
Abstract
Blood vessels span throughout the body to nourish tissue cells and to provide gateways for immune surveillance. Endothelial cells that line capillaries have the remarkable capacity to be quiescent for years but to switch rapidly into the activated state once new blood vessels need to be formed. In addition, endothelial cells generate niches for progenitor and tumor cells and provide organ-specific paracrine (angiocrine) factors that control organ development and regeneration, maintenance of homeostasis and tumor progression. Recent data indicate a pivotal role for blood vessels in responding to metabolic changes and that endothelial cell metabolism is a novel regulator of angiogenesis. The Notch pathway is the central signaling mode that cooperates with VEGF, WNT, BMP, TGF-β, angiopoietin signaling and cell metabolism to orchestrate angiogenesis, tip/stalk cell selection and arteriovenous specification. Here, we summarize the current knowledge and implications regarding the complex roles of Notch signaling during physiological and tumor angiogenesis, the dynamic nature of tip/stalk cell selection in the nascent vessel sprout and arteriovenous differentiation. Furthermore, we shed light on recent work on endothelial cell metabolism, perfusion-independent angiocrine functions of endothelial cells in organ-specific vascular beds and how manipulation of Notch signaling may be used to target the tumor vasculature.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Angiogenesis
- Notch signaling
- Arteriovenous differentiation
- Tumor angiogenesis
- Angiocrine signaling
- Endothelial metabolism
- Endothelial cells
- ADAM:
-
A disintegrin and metalloprotease
- ALK:
-
Activin receptor-like kinase
- BMP:
-
Bone morphogenetic protein
- CADASIL:
-
Cerebral Autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- DLL:
-
Delta-like
- EC:
-
Endothelial cell
- FGF:
-
Fibroblast growth factor
- FOX:
-
Forkhead box protein
- HES:
-
Hairy and enhancer of split
- HEY:
-
Hairy/enhancer-of-split related with YRPW motif
- IL:
-
Interleukin
- NICD:
-
Notch intracellular domain
- NRARP:
-
Notch-regulated ankyrin repeat-containing protein
- NRP:
-
Neuropilin
- PFKFB3:
-
6-Phosphofructo-2-kinase
- PI3K:
-
Phosphatidylinositol 4,5-Bisphosphate 3-kinase
- RBPJ:
-
Recombining binding protein suppressor of hairless
- SHH:
-
Sonic hedgehog
- SMAD:
-
Mothers against decapentaplegic
- TGF:
-
Transforming growth factor
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
1 Introduction
The vasculature comprises one of the largest organs in mammals. Blood vessels nourish all tissues in the body and provide gateways for immune surveillance . In addition, vascular cells provide organ-specific paracrine factors, also termed angiocrine factors, which instruct the behavior of neighboring cells. Angiocrine signaling is essential for the maintenance of homeostasis and metabolism, stem cell differentiation , organ regeneration and tumor progression (Rafii et al. 2016). The importance of the vasculature becomes apparent by studying vascular dysfunction, which is the major contributor to human mortality. Abnormalities in vessel functionality are causative for heart infarction, stroke, neurodegenerative diseases, dementia, diabetic complications and obesity-associated disorders, while excessive blood vessel formation is a hallmark of cancer , chronic inflammation and eye diseases such as wet macular degeneration (Folkman 2007). Drugs that inhibit blood vessel growth have recently become first-line therapies for certain eye and tumor diseases (Carmeliet and Jain 2011) .
Blood vessels are formed by endothelial cells (ECs) , which provide an anti-thrombotic surface, and by mural cells (vascular smooth muscle cells and pericytes). In mature vessels, ECs are in a quiescent state, divide rarely and form barriers between blood and surrounding tissues. ECs have the remarkable capacity to switch between the quiescent and the activated state during injuries, hypoxia, inflammation or tissue growth, when the formation of new blood vessels is required (Potente et al. 2011).
The de novo formation of blood vessels from mesodermal-derived endothelial precursor cells is called vasculogenesis (Risau and Flamme 1995). It occurs predominantly during early development to generate a primordial vascular plexus and the first large vessels such as the dorsal aorta. The vascular plexus is further remodeled and new vessels are formed from the pre-existing ones in a process called angiogenesis (Herbert and Stainier 2011). Similarly to new branches growing on a tree, angiogenesis occurs primarily by sprouting of new branches from existing microvessels. Angiogenesis occurs throughout life as capillaries grow and regress accordingly to functional demands. For example, physical exercise stimulates angiogenesis in skeletal muscle (Hellsten and Hoier 2014) and expansion of adipose tissue is also associated with the formation of new blood vessels (Cao 2010). Intussusception (vessel splitting) is another way of generating new vessels. During this process blood vessels develop transluminal tissue pillars which subsequently fuse resulting in new vascular entities (Makanya et al. 2009). Once the new vessels establish nutrient and oxygen supplies that meet the metabolic tissue demand, the ECs will turn quiescent (Risau 1997).
Notch signaling is of utmost importance for vessel morphogenesis and function. Based on a series of previously published excellent review articles (Blanco and Gerhardt 2013; Carmeliet and Jain 2011; Eilken and Adams 2010; Gridley 2010; Siekmann et al. 2013; Potente et al. 2011), this chapter will summarize the current view about Notch signaling in the vasculature with a focus on vessel sprouting, arteriovenous differentiation , EC metabolism and tumor angiogenesis . We will also highlight recent work showing the tight interconnections of the Notch pathway with other core signaling pathways and its roles for organ-specific angiocrine signaling .
2 Notch Signaling in Endothelial Cells
Canonical Notch signaling requires the interaction of membrane-bound Notch ligands on the signal-sending cell with Notch receptors on the signal-receiving cell to trigger proteolytic cleavages of the Notch receptors. γ-secretase releases the active Notch intracellular domain (NICD) from the cell membrane, which translocates to the nucleus, binds to the transcription factor Rbpj [also known as CSL , CBF1, Su(H) or Lag2] and activates gene expression (Kopan and Ilagan 2009). In principle, expression of the Notch ligands Dll1, Dll4, Jag1 and Jag2 and the Notch receptors Notch1 and Notch4 on ECs has been reported (Hofmann and Luisa Iruela-Arispe 2007). However, one needs to keep in mind that the individual endothelial expression patterns are quite variable in different vascular beds (e.g. Notch signaling is much higher in arterial than venous ECs), and are depending on the developmental state. Compared to the normal, quiescent vasculature in tissues of the adult, the expression of Notch ligands is typically stronger in tumor blood vessels (Patel et al. 2005; Lu et al. 2007; Jubb et al. 2012; Gale et al. 2004; Mailhos et al. 2001; Scehnet et al. 2007). Prototypical Notch1 target genes in ECs are Hey1, Hey2, Hes1, Nrarp, EphrinB2, but also the Notch ligand-encoding gene Dll4 (Dou et al. 2008; Fischer et al. 2004; Taylor et al. 2002; Liu et al. 2006; Krebs et al. 2001; Phng et al. 2009; Lawson et al. 2002; Ridgway et al. 2006; Lobov et al. 2007; Iso et al. 2006; Patel et al. 2005). The latter is quite unusual and suggests a positive Dll4-Notch1 feedback loop in ECs (Diez et al. 2007; Lanner et al. 2013). Notch ligands are also cleaved by the γ-secretase and their intracellular domain enters the nucleus. However, no functional role for a potential “Notch reverse signaling” during angiogenesis could be detected (Liebler et al. 2012; Redeker et al. 2013).
Gene targeting studies in mice revealed that deletion of Dll4 (Duarte et al. 2004; Krebs et al. 2004; Gale et al. 2004), Jag1 (Xue et al. 1999), Notch1 (Huppert et al. 2000; Krebs et al. 2000; Limbourg et al. 2005), Notch1/Notch4 (Krebs et al. 2000), the Notch S2 cleavage enzyme Adam10 (Glomski et al. 2011), components of the γ-secretase complex (Herreman et al. 1999; Li et al. 2003), Rbpj (Krebs et al. 2004), Hey1/Hey2 (Kokubo et al. 2005; Fischer et al. 2004), or a constitutive endothelium-specific expression of activated alleles for Notch1 (Krebs et al. 2010) or Notch4 (Uyttendaele et al. 2001) lead to embryonic lethality with severe vascular remodeling abnormalities and defects in arteriovenous specification. Besides embryonic development, Notch signaling coordinates vascular remodeling also in the adult (Limbourg et al. 2007; Takeshita et al. 2007). Interestingly, the loss of a single Dll4 allele already results in severe angiogenesis defects (Duarte et al. 2004; Gale et al. 2004; Krebs et al. 2004). Dll4 and Vegf-a belong to the very few genes, of which heterozygosity results in a lethal embryonic phenotype.
