbol esters is potentiated in the presence of cadmium by ~2-fold [70]. Astonishingly, PKC binding to nuclear proteins is enhanced by 0.1 nM free Cd2+ compared to 1 nM free Zn2+ in rat liver nuclei [70]. These observations could affect the phosphorylation status of histones and therefore chromatin organization. Lastly, it was proposed that inhibition of 8-oxo-dGTPase by CdAc (20 µmol/kg/bw, s.c., 2–48 h) in rat testis may lead to incorporation of promutagenic 8-oxo-2’-deoxyguanosine, promoting carcinogenesis, but time course inconsistencies question a causal relationship [71].
4.4.4 Transcriptional Regulation
Typically, activated cytosolic transcription factor protein is stabilized and shuttles to the nucleus wherein transcription is initiated after promoter binding. A multitude of factors determines whether a gene is “switched on” or “turned off” such as nuclear abundance and activating modifications of transcription factor protein, levels of accessory regulatory proteins, accessibility of response elements, and the presence of metal ions, in particular zinc. Extensive reports in the literature evidence altered gene expression upon cadmium exposure that involves classical transcription factors, such as nuclear factor kappa B, AP-1, c-Myc, and Nrf2 (reviewed in [14]). It is important to note that most of these studies allude to regulation by upstream signaling pathways initiated by cadmium, and not a direct effect. However, nuclear export of transcription factors [72] or mRNA [73] can also be affected by cadmium. Additional putative mechanisms are displacement of zinc (reviewed in [74]), changes in nuclear architecture that prevent access to response elements, and modification of accessory transcription complex proteins or transcriptional machinery.
To date, only a single study has identified the direct participation of cadmium on transcription factor activity. The tumor suppressor gene p53 is often quoted as the guardian of the cell, overseeing genotoxic and non-genotoxic-induced stress responses, primarily by DNA quality control, and is mutated in over 50% of cancers. In a set of elegant experiments, Meplan et al. utilized recombinant wildtype p53 and conformation-specific antibodies to show that cadmium induces a conformational change in p53, dissipating its DNA binding capacity, preventing activation of p53 target genes [75], and thus contributing to cadmium-induced carcinogenesis.
4.5 Cadmium and Lysosomes (Fig. 4.2)
Cargo internalized into the cell by endocytosis, macrocytosis or phagocytosis as well as cargo from within the cell, taken up into autophagosomes (see Sect. 4.7.1), are trafficked to the lysosome for degradation by acidic digestive hydrolase enzymes followed by expulsion or recycling.

Cadmium effects on vesicular trafficking. Through molecular mimicry, cadmium inhibits calcium-dependent processes, such as regulated exocytosis (EX) and microvesicle (MV) formation. Blockade of the vacuolar H+-ATPase prevents late endosome (LE) and lysosome (lyso) acidification and thus endocytosis (EN). Further, cadmium prevents recycling endosome (RE) formation from early endosomes (EE) or RE exocytosis. Cadmium expands the trans-Golgi network (TGN) and lysosome number, possibly due to increased secretory vesicles (sec) containing lysosomal proteins. Lysosomal membrane permeabilization by cadmium could lead to cell death. Autophagopore (PP) formation precedes autophagosomes (PS), which are prevented from fusing with lysosomes by cadmium and hindering autophagy. Cadmium could be involved in mobilizing multivesicular bodies (MVB) during exosome (exo) secretion. See Sects. 4.5–4.7 for further details
4.5.1 Lysosome Maturation
Lysosomes originating from the endocytic/endolysosomal pathway have been best studied. Early endosomes (EEs) bud off the PM and can either return to the PM as a recycling endosome (RE) or enter the late endosomal (LE) pathway. LEs are acidified to pH ~ 6 by the vacuolar H+-ATPase (V-ATPase) and acquire lysosomal proteins through fusion with lysosome-targeting mannose-6-phosphate transport vesicles from the trans-Golgi. Further acidification of lysosomes to pH ~ 4.5 depends on V-ATPase driven accumulation of protons in the intralysosomal space in concert with extrusion of a counterion. The final step in lysosome maturation is activation of acid phosphatase. In rat RPTCs, cadmium inhibits the V-ATPase and perturbs the endocytic trafficking pathway culminating in proteinuria [76] (see Sect. 4.6.1). Intriguingly, micromolar cadmium inhibits phosphomannose isomerase by competition with the substrate mannose-6-phosphate [77] thereby increasing its levels and promoting lysosome biogenesis. To this end, increases in heteromorphous lysosome size and number by cadmium have been observed using TEM [17, 18] and identified by neutral red uptake and acid phosphatase staining [17], as well as fluorescent dye labeling of acidic compartments [78]. Lysosomal system expansion could be resultant of: (1) lysosomal homotypic fusion and fission; (2) inhibition of lysosomal trafficking; (3) inhibition of RE formation forcing endosomes into lysosomal maturation; and (4) increased delivery of lysosomal proteins from Golgi to LEs to drive lysosomal maturation. Enlargement of the Golgi apparatus by cadmium [19, 79] could contribute to increased lysosome biogenesis, however, how cadmium affects these processes is unknown (Fig. 4.2).
