
Abstract
Loss of mitochondrial function not only causes specific mitochondrial diseases but also contributes to serious conditions such as neurodegeneration and diabetes. Since mitochondrial DNA is transcribed as a polycistronic message comprised of three forms of RNA (rRNA, mRNA, and tRNA), proper 5′- and 3′-end cleavage is essential. In the nucleus, tRNA 5′-end processing is carried out by the first identified ribozyme, RNase P. In contrast, mitochondrial tRNAs are processed by a three-protein complex, mitochondrial RNase P, which does not have an RNA component. An accessory subcomplex made of the m1A9 methyltransferase MRPP1 and the dehydrogenase MRPP2 binds to the metallonuclease MRPP3 that cleaves the RNA phosphodiester backbone. Each protein has been shown to be essential in model organisms, and loss of each gives rise to human multisystemic diseases with many characteristics of mitochondrial disease. In this review, we discuss what is known about the mitochondrial RNase P complex, the molecular mechanism of 5′-end mitochondrial tRNA processing, and how loss of this activity causes human disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


3.1 The Polycistronic Nature of Mitochondrial DNA Transcription
3.1.1 The Mitochondrial Genome
Mitochondria have evolved from alphaproteobacteria that resided within the eukaryotic cell, maintaining an endosymbiotic relationship (Gray 2012; Margulis 1970). Owing to their bacterial origin, mitochondria have their own genetic material, but during evolution, most of the bacterial genes were either lost or transferred to the nucleus, with the actual mitochondrial DNA (mtDNA) being much smaller, about 16.6 kb, as compared to the bacterial genome (>5 Mb) (Gray 2012; Land et al. 2015). Even though the mitochondrial genome is replicated and maintained independently of the nuclear DNA, numerous nucleus-encoded proteins are required for replication, transcription, posttranscriptional RNA processing, and mitochondrial translation. Around 200–300 nucleus-encoded proteins are translocated into the mitochondrion to bring about mitochondrial gene expression (Powell et al. 2015).
3.1.2 The Punctuation Model
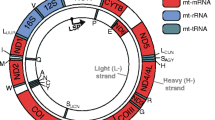
Mitochondrial DNA (mtDNA) is highly conserved across metazoans, encoding the same products often organized differently on the two (heavy and light) strands (Fig. 3.1). Drosophila mtDNA is similar to human in its size and with respect to the products it encodes, making Drosophila a good model to study human mitochondrial function and disease (Lewis et al. 1995; Sen and Cox 2017) (Fig. 3.1b). mtDNA exercises a complete economy of organization, with no introns, only one intergenic sequence, and smaller rRNAs and tRNAs as compared to their nuclear counterparts. mtDNA encodes 13 proteins that are components of Complexes I (NADH dehydrogenase), III (cytochrome b), IV (cytochrome c oxidase), and V (ATP synthase) of the OXPHOS pathway. It also encodes 2 ribosomal RNAs (rRNAs) and the complete suite of 22 transfer RNAs (tRNAs) required for the translation of these mitochondria-encoded proteins.

Human and Drosophila mtDNA. (a) Human mtDNA. Three polycistronic messages are transcribed for human mtDNA (arrows): two promoters initiate on the heavy strand (HSP, top) and one on the light strand (LSP, bottom). With the exception of ND6, all mRNAs and both rRNAs are encoded on the heavy strand. All mt-RNAs are flanked 5′ and 3′ by mt-tRNAs except for the following noncanonical junctions: (1) 5′-end of CoI, (2) ATP8/6 – CoIII, (3) mt-tRNALeu(CUN)—ND5, (4) 3′-end of ND5, (5) 5′-end of Cytb, (6) 3′-end of ND6. (b) Drosophila mtDNA. The combined five polycistronic messages (arrows) transcribe the same products as human mtDNA. The expanded length is due to the A + T-rich region (not to scale). Single letters (black boxes) represent mt-tRNAs. ND, NADH dehydrogenase (Complex I); Cytb, Cytochrome b (Complex III); Co, cytochrome c oxidase (Complex IV); and ATP, ATP synthase (Complex V)
To meet the continuous energy demands of the cells, correct transcription and translation of mitochondria-encoded peptides are obligatory. As in bacteria, mtDNA is transcribed as a polycistronic message. The arrangement and the number of transcripts differ in various species. In humans, mtDNA is transcribed as three polycistronic transcripts, two from the heavy and one from the light strand, whereas in Drosophila mtDNA encodes five polycistronic transcripts, three on the heavy and two on the light strand (Fig. 3.1). In humans, the heavy-strand transcripts are considered more informationally rich as they together encode 12 proteins, 2 rRNAs, and 14 tRNAs, while the light-strand transcript encodes 8 tRNAs and only 1 protein (Taanman 1999). The mature RNA products are formed from the polycistronic transcripts following endonucleolytic cleavages at the 5′- and 3′-ends. In most pre-RNA transcripts, the junctions between mitochondrial rRNAs (mt-rRNAs) and mitochondrial mRNAs (mt-mRNAs) are punctuated by tRNAs (Ojala et al. 1981). The secondary cloverleaf structure of mitochondrial tRNAs (mt-tRNAs) is thought to define the cleavage sites for endonucleases to release the individual mature RNA products. Cleavage at the 5′-end of the mt-tRNA molecule is brought about by the three-protein mitochondrial RNase P complex (mtRNase P; Holzmann et al. 2008), whereas a ribonuclease Z (RNase Z)-like protein, ELAC2 in humans and RNase ZL in Drosophila, is responsible for the cleavage at the 3′-end of the mt-tRNAs (Brzezniak et al. 2011; Sanchez et al. 2011).
Most noncanonical mt-mRNAs are flanked by tRNAs [Fig. 3.1a, (1)–(6)]. For 5′-end cleavage of cytochrome c oxidase I (CoI), there seems to be a noncoding RNA which adopts a tRNA-like conformation that is recognized and cleaved by mtRNase P (Mercer et al. 2011; Sanchez et al. 2011). The NADH dehydrogenase 5 (ND5)-cytochrome b (Cyt b) junction is known to be processed by PTCD2, a pentatricopeptide repeat protein (Xu et al. 2008), whereas the processing of the NADH dehydrogenase 6 (ND6)-noncoding (nc) RNA and the ATP6-cytochrome c oxidase III (CoIII) junctions is still not clear. Recently, FASTKD5 was shown to be necessary for processing the 5′-end of CoI, ATP8/6-CoIII, and ND5-Cytb junctions, but the exact mechanism is still not well understood (Antonicka and Shoubridge 2015).
3.2 RNase P Function in Mitochondria
3.2.1 Mitochondrial RNase P Cleaves the 5′-End of Mt-tRNA
RNase P is the endonuclease that catalyzes hydrolysis of the phosphodiester bond at the 5′-end of pre-tRNAs to generate tRNAs with a mature 5′-end and a 5′-leader sequence (Robertson et al. 1972). RNase P is found in all domains of life and in nearly all species. It is a ribozyme with a catalytically active RNA subunit and a variable number of proteins depending on the organism (Klemm et al. 2016). In eukaryotic land plants, algae, and protists, single-peptide protein-only ribonuclease P proteins (PRORPs) are sufficient to bring about 5′-end pre-tRNA processing (Gobert et al. 2010; Klemm et al. 2016; Taschner et al. 2012). In Arabidopsis, PRORPs carry out this function in the nucleus, chloroplasts, and mitochondria. However, in human mitochondria, this reaction is carried out by a three-protein mtRNase P complex made up of mitochondrial RNase P protein 1 (MRPP1), mitochondrial RNase P protein 2 (MRPP2), and mitochondrial RNase P protein 3 (MRPP3). In this three-protein complex, MRPP3 is also sometimes referred to as protein-only ribonuclease P (PRORP) (Holzmann et al. 2008).