One could assume that endothelial Notch ligands act in a redundant manner. However, it was shown that they play distinct roles in blood vessel morphogenesis and do not act redundantly (Preuße et al. 2015). Expression of Dll1 on ECs begins later than that of Dll4 during fetal mouse development. While Dll4 is needed to establish arterial cell fate (see below), Dll1 is required for maintenance of arterial cell fate (Sörensen et al. 2009). On the other hand, Jag1 can even antagonize Dll4/Notch1 signaling in ECs during tip/stalk cell selection depending on the glycosylation pattern of Notch1 receptor (Benedito et al. 2009).
3 Sprouting Angiogenesis
The outgrowth of a new vessel branch is stimulated by proangiogenic growth factors, which are released during hypoxia, inflammation, nutrient starvation or from oncogene-transformed cells. These shift the balance between proangiogenic (e.g. VEGF, FGF) and antiangiogenic (e.g. endostatin, angiostatin, tumstatin, soluble VEGFR1) factors towards a proangiogenic outcome, an event termed the “angiogenic switch” (Folkman 1995; Folkman 2007). The most important proangiogenic protein is vascular endothelial growth factor (VEGF-A; hereafter called VEGF). The complex signaling biology of VEGF family members [VEGF-A, -B, -C, −D, −E and placenta growth factor (PlGF)] and VEGF-A splice isoforms has been reviewed elsewhere (Simons et al. 2016). Deletion of Vegf or its receptors in mice leads to embryonic death as consequence of abnormal vascular development (Fong et al. 1995; Dumont et al. 1998; Shalaby et al. 1995; Carmeliet et al. 1996; Ferrara et al. 1996). In the postnatal mouse retina, a Vegf gradient is generated by the already existing astrocyte network that serves as a guiding scaffold for the developing blood vessels (Ruhrberg et al. 2002; Gerhardt et al. 2003).
Angiogenesis is induced by VEGF, which signals through VEGFR2 and VEGFR3 to activate quiescent ECs. Activated ECs protrude filopodia , secrete matrix metalloproteinases to degrade the basement membrane and become invasive (Arroyo and Iruela-Arispe 2010). The breakdown of basement membrane is in particular mediated by EC podosome rosettes (Seano et al. 2014). Podosomes are specialized actin-based structures that degrade extracellular matrix and promote invasive cell migration (Murphy and Courtneidge 2011). The formation of EC podosomes is controlled by VEGF and Notch signaling (Spuul et al. 2016). Furthermore, stimulated ECs release angiopoietin-2 leading to detachment of pericytes. This further allows ECs to invade the surrounding tissue (Augustin et al. 2009). During invasion ECs usually remain connected to the vessel network (Blanco and Gerhardt 2013).
The nascent sprout contains two different cell phenotypes: tip and stalk cells (Fig. 1). The leading tip cell is characterized by its position, its long and dynamic filopodia and its pro-invasive and migratory behavior (Gerhardt et al. 2003), but also its highly glycolytic metabolic activity (De Bock et al. 2013). Similar to axonal growth cones, tip cells integrate attractive and repellent guidance cues (e.g. Semaphorin, Netrin, VEGF or Slit proteins) to define the route in which the new sprout grows (Adams and Eichmann 2010). Guidance is facilitated by actin-rich filopodia on the tip cells, whose formation is driven by VEGF via RhoGTPase signaling. Interestingly, filopodia are not absolutely necessary for migration of ECs as lamellipodia can partially compensate for their function (Phng et al. 2013). It was reported that there can be two cells that extend filopodia and have significant overlap in space and time at the tip of angiogenic sprouts (Pelton et al. 2014). This surprising observation challenges the model of a single EC at the sprout tip. The trailing stalk cells are proliferative, less migratory than tip cells and form the nascent vascular lumen (Gerhardt et al. 2003). Furthermore, tip and stalk cells possess distinct gene expression profiles (e.g. higher expression of Dll4, Vegfr2, Vegfr3, Pdgfb, Unc5b, Cxcr4, Nidogen-2, Esm1, Angiopoietin-2, Apelin in tip cells) (Del Toro et al. 2010; Blanco and Gerhardt 2013). For cell proliferation, stalk cells have to generate biomass (nucleotides, protein, lipids). Therefore, cell metabolism differs between tip and stalk cells (see 3.4). Stalk cells produce extracellular matrix and recruit pericytes that attach to the new vessel sprout (Fig. 1). ECs in new vessel loops that are well covered by mural cells and have again become quiescent were named “phalanx cells” (Mazzone et al. 2009).

Model of tip/stalk cell phenotypes. The leading tip cell protrudes many filopodia and guides the new vessel sprout towards the VEGF gradient. Tip cells are highly invasive and migratory and require high ATP amounts, which are predominantly generated by glycolysis. The trailing stalk cells proliferate and form a new vessel lumen. The newly formed vessel sprout gets covered by extracellular matrix proteins and by pericytes. However, this is a dynamic process and stalk cells battle for the tip position to take over the lead
3.1 VEGF and Notch Signaling Control Tip/Stalk Cell Selection
The ability of ECs to lead a nascent sprout is strongly dependent on their VEGF receptor expression profile and their competence to respond to VEGF (Jakobsson et al. 2010; Gerhardt et al. 2003). While tip cells are characterized by high expression levels of Vegfr2 and also Vegfr3 (Tammela et al. 2008; Tammela et al. 2011; Zarkada et al. 2015; Blanco and Gerhardt 2013), the role of the Vegfr1, which acts as a VEGF trap, is less clear (Siekmann et al. 2013). In zebrafish, Notch-driven Vegfr1 expression acts as a negative regulator of tip cell differentiation (Krueger et al. 2011) and neuronal-derived soluble Vegfr1 is critical for guiding the direction of vessel growth (Wild et al. 2017).
VEGF signaling acts upstream of the Notch pathway and induces Dll4 expression (Lawson et al. 2002; Ridgway et al. 2006; Lobov et al. 2007; Patel et al. 2005). It has been suggested that Vegf acts via the PI3K pathway activating the Forkhead family transcription factors Foxc1 and Foxc2, which then bind to a Dll4 enhancer element, or alternatively via the disassembly of a repressor complex at the Dll4 promoter (Seo et al. 2006; Hayashi and Kume 2008). Subsequently, Dll4 binds and signals to Notch1 receptors on adjacent ECs. The Notch-induced transcription factors Hey1 and Hey2 decrease expression of Vegfr2/3 and thereby reduce responsiveness to VEGF. Such cells will most likely behave as stalk cells (Blanco and Gerhardt 2013). Therefore the nascent sprout is guided by a tip cell with high Dll4 expression and low Notch signaling activity followed by stalk cells with high Notch signaling output (Fig. 2).

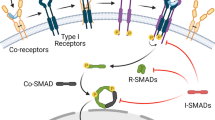
Core signaling pathways during tip/stalk cell selection. VEGF induces tip cell behavior and expression of the Notch ligand DLL4. This leads to NOTCH1 activation in adjacent cells which adopt the stalk cell phenotype. In stalk cells, Notch signaling represses expression of tip cell-enriched genes like VEGFR2/3 and thereby suppress responsiveness to the pro-angiogenic VEGF. Notch inhibits expression of PFKFB3, an activator of glycolysis, which is required to adopt the tip cell phenotype. Moreover, Notch inhibits proliferation via inhibition of p21 but this is counteracted via WNT signaling since stalk cells need to proliferate. In addition, Notch activates expression of the inhibitory SMAD6 proteins to counteract pro-angiogenic BMP2/6 signaling. Notch inhibits NRP1 expression, which suppresses the stalk cell phenotype by limiting SMAD2/3 activation
Studies with genetic or pharmacologic inhibition of Notch signaling underlined the importance of this pathway during sprouting angiogenesis and tip/stalk cell selection. Notch inhibition leads to the formation of excessive tip cell numbers and vessel branches, a process called hypersprouting (Noguera-Troise et al. 2007; Ridgway et al. 2006; Hellström et al. 2007; Lobov et al. 2007; Siekmann and Lawson 2007; Suchting et al. 2007; Sainson et al. 2005; Leslie et al. 2007). Accordingly, ECs with low Notch signaling activity dominate at the tip cell position, whereas Notch-active ECs are mostly excluded (Jakobsson et al. 2010; Hellström et al. 2007; Siekmann and Lawson 2007; Benedito et al. 2009).