4.5.2 Lysosomal Membrane Permeabilization (LMP)
Loss of lysosomal membrane integrity results in leakage of lysosomal digestion enzymes and consequent lysosomal cell death. LMP is most likely caused by alterations in lysosomal membrane lipid composition since sphingomyelin increases lysosomal instability and LMP [80]. In RPTCs, defective lysosomes were observed with 5 µM CdCl2 after 6 h as assessed by decreased lysosomal LAMP1 and cathepsin B [5], suggesting cadmium induces LMP. Furthermore, increased sphingomyelin levels were associated with increased membrane fluidity, which could alter protein interactions with the lysosomal membrane, such that they are degraded and leaked out. Cadmium-induced LMP has also been indicated by loss of acidic compartment labeling [78, 81], leakage of lysosomal DNase II in necrotic endothelial cells [78], and selective leakage of β-glucuronidase but not acid phosphatase, which was inhibited by cadmium, from isolated lung lysosomes [82].
4.6 Cadmium and Vesicular Trafficking (Fig. 4.2)
Through endocytosis, transcytosis, and exocytosis, intracellular trafficking vesicles exist in many different interconnected pools and are multifunctional, such as enabling the cell interior to communicate with the external environment, directing cargo to its final destination, regulating transmembrane proteins, such as receptors and transporters, and regulating lipid balance.
4.6.1 Endocytosis
Invagination and budding from the membrane with the aid of clathrin or caveolin during endocytosis results in the formation of EEs that develop into REs or into acidic LEs and subsequently, lysosomes in a GTPase-dependent manner.
Cadmium impairs endocytosis. In the RPTC, which is the primary site of cadmium accumulation and has high endocytic turnover at its apical brush border membrane (BBM) because of its role in mass reabsorption, cadmium exposure diminished receptor-mediated endocytosis of fluorescently labeled ligands. In opossum kidney (OK) proximal tubule cells, FITC-albumin receptor binding, Bmax, and uptake were maximally attenuated by 100 µM CdCl2 for 1 h, whereas no effect was seen on fluid-phase endocytosis. In more intricate studies in a rat model (2 mg/kg/day s.c. CdCl2, 14 days), BBM was damaged and isolated BBM vesicles harbored ~40% less V-ATPase protein expression and bafilomycin-sensitive ATPase activity [76]. Since acidification is essential for endocytic trafficking, cadmium inhibition of vesicular acidification would impair endocytosis. Indeed, using BBM vesicles from non-treated rat kidneys, 10 µM CdCl2 significantly attenuated bafilomycin-sensitive ATPase activity, quenching of acridine orange (which accumulates in acidic compartments), and uptake of FITC-dextran [76]. Furthermore, cadmium-metallothionein exposed rats exhibited redistribution of apical membrane proteins, such as megalin and Na+/H+-exchanger, into vesicles suggesting that REs are also perturbed by cadmium inhibition of vesicular acidification [83].
In addition to fluid-phase and receptor-mediated endocytosis, cells with high rates of exocytosis, such as developing, endocrine, secretory epithelial, and nerve cells, must retrieve exocytosed membrane by compensatory endocytosis to prevent significant changes in membrane surface area. Compensatory endocytosis in response to triggered exocytosis appears to be calcium-dependent thus cadmium inhibits the influx of extracellular calcium and, in turn, compensatory endocytosis in sea urchin embryos [84].
4.6.2 Secretory Vesicles and Exocytosis
Secretory vesicles derived from the trans-Golgi network migrate along microtubules to the PM for exocytosis, whereby their contents are either incorporated into the PM or released into the extracellular space.