Unlike its nuclear ribozyme counterpart, mtRNase P lacks a catalytic RNA. Instead, MRPP3, a metallonuclease-like protein, is the catalytic endonuclease that performs the 5′-end cleavage of mitochondrial pre-tRNAs. MRPP1 and 2 have each been recruited from other biological pathways during evolution. MRPP1, or TRMT10C/RG9MTD1, is a methyltransferase required for methylation of the ninth position of certain tRNAs (Jackman et al. 2003), and MRPP2, also known as HDS10 or SDR5C1, is a member of the short-chain dehydrogenase/reductase family which is involved in isoleucine and lipid metabolism (Moeller and Adamski 2009; Shafqat et al. 2003; Yang et al. 2014). MRPP2 acts as a scaffold to form a MRPP1/2 subcomplex and is required for the methyltransferase activity of MRPP1. The catalytic activity of MRPP3 is independent of the methyltransferase activity of the MRPP1/2 subcomplex (Vilardo et al. 2012). The MRPP1/2 subcomplex increases the affinity of MRPP3 for its substrate, likely through a structural rearrangement of mt-tRNA or by altering the active site of MRPP3 (Shafqat et al. 2003). Recently, Reinhard et al. showed that MRPP1/2 subcomplex plays a more central role in coordinating the activities of mt-tRNA processing. They observed that the mt-tRNAs remained bound to the subcomplex after the 5′-end cleavage and that the subcomplex enhances the 3′-end cleavage activity by ELAC2. Furthermore, they observed that MRPP1/2 remained complexed with mt-tRNAs before and after 3′–CCA addition, orchestrating the events leading to the formation of mature mt-tRNA formation (Reinhard et al. 2017).
3.2.2 What Is Known About mtRNase P Mechanism In Vivo
The Drosophila homologs of mtRNase P, called Roswell (MRPP1), Scully (MRPP2), and Mulder (MRPP3), have been identified and characterized (Sen et al. 2016). These three proteins share a high degree of sequence similarity with human mtRNase P proteins, containing all the same recognizable domains (Fig. 3.2). They also appear to function in the same way (see below). Sen et al. showed that mutations or knockdown of Mulder (Mldr), Roswell (Rswl), or Scully (Scu) causes lethality in Drosophila. Loss or knockdown of each component disrupts mt-RNA processing. Northern blots probed with four mt-tRNAs, each in a different transcript environment, showed the accumulation of larger mt-RNA species in mutants. Additionally, overexpressing Mldr and Rswl caused larval lethality with highly reduced levels of ATP as compared to wild type. Lethality was not observed upon overexpressing Scu. Ectopically increasing one of the enzymes of this multiprotein complex could be a dominant negative effect due to sequestration of essential components or mis-regulation of the active enzyme complex formation. Whether the lethality was associated with mis-processing of mt-tRNA remains to be determined.

Mitochondrial RNase P complex homologies. Schematics representing the domains of MRPP1, MRPP2, and MRPP3 from Drosophila, mouse, and humans are shown in (a), (c), and (f), respectively. The N-terminal mitochondrial targeting peptide (MTS, yellow) was predicted using the MitoProt server (https://ihg.gsf.de/ihg/mitoprot.html; Claros and Vincens 1996). The domain boundaries are predicted through homology-based alignments performed using Clustal Omega and ExPASy (PROSITE) servers. The percent identity and similarity of individual domains as compared to the human counterparts were calculated using BLAST. The Drosophila Roswell (b), Scully (d, e), and Mulder (g) protein structural models were created using the I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/, Zhang 2008). The Roswell model is based on the structure of tRNA m1G9 methyltransferase Trm10 from Schizosaccharomyces pombe (PDB ID: 4jwf), whereas the modeled Scully structure is based on human MRPP2/HSD10 (PDB ID: 1u7t). The expected Drosophila Scully tetramer assembly is shown in (e). The Mulder model was based on Arabidopsis thaliana PRORP1, PRORP2, and human MRPP3 structures (PDB IDs: 4G24, 4G23, 5DIZ, 4XGL)
Recently, MRPP3 was shown to be essential in mouse (see below; Rackham et al. 2016). Using tissues from conditional MRPP3 knockout mice, Northern blots, and q-RT PCR analysis, Rackham et al. showed that MRPP3 loss led to the loss of 5′-end processing of the precursor RNA, resulting in the depletion of mature mt-mRNA over time and to the accumulation of unprocessed, higher molecular weight transcripts. Additionally, they observed an increase in the rate of mtDNA transcription accompanied by a decrease in the synthesis of mitochondria-encoded respiratory complex proteins. The increase in transcription was most likely needed to compensate for the lack of proteins but was unable to rescue the deficient RNA processing. Rackham et al. also demonstrated that previously known canonical RNA junctions depended on MRPP3 for their 5′-end processing. However, the noncanonical junctions, like ND5, which is bordered by tRNALeu(CUN) at its 5′-end and a noncoding RNA on its 3′-end, are dependent on cleavage by ELAC2 exclusively. Using Northern blot analysis, they demonstrated that the loss of MRPP3 did not have a significant effect on the overall levels of mature ND5 mRNA over time, suggesting that ND5 mRNA maturation is in fact independent of MRPP3 cleavage. Similar independence was also observed for ND6-tRNAGlu and ATP8/6-CoIII junctions, which require ELAC2 and FASTKD5, respectively, for maturation (Sanchez et al. 2011). Interestingly, for some junctions, ELAC2 alone was unable to process the 3′-ends of tRNAs in the absence of 5′-end processing by MRPP3, which was shown by a decrease in the processing at tRNAVal-16S rRNA, tRNALeu(UUR)-ND1, and tRNAMet-ND2 junctions. This overall pattern of the endonucleolytic cleavages suggests that mt-tRNA maturation happens in a sequential manner, where in most cases 5′-end processing is required for 3′-end cleavage to happen. Furthermore, Rackham et al. also demonstrated that in MRPP3 knockout mice, the expression levels of nucleus-encoded mitochondrial proteins involved in mitochondrial gene expression and mitochondrial ribosome biogenesis were affected. Taken together their results suggested that 5′-end processing by MRPP3 is an indispensable step in mitochondrial RNA maturation, translation, and correct mitoribosome biogenesis.