Dll4/Notch1 is the most important ligand and receptor pair in coordinating angiogenesis. However, the situation is more complex. For example, stalk cells also express few Dll4 ligands on their membrane and this could potentially lead to signaling back to Notch receptors on tip cells. This is antagonized by the Notch ligand Jag1, which is strongly expressed on stalk cells (Hofmann and Luisa Iruela-Arispe 2007; Benedito et al. 2009) and inhibits Dll4/Notch1 signaling. Thereby, Jag1 antagonizes signaling from the stalk back to the tip cell (Benedito et al. 2009) and it may also prevent Notch over-activation in the stalk cell plexus.
3.2 Crosstalk Between Notch and Other Signaling Pathways to Control Tip/Stalk Cell Selection
Numerous additional molecules influence tip or stalk cell fate selection through interactions with Notch signaling. In brief, WNT/β-catenin signaling promotes transcription of Dll4 by binding to an enhancer element (Corada et al. 2010) or through protein interaction of β-catenin with Rbpj (Yamamizu et al. 2010). Furthermore, WNT signaling induces expression of the transcription factor Sox17, which can activate Notch signaling and promote expression of tip cell-enriched genes (Lee et al. 2014; Corada et al. 2013). On the other hand, Sox17 expression is repressed by Notch signaling in stalk cells (Lee et al. 2014). It was demonstrated, that the mRNA level of Sox17 is not altered by Notch whereas the protein level of Sox17 is. This shows that Sox17 is post-transcriptionally regulated by the Notch pathway. Taken together this indicates that through a negative feedback loop, hypersprouting is prevented. Similarly, Notch and WNT signaling are linked via Nrarp to control the stability of new vessels. Notch induces Nrarp expression, which in turn limits Notch signaling and promotes WNT signaling in stalk cells (Phng et al. 2009).
The competence of ECs to become a tip cell is also influenced by bone morphogenetic proteins (BMPs) and TGF-β signaling. Bmp9 signals through Alk1 in stalk cells to induce Smad1/5/8 phosphorylation. These Smads synergize with activated Notch receptors to induce expression of Notch targets Hey1 and Hey2, which inhibit VEGF receptor expression (Larrivée et al. 2012; Moya et al. 2012). This is further promoted by Smad1/5-mediated induction of Id proteins which augment Hes1 protein levels (Moya et al. 2012). However, the roles of BMP signaling for tip/stalk selection and angiogenesis are not fully defined yet and still controversial. Very recently, it was reported that Notch promotes expression of the inhibitory Smad6 protein and thereby limits the responsiveness of stalk cells towards the proangiogenic Bmp2 and Bmp6 (Mouillesseaux et al. 2016). Lastly, it was reported that the stalk cell phenotype has to be actively repressed to allow tip cell formation. Neuropilin-1 (Nrp1) plays a key role in suppressing the stalk cell phenotype through limiting Smad2/3 activation. Nrp1 promotes tip cell behavior and the formation of filopodia (Fantin et al. 2013; Fantin et al. 2015). Notch downregulates Nrp1 expression and thus promotes stalk cell behavior (Aspalter et al. 2015).
The Notch-dependent acquisition of the stalk cell phenotype also requires the phosphatase Pten (Serra et al. 2015). Furthermore, Dll4 expression in tip cells is regulated via laminin/integrin signaling (Stenzel et al. 2011). Besides crosstalk of Notch signaling with other signaling pathways, direct protein-protein interactions influence tip-stalk-cell selection. Synaptojanin-2-binding protein (Synj2bp) stabilizes Delta-like protein expression in stalk cells to allow continuous Notch signaling within the stalk cell plexus and to prevent formation of ectopic vessel branches (Adam et al. 2013).
3.3 The Dynamic Nature of Tip/Stalk Cell Differentiation
EC tip and stalk cell specification does not represent permanent cell fate decisions but rather dynamic fluctuations in cell phenotypes (Blanco and Gerhardt 2013). The Gerhardt laboratory has shown that stalk cells compete in a highly dynamic manner for the tip position leading to frequent exchange of the tip cells (Jakobsson et al. 2010). Such EC shuffling occurs every few hours (Ubezio et al. 2016). Mechanistically, the VEGF-Dll4/Notch feedback system drives the competition for the tip/stalk cell selection. This is facilitated by the oscillatory output strength of Notch signaling (Kageyama et al. 2007). As such, the expression of Dll4 fluctuates in individual ECs within sprouting vessels (Ubezio et al. 2016). Therefore, one can assume that concomitantly the levels of Vegfrs, Dll4 and Notch target genes change constantly as ECs interact with each other. As a result, the competence of acting as a tip cell changes constantly, certain stalk cells are relieved from tip cell inhibition and overtake the lead position (Blanco and Gerhardt 2013). This leads to a dynamic position shuffle in the growing sprout.
The tip cell competence concept is further strengthened by the finding that the continual flux in Notch signaling output strength in individual ECs results in differential VE-cadherin turnover to generate spatial differentials in cell-cell adhesions and polarized junctional protrusions. These permanent switches between active and inactive cell junctions allow EC rearrangements during sprout elongation (Bentley et al. 2014).
3.4 Control of Angiogenesis by Metabolism
The vasculature contributes to systemic metabolism control. On the one hand the endothelium controls the shuttling of nutrients from blood to tissue cells in an organ-specific manner (Robciuc et al. 2016; Jais et al. 2016; Hagberg et al. 2010; Corvera and Gealekman 2014) and therefore plays a critical, but poorly understood role, for organ homeostasis. On the other hand, metabolism controls angiogenesis. For example, the expansion of adipose tissue requires angiogenesis, which is stimulated by proangiogenic factors released from adipocytes (Corvera and Gealekman 2014). ECs contain metabolic sensors and their effectors (Sirtuins, mTOR, Pgc1α, Lkb1, Ampk, Foxos and Sirt1) (Potente and Carmeliet 2017) and respond to alteration in nutrient supply. To understand how cellular metabolism affects angiogenesis, one needs to consider how ECs generate ATP. Research from the Carmeliet laboratory revealed that ECs are very glycolytic and produce the majority of ATP by metabolizing glucose into lactate rather than by oxidative phosphorylation, even if plenty of oxygen is available (De Bock et al. 2013). As such, ECs behave similar to cancer cells, which consume high amount of glucose for aerobic glycolysis (Schulze and Harris 2012). Although much less ATP is gained compared to oxidative phosphorylation, glycolysis has the advantage of generating ATP in a very rapid manner and glycolysis allows energy production in hypoxic areas, into which angiogenic ECs need to migrate (Potente and Carmeliet 2017).
Activated ECs require in particular high glycolytic flux for migration and invasion (De Bock et al. 2013; Cruys et al. 2016). This is facilitated by VEGF and hypoxia signaling that together increase the uptake and breakdown of glucose by up-regulating glucose transporter type-1 and glycolytic enzymes, such as 6-Phosphofructo-2-kinase (Pfkfb3) and lactate dehydrogenase-A (Yeh et al. 2008; Peters et al. 2009; Nakazawa et al. 2016; De Bock et al. 2013). Even in ECs with constitutive Notch1 signaling, which are genetically determined to become stalk cells, enhanced glycolysis by Pfkfb3 activation induces tip cell behavior (De Bock et al. 2013). This indicates that EC metabolism can exert control over genetic circuits (Potente and Carmeliet 2017).
In stalk cells, Notch signaling reduces but not eliminates the expression of Pfkfb3 and Pfkfbp3-driven glycolysis, as it is also essential for stalk cells (De Bock et al. 2013). Moreover, stalk cells must synthesize all cellular components (e.g. nucleotides, proteins and lipids) for cell division and cell growth. Therefore, ECs also break down fatty acids to generate carbons for the de novo nucleotide synthesis and not only for energy production (Schoors et al. 2015).