Cytosolic calcium increase is prerequisite to stimulated exocytosis. In addition to neutralizing opposing surface negative charges between the cell and vesicle membranes, calcium is required for activation of membrane fusion proteins, such as synaptotagmin, which mechanically pull the membranes together. Based on the interplay of calcium and cadmium [29], an effect of cadmium on exocytosis is assumed. In fact, isolated single synaptotagmin C2 domains, which harbor calcium-binding sites, do not associate with lipid membranes when complexed with cadmium, in contrast to full-length synaptotagmin containing multiple C2 domains, indicating the avidity of C2 domains [85]. However, the association of cadmium-complexed full-length synaptotagmin with lipid membranes was shallower than when calcium was used [85] and could have an impact on the fusogenicity of membranes during exocytosis events.
4.7 Cadmium and Other Cell Organelles (Fig. 4.2)
4.7.1 Autophagosomes
During autophagy, the mechanism of self-digestion that fuels that cell during periods of starvation, membranes are pinched off from intracellular organelles, such as the ER or mitochondria, and used to form double-membraned autophagosomes [86], which link cytosolic LC3 to phosphotidylethanolamine. Lipidated LC3, also known as LC3-II, serves as an anchorage point for autophagosomal chaperones that direct cargo destined for degradation but is also degraded itself. Actually, LC3 degradation is a measure of autophagic flux. Autophagosomes engulf cytosolic constituents in either a targeted (ERphagy, mitophagy) or nontargeted (macroautophagy) manner and subsequently fuse with lysosomes to generate acidic autophagolysosomes, wherein cargo is degraded by lysosomal enzymes.
Without a doubt, cadmium induces autophagy. However, the outcome is disputed. The self-preservation function of autophagy has led to the hypothesis that cadmium autophagy is protective but use of the multi-target autophagy inducer rapamycin hampers correct interpretation [87]. Nevertheless, numerous studies have reported that autophagy protects against cadmium toxicity (reviewed in [14, 88]). Autophagy has been observed in kidney cortex of rats exposed to sublethal cadmium (0.3 mg/kg/bw CdCl2, i.p. 1–5 days) suggesting autophagy as a stress or protective response [89]. Unfortunately, the contribution of autophagy in protection against kidney damage by cadmium was not investigated. In cell culture studies, cadmium-induced morphological changes and LC3-II were reversed using 3-methyladenine but not bafilomycin A1 [89]. The authors suggested cadmium inhibits the V-ATPase (see above and [76]), thus accounting for the lack of effect by bafilomycin A1, but indicate cadmium hinders both autophagosome formation and autophagic flux.
More recent evidence clearly shows that cadmium negates autophagy execution. Colocalization of LAMP proteins with LC3 was significantly inhibited by cadmium (2.5–50 µM, 12–24 h) [90, 91], whereas 3-methyladenine and Atg5 siRNA had no effect on LC3-II, p62 or cadmium-induced loss of cell viability [91]. Moreover, cadmium increased lysosomal pH and lysosome biogenesis-related genes and decreased protease activity [91]. Melatonin reversed detrimental lysosomal effects by cadmium, but melatonin, a powerful antioxidant, also binds cadmium [92]. In RTPCs, 5 µM CdCl2 induces transient protective autophagy within 3 h (↓LC3-II/p62) but disrupts autophagic flux at 6–8 h (↑LC3-II/p62) [5]. Conversely to previous reports, rapamycin was ineffective against cadmium cell death despite inducing autophagy [5, 87]. Low cadmium stress-induced autophagy delayed the onset of apoptosis, which could be reversed by autophagosome–lysosome fusion inhibition, whereas accrual of cadmium stress decreased the effectiveness of lysosomal inhibitors on LC3-II/p62, and increased membrane fluidity and instability of isolated lysosomes [5]. These changes elicited by cadmium could alter the fusion capacity of lysosomes, thereby preventing autophagy execution, which is in agreement with other reports [90, 91]. Blockade of autophagic flux by cadmium concurs with decreased prosurvival signaling as well as with accumulation of autophagosomes resulting in cell death [93].
4.7.2 Extracellular Vesicles
Communication between cells can occur through secreted extracellular vesicles that pass on information with mRNA/microRNA, proteins and signaling molecules. Microvesicles (MVs) are derived from PM budding whereas exosomes are produced in multivesicular endosomes. Exosomes are enriched in tetraspanins, flotillin and lipids, including sphingomyelin and cholesterol [94], and tend to be smaller in size (40–100 nm) than MVs (100–1000 nm).
Cadmium has not been documented to affect exosome formation or secretion though it would be a plausible hypothesis since cadmium increases ceramide [6, 95] which promotes exosome release [96], and cadmium-containing cigarette smoke induces the release of ceramide-rich exosome-containing microparticles from lung endothelial cells [97]. Budding of the PM in MV formation is calcium-dependent thus, unsurprisingly, cadmium prevents MV secretion of glutamate from rat pineal gland cells [98].