3.3 RNA Processing Centers in Mitochondria
3.3.1 Mitochondrial RNA Processing Granules
Discrete foci containing newly transcribed mt-RNA were first observed a decade ago by pulse-labeling cells with bromouridine (BrdU). However, the other components of these granules were largely unknown until recently (Fig. 3.3; Iborra et al. 2004). G-rich sequence factor 1 isoform (GRSF1), an RNA-binding protein that binds the G-rich sequence motif AGGGD (where D is either A, U or G), has been previously implicated in posttranscriptional processing of cellular and viral mRNAs (Jablonski and Caputi 2009; Kash et al. 2002; Schaub et al. 2007). GRSF1 was recently shown to accumulate in most BrdU-labeled granules in the mitochondrial matrix (Antonicka et al. 2013; Jourdain et al. 2013). GRSF1 was also shown to play a role in RNA processing even though the exact role of the protein remains unclear. Downregulation of GRSF1 causes a delay in clearing RNA from the mitochondrial RNA granules and affects the steady-state levels of mature RNA transcripts, with an associated decrease in mitochondrial protein levels. It has also been implicated in mitochondrial ribosome biogenesis and stability. Through affinity purification, Jourdain et al. identified several interacting proteins of FLAG-GRSF1 in HEK293T cells. MRPP1 and 2 were the top hits and this interaction was not mediated by RNA. In contrast, while MRPP3 was not identified as a binding partner for GRSF1 through affinity purification, C-terminally tagged MRPP3 was shown to co-localize with GRSF1 in mitochondrial granules using immunocytochemistry (Jourdain et al. 2013). The GRSF1-rich granules were shown to localize newly synthesized precursor RNA transcripts, thus potentially representing sites for mitochondrial RNA processing (Antonicka et al. 2013; Jourdain et al. 2013). In addition, the granules were sensitive to the presence of transcription inhibitors. Jourdain et al. did not observe accumulation of RNase Z in the granules, which could support the sequential cleavage of the 5′- and 3′-ends of mt-tRNAs by mtRNase P and ELAC2, respectively, with the argument that only the first step of mt-tRNA processing happens in the RNA granules.

RNA processing granules. mtDNA replication and repair take place in nucleoids. The newly synthesized mt-RNA is processed and matured in mitochondrial RNA granules. The mtRNase P complex performs 5′-end cleavage, and ELAC2 performs 3′-end cleavage of the polycistronic message. Polyadenylation, mt-tRNA modification, and mitoribosome biogenesis also take place in the RNA granules. Mis-processed mt-RNAs are degraded in the associated degradation granules
Nucleoids are the centers in which the replication and transcription of the mitochondrial genome take place. mtRNase P complex proteins and ELAC2 were shown to accumulate in distinct foci adjacent to the nucleoids in mouse 3 T3 cells, and some studies propose that RNA processing granules are extensions of dynamic nucleoids (Borowski et al. 2013). mtRNase P and ELAC2 proteins were shown to be enriched in the nucleoid granule fractions, even though they are not always co-localized within the nucleoid as judged by immunofluorescence. Thus, it seems likely that the processing of the newly emerged RNA transcript happens either within or very close to the nucleoid structure. Borowski et al. demonstrated a small proportion of the foci co-localize with SUPV3L1/PNPase complex proteins to form a mitochondrial degradosome (Borowski et al. 2013) (Fig. 3.3). As the RNA processing granule research matures, many questions remain regarding the exact order of events. More specifically, does GRSF1 act upstream or downstream of the mtRNase P complex? How would mtRNase P protein inhibition affect granule formation? Will mtRNase P protein downregulation lead to GRSF1 accumulation in mitochondrial RNA processing granules?
3.4 Mitochondrial tRNA Processing and Disease
Since mitochondria play a fundamentally important role in tissues, mutations in mt-tRNAs and defects in mt-tRNA processing lead to diseases that are often severe, sometimes fatal. Currently, treatments are not very effective and there are no cures for these diseases. One unusual trait of mitochondrial diseases is that different mutations in the same mt-tRNA can give rise to very different clinical presentations (Chinnery 2000). There are approximately 275 disease-associated mutations in mt-tRNAs, which often do not affect the anticodon region (Brandon et al. 2005). Mitochondrial diseases can also occur due to defects in the nucleus-encoded enzymes required to carry out the up to 20 mt-tRNA modifications needed to produce mature and fully functional mt-tRNAs (Van Haute et al. 2015).
In metazoans, the full-suite of mt-tRNAs (20) is encoded in mtDNA. However, in contrast to nuclear tRNAs, there are only 2 sets of mitochondrial isoacceptors (different tRNAs species that carry the same amino acids but have different anticodon sequences), whereas there are 513 identified tRNA-encoding genes in the human genome. Thus in mitochondria, there is only 1 mt-tRNA for each of 18 of the amino acids and 2 for serine and leucine. This means that for the majority of mt-tRNAs, if a mt-tRNA becomes nonfunctional due to mutation, the corresponding amino acid will simply not be available for translation. This situation can have differential effects on mt-mRNAs depending on the amino acid requirements for their synthesis.
Due to the polycistronic nature of mtDNA and the mt-tRNA “punctuation model,” teasing out the effect of abnormal mt-tRNA levels is challenging. Loss of normal mt-tRNA processing can lead to a decrease in mt-tRNA levels, mitochondrial protein levels, and mt-rRNAs. If available, fully functional mt-tRNAs are too scarce, translation will not occur at normal levels. For example, translation is reduced if enzymes that carry out specific mt-tRNA modifications are defective. However, if mt-tRNAs are not cleaved properly from the polycistronic message, this will have detrimental effects on mt-mRNA processing and mt-rRNA abundance irrespective of mt-tRNA levels. Diseases arising from mt-tRNA processing defects can therefore be classified into two groups: those that arise from mutations in mt-tRNAs and those due to mutations in the nucleus-encoded processing enzymes.
3.4.1 Point Mutations in mt-tRNAs Affecting mt-RNA Processing
Various point mutations in mt-tRNAs have been shown to alter 5′- and 3′-end processing (Table 3.1). The difference thus far appears to be that the mutation must be at the mtRNase P site in order for 5′-end processing to be disrupted, whereas there is evidence that mutations in various regions of the mt-tRNA can reduce the level of 3′-end processing. This may be due to the different mechanisms by which mtRNase P and RNase Z interact with their substrates. As mentioned above, mtRNase P appears to cleave mt-tRNAs first from mt-RNAs and thus may require less structural information from the folded mt-tRNAs compared to RNase Z. Indeed, nucleus-encoded tRNAs and mt-tRNAs can form alternative structures before base modification occurs, and base modifications are required for normal tertiary structure. The initial extended hairpin structures that form after transcription may be efficiently recognized by RNase P, but not RNase Z. Also, MRPP2/TRMT10C is responsible for the m1A9 methylation in mitochondria, which has been shown to be important for tertiary folding for some mt-tRNAs. Therefore, this modification by the MRRP1/MRPP2 subcomplex may be required before RNase Z is able to recognize the mt-tRNA. Adding further complexity, mt-tRNA ends are in different contexts. The secondary structure each mt-tRNA assumes after transcription presumably affects the directly neighboring mt-RNA. For example, mt-tRNALeu(UUR) is between mt-rRNA and ND1, whereas mt-tRNASer(AGY) is directly between two mt-tRNAs (Fig. 3.1).