It will have to be determined how exactly the metabolic status influences the EC genetic program and vice versa. Fluctuations of Notch and VEGF signaling outputs alter glycolysis rates and ATP production in ECs and thereby change the fitness of ECs to battle for the tip position (Spuul et al. 2016; Potente and Carmeliet 2017; De Bock et al. 2013). The energy status also controls the activity of Foxo1 by Sirt1 and the latter inhibits Notch signaling through deacetylation of the Nicd1 resulting in increased angiogenesis (Guarani et al. 2011). Latest research showed that Foxo1 is an essential regulator of vascular growth by coupling metabolic and proliferative activities in ECs via inhibition of Myc , which fuels glycolysis and mitochondrial metabolism (Wilhelm et al. 2016). In addition, the Notch signaling activity in ECs is influenced by plasma glucose levels (Yoon et al. 2014) and by the presence of certain pro-inflammatory fatty acids (Briot et al. 2015). Taken together, these reports show that Notch signaling integrates angiogenic signaling with the metabolic status.
3.5 Anastomosis of Vessel Sprouts and Remodeling of the New Vessel Network
Newly formed sprouts need to connect with other sprouts or existing vessels to generate a new circulatory loop. Anastomosis is a complex process that has not yet been fully resolved (Betz et al. 2016). Tip cells contact other tip cells to initiate fusing of two sprouts (Isogai et al. 2003), which is supported by tissue-resident macrophages (Tammela et al. 2011; Fantin et al. 2010; Outtz et al. 2011). Anastomosis requires the formation of new VE-Cadherin-containing EC junctions to consolidate the connection (Bentley et al. 2014). Such junctions are essential for EC polarization and lumen formation. After formation of a patent lumen, blood flow contributes to stabilize the new vascular loop. Increasing oxygen tension decreases VEGF production and helps to switch the activated EC status into a quiescent one. Further vessel maturation includes production of extracellular matrix, recruitment of mural cells, remodeling into a hierarchical network and the pruning of excessive vessel branches (Potente et al. 2011). Notch signaling is critically involved in the recruitment and the tight interactions of ECs with pericytes and smooth muscle cells (Fouillade et al. 2012). Further research is required to elucidate the detailed mechanisms of how Notch signaling is involved in vessel pruning.
4 Arteriovenous Differentiation
After the assembly of the first primitive vessels in the embryo or in a growing tissue of the adult (e.g. muscle or adipose tissue) a rapid differentiation into a hierarchically organized network of arteries, capillaries, veins and lymphatic vessels occurs. The specification of lymphatics has been reviewed elsewhere (Yang and Oliver 2014). Arteries transport blood away from the heart towards the capillaries. As such, arterial vessels are subjected to high blood pressure and pulsatile shear stress, whereas veins face low-pressure gradients can contain valves to prevent backflow and are more distensible than arteries (Corada et al. 2014).
Several studies indicated that vascular progenitor cells, which form the first large vessels in the embryo, are already committed for arterial or venous cell fate (Quillien et al. 2014; Kohli et al. 2013). On the other hand, it was shown that venous-fated EphB4-positive ECs migrate away from arterial-fated EphrinB2-positive ECs in mixed vessels to establish the first artery and vein (Lindskog et al. 2014; Herbert et al. 2009). Subsequently, new branches sprout out of the first arteries and veins. Time-lapse movies of zebrafish embryos demonstrated that vessel sprouts can disconnect from the originating vein and reconnect with the adjacent artery (Betz et al. 2016). Also tip cells from venous sprouts can migrate backwards and incorporate into newly formed arteries in mice and fish (Xu et al. 2014). This suggests that the arteriovenous cell fate is not terminally defined in the early stage of development.
4.1 Arterial Differentiation
Vascular remodeling can occur in absence of blood flow and is largely determined by genetic factors whereby the VEGF and Notch pathways play key roles. Arterial and venous ECs possess specific molecular identities such as EphrinB2 expression exclusively in arterial and EphB4 exclusively in venous beds (Wang et al. 1998). Notch pathway components are expressed at much higher levels in arterial than venous ECs (Villa et al. 2001; Claxton and Fruttiger 2004) and are major players during embryonic arterial differentiation (Gridley 2010; Swift and Weinstein 2009). This was demonstrated by gene targeting approaches in mouse and zebrafish, which revealed that disruption of the Notch pathway does not only lead to impaired vessel sprouting but also to poorly formed arterial vessels, loss of arterial markers (e.g. EphrinB2, Hey2, Cxcr4, Cx40, Nrp1) and/or ectopic expression of venous markers (e.g. EphB4, COUP-TFII (Nr2f2), Nrp2) (Lawson et al. 2001; Zhong et al. 2001; Zhong et al. 2000; Fischer et al. 2004; Lawson et al. 2002; Duarte et al. 2004; Krebs et al. 2004; Sörensen et al. 2009).
Dll4-mediated Notch signaling induces expression of arterial-specific genes (Kim et al. 2008; Iso et al. 2006) and suppresses the expression of the master regulator of venous specification, COUP-TFII (Swift et al. 2014). Dll1 plays a distinct role. Dll1 is expressed selectively on fetal mouse arteries and is not required for the establishment but for the maintenance of arterial identity and VEGF receptor expression (Sörensen et al. 2009). It should be taken into account that blood pressure, blood flow dynamics and hypoxia are also important for the proper differentiation and the maintenance of arteriovenous identity (Le Noble et al. 2005; Lanner et al. 2013; Diez et al. 2007).
Once the circulatory system is formed and fully functional, the arteriovenous fate needs to be actively maintained to prevent the formation of arteriovenous shunts. Arteriovenous malformations in the brain are an important cause of intracerebral hemorrhage in young adults (Lawton et al. 2015). Increased NOTCH1 activity has been observed in human arteriovenous malformations (Murphy et al. 2009; Zhuge et al. 2009). Based on gene targeting approaches, Notch signaling appears to be involved in its pathogenesis. Interestingly, both endothelial-specific inhibition and over-activation of Notch signaling can lead to the formation of arteriovenous malformations at least in certain vascular beds (Trindade et al. 2008; Carlson et al. 2005; Miniati et al. 2010; Murphy et al. 2012; Murphy et al. 2014; Murphy et al. 2008; Gale et al. 2004; Murphy et al. 2009).
Besides maintaining arterio-venous identity, Notch signaling is required to maintain integrity of vascular smooth muscle cells. Neomorphic mutations in NOTCH3 , which often lead to unequal numbers of cysteine residues in the extracellular domain , cause cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). This leads to degeneration of vascular smooth muscle cells in small-sized arteries, changes in brain blood perfusion that cause migraine attacks, stroke and dementia. Gene targeting experiments have shown that mice carrying a CADASIL-causing Notch3 point mutation displayed attenuated myogenic responses and reduced caliber of brain arteries as well as impaired cerebrovascular autoregulation and functional hyperemia (Chabriat et al. 2009; Joutel et al. 2010).
4.2 Venous Specification
It was previously believed that venous differentiation is the default differentiation pathway in the absence of Notch activation. However, mouse knockout studies revealed a pivotal role for the transcription factor COUP-TFII, which is exclusively expressed in venous and lymphatic ECs to establish venous fate (You et al. 2005). Interestingly, Notch signaling suppresses COUP-TFII expression, most likely via Hey transcriptional repressors , and thereby allows arterial fate specification (Swift et al. 2014). In summary, it appears likely that Notch and COUP-TFII repress each other to allow the establishment of the arterial or venous gene expression programs, respectively.
4.3 Upstream Regulators of Notch During Arteriovenous Differentiation
It still remains unclear what mechanisms act upstream of Notch signaling in early phases of arteriovenous differentiation. Hypoxia might play an important role by inducing DLL4 expression (Diez et al. 2007; Patel et al. 2005). In zebrafish, Shh and Vegf-a act upstream of Notch to promote arterial differentiation. Alternatively, Shh might promote arterial differentiation independently of VEGF signaling via the calcitonin receptor-like receptor (Wilkinson et al. 2012). In mammals, neurons or glial cells release VEGF to support arterial differentiation. VEGF signaling via Erk induces transcription of Dll4 and arterial-specific genes (Deng et al. 2013; Ren et al. 2010). However, VEGF signaling can also induce Pi3k activity, which has an opposite effect on arterial morphogenesis (Hong et al. 2008; Ren et al. 2010), indicating that other factors are needed to fine-tune VEGF signaling branches . Neuropilin-1, which is more abundantly expressed on arterial than venous ECs, could be one of these factors as it promotes Vegfr2 trafficking and Erk signaling (Lanahan et al. 2013).