4.7.3 Peroxisomes
Surrounded by a single membrane, peroxisomes are characterized by the presence of H2O2-producing and -degrading enzymes, present in virtually all eukaryotic cells and largely associated with oxidative status [99]. They execute similar biochemical reactions as those found in mitochondria though the enzymatic machinery employed by each organelle is entirely different. Peroxisomes do not possess an ETC, therefore electrons from FADH2 are passed onto O2 to form H2O2. Though H2O2 is metabolized to water and oxygen by intraperoxisomal H2O2-metabolizing enzymes, of which catalase is the most abundant, this is insufficient to prevent H2O2 release into the cytosol where it could serve as a messenger or cause imbalance in cellular oxidative status.
Peroxisomes have been largely overlooked in cadmium toxicity in spite of their well-described ROS/RNS generating capacity probably due to their functional similarity to mitochondria (see Sect. 4.2). Only a single study has examined the effect of cadmium directly on peroxisomes. In yeast, 1 mM CdCl2 for 24 h inhibited cell growth but increased peroxisome number and catalase activity [100]. Despite catalase’s detoxifying function, oxidative stress has been associated with peroxisome proliferation because H2O2-producing enzymes are augmented by >10 times over increase in H2O2-metabolizing enzymes [101] suggesting that cadmium-induced peroxisome proliferation and catalase activity would contribute to oxidative stress. In RTPCs, 10–50 µM CdCl2 increased catalase activity (Lee, W. K. and Thévenod, F., unpublished data) but peroxisome number was not determined. In contrast, a study in mussels found cadmium had no effect on catalase activity and decreased peroxisome volume density [102]. Moreover, cadmium (10–40 µM Cd(NO3)2, 24 h) weakly interacts and inhibits catalase activity in zebrafish [103]. These conflicting data of cadmium on catalase activity and peroxisomes could lie in the diverse model systems used.
4.7.4 Specialized Organelles
The effect of cadmium on other more specialized organelles, such as melanosomes, phagosomes, and secretory lysosomes has not been considerably documented.
4.8 Summary and Conclusions
Compartmentalization by lipid membranes to create organelles with specialized functions is essential to normal cell physiology. Cadmium is a promiscuous non-native metal and disrupts organelle function through a few fundamental mechanisms: (1) altered biophysical properties of membranes [4]; (2) ionic mimicry [29, 74]; and (3) direct macromolecular interactions [104]. With chronic low cadmium, organelle function could initially diminish but stress adaptive responses, which include organelle biogenesis and strengthened interorganellar communication, would strive to restore this loss of function. In acute and/or high cadmium, organelle function is compromised such that it is irreversible and detrimental to the cell. Our current knowledge clearly evidences cadmium effects on organelles as single entities in cellular processes but further understanding of how organelles interact and communicate with each other to coordinate the adaptive response under cadmium stress would be crucial to delineating and predicting heavy metal toxicity.
References
Voeltz GK, Barr FA (2013) Cell organelles. Curr Op Cell Biol 25(4):403–405
Elbaz-Alon Y (2017) Mitochondria-organelle contact sites: the plot thickens. Biochem Soc Trans 45(2):477–488
Le MT, Gailer J, Prenner EJ (2009) Hg2+ and Cd2+ interact differently with biomimetic erythrocyte membranes. Biometals 22(2):261–274
Payliss BJ, Hassanin M, Prenner EJ (2015) The structural and functional effects of Hg(II) and Cd(II) on lipid model systems and human erythrocytes: a review. Chem Phys Lipids 193:36–51
Lee WK, Probst S, Santoyo-Sanchez MP, Al-Hamdani W, Diebels I, von Sivers JK, Kerek E, Prenner EJ, Thevenod F (2017) Initial autophagic protection switches to disruption of autophagic flux by lysosomal instability during cadmium stress accrual in renal NRK-52E cells. Arch Toxicol 91(10):3225–3245
Lee WK, Torchalski B, Kohistani N, Thevenod F (2011) ABCB1 protects kidney proximal tubule cells against cadmium-induced apoptosis: roles of cadmium and ceramide transport. Toxicol Sci 121(2):343–356