Defects in 5′-end processing have been identified in patients suffering from maternally inherited hypertension (Guo et al. 2016; Jiang et al. 2016; Li et al. 2009; Wang et al. 2011; Zhu et al. 2009). Since mitochondria are loaded into the oocyte, this non-Mendelian form of inheritance is the first indication that the phenotype is due to mutations in mtDNA. Numerous mtDNA mutations are now associated with maternally inherited hypertension. Wang et al. were the first to identify a novel mtDNA mutation 4263A → G that altered 5′-end mt-tRNA processing (Wang et al. 2011). The causes of hypertension are not well understood but are thought to arise from complex changes in different tissues (Page 1967). However, the production of reactive oxygen species (ROS) and inflammation appear to be two underlying causes, and damaged mitochondria are in general frequently the source of abnormal levels of ROS. Wang et al. identified homoplasmic females harboring the 4263A → G mutation, which occurs in position 1 of mt-tRNAIle (Fig. 3.4b, Wang et al. 2011). This mutation results in decreased levels of mt-tRNAIle, mt-RNA translation, and respiration. Using an in vitro mtRNase P reconstitution assay, the authors demonstrated that the 4263A → G mutation decreased 5′-end processing efficiency by ~30% compared to wild type. Recently, Jiang et al. identified an additional mutation in mt-tRNAAla associated with hypertension in three unrelated individuals whose families exhibited maternally inherited hypertension (Jiang et al. 2016). The 5655A → G mutation is in the same relative position in the tRNA as the 4263A → G mutation and causes the same mitochondrial deficits, such as reduced mt-tRNAAla levels, decreased respiration, and a 35% decrease in 5′-end processing (Fig. 3.4f). In addition, cybrids derived from patient lymphocytes exhibited reduced oxygen consumption and ATP levels, increased ROS, and improperly aminoacylated mt-tRNAAla (Jiang et al. 2016).

Human pathogenic mt-tRNA mutations. (a–g) Human mt-tRNA cloverleaf structures encoded from left to right of human mtDNA (Fig. 3.1a). Blue nucleotides represent pathogenic mutations (Table 3.1). The numbering system is according to (Anderson et al. 1981). Arrows at the 5′- and 3′-ends indicate the mtRNase P and RNase Z sites, respectively, impacted by mutation. Red nucleotides indicate the neighboring 5′ and 3′ transcripts. The arrows above and below the red nucleotides indicate the start (beginning of arrow) or stop (arrowhead) of the transcript close to the cleavage sites. The flanking gene may overlap with the mt-tRNA (e.g., mt-tRNALeu(UUR), a) or there may be intervening nucleotides (e.g., mt-tRNATrp, e). The conserved CCA sequence (green) found on the acceptor stem at the 3′-end of all tRNAs acts as the amino acid attachment site and is uncoded; tRNA-nucleotidyltransferase is responsible for CCA addition. (h) Tertiary structure of yeast tRNAPhe color-coded for the D loop, anticodon loop, variable loop, TψC loop, acceptor stem, and CAA tail as indicated by the cloverleaf cartoon
Three reported mt-tRNA mutations in the literature have not been directly tested for loss of 5′-end processing, but they are in a location that strongly suggests a causal effect on a processing defect. Two groups independently identified 4401A → G in maternally hypertensive patients (Li et al. 2009; Zhu et al. 2009). This mutation is particularly interesting as it affects both mt-tRNAGln (encoded on the L-strand) and mt-tRNAMet (encoded on the H-strand) at their RNase P cleavage sites (Fig. 3.4c, d). Using patient-derived cybrid cells, both groups showed decreased mt-tRNA levels and respiration, and Li et al. also showed a decrease in mitochondrial protein synthesis. More recently, Guo et al. identified a 5512A → G mutation in mt-tRNATrp in a family of maternally inherited hypertensives (Guo et al. 2016). This mutation is at the analogous position to the 4263A → G mutation in mt-tRNAIle and thus likely also affects 5′-end processing (Fig. 3.4e).
The investigators who uncovered mtDNA mutations that affect 5′-end processing were interested in causes of maternally inherited hypertension. To identify mt-tRNA mutations affecting 3′-end processing, Levinger et al. instead started with mt-tRNA mutations known to cause mitochondrial diseases and syndromes (Levinger et al. 2001, 2003, 2004). The U7445 U → C mutation in mt-tRNASer(UCN) was known to cause non-syndromic deafness, reduced mt-tRNA Ser(UCN) levels and mitochondrial protein synthesis (Guan et al. 1998; Reid et al. 1997). This mutation is located precisely at the RNase Z cleavage site (Fig.3.4g). Using in vitro-labeled mt-tRNA Ser(UCN) and mitoplast extract from HeLa cells as a source of RNase Z, Levinger et al. found the mutant mt-tRNA was unable to undergo any 3′-end processing in contrast to wild-type mt-tRNA even though they both appeared to form normal structures (Levinger et al. 2001). To extend these studies, the authors examined the effect on 3′-end processing of mutations in mt-tRNAIle and mt-tRNALeu(UUR), two mt-tRNAs linked to cardiomyopathies, ophthalmoplegia, and mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) (Levinger et al. 2003, 2004). Using experiments similar to their previous work, the authors found four mutations in mt-tRNAIle and one in mt-tRNALeu(UUR) that substantially reduced 3′-end processing (Fig. 3.4a, b). Some of these mutations also resulted in structural and aminoacylation changes (Table 3.1).
An additional mt-tRNA mutation was recently published that was not analyzed for defective 3′-end processing but that may interfere based on its position. Liu et al. identified the 4467C → A mutation in mt-tRNAMet in a family with maternally inherited hypertension (Liu et al. 2017). This mutation is located near the RNase Z cleavage site (Fig. 3.4d). Using Lymphocyte cell lines derived from patients, Liu et al. demonstrated decreased ATP in cell extract, increased oxidative damage, and lower oxygen consumption, indicating disrupted mitochondrial function.
3.4.2 Mutations in Mitochondrial RNase P
Human diseases due to mitochondrial deficiencies have now been linked to mutations in all three members of the mtRNase P complex (Table 3.2). They are rare diseases and many of the mutations occur in highly conserved residues. MRPP2/HSD10 (encoded by the HSD17B10 gene) has the largest number of identified mutations, referred to as HSD10, and was the first to be described [OMIM #300438, 300,256 (reviewed in Zschocke 2012)]. For consistency, we will refer to the protein as MRPP2 in this section; however, HSD10 disease is the name of diseases caused by mutations in HSD17B10. As described above, MRPP2 is a multifunctional protein. It functions in isoleucine metabolism but has been shown to be a promiscuous dehydrogenase in vitro (reviewed in Moeller and Adamski 2009). Because MRPP2 was identified as a member of the mtRNase P complex and plays a role in mt-tRNA 5′-end processing, ascribing its molecular role in disease has been complicated (Holzmann et al. 2008). However, the primary cause of symptoms experienced by patients with MRPP2 mutations may be due to loss of mitochondrial function and mt-RNA processing (Zschocke 2012). For example, no clinical correlation has been found between loss of the dehydrogenase activity and symptoms. The mutation Q165H, which abolishes this activity, does not cause the classical, more severe disease onset, and those mutations that are not near the enzyme’s active center cause the neonatal form of the disease (Rauschenberger et al. 2010). In addition, reduced dietary isoleucine does not improve HSD10 symptoms (Korman 2006; Sutton et al. 2003; Zschocke et al. 2000). In vitro work has also shown dehydrogenase-dead protein can still rescue vital cellular functions and functions in the mtRNase P complex (Rauschenberger et al. 2010; Vilardo et al. 2012).
HSD10 disease is rare with no effective treatment and exhibits variable ages of onset. Symptoms are usually multisystemic, consistent with mitochondrial disease. Patients often have a progressive loss of cognitive and motor functions, with epilepsy and blindness. The most severe forms result in cardiomyopathy and severe neurodegeneration (Zschocke 2012). Mutations can affect homotetramization, the dehydrogenase activity or its ability to function in the mtRNase P complex (Vilardo and Rossmanith 2015). As HSD17B10 is X-linked, mutations mostly affect males, but heterozygous females can also have disease symptoms, making the inheritance pattern X-linked dominant.