Besides VEGF signaling, the expression of Dll4 during arterial differentiation is also promoted by SoxF transcription factors (Corada et al. 2013; Sacilotto et al. 2013), WNT/β-catenin signaling (Corada et al. 2010; Yamamizu et al. 2010), angiopoietin-1 (Zhang et al. 2011) and the transcription factors Foxc1 and Foxc2 (Seo et al. 2006; Hayashi and Kume 2008). Lastly, it should be noted that also blood flow dynamics induce the expression of Notch pathway components and other arterial-specific genes in cultured ECs (Lehoux and Jones 2016) and endothelial cells in mice (Ramasamy et al. 2016). Furthermore, studies using cultured cells have shown that such physiologic forces can sensitize the negative regulatory region of Notch1 to ADAM-mediated cleavage (Gordon et al. 2015). As such, a large amount of genetic and environmental factors promote EC Notch signaling to enable and maintain arterial morphogenesis.
5 Organ-Specific Vascular Beds and Angiocrine Signaling
A major challenge for the research field will be the analysis of organ-specific vascular beds. Blood vessel anatomy and function differs dramatically between organs and even within the same organ (e.g. the fenestrated endothelium in kidney glomeruli vs. the continuous endothelium in peritubular capillaries). The tightness of vessels is adapted to the organ-specific requirements with e.g. tight EC connections in the central nervous system and gaps (fenestrations) in the sinusoidal endothelium of liver, endocrine organs or bone marrow (Aird 2007). Little is known so far regarding how these differences are established during development and maintained throughout life. This is, however, of utmost importance. For example, treatment of mice with tyrosine kinase inhibitors targeting VEGF receptors led to pronounced regression of fenestrated capillaries, that are typically present in endocrine organs and that under normal conditions express high levels of Vegfr2/3 (Kamba et al. 2006). Similar data were obtained in pancreatic islets by a genetic approach (Lammert et al. 2003), indicating that VEGF acts as a survival factor for fenestrated capillaries in endocrine organs.
Several angiocrine functions have recently been described in which ECs control organ development and regeneration by secreting e.g. growth factors or by providing niches and cell surface molecules for hematopoietic stem cells or tumor cells (Rafii et al. 2016). Here we focus on such examples in which the Notch pathway is critically involved.
Work from the Adams laboratory gave fascinating insights on how blood vessels orchestrate the formation, function and remodeling of bone (Kusumbe et al. 2014). In contrast to other organs, active Notch signaling in bone ECs promotes blood vessel growth. Furthermore, Notch regulates the angiocrine release of Noggin, which is involved in bone growth, mineralization and chrondrocyte maturation (Ramasamy et al. 2014). It is known that many diseases lead to impaired skeletal blood flow. Interestingly, flow-responsive genes induce endothelial Notch signaling in bone. Therefore, impaired blood flow hampers osteogenesis and rejuvenation of bone through impaired EC Notch signaling and decreased angiogenesis (Ramasamy et al. 2016).
In the liver, Notch1 is important to maintain quiescence and morphology of the specialized sinusoidal vasculature. Disruption of Notch1 using the rather tissue-unspecific Mx-Cre line led to de-differentiation of sinusoidal ECs, vascular remodeling, detachment of mural cells and intussusceptive angiogenesis (Dill et al. 2012; Dimova et al. 2013). In the bone marrow, Jag1 expression on ECs is important for hematopoietic stem cell differentiation (Poulos et al. 2013) and niche-forming vessels can be restored by activation of EC Notch signaling (Kusumbe et al. 2016). In the lung, Jag1 expressed on pulmonary capillary ECs induces Notch signaling in perivascular fibroblasts and thereby enhances lung fibrosis (Cao et al. 2016).
Lastly, we want to emphasize that aside from their role in angiogenesis, tumor ECs possess additional roles. ECs within a solid tumor mass are in close contact with tumor cells and many immune cells and their released cytokines. As such, tumor ECs often do not form tight barriers any more, exhibit altered gene expression programs and also actively alter the behavior of adjacent cells in the tumor microenvironment. In this regard, ECs can provide several membrane-bound and secreted factors that promote tumor progression (Butler et al. 2010). Notch ligands of the Delta-like and Jagged families are frequently present on tumor ECs and can promote Notch signaling in adjacent tumor cells. This increased aggressiveness of lymphoma cells (Cao et al. 2014), promotes the cancer stem cell phenotype (Lu et al. 2013; Zhu et al. 2011), increases tumor cell survival (Pedrosa et al. 2015) and facilitates metastasis (Sonoshita et al. 2011). Interestingly, Notch ligands can also be secreted by tumor cells via exosomes and be incorporated in EC membranes at distant sites to either activate or inhibit Notch signaling (Sharghi-Namini et al. 2014; Sheldon et al. 2010). Furthermore, Notch activation in ECs can be driven by inflammation and this in turn contributes to increased expression of leukocyte adhesion molecules (Liu et al. 2012; Verginelli et al. 2014). Work from our group showed that sustained NOTCH1 activation in ECs leads to senescence, expression of adhesion molecules and weakening of cell junctions that promote transmigration and homing of circulating tumor cells (Wieland et al. 2017).
6 Tumor Angiogenesis and Notch Targeting Agents
Angiogenesis is a hallmark of cancer (Hanahan and Weinberg 2011). The growth of small tumor cell clumps into a clinically relevant tumor is only possible by the induction of blood vessel growth into the tumor mass. Tumor vessels have an abnormal structure and often function poorly. The endothelial lining contains gaps and disorganized cell-cell junction integrity. Also the coverage with pericytes is frequently impaired making vessels leaky. This increases interstitial pressure what impairs the transport of nutrients and drugs towards tumor cells. Moreover, vascular leakiness facilitates intravasation of tumor cells and dissemination (Goel et al. 2011). The tumor vasculature lacks a strict hierarchical structure, arteriovenous identity is poorly defined, vessels have irregular lumen sizes, are often tortuous shaped and thin-walled. Both hyper-vascularized and poorly vascularized tumor areas accompany tumor heterogeneity . Irregular vessel branches, shunts, blind-ended branches, weak vessel contractility and irregular lumen sizes together lead to abnormal and very heterogeneous perfusion rates. Irregular perfusion impairs oxygen, nutrient and drug delivery, thereby limiting the efficiency of chemotherapy and radiation. Impaired perfusion causes aggravation, as hypoxic tumor areas secrete even higher amounts of proangiogenic factors leading to the formation of even more chaotic vessel structures with increased permeability (Carmeliet and Jain 2011; Potente et al. 2011).
VEGF targeting substances are in clinical use but show limited efficiency (Carmeliet and Jain 2011; Potente et al. 2011). Anti-VEGF drugs inhibit the formation of new vessel sprouts and also induce regression of pre-existing tumor vessels, in particular immature vessels. It is assumed that the mode of action is not starving the tumor to death but rather to normalize the tumor vasculature by regression of immature vessels and maturation of the remaining ones. The normalized tumor vasculature is better perfused and enables better delivery of cytotoxic agents to tumor cells (Goel et al. 2011). It is assumed that many initially sensitive tumors develop resistance against VEGF-targeting drugs by secretion of other proangiogenic proteins (e.g. FGF2, PDFG, PlGF, IL-8, ANG2) and by other means of vessel formation (e.g. cooption of already existing vessels) (Bergers and Hanahan 2008). This indicates that better combination therapies are required to target the tumor vasculature.
Besides VEGF, Notch signaling is an interesting target . As in physiological angiogenesis, Notch signaling is involved in tumor angiogenesis (Noguera-Troise et al. 2006; Ridgway et al. 2006; Lobov et al. 2007). However, the pathological high VEGF concentrations may disrupt oscillatory Notch signaling outputs and thereby impair the formation of proper cell junctions and promote vessel expansion (Bentley et al. 2014; Ubezio et al. 2016). Dll4 and Jag1 are abundantly expressed on tumor vessels (Patel et al. 2005; Lu et al. 2007; Jubb et al. 2012; Gale et al. 2004; Mailhos et al. 2001; Scehnet et al. 2007) and tumor vessels often exhibit strong Notch1 activity (Fig. 3). By computational modeling, it was suggested that the higher production levels of the antagonistic ligand Jag1 give rise to a hybrid tip/stalk phenotype that leads to poorly perfused vessels (Boareto et al. 2015).