Modi HR, Katyare SS (2009) Effect of treatment with cadmium on structure-function relationships in rat liver mitochondria: studies on oxidative energy metabolism and lipid/phospholipids profiles. J Membr Biol 232(1–3):47–57
Modi HR, Katyare SS (2009) Cadmium exposure-induced alterations in the lipid/phospholipids composition of rat brain microsomes and mitochondria. Neurosci Lett 464(2):108–112
Lee WK, Kolesnick RN (2017) Sphingolipid abnormalities in cancer multidrug resistance: chicken or egg? Cell Signal 38:134–145
Ribas V, Garcia-Ruiz C, Fernandez-Checa JC (2016) Mitochondria, cholesterol and cancer cell metabolism. Clin Trans Med 5(1):22
Thevenod F, Lee WK (2013) Toxicology of cadmium and its damage to mammalian organs. Met Ions Life Sci 11:415–490
Hartwig A (2013) Cadmium and cancer. Met Ions Life Sci 11:491–507
Kerek EM, Prenner EJ (2016) Inorganic cadmium affects the fluidity and size of phospholipid based liposomes. Biochim Biophys Acta 1858(12):3169–3181
Thevenod F, Lee WK (2013) Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Arch Toxicol 87(10):1743–1786
Yang XF, Han QG, Liu DY, Zhang HT, Fan GY, Ma JY, Wang ZL (2016) Microstructure and ultrastructure alterations in the pallium of immature mice exposed to cadmium. Biol Trace Elem Res 174(1):105–111
Early JL 2nd, Nonavinakere VK, Weaver A (1992) Effect of cadmium and/or selenium on liver mitochondria and rough endoplasmic reticulum in the rat. Toxicol Lett 62(1):73–83
Braeckman B, Brys K, Rzeznik U, Raes H (1999) Cadmium pathology in an insect cell line: ultrastructural and biochemical effects. Tissue Cell 31(1):45–53
Asar M, Kayisli UA, Izgut-Uysal VN, Akkoyunlu G (2004) Immunohistochemical and ultrastructural changes in the renal cortex of cadmium-treated rats. Biol Trace Elem Res 97(3):249–263
Ord MJ, Bouffler SD, Chibber R (1988) Cadmium induced changes in cell organelles: an ultrastructural study using cadmium sensitive and resistant muntjac fibroblast cell lines. Arch Toxicol 62(2–3):133–145
Takaki A, Jimi S, Segawa M, Hisano S, Takebayashi S, Iwasaki H (2004) Long-term cadmium exposure accelerates age-related mitochondrial changes in renal epithelial cells. Toxicology 203(1–3):145–154
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337(6098):1062–1065
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang H, Tang J, Li H, Feng M, Deng P, Guo P, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Wang W, Reiter RJ, Yu Z, Zhou Z (2016) Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J Pineal Res 60(3):291–302
Pi H, Xu S, Zhang L, Guo P, Li Y, Xie J, Tian L, He M, Lu Y, Li M, Zhang Y, Zhong M, Xiang Y, Deng L, Zhou Z, Yu Z (2013) Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy 9(11):1780–1800
Luz AL, Godebo TR, Smith LL, Leuthner TC, Maurer LL, Meyer JN (2017) Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology 387:81–94
Jones AW, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G (2012) PGC-1 family coactivators and cell fate: roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 12(1):86–99
Nair AR, Lee WK, Smeets K, Swennen Q, Sanchez A, Thevenod F, Cuypers A (2015) Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch Toxicol 89(12):2273–2289
Guo P, Pi H, Xu S, Zhang L, Li Y, Li M, Cao Z, Tian L, Xie J, Li R, He M, Lu Y, Liu C, Duan W, Yu Z, Zhou Z (2014) Melatonin improves mitochondrial function by promoting MT1/SIRT1/PGC-1 alpha-dependent mitochondrial biogenesis in cadmium-induced hepatotoxicity in vitro. Toxicol Sci 142(1):182–195
Wolff NA, Ghio AJ, Garrick LM, Garrick MD, Zhao L, Fenton RA, Thevenod F (2014) Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J 28(5):2134–2145
Choong G, Liu Y, Templeton DM (2014) Interplay of calcium and cadmium in mediating cadmium toxicity. Chem Biol Interact 211:54–65
Lee WK, Bork U, Gholamrezaei F, Thevenod F (2005) Cd2+-induced cytochrome c release in apoptotic proximal tubule cells: role of mitochondrial permeability transition pore and Ca2+ uniporter. Am J Physiol Renal Physiol 288(1):F27–39
Lee WK, Spielmann M, Bork U, Thevenod F (2005) Cd2+-induced swelling-contraction dynamics in isolated kidney cortex mitochondria: role of Ca2+ uniporter, K+ cycling, and protonmotive force. Am J Physiol Cell Physiol 289(3):C656–664