After MRPP2 was identified as a member of the mtRNase P complex, researchers began testing samples from patients with HSD10 disease, as well as recombinant pathogenic MRPP2 mutations, for mitochondrial deficits and decreased mt-RNA processing, along with dehydrogenase and methyltransferase activities (Chatfield et al. 2015; Deutschmann et al. 2014; Falk et al. 2016; Oerum et al. 2017; Vilardo and Rossmanith 2015). Deutschmann et al. first showed that patient fibroblast cells harboring R130C, the most prevalent mutation, significantly accumulate unprocessed mt-RNAs encoded on the mtDNA heavy strand, but not the light strand (Deutschmann et al. 2014). Chatfield et al. examined post-autopsy tissues from a patient with the N247S mutation and found increased unprocessed mt-RNAs and decreased mitochondrial protein synthesis (Chatfield et al. 2015). Vilardo and Rossmanith took an in vitro approach, examining four different mutations, R226Q, N247S, P210S, and R130C. All four mutations greatly impair dehydrogenase activity, mt-tRNA processing, and methylation. R226Q and N247S mutations also disrupted homotetramer formation and interaction with MRPP1 (Vilardo and Rossmanith 2015). The MRPP1/2 subcomplex is highly stable. There is evidence that reducing the amount of MRPP2 can affect the stability of MRPP1. Deutschmann et al. examined protein levels in patient fibroblasts and found the reduced level of MRPP2 resulted in lower MRPP1 levels, but not MRPP3 levels, whereas loss of MRPP1 had no effect on MRPP2 levels (Deutschmann et al. 2014). In addition, Falk et al. identified a novel K212E mutation and showed that while it has only a modest effect on dehydrogenase activity, it reduced methylation and 5′-end processing activities, as well as binding to MRPP1 (Falk et al. 2016). Most recently, Oerum et al. identified the novel mutations V12 L and V176 M. Both reduced dehydrogenase, methyltransferase, and mtRNase P activities, with V176 M being much more severe, in agreement with patient symptoms (Oerum et al. 2017).
As with MRPP2, the dual function of MRPP1 as a methyltransferase and member of the mtRNase P complex makes it challenging to ascribe the primary defect of mitochondrial loss of function. Disease-causing mutations in MRPP1/TRMT10C have recently been identified (Metodiev et al. 2016). As mentioned above, the MRPP1/2 subcomplex is an active methyltransferase that can methylate adenosine and guanine nucleotides at position 9 (Vilardo et al. 2012). Nineteen out of the 22 mt-tRNAs have an A or G at position 9, and thus, they are likely acted upon by MRPP2. Metodiev et al. described two unrelated patients suffering from lactic acidosis, hypotonia, feeding difficulties, and deafness. The patients’ symptoms were so severe that they died after only 5 months of age. After whole-exome sequencing, they were found to harbor mutations in MRPP1 (Table 3.2). Both patients exhibited clear signs of mitochondrial disease, with deficits in Complexes I and IV. MRPP1 protein levels were reduced in patient-derived fibroblast cell lines, while MRPP2 and MRPP3 levels were unchanged. The amount of m1R9 methyltransferase activity was also unchanged. The steady-state levels of mt-tRNAs and mt-mRNAs remained mostly unchanged in patient-derived fibroblasts; however there was an increase in unprocessed mt-RNAs that was rescued by transfecting the cells with wild-type MRPP1. These results led the authors to speculate that the MRPP1 mutations may affect interaction with MRPP3 rather than MRPP2 and the increased amount of unprocessed mt-RNAs may interfere with translation.
Hochberg et al. have identified a disease-causing mutation in MRPP3 (Hochberg et al. 2017). Perrault syndrome is a rare, genetically heterogeneous disease characterized by sensorineural hearing loss in males and females and primary ovarian insufficiency (Jenkinson et al. 2012). Most of the genes mutated in Perrault syndrome are involved in mitochondrial translation (OMIM #233400). Hochberg et al. identified a family affected by Perrault syndrome of which three individuals harbored a A485V mutation in MRPP3. Located in the metallonuclease domain, A485V does not cause reduced protein levels in patient-derived fibroblasts but does decrease the level of mtDNA-encoded respiratory chain complex proteins. In addition, the authors detected multiple unprocessed mt-RNAs. The authors went on to test the A485V mutation in an in vitro mtRNase P reconstitution assay and found the mutated MRPP3 possessed significantly less 5′-end processing activity compared to wild type.
Since mutations in mtRNase P clearly affect mt-RNA processing, causing mitochondrial dysfunction and disease, it is possible that defects in 3′-end mt-RNA processing could do the same. Haack et al. performed whole-exome sequencing on patients suffering from oxidative phosphorylation deficiencies and identified four disease-associated alleles of ELAC2 in three families (Table 3.2, Haack et al. 2013). Using qPCR, they found an increase in unprocessed mt-RNA in patient tissue samples. The processing defect could be rescued in patient fibroblasts by lentivirus-mediated infection with wild-type ELAC2. However, mt-tRNA levels and at least four mt-mRNAs were unchanged, and there was no evidence of 3′-end processing defects. Despite this, mitochondrial translation and protein levels were reduced, suggesting that the increase in unprocessed mt-RNA intermediates may hamper mitochondrial translation. Akawi et al. recently identified a splicing mutation that reduces ELAC2 levels (Akawi et al. 2016). Using real-time PCR, they found increased levels of unprocessed mt-RNAs in fibroblasts of one patient.
3.5 Loss of Mt-tRNA Processing in Model Organisms
Model organisms are powerful systems for studying the cellular and molecular bases underlying human disease. Human mutations in MRPP1, MRPP2, and MRPP3 are rarely null alleles. This is because complete absence of mtRNase P activity is likely incompatible with life. Because we can carry out conditional knockouts and rigorous genetics, model organisms offer the opportunity to study the in vivo effects of complete loss of mtRNase P activity on mt-RNA processing and mitochondrial function, as well as the dehydrogenase and methyltransferase activities of MRPP1 and MRPP2. Single orthologs for each of the three mtRNase P complex members have been identified in mouse and Drosophila. MRPP2 is the smallest protein and has very high amino acid identity among human, mouse, and Drosophila (Fig. 3.2). The crystal structure of human HSD10 was determined as a homotetramer complexed with NAD+ and an inhibitor (Kissinger et al. 2004). Given the high amino acid identity, this structure was used to model the structure for the Drosophila MRPP2 Scully (Fig. 3.2d, e). MRPP3 contains seven pentatricopeptide repeats and a metallonuclease domain (Fig. 3.2; Howard et al. 2012). There is only one MRPP3 in all three species, with the highest identity in the metallonuclease domain (Fig. 3.2). MRPP1/TRMT10C also has only one ortholog in humans, mouse, and Drosophila. Shao et al. solved the structure for yeast TRM10 in the presence and absence of a methyl donor and showed that the catalytic domain displays the typical SpoU-TrmD fold found in SPOUT family methyltransferases (Fig. 3.2; Shao et al. 2014).