Notch signaling is active in blood vessels of adult and tumor blood vessels. Sections of normal lung and lung adenocarcinoma were stained against the endothelial marker CD34 (brown color) or the cleaved NOTCH1 receptor (NOTCH1-ICD). Cell nuclei were counterstained with hematoxylin (blue color). Magnification 400-fold
Manipulation of EC Notch signaling appears to be an attractive target to interfere with tumor progression. Notch signaling is often hyperactive in cancer cells (in particular in the cancer stem cells) and acts as an oncogene in many tumor entities. Therefore, Notch inhibition could target tumor cells and tumor vessels simultaneously. Many academic groups and pharmaceutical companies have developed Notch inhibiting substances and several ones are in phase I/II trials (Andersson and Lendahl 2014). In rodent models, blockade of Dll4, Notch1 or γ-secretase leads to a non-productive hypersprouting phenotype resulting in central tumor necrosis (Noguera-Troise et al. 2006; Ridgway et al. 2006; Scehnet et al. 2007). This may sound paradoxical, but the excessive vessel branches generate such a chaotic network that dramatically diminishes tumor perfusion . Whether this can also be achieved in human cancer patients is not clear yet. γ-secretase inhibitors, which block the activity of all four Notch receptors, have quite profound adverse effects (e.g. gastrointestinal toxicity) in clinical trials (Andersson and Lendahl 2014) but neutralizing antibodies against individual Notch receptors might be able to overcome this (Wu et al. 2010). In addition, antibodies targeting individual Notch ligands have also been developed (Andersson and Lendahl 2014). Nevertheless, DLL4-neutralizing antibody can also cause severe adverse effects (Yan et al. 2010), e.g. development of congestive heart failure was observed in clinical phase I studies (Chiorean et al. 2015; Falchook et al. 2015; Smith et al. 2014). It will be important to study the underlying mechanisms to overcome this problem.
As outlined above, it appears to be more reasonable to induce tumor vessel normalization instead of tumor vessel regression. A novel approach to achieve this might be targeting EC metabolism . ECs are highly glycolytic and high rates of glucose breakdown are instrumental for adopting the tip cell phenotype during sprouting. A rather mild inhibition of glycolysis can be achieved by targeting its activator Pfkfb3. In mouse cancer models, Pfkfb3 inhibition tightened the vascular barrier, improved adhesion of pericytes and reduced the pro-inflammatory phenotype of tumor ECs that facilitates metastasis (Cantelmo et al. 2016). Another option to normalize the tumor vasculature could be the activation of EC Notch signaling. As shown by genetic approaches in mice, this reduced tumor angiogenesis, but increased vessel diameter and improved perfusion and oxygenation (Li et al. 2007). Notch activation could also help to reduce glycolysis in ECs as Notch signaling reduces expression of Pfkfb3 (De Bock et al. 2013). While Notch-inhibiting substances are in clinical trials, we still lack fully validated drugs to activate Notch signaling in a therapeutic manner. The Kitajewski laboratory has generated soluble Notch1 extracellular domain proteins fused to IgG-Fc (Notch decoys) that bind and inhibit selectively either the stimulatory Delta-like or the inhibitory Jagged ligands (Funahashi et al. 2008; Kangsamaksin et al. 2015). These Notch decoys inhibit sprouting angiogenesis and also target pericytes in the vessel wall (Klose et al. 2015; Funahashi et al. 2008; Kangsamaksin et al. 2015). Future experiments will determine whether Notch-activating substances can be used successfully in combination with chemotherapy to better target tumor cells.
7 Perspectives
In recent years there has been a significant progress in the understanding of Notch signaling during sprouting angiogenesis. However, much remains to be learned. As the tip/stalk cell selection is tightly dependent on subtle fluctuations in Notch signal output strengths, it is necessary to determine multiple genetic and environmental factors, such as hemodynamics and metabolites that fine-tune ligand expression and localization at the cell surface, receptor glycosylation, NICD protein stability, nuclear NICD complex formation and the dynamic control of Notch target gene expression.
Inducible tissue-specific transgene models and therapeutic antibodies will be key to determine how VEGF and Notch signaling are involved in organ-specific angiogenesis , maintenance of EC quiescence, as well as barrier and transport functions throughout life. There is already solid evidence that VEGF does not only control blood vessel formation, but also acts as a survival factor for ECs (Domigan et al. 2015) and non-vascular cells (Mackenzie and Ruhrberg 2012). Similar to this, basal Dll4/Notch activity has been detected in quiescent ECs (Zhang et al. 2011) and is important to maintain vascular integrity and function (Liu et al. 2011; Yan et al. 2010). Lastly, angiocrine functions of ECs have attracted enormous attention (Rafii et al. 2016). It will be fascinating to see how ECs control the function of parenchymal cells through the secretion of signaling molecules or through providing membrane-bound factors that orchestrate the behavior of its neighboring cells in organ-specific vascular beds.
References
Adam MG et al (2013) Synaptojanin-2 binding protein stabilizes the Notch ligands DLL1 and DLL4 and inhibits sprouting angiogenesis. Circ Res 113(11):1206–1218
Adams RH, Eichmann A (2010) Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol 2(5):a001875
Aird WC (2007) Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 100(2):158–173
Andersson ER, Lendahl U (2014) Therapeutic modulation of Notch signalling--are we there yet? Nat Rev Drug Discov 13(5):357–378
Arroyo AG, Iruela-Arispe ML (2010) Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc Res 86(2):226–235
Aspalter IM et al (2015) Alk1 and Alk5 inhibition by Nrp1 controls vascular sprouting downstream of Notch. Nat Commun 6:7264
Augustin HG et al (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin - Tie system. Nat Rev Mol Cell Biol 10(3):165–177
Benedito R et al (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137(6):1124–1135
Bentley K et al (2014) The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol 16(4):309–321
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8):592–603
Betz C et al (2016) Cell behaviors and dynamics during angiogenesis. Development 143(13):2249–2260
Blanco R, Gerhardt H (2013) VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med 3(1):a006569
Boareto M et al (2015) Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. PNAS 112(29):E3836–E3844
Briot A et al (2015) Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J Exp Med 212(12):2147–2163
Butler JM, Kobayashi H, Rafii S (2010) Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 10(2):138–146
Cantelmo AR et al (2016) Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30(6):968–985
Cao Y (2010) Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov 9(2):107–115
Cao Z et al (2014) Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 25(3):350–365
Cao Z et al (2016) Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat Med 22(2):154–162
Carlson TR et al (2005) Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. PNAS 102(28):9884–9889
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Carmeliet P et al (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380(6573):435–439
Chabriat H et al (2009) CADASIL. Lancet Neurol 8(7):643–653
Chiorean EG et al (2015) A phase I first-in-human study of Enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res 21(12):2695–2703
Claxton S, Fruttiger M (2004) Periodic Delta-like 4 expression in developing retinal arteries. Gene Expr Patterns 5(1):123–127
Corada M et al (2010) The Wnt/β-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/notch signaling. Dev Cell 18(6):938–949
Corada M et al (2013) Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun 4:2609
Corada M, Morini MF, Dejana E (2014) Signaling pathways in the specification of arteries and veins. Arterioscler Thromb Vasc Biol 34(11):2372–2377
Corvera S, Gealekman O (2014) Adipose tissue angiogenesis: impact on obesity and type-2 diabetes. Biochim Biophys Acta Mol basis Dis 1842(3):463–472
Cruys B et al (2016) Glycolytic regulation of cell rearrangement in angiogenesis. Nat Commun 7:12240
De Bock K et al (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663
Del Toro R et al (2010) Identification and functional analysis of endothelial tip cell-enriched genes. Blood 116(19):4025–4033
Deng Y et al (2013) Endothelial RAF1 / ERK activation regulates arterial morphogenesis Endothelial RAF1 / ERK activation regulates arterial morphogenesis. Blood 121(19):3988–3997
Diez H et al (2007) Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res 313(1):1–9
Dill MT et al (2012) Disruption of Notch1 induces vascular remodeling, intussusceptive angiogenesis, and angiosarcomas in livers of mice. Gastroenterology 142(4):967–977.e2
Dimova I et al (2013) Inhibition of Notch signaling induces extensive intussusceptive neo-angiogenesis by recruitment of mononuclear cells. Angiogenesis 16(4):921–937
Domigan CK et al (2015) Autocrine VEGF maintains endothelial survival through regulation of metabolism and autophagy. J Cell Sci 128(12):2236–2248
Dou G-R et al (2008) RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J 22(5):1606–1617
Duarte A et al (2004) Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev 18(20):2474–2478
Dumont DJ et al (1998) Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282(5390):946–949