Li M, Xia T, Jiang CS, Li LJ, Fu JL, Zhou ZC (2003) Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 194(1–2):19–33
Dorta DJ, Leite S, DeMarco KC, Prado IM, Rodrigues T, Mingatto FE, Uyemura SA, Santos AC, Curti C (2003) A proposed sequence of events for cadmium-induced mitochondrial impairment. J Inorg Biochem 97(3):251–257
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36(11):1434–1443
Miccadei S, Floridi A (1993) Sites of inhibition of mitochondrial electron transport by cadmium. Chem Biol Interact 89(2–3):159–167
Petrosillo G, Ruggiero FM, Paradies G (2003) Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J 17(15):2202–2208
Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9(5):550–555
Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427(6973):461–465
Belyaeva EA, Glazunov VV, Korotkov SM (2002) Cyclosporin A-sensitive permeability transition pore is involved in Cd2+-induced dysfunction of isolated rat liver mitochondria: doubts no more. Arch Biochem Biophys 405(2):252–264
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434(7033):658–662
Lizana L, Bauer B, Orwar O (2008) Controlling the rates of biochemical reactions and signaling networks by shape and volume changes. Proc Nat Acad Sci USA 105(11):4099–4104
Kaasik A, Kuum M, Joubert F, Wilding J, Ventura-Clapier R, Veksler V (2010) Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovasc Res 87(1):83–91
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362
Sanchez-Wandelmer J, Ktistakis NT, Reggiori F (2015) ERES: sites for autophagosome biogenesis and maturation? J Cell Sci 128(2):185–192
Balla T (2018) Ca2+ and lipid signals hold hands at ER-plasma membrane contact sites. J Physiol 596(14):2709–2716
Woehlbier U, Hetz C (2011) Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci 36(6):329–337
Margittai E, Enyedi B, Csala M, Geiszt M, Banhegyi G (2015) Composition of the redox environment of the endoplasmic reticulum and sources of hydrogen peroxide. Free Radic Biol Med 83:331–340
Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, Pinton P (2008) Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 43(2):184–195
Liu F, Inageda K, Nishitai G, Matsuoka M (2006) Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ Health Perspect 114(6):859–864
Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M (2008) Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem 283(7):4252–4260
Gardarin A, Chedin S, Lagniel G, Aude JC, Godat E, Catty P, Labarre J (2010) Endoplasmic reticulum is a major target of cadmium toxicity in yeast. Mol Microbiol 76(4):1034–1048
Hechtenberg S, Beyersmann D (1994) Interference of cadmium with ATP-stimulated nuclear calcium uptake. Environ Health Perspect 102(Suppl 3):265–267
Fighetti MA, Miele M, Montella A, Desole MS, Congiu AM, Anania V (1988) Possible involvement of nuclei in cadmium-induced modifications of cultured cells. Arch Toxicol 62(6):476–478
Bryan SE, Hidalgo HA (1976) Nuclear 115cadmium: uptake and disappearance correlated with cadmium-binding protein synthesis. Biochem Biophys Res Commun 68(3):858–866
Goering PL, Klaassen CD (1983) Altered subcellular distribution of cadmium following cadmium pretreatment: possible mechanism of tolerance to cadmium-induced lethality. Toxicol Appl Pharmacol 70(2):195–203
Peereboom-Stegeman JH, Morselt AF (1981) Increase in liver cell nuclear size after chronic cadmium treatment. Arch Toxicol 48(2–3):209–211
Matsuura K, Takasugi M, Kunifuji Y, Horie A, Kuroiwa A (1991) Morphological effects of cadmium on proximal tubular cells in rats. Biol Trace Elem Res 31(2):171–182
Banfalvi G, Gacsi M, Nagy G, Kiss ZB, Basnakian AG (2005) Cadmium induced apoptotic changes in chromatin structure and subphases of nuclear growth during the cell cycle in CHO cells. Apoptosis 10(3):631–642
Puvion E, Lange M (1980) Functional significance of perichromatin granule accumulation induced by cadmium chloride in isolated rat liver cells. Exp Cell Res 128(1):47–58
Cervera J, Alamar M, Martinez A, Renau-Piqueras J (1983) Nuclear alterations induced by cadmium chloride and l-canavanine in HeLa S3 cells. Accumulation of perichromatin granules. J Ultrastruc Res 82(3):241–263