3.5.1 Drosophila Mitochondrial RNase P
Drosophila is the only model organism in which all three members of the mtRNase P complex have been studied and in which MRPP3’s essential role in vivo was first demonstrated (Table 3.3; Sen et al. 2016). Sen et al. identified the MRPP3 and MRPP1 orthologs, called Mulder (Mldr) and Roswell (Rswl), respectively, and used genetics and cell biology to determine the role of mtRNase P in mitochondrial function and mt-RNA processing in vivo. All three Drosophila orthologs localize to mitochondria and associate with each other (Sen et al. 2016). Torroja et al. characterized mutations in scu and found they were lethal and had reduced dehydrogenase activity (Torroja et al. 1998). By examining EMS-induced point mutations, as well as RNAi knockdown, Sen et al. showed that loss of each mtRNase P complex member delayed larval development and was pupal-lethal. Tissue extract from mutants had very low ATP levels, and immunofluorescence showed swollen mitochondria. Using Northern blots probed with four different mt-tRNAs in different RNA contexts, the authors found unprocessed mt-RNAs in extract from rswl RNAi knockdown and mldr and scu mutants. While Sen et al. only had RNAi available for rswl, we can show that a transposable element-induced mutation in rswl is also lethal, with delayed pupal development consistent with our previous observations (Fig. 3.5, M.S. and R.T.C., unpublished data).

A mutation in roswell causes pupal lethality. (a, b) A graph showing rswl 07838 mutant larvae have delayed development and only approximately 30% eventually pupate compared to sibling controls. No rswl 07838 mutant adults emerge from the pupal cases (eclosion) (b). rswl 07838 is induced by a transposable element insertion (Bellen et al. 2004; Spradling et al. 1999). (c) After 5 days, rswl 07838 mutant larvae (left) are much smaller than their wild-type siblings (right). These phenotypes are consistent with rswl RNAi knockdown and loss of Mulder and Scully (Sen et al. 2016). Pupation and eclosion rates were performed as described in Sen et al. (2016)
Mouse and human contain two proteins, ELAC1 and ELAC2, that perform 3′-end processing of nuclear and mitochondrial tRNAs, respectively (see above). ELAC2 encodes two products of which only one is targeted to mitochondria. In contrast, Drosophila contains a single gene, dRNaseZ, which encodes an N-terminal mitochondrial targeting sequence and two nuclear localization signals (NLS). It is not clear how the protein gets differentially targeted to the two organelles, though there are other examples of this situation in the literature (Yogev and Pines 2011). Dubrovsky et al. cloned the Drosophila homolog and showed that it can cleave tRNAs in vitro (Dubrovsky et al. 2004). They also demonstrated that RNAi knockdown and deletion of the gene causes growth defects and defects in tRNA and mt-tRNA processing (Table 3.3; Xie et al. 2011, 2013). However, it is not clear whether the lethality due to loss of the gene is caused by mitochondria-specific disruptions, nucleus disruptions, or both.
3.5.2 Mouse Mitochondrial RNase P
Rackham et al. created knockout mice for MRPP3 (Table 3.3; Rackham et al. 2016). The authors not only clearly showed mt-tRNA processing defects but were also able to show that mt-RNA processing links transcription to translation through mitoribosome assembly (described above). Using Cre recombinase, the authors removed the third exon of MRPP3 to create a knockout allele. The MRPP3 full-body conditional knockout mice died at day E8.5. This is consistent with mouse knockouts for proteins involved in mitochondrial gene expression. Since the mice died so young, Rackham et al. used muscle-specific Cre to produce mice lacking MRPP3 in heart and skeletal muscle. The mice had reduced muscle fibers with reduced Complex I and Complex IV staining, and died at week 11 from cardiomyopathy, a phenotype frequently seen in mitochondrial disease.
MRPP2/HSD10 is the other member of the mtRNase P complex that has been studied in mouse. Rauschenberger et al. created a conditional knockout of MRPP2 (Table 3.3; Rauschenberger et al. 2010). They reported that the mouse knockout resulted in early embryonic lethality. In order to study MRPP2 function, they established conditional knockouts, one using a Cre recombinase to eliminate the protein in endothelial and immune cells and one using Cre to affect noradrenergic neurons. The endothelial knockout mice died at week 25 with defects in spleen and vasculature. The noradrenergic knockout mice died at week 26. Mitochondria in the loci coerulei of the brain, which contain noradrenergic neurons, lacked normal, dense cristae. To circumvent the early embryonic lethality in mice, Rauschenberger et al. also examined MRPP2 function in another vertebrate, Xenopus laevis, using morpholinos to knock down protein expression in the animal cap (Rauschenberger et al. 2010). Explants from these cells had decreased mitochondrial function inferred by a decrease in pyruvate turnover. Mitochondrial ultrastructure was also disrupted as judged by transmission electron microscopy. In addition, the authors examined morpholino-induced loss of MRPP2 on neural tissue patterning. They found patterning was not disrupted, but tissue size was reduced. This may have been due to an increase in apoptosis, as shown with increased TUNEL labeling.
3.6 Concluding Remarks and Future Directions
Processing of mtDNA transcripts presents unique challenges due to the constraints of the very small genome encoding all three major RNA types encoded in polycistronic messages. mtDNA supplies the mt-rRNAs for the mitoribosome and all the mt-tRNAs. Both types of mt-RNA are required in order to translate the 13 proteins encoded by mtDNA. As such, properly cleaving each product becomes essential in order for the organelle to maintain its function. The suite of mt-tRNAs is also small relative to the number encoded in the nucleus. There is only one cognate mt-tRNA per amino acid, with the exception of serine and leucine; thus, disrupting processing of a single mt-tRNA transcript is problematic.
We now know that point mutations in mt-tRNAs can affect processing, leading to maternally inherited hypertension and mitochondrial disease. In addition, pathogenic mutations in MRPP1, MRPP2, and MRPP3 have been identified. Because null alleles of mtRNase P are likely to be lethal, they have not appeared in humans. Given the diverse nature of symptoms resulting from defects in mt-tRNA processing, especially the link to hypertension, there may be many more as yet undetected mild mutations that could predispose individuals to a variety of skeletal, neural, and cardiovascular problems.
This is an exciting time to study mt-tRNA processing. Mutations in mt-tRNAs have long been known to cause mitochondrial diseases, but we are still parsing the details of how mtDNA transcription, processing, and translation control feedback to each other, the sequential steps of regulation, and how defects in each process lead to different diseases and symptoms. A better understanding of the molecular mechanisms underlying mt-RNA processing may open avenues to more effective treatments and cures of those with mitochondrial diseases.