Eilken HM, Adams RH (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22(5):617–625
Falchook G et al (2015) Phase 1 study of MEDI0639 in patients with advanced solid tumors. J Clin Oncol 33(suppl):abstr 3024
Fantin A et al (2010) Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116(5):829–840
Fantin A et al (2013) NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 121(12):2352–2362
Fantin A et al (2015) NRP1 regulates CDC42 activation to promote Filopodia formation in endothelial tip cells. Cell Rep 11(10):1577–1590
Ferrara N et al (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380(6573):439–442
Fischer A et al (2004) The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev 18(8):901–911
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1(1):27–31
Folkman J (2007) Is angiogenesis an organizing principle in biology and medicine? J Pediatr Surg 42(1):1–11
Fong G-HH et al (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376(6535):66–70
Fouillade C et al (2012) Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res 95(2):138–146
Funahashi Y et al (2008) A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res 68(12):4727–4735
Gale NW et al (2004) Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. PNAS 101(45):15949–15954
Gerhardt H et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161(6):1163–1177
Glomski K et al (2011) Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood 118(4):1163–1174
Goel S et al (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121
Gordon WR et al (2015) Mechanical Allostery: evidence for a force requirement in the proteolytic activation of notch. Dev Cell 33(6):729–736
Gridley T (2010) Notch signaling in the vasculature. Curr Top Dev Biol 92:277–309
Guarani V et al (2011) Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473(7346):234–238
Hagberg CE et al (2010) Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464(7290):917–921
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hayashi H, Kume T (2008) Foxc transcription factors directly regulate DLL4 and hey2 expression by interacting with the VEGF-notch signaling pathways in endothelial cells. PLoS One 3(6):1–9
Hellsten Y, Hoier B (2014) Capillary growth in human skeletal muscle: physiological factors and the balance between pro-angiogenic and angiostatic factors. Biochem Soc Trans 42(6):1616–1622
Hellström M et al (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 1(3):133–136
Herbert SP, Stainier DYR (2011) Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12(9):551–564
Herbert SP et al (2009) Arterial-venous segregation by selective cell sprouting: an alternative mode of blood vessel formation. Science 326(5950):294–298
Herreman A et al (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. PNAS 96(21):11872–11877
Hofmann JJ, Luisa Iruela-Arispe M (2007) Notch expression patterns in the retina: an eye on receptor-ligand distribution during angiogenesis. Gene Expr Patterns 7(4):461–470
Hong CC, Kume T, Peterson RT (2008) Role of crosstalk between phosphatidylinositol 3-kinase and extracellular signal-regulated kinase/mitogen-activated protein kinase pathways in artery-vein specification. Circ Res 103(6):573–579
Huppert SS et al (2000) Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature 405(June):966–970
Iso T et al (2006) Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem Biophys Res Commun 341(3):708–714
Isogai S et al (2003) Angiogenic network formation in the developing vertebrate trunk. Development 130(21):5281–5290
Jais A et al (2016) Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 166(5):1338–1340
Jakobsson L et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12(10):943–953
Joutel A et al (2010) Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Investig 120(2):433–445
Jubb AM et al (2012) Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology 60(5):740–747
Kageyama R, Masamizu Y, Niwa Y (2007) Oscillator mechanism of Notch pathway in the segmentation clock. Dev Dyn 236(6):1403–1409
Kamba T et al (2006) VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol 290(2):H560–H576
Kangsamaksin T et al (2015) NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov 5(2):182–197
Kim YH et al (2008) Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 135(22):3755–3764
Klose R et al (2015) Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiovasc Res 107(1):153–163
Kohli V et al (2013) Arterial and venous progenitors of the major axial vessels originate at distinct locations. Dev Cell 25(2):196–206
Kokubo H et al (2005) Mouse hesr1 and hesr2 genes are redundantly required to mediate Notch signaling in the developing cardiovascular system. Dev Biol 278(2):301–309
Kopan R, Ilagan MXG (2009) The canonical notch signaling pathway: unfolding the activation mechanism. Cell 137(2):216–233
Krebs LT et al (2000) Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14(11):1343–1352
Krebs LT et al (2001) The Nrarp gene encodes an ankyrin-repeat protein that is transcriptionally regulated by the notch signaling pathway. Dev Biol 238(1):110–119
Krebs LT et al (2004) Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev 18(20):2469–2473
Krebs LT et al (2010) Notch1 activation in mice causes arteriovenous malformations phenocopied by EphrinB2 and EphB4 mutants. Genesis 48(3):146–150
Krueger J et al (2011) Flt1 acts as a negative regulator of tip cell formation and branching morphogenesis in the zebrafish embryo. Development 138(10):2111–2120
Kusumbe AP, Ramasamy SK, Adams RH (2014) Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507(7492):323–328
Kusumbe AP et al (2016) Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532(7599):380–384
Lammert E et al (2003) Role of VEGF-A in vascularization of pancreatic islets. Curr Biol 13(12):1070–1074
Lanahan A et al (2013) The Neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent Arteriogenesis. Dev Cell 25(2):156–168
Lanner F et al (2013) Hypoxia-induced arterial differentiation requires adrenomedullin and notch signaling. Stem Cells Dev 22(9):1360–1369
Larrivée B et al (2012) ALK1 signaling inhibits angiogenesis by cooperating with the notch pathway. Dev Cell 22(3):489–500
Lawson ND et al (2001) Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128(19):3675–3683
Lawson ND, Vogel AM, Weinstein BM (2002) Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3(1):127–136
Lawton MT et al (2015) Brain arteriovenous malformations. Nat Rev Dis Prim 1(Table 1):15008
Le Noble F et al (2005) Control of arterial branching morphogenesis in embryogenesis: go with the flow. Cardiovasc Res 65(3):619–628
Lee SH et al (2014) Notch pathway targets proangiogenic regulator Sox17 to restrict angiogenesis. Circ Res 115(2):215–226
Lehoux S, Jones EA (2016) Shear stress, arterial identity and atherosclerosis. Thromb Haemost 115(3):467–473
Leslie JD et al (2007) Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 134(5):839–844
Li T et al (2003) Nicastrin is required for assembly of Presenilin/γ-Secretase complexes to mediate notch signaling and for processing and trafficking of β-amyloid precursor protein in mammals. J Neurosci 23(8):3272–3277
Li JL et al (2007) Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res 67(23):11244–11253
Liebler SS et al (2012) No evidence for a functional role of bi-directional notch signaling during angiogenesis. PLoS One 7(12):1–10
Limbourg FP et al (2005) Essential role of endothelial Notch1 in angiogenesis. Circulation 111(14):1826–1832
Limbourg A et al (2007) Notch ligand Delta-like 1 is essential for postnatal arteriogenesis. Circ Res 100:363–371
Lindskog H et al (2014) Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development 141(5):1120–1128
Liu ZJ et al (2006) Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J 20(7):1009–1011
Liu Z et al (2011) Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. J Clin Investig 121(2):800–808
Liu Z-J et al (2012) Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis 225(2):296–303
Lobov IB et al (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. PNAS 104(9):3219–3224
Lu C et al (2007) Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res 67(4):1757–1768
Lu J et al (2013) Endothelial cells promote the colorectal Cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 23(2):171–185
Mackenzie F, Ruhrberg C (2012) Diverse roles for VEGF-A in the nervous system. Development 139(8):1371–1380
Mailhos C et al (2001) Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differ Res Biol Divers 69(2–3):135–144
Makanya AN, Hlushchuk R, Djonov VG (2009) Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 12(2):113–123
Mazzone M et al (2009) Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136(5):839–851
Miniati D et al (2010) Constitutively active endothelial Notch4 causes lung arteriovenous shunts in mice. Am J Physiol Lung Cell Mol Physiol 298(2):L169–L177
Mouillesseaux KP et al (2016) Notch regulates BMP responsiveness and lateral branching in vessel networks via SMAD6. Nat Commun 7:13247
Moya IM et al (2012) Stalk cell phenotype depends on integration of notch and Smad1/5 signaling cascades. Dev Cell 22(3):501–514
Murphy DA, Courtneidge SA (2011) The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 12(7):413–426
Murphy P a et al (2008) Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. PNAS 105(31):10901–10906
Murphy P a et al (2009) Endothelial notch signaling is upregulated in human brain arteriovenous malformations and a mouse model of the disease. Lab Investig 89(9):971–982
Murphy PA et al (2012) Notch4 normalization reduces blood vessel size in arteriovenous malformations. Sci Transl Med 4(117):117ra8