Neri LM, Raymond Y, Giordano A, Borgatti P, Marchisio M, Capitani S, Martelli AM (1999) Spatial distribution of lamin A and B1 in the K562 cell nuclear matrix stabilized with metal ions. J Cell Biochem 75(1):36–45
Ungricht R, Kutay U (2017) Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol 18(4):229–245
Neri LM, Bortul R, Zweyer M, Tabellini G, Borgatti P, Marchisio M, Bareggi R, Capitani S, Martelli AM (1999) Influence of different metal ions on the ultrastructure, biochemical properties, and protein localization of the K562 cell nuclear matrix. J Cell Biochem 73(3):342–354
Mathers JC, Strathdee G, Relton CL (2010) Induction of epigenetic alterations by dietary and other environmental factors. Adv Genet 71:3–39
Humphries B, Wang Z, Yang C (2016) The role of microRNAs in metal carcinogen-induced cell malignant transformation and tumorigenesis. Food Chem Toxicol 98(Pt A):58–65
Gadhia SR, O’Brien D, Barile FA (2015) Cadmium affects mitotically inherited histone modification pathways in mouse embryonic stem cells. Toxicol In Vitro 30(1 Pt B):583–592
Xiao C, Liu Y, Xie C, Tu W, Xia Y, Costa M, Zhou X (2015) Cadmium induces histone H3 lysine methylation by inhibiting histone demethylase activity. Toxicol Sci 145(1):80–89
Gaido ML, Cidlowski JA (1991) Identification, purification, and characterization of a calcium-dependent endonuclease (NUC18) from apoptotic rat thymocytes. NUC18 is not histone H2B. J Biol Chem 266(28):18580–18585
Lohmann RD, Beyersmann D (1994) Effects of zinc and cadmium on apoptotic DNA fragmentation in isolated bovine liver nuclei. Environ Health Perspect 102(Suppl 3):269–271
Beyersmann D, Block C, Malviya AN (1994) Effects of cadmium on nuclear protein kinase C. Environ Health Perspect 102(Suppl 3):177–180
Bialkowski K, Bialkowska A, Kasprzak KS (1999) Cadmium(II), unlike nickel(II), inhibits 8-oxo-dGTPase activity and increases 8-oxo-dG level in DNA of the rat testis, a target organ for cadmium(II) carcinogenesis. Carcinogenesis 20(8):1621–1624
Suzuki H, Tashiro S, Sun J, Doi H, Satomi S, Igarashi K (2003) Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J Biol Chem 278(49):49246–49253
Topisirovic I, Capili AD, Borden KL (2002) Gamma interferon and cadmium treatments modulate eukaryotic initiation factor 4E-dependent mRNA transport of cyclin D1 in a PML-dependent manner. Mol Cell Biol 22(17):6183–6198
Petering DH (2017) Reactions of the Zn proteome with Cd2+ and other xenobiotics: trafficking and toxicity. Chem Res Toxicol 30(1):189–202
Meplan C, Mann K, Hainaut P (1999) Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem 274(44):31663–31670
Herak-Kramberger CM, Brown D, Sabolic I (1998) Cadmium inhibits vacuolar H+-ATPase and endocytosis in rat kidney cortex. Kidney Int 53(6):1713–1726
Wells TN, Coulin F, Payton MA, Proudfoot AE (1993) Phosphomannose isomerase from Saccharomyces cerevisiae contains two inhibitory metal ion binding sites. Biochemistry 32(5):1294–1301
Messner B, Ploner C, Laufer G, Bernhard D (2012) Cadmium activates a programmed, lysosomal membrane permeabilization-dependent necrosis pathway. Toxicol Lett 212(3):268–275
Ferri S (1980) Effect of cadmium on Golgi complex of freshwater teleost (Pimelodus maculatus) hepatocytes. Protoplasma 103(1):99–103
Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lonborg A, Vindelov SD, Hanahan D, Arenz C, Ejsing CS, Kirkegaard T, Rohde M, Nylandsted J, Jaattela M (2013) Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24(3):379–393
Fotakis G, Cemeli E, Anderson D, Timbrell JA (2005) Cadmium chloride-induced DNA and lysosomal damage in a hepatoma cell line. Toxicol In Vitro 19(4):481–489
Giri SN, Hollinger MA (1995) Effect of cadmium on lung lysosomal enzymes in vitro. Arch Toxicol 69(5):341–345
Sabolic I, Ljubojevic M, Herak-Kramberger CM, Brown D (2002) Cd-MT causes endocytosis of brush-border transporters in rat renal proximal tubules. Am J Physiol Renal Physiol 283(6):F1389–1402
Covian-Nares JF, Smith RM, Vogel SS (2008) Two independent forms of endocytosis maintain embryonic cell surface homeostasis during early development. Dev Biol 316(1):135–148