References
Akagawa S et al (2017) Japanese male siblings with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) without neurological regression. JIMD Rep 32:81–85. https://doi.org/10.1007/8904_2016_570
Akawi NA et al (2016) A homozygous splicing mutation in ELAC2 suggests phenotypic variability including intellectual disability with minimal cardiac involvement. Orphanet J Rare Dis 11:139. https://doi.org/10.1186/s13023-016-0526-8
Anderson S et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Antonicka H, Shoubridge EA (2015) Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. https://doi.org/10.1016/j.celrep.2015.01.030
Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA (2013) The mitochondrial RNA-binding protein GRSF1 localizes to RNA granules and is required for posttranscriptional mitochondrial gene expression. Cell Metab 17:386–398. https://doi.org/10.1016/j.cmet.2013.02.006
Bellen HJ et al (2004) The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167:761–781. https://doi.org/10.1534/genetics.104.026427
Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ (2013) Human mitochondrial RNA decay mediated by PNPase–hSuv3 complex takes place in distinct foci. Nucleic Acids Res 41:1223–1240. https://doi.org/10.1093/nar/gks1130
Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC (2005) MITOMAP: a human mitochondrial genome database—2004 update. Nucleic Acids Res 33:D611–D613. https://doi.org/10.1093/nar/gki079
Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP (2011) Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol 8:616–626. https://doi.org/10.4161/rna.8.4.15393
Chatfield KC et al (2015) Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 21:1–10. https://doi.org/10.1016/j.mito.2014.12.005
Chinnery PF (2000) Mitochondrial disorders overview. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews® [Internet]. University of Washington, Seattle, pp 1993–2017
Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241:779–786
Deutschmann AJ et al (2014) Mutation or knock-down of 17beta-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum Mol Genet 23:3618–3628. https://doi.org/10.1093/hmg/ddu072
Dubrovsky EB, Dubrovskaya VA, Levinger L, Schiffer S, Marchfelder A (2004) Drosophila RNase Z processes mitochondrial and nuclear pre-tRNA 3′ ends in vivo. Nucleic Acids Res 32:255–262. https://doi.org/10.1093/nar/gkh182
Falk MJ et al (2016) A novel HSD17B10 mutation impairing the activities of the mitochondrial RNase P complex causes X-linked intractable epilepsy and neurodevelopmental regression. RNA Biol 13:477–485. https://doi.org/10.1080/15476286.2016.1159381
Fukao T et al (2014) The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation. J Hum Genet 59:609–614. https://doi.org/10.1038/jhg.2014.79
Gobert A et al (2010) A single Arabidopsis organellar protein has RNase P activity. Nat Struct Mol Biol 17:740. doi:https://doi.org/10.1038/nsmb.1812. https://www.nature.com/articles/nsmb.1812—supplementary-information
Gray MW (2012) Mitochondrial evolution. Cold Spring Harb Perspect Biol 4:a011403. https://doi.org/10.1101/cshperspect.a011403
Guan MX, Enriquez JA, Fischel-Ghodsian N, Puranam RS, Lin CP, Maw MA, Attardi G (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol Cell Biol 18:5868–5879
Guo L, Yuan Y, Bi R (2016) Mitochondrial DNA mutation m.5512A > G in the acceptor-stem of mitochondrial tRNATrp causing maternally inherited essential hypertension. Biochem Biophys Res Commun 479:800–807. https://doi.org/10.1016/j.bbrc.2016.09.129
Haack TB et al (2013) ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet 93:211–223. https://doi.org/10.1016/j.ajhg.2013.06.006
Hochberg I et al (2017) A homozygous variant in mitochondrial RNase P subunit PRORP is associated with Perrault syndrome characterized by hearing loss and primary ovarian insufficiency. BioRxiv. https://doi.org/10.1101/168252
Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135:462–474. https://doi.org/10.1016/j.cell.2008.09.013
Howard MJ, Lim WH, Fierke CA, Koutmos M (2012) Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci U S A 109:16149–16154. https://doi.org/10.1073/pnas.1209062109
Iborra FJ, Kimura H, Cook PR (2004) The functional organization of mitochondrial genomes in human cells. BMC Biol 2:9. https://doi.org/10.1186/1741-7007-2-9
Jablonski JA, Caputi M (2009) Role of cellular RNA processing factors in human immunodeficiency virus type 1 mRNA metabolism, replication, and infectivity. J Virol 83:981–992. https://doi.org/10.1128/JVI.01801-08
Jackman JE, Montange RK, Malik HS, Phizicky EM (2003) Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA 9:574–585. https://doi.org/10.1261/rna.5070303
Jenkinson EM et al (2012) Perrault syndrome: further evidence for genetic heterogeneity. J Neurol 259:974–976. https://doi.org/10.1007/s00415-011-6285-5
Jiang P et al (2016) A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol Cell Biol 36:1920–1930. https://doi.org/10.1128/MCB.00199-16
Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, Martinou JC (2013) GRSF1 regulates RNA processing in mitochondrial RNA granules. Cell Metab 17:399–410. https://doi.org/10.1016/j.cmet.2013.02.005
Kash JC, Cunningham DM, Smit MW, Park Y, Fritz D, Wilusz J, Katze MG (2002) Selective translation of eukaryotic mRNAs: functional molecular analysis of GRSF-1, a positive regulator of influenza virus protein synthesis. J Virol 76:10417–10426. https://doi.org/10.1128/JVI.76.20.10417-10426.2002
Kissinger CR et al (2004) Crystal structure of human ABAD/HSD10 with a bound inhibitor: implications for design of Alzheimer’s disease therapeutics. J Mol Biol 342:943–952. https://doi.org/10.1016/j.jmb.2004.07.071
Klemm BP, Wu N, Chen Y, Liu X, Kaitany KJ, Howard MJ, Fierke CA (2016) The diversity of ribonuclease P: protein and RNA catalysts with analogous biological functions. Biomol Ther 6. https://doi.org/10.3390/biom6020027
Korman SH (2006) Inborn errors of isoleucine degradation: a review. Mol Genet Metab 89:289–299. https://doi.org/10.1016/j.ymgme.2006.07.010
Land M et al (2015) Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15:141–161. https://doi.org/10.1007/s10142-015-0433-4
Levinger L, Jacobs O, James M (2001) In vitro 3′-end endonucleolytic processing defect in a human mitochondrial tRNASer(UCN) precursor with the U7445C substitution, which causes non-syndromic deafness. Nucleic Acids Res 29:4334–4340
Levinger L, Giege R, Florentz C (2003) Pathology-related substitutions in human mitochondrial tRNAIle reduce precursor 3′ end processing efficiency in vitro. Nucleic Acids Res 31:1904–1912
Levinger L, Oestreich I, Florentz C, Mörl M (2004) A pathogenesis-associated mutation in human mitochondrial tRNALeu(UUR) leads to reduced 3′-end processing and CCA addition. J Mol Biol 337:535–544. https://doi.org/10.1016/j.jmb.2004.02.008
Lewis OL, Farr CL, Kaguni LS (1995) Drosophila melanogaster Mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol 4:263–278. https://doi.org/10.1111/j.1365-2583.1995.tb00032.x
Li R, Liu Y, Li Z, Yang L, Wang S, Guan MX (2009) Failures in mitochondrial tRNAMet and tRNAGln metabolism caused by the novel 4401A>G mutation are involved in essential hypertension in a Han Chinese family. Hypertension 54:329–337. https://doi.org/10.1161/HYPERTENSIONAHA.109.129270
Liu Y, Li Y, Zhu C, Tian L, Guan M, Chen Y (2017) Mitochondrial biogenesis dysfunction and metabolic dysfunction from a novel mitochondrial tRNAMet 4467 C>A mutation in a Han Chinese family with maternally inherited hypertension. Sci Rep 7:3034. https://doi.org/10.1038/s41598-017-03303-w
Margulis L (1970) Origin of eukaryotic cells. Yale University Press, New Haven
Mercer TR et al (2011) The human mitochondrial transcriptome. Cell 146:645–658. https://doi.org/10.1016/j.cell.2011.06.051