Murphy PA et al (2014) Constitutively active Notch4 receptor elicits brain arteriovenous malformations through enlargement of capillary-like vessels. PNAS 111(50):18007–18012
Nakazawa MS, Keith B, Simon MC (2016) Oxygen availability and metabolic adaptations. Nat Rev Cancer 16(10):663–673
Noguera-Troise I et al (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444(7122):1032–1037
Noguera-Troise I et al (2007) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Novartis Found Symp 283:106–120
Outtz HH et al (2011) Notch1 controls macrophage recruitment and Notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood 118(12):3436–3439
Patel NS et al (2005) Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res 65(19):8690–8697
Pedrosa A-R et al (2015) Endothelial Jagged1 promotes solid tumor growth through both pro-angiogenic and angiocrine functions. Oncotarget 6(27):24404–24423
Pelton JC et al (2014) Multiple endothelial cells constitute the tip of developing blood vessels and polarize to promote lumen formation. Development 141(21):4121–4126
Peters K et al (2009) Changes in human endothelial cell energy metabolic capacities during in vitro cultivation. The role of “aerobic glycolysis” and proliferation. Cell Physiol Biochem 24(5–6):483–492
Phng LK et al (2009) Nrarp coordinates endothelial notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell 16(1):70–82
Phng L-K, Stanchi F, Gerhardt H (2013) Filopodia are dispensable for endothelial tip cell guidance. Development 140(19):4031–4040
Potente M, Carmeliet P (2017) The link between angiogenesis and endothelial metabolism. Annu Rev Physiol 79(1). p.annurev-physiol-021115-105134
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146(6):873–887
Poulos MG et al (2013) Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep 4(5):1022–1034
Preuße K et al (2015) Context-dependent functional divergence of the notch ligands DLL1 and DLL4 in vivo. PLoS Genet 11(6):e1005328
Quillien A et al (2014) Distinct Notch signaling outputs pattern the developing arterial system. Development 141(7):1544–1552
Rafii S, Butler JM, Ding B-S (2016) Angiocrine functions of organ-specific endothelial cells. Nature 529(7586):316–325
Ramasamy SK et al (2014) Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507(7492):376–380
Ramasamy SK et al (2016) Blood flow controls bone vascular function and osteogenesis. Nat Commun 7:13601
Redeker C et al (2013) Normal development in mice over-expressing the intracellular domain of DLL1 argues against reverse signaling by DLL1 in vivo. PLoS One 8(10):e79050
Ren B et al (2010) ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J Clin Investig 120(4):1217–1228
Ridgway J et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444(7122):1083–1087
Risau W (1997) Mechanisms of angiogenesis. Nature 386(6626):671–674
Risau W, Flamme I (1995) Vasculogenesis. Annu Rev Cell Dev Biol 11:73–91
Robciuc MR et al (2016) VEGFB/VEGFR1-induced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab 23(4):712–724
Ruhrberg C et al (2002) Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev 16(20):2684–2698
Sacilotto N et al (2013) Analysis of Dll4 regulation reveals a combinatorial role for Sox and Notch in arterial development. PNAS 110(29):11893–11898
Sainson RC et al (2005) Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J 19(8):1027–1029
Scehnet JS et al (2007) Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 109(11):4753–4760
Schoors S et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520(7546):192–197
Schulze A, Harris AL (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491(7424):364–373
Seano G et al (2014) Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat Cell Biol 16(10):931–941
Seo S et al (2006) The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol 294(2):458–470
Serra H et al (2015) PTEN mediates Notch-dependent stalk cell arrest in angiogenesis. Nat Commun 6:7935
Shalaby F et al (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376:62–66
Sharghi-Namini S et al (2014) Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci Rep 4:4031
Sheldon H et al (2010) New mechanism for Notch signaling to endothelium at a distance by delta-like 4 incorporation into exosomes. Blood 116(13):2385–2394
Siekmann AF, Lawson ND (2007) Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445(7129):781–784
Siekmann AF, Affolter M, Belting HG (2013) The tip cell concept 10 years after: new players tune in for a common theme. Exp Cell Res 319(9):1255–1263
Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 10(10):611–625
Smith DC et al (2014) A phase I dose escalation and expansion study of the anticancer stem cell agent Demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res 20(24):6295–6303
Sonoshita M et al (2011) Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 19(1):125–137
Sörensen I, Adams RH, Gossler A (2009) DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood 113(22):5680–5688
Spuul P et al (2016) VEGF-A/notch-induced Podosomes Proteolyse basement membrane collagen-IV during retinal sprouting angiogenesis. Cell Rep 17(2):484–500
Stenzel D et al (2011) Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep 12(11):1135–1143
Suchting S et al (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. PNAS 104(9):3225–3230
Swift MR, Weinstein BM (2009) Arterial-venous specification during development. Circ Res 104(5):576–588
Swift MR et al (2014) SoxF factors and Notch regulate nr2f2 gene expression during venous differentiation in zebrafish. Dev Biol 390(2):116–125
Takeshita K et al (2007) Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ Res 100(1):70–78
Tammela T et al (2008) Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454(7204):656–660
Tammela T et al (2011) VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13(10):1202–1213
Taylor KL, Henderson AM, Hughes CCW (2002) Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and Downregulates VEGFR-2/KDR expression. Microvasc Res 64(3):372–383
Trindade A et al (2008) Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112(5):1720–1729
Ubezio B et al (2016) Synchronization of endothelial Dll4-Notch dynamics switch blood vessels from branching to expansion. eLife 5:e12167
Uyttendaele H et al (2001) Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. PNAS 98(10):5643–5648
Verginelli F et al (2014) Activation of an endothelial Notch1-Jagged1 circuit induces VCAM1 expression, an effect amplified by interleukin-1β. Oncotarget 6(41):43216–43229
Villa N et al (2001) Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev 108(1–2):161–164
Wang HU, Chen ZF, Anderson DJ (1998) Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93(5):741–753
Wieland E et al (2017) Endothelial Notch1 activity facilitates metastasis. Cancer Cell 31(3):355–367
Wild R et al (2017) Neuronal sFlt1 and Vegfaa determine venous sprouting and spinal cord vascularization. Nat Commun 10(8)
Wilhelm K et al (2016) FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529(7585):216–220
Wilkinson RN et al (2012) Hedgehog signaling via a calcitonin receptor-like receptor can induce arterial differentiation independently of VEGF signaling in zebrafish. Blood 120(2):477–488
Wu Y et al (2010) Therapeutic antibody targeting of individual Notch receptors. Nature 464(7291):1052–1057
Xu C et al (2014) Arteries are formed by vein-derived endothelial tip cells. Nat Commun 5:5758
Xue Y et al (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8(5):723–730
Yamamizu K et al (2010) Convergence of Notch and β-catenin signaling induces arterial fate in vascular progenitors. J Cell Biol 189(2):325–338
Yan M et al (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463(7282):E6–E7
Yang Y, Oliver G (2014) Development of the mammalian lymphatic vasculature. J Clin Investig 124(3):888–897
Yeh WL, Lin CJ, Fu WM (2008) Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol 73(1):170–177
Yoon CH et al (2014) High glucose-induced jagged 1 in endothelial cells disturbs notch signaling for angiogenesis: a novel mechanism of diabetic vasculopathy. J Mol Cell Cardiol 69:52–66
You L-R et al (2005) Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435(7038):98–104
Zarkada G et al (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. PNAS 112(3):761–766
Zhang J et al (2011) Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing Delta-like 4 expression through AKT-mediated activation of β-catenin. J Biol Chem 286(10):8055–8066
Zhong TP et al (2000) Gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science 287(5459):1820–1824
Zhong TP et al (2001) Gridlock signalling pathway fashions the first embryonic artery. Nature 414(6860):216–220
Zhu TS et al (2011) Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res 71(18):6061–6072
Zhuge Q et al (2009) Notch-1 signalling is activated in brain arteriovenous malformations in humans. Brain 132(12):3231–3241
Acknowledgments
We thank all members of the Fischer laboratory for critical discussion and apologize to all colleagues whose work could not be cited in this chapter. Work in our laboratory on these topics has been supported by the Deutsche Forschungsgemeinschaft (DFG-FI1568/3-1; FI1568/5-1; SFB-TR23), the Helmholtz Society and the German Cancer Research Center Heidelberg.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Tetzlaff, F., Fischer, A. (2018). Control of Blood Vessel Formation by Notch Signaling. In: Borggrefe, T., Giaimo, B. (eds) Molecular Mechanisms of Notch Signaling. Advances in Experimental Medicine and Biology, vol 1066. Springer, Cham. https://doi.org/10.1007/978-3-319-89512-3_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-89512-3_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89511-6
Online ISBN: 978-3-319-89512-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)