Katti S, Nyenhuis SB, Her B, Srivastava AK, Taylor AB, Hart PJ, Cafiso DS, Igumenova TI (2017) Non-native metal ion reveals the role of electrostatics in synaptotagmin 1-membrane interactions. Biochemistry 56(25):3283–3295
Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15(7):713–720
Thevenod F, Lee WK, Wolff NA (2015) Rapamycin: a therapy of choice for endoplasmic reticulum stress-induced renal proximal tubule toxicity? Toxicology 330:41–43
Thevenod F, Lee WK (2015) Live and let die: roles of autophagy in cadmium nephrotoxicity. Toxics 3(2):130–151
Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, Duranton C, Tauc M, Hofman P, Poujeol P, El May MV, Mograbi B (2011) Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 121(1):31–42
Liu F, Wang XY, Zhou XP, Liu ZP, Song XB, Wang ZY, Wang L (2017) Cadmium disrupts autophagic flux by inhibiting cytosolic Ca2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 383:13–23
Li M, Pi H, Yang Z, Reiter RJ, Xu S, Chen X, Chen C, Zhang L, Yang M, Li Y, Guo P, Li G, Tu M, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Yu Z, Zhou Z (2016) Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J Pineal Res 61(3):353–369
Limson J, Nyokong T, Daya S (1998) The interaction of melatonin and its precursors with aluminium, cadmium, copper, iron, lead, and zinc: an adsorptive voltammetric study. J Pineal Res 24(1):15–21
Choi J, Jo M, Lee E, Choi D (2011) Induction of apoptotic cell death via accumulation of autophagosomes in rat granulosa cells. Fertil Steril 95(4):1482–1486
Maas SL, Breakefield XO, Weaver AM (2017) Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol 27(3):172–188
Lee WK, Torchalski B, Thevenod F (2007) Cadmium-induced ceramide formation triggers calpain-dependent apoptosis in cultured kidney proximal tubule cells. Am J Physiol Cell Physiol 293(3):C839–847
Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319(5867):1244–1247
Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, Patel M, Tsvetkova I, Kamocki K, Mikosz A, Schweitzer KS, Jacobson S, Cardoso A, Carlesso N, Hubbard WC, Kechris K, Dragnea B, Berdyshev EV, McClintock J, Petrache I (2016) Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep 6:31596
Yamada H, Yamamoto A, Yodozawa S, Kozaki S, Takahashi M, Morita M, Michibata H, Furuichi T, Mikoshiba K, Moriyama Y (1996) Microvesicle-mediated exocytosis of glutamate is a novel paracrine-like chemical transduction mechanism and inhibits melatonin secretion in rat pinealocytes. J Pineal Res 21(3):175–191
Lodhi IJ, Semenkovich CF (2014) Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab 19(3):380–392
Chen T, Li W, Schulz PJ, Furst A, Chien PK (1995) Induction of peroxisome proliferation and increase of catalase activity in yeast, Candida albicans, by cadmium. Biol Trace Elem Res 50(2):125–133
Rao MS, Reddy JK (1991) An overview of peroxisome proliferator-induced hepatocarcinogenesis. Environ Health Perspect 93:205–209
Orbea A, Ortiz-Zarragoitia M, Cajaraville MP (2002) Interactive effects of benzo(a)pyrene and cadmium and effects of di(2-ethylhexyl) phthalate on antioxidant and peroxisomal enzymes and peroxisomal volume density in the digestive gland of mussel Mytilus galloprovincialis Lmk. Biomarkers 7(1):33–48
Wang J, Zhang H, Zhang T, Zhang R, Liu R, Chen Y (2015) Molecular mechanism on cadmium-induced activity changes of catalase and superoxide dismutase. Int J Biol Macromol 77:59–67
Jacobson KB, Turner JE (1980) The interaction of cadmium and certain other metal ions with proteins and nucleic acids. Toxicology 16(1):1–37
Acknowledgements
W. -K. L is financially supported by the Intramural Research Program at Witten/Herdecke University (IFF2016-20, IFF2017-14).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lee, WK. (2018). Cell Organelles as Targets of Cadmium Toxicity. In: Thévenod, F., Petering, D., M. Templeton, D., Lee, WK., Hartwig, A. (eds) Cadmium Interaction with Animal Cells. Springer, Cham. https://doi.org/10.1007/978-3-319-89623-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-89623-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89622-9
Online ISBN: 978-3-319-89623-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)