Metodiev MD et al (2016) Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet 98:993–1000. https://doi.org/10.1016/j.ajhg.2016.03.010
Moeller G, Adamski J (2009) Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol 301:7–19. https://doi.org/10.1016/j.mce.2008.10.040
Oerum S et al (2017) Novel patient missense mutations in the HSD17B10 gene affect dehydrogenase and mitochondrial tRNA modification functions of the encoded protein. Biochim Biophys Acta 1863(12):3294–3302. https://doi.org/10.1016/j.bbadis.2017.09.002
Ofman R et al (2003) 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am J Hum Genet 72:1300–1307
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290:470–474
Page IH (1967) The mosaic theory of arterial hypertension—its interpretation. Perspect Biol Med 10:325–333
Park H, Davidson E, King MP (2003) The pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR) decreases the efficiency of aminoacylation. Biochemistry 42:958–964. https://doi.org/10.1021/bi026882r
Perez-Cerda C et al (2005) 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: an X-linked inborn error of isoleucine metabolism that may mimic a mitochondrial disease. Pediatr Res 58:488–491. https://doi.org/10.1203/01.pdr.0000176916.94328.cd
Powell CA, Nicholls TJ, Minczuk M (2015) Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front Genet 6:79. https://doi.org/10.3389/fgene.2015.00079
Rackham O et al (2016) Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16:1874–1890. https://doi.org/10.1016/j.celrep.2016.07.031
Rauschenberger K et al (2010) A non-enzymatic function of 17beta-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol Med 2:51–62. https://doi.org/10.1002/emmm.200900055
Reid FM, Rovio A, Holt IJ, Jacobs HT (1997) Molecular phenotype of a human lymphoblastoid cell-line homoplasmic for the np 7445 deafness-associated mitochondrial mutation. Hum Mol Genet 6:443–449
Reinhard L, Sridhara S, Hällberg BM (2017) The MRPP1/MRPP2 complex is a tRNA-maturation platform in human mitochondria. Nucleic Acids Res 45(21):12469–12480. https://doi.org/10.1093/nar/gkx902
Richardson A, Berry GT, Garganta C, Abbott MA (2016) Hydroxysteroid 17-beta dehydrogenase type 10 disease in siblings. JIMD Rep 32:25–32. https://doi.org/10.1007/8904_2016_547
Robertson HD, Altman S, Smith JD (1972) Purification and properties of a specific Escherichia coli ribonuclease which cleaves a tyrosine transfer ribonucleic acid precursor. J Biol Chem 247:5243–5251
Sanchez MI et al (2011) RNA processing in human mitochondria. Cell Cycle 10:2904–2916. https://doi.org/10.4161/cc.10.17.17060
Schaub MC, Lopez SR, Caputi M (2007) Members of the heterogeneous nuclear ribonucleoprotein H family activate splicing of an HIV-1 splicing substrate by promoting formation of ATP-dependent spliceosomal complexes. J Biol Chem 282:13617–13626
Seaver LH et al (2011) A novel mutation in the HSD17B10 gene of a 10-year-old boy with refractory epilepsy, choreoathetosis and learning disability. PLoS One 6:e27348. https://doi.org/10.1371/journal.pone.0027348
Sen A, Cox RT (2017) Fly models of human diseases: Drosophila as a model for understanding human mitochondrial mutations and disease. Curr Top Dev Biol 121:1–27. https://doi.org/10.1016/bs.ctdb.2016.07.001
Sen A, Karasik A, Shanmuganathan A, Mirkovic E, Koutmos M, Cox RT (2016) Loss of the mitochondrial protein-only ribonuclease P complex causes aberrant tRNA processing and lethality in Drosophila. Nucleic Acids Res 44:6409–6422. https://doi.org/10.1093/nar/gkw338
Shafqat N et al (2003) Expanded substrate screenings of human and Drosophila type 10 17β-hydroxysteroid dehydrogenases (HSDs) reveal multiple specificities in bile acid and steroid hormone metabolism: characterization of multifunctional 3α/7α/7β/17β/20β/21-HSD. Biochem J 376:49–60
Shao Z et al (2014) Crystal structure of tRNA m1G9 methyltransferase Trm10: insight into the catalytic mechanism and recognition of tRNA substrate. Nucleic Acids Res 42:509–525. https://doi.org/10.1093/nar/gkt869
Spradling AC et al (1999) The Berkeley Drosophila genome project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153:135–177
Sutton VR, O'Brien WE, Clark GD, Kim J, Wanders RJ (2003) 3-Hydroxy-2-methylbutyryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 26:69–71
Taanman J-W (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123. https://doi.org/10.1016/S0005-2728(98)00161-3
Taschner A, Weber C, Buzet A, Hartmann Roland K, Hartig A, Rossmanith W (2012) Nuclear RNase P of Trypanosoma brucei: a single protein in place of the multicomponent RNA-protein complex. Cell Rep 2:19–25. https://doi.org/10.1016/j.celrep.2012.05.021
Torroja L, Ortuno-Sahagun D, Ferrus A, Hammerle B, Barbas JA (1998) scully, an essential gene of Drosophila, is homologous to mammalian mitochondrial type II L-3-hydroxyacyl-CoA dehydrogenase/amyloid-beta peptide-binding protein. J Cell Biol 141:1009–1017
Van Haute L, Pearce SF, Powell CA, D'Souza AR, Nicholls TJ, Minczuk M (2015) Mitochondrial transcript maturation and its disorders. J Inherit Metab Dis 38:655–680. https://doi.org/10.1007/s10545-015-9859-z
Vilardo E, Rossmanith W (2015) Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res 43:5112–5119. https://doi.org/10.1093/nar/gkv408
Vilardo E, Nachbagauer C, Buzet A, Taschner A, Holzmann J, Rossmanith W (2012) A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase—extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res 40:11583–11593. https://doi.org/10.1093/nar/gks910
Wang S et al (2011) Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res 108:862–870. https://doi.org/10.1161/CIRCRESAHA.110.231811
Xie X, Dubrovskaya VA, Dubrovsky EB (2011) RNAi knockdown of dRNaseZ, the Drosophila homolog of ELAC2, impairs growth of mitotic and endoreplicating tissues. Insect Biochem Mol Biol 41:167–177. https://doi.org/10.1016/j.ibmb.2010.12.001
Xie X, Dubrovskaya V, Yacoub N, Walska J, Gleason T, Reid K, Dubrovsky EB (2013) Developmental roles of Drosophila tRNA processing endonuclease RNase ZL as revealed with a conditional rescue system. Dev Biol 381:324–340. https://doi.org/10.1016/j.ydbio.2013.07.005
Xu F et al (2008) Disruption of a mitochondrial RNA-binding protein gene results in decreased cytochrome b expression and a marked reduction in ubiquinol–cytochrome c reductase activity in mouse heart mitochondria. Biochem J 416:15
Yang SY et al (2009) Mental retardation linked to mutations in the HSD17B10 gene interfering with neurosteroid and isoleucine metabolism. Proc Natl Acad Sci U S A 106:14820–14824. https://doi.org/10.1073/pnas.0902377106
Yang S-Y, He X-Y, Isaacs C, Dobkin C, Miller D, Philipp M (2014) Roles of 17β-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J Steroid Biochem Mol Biol 143:460–472. https://doi.org/10.1016/j.jsbmb.2014.07.001
Yogev O, Pines O (2011) Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim Biophys Acta 1808:1012–1020. https://doi.org/10.1016/j.bbamem.2010.07.004
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. https://doi.org/10.1186/1471-2105-9-40
Zhu HY, Wang SW, Liu L, Li YH, Chen R, Wang L, Holliman CJ (2009) A mitochondrial mutation A4401G is involved in the pathogenesis of left ventricular hypertrophy in Chinese hypertensives. Eur J Hum Genet 17:172–178. https://doi.org/10.1038/ejhg.2008.151
Zschocke J (2012) HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis 35:81–89. https://doi.org/10.1007/s10545-011-9415-4
Zschocke J, Ruiter JP, Brand J, Lindner M, Hoffmann GF, Wanders RJ, Mayatepek E (2000) Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr Res 48:852–855. https://doi.org/10.1203/00006450-200012000-00025
Funding
This work was supported by the National Institutes of Health/Department of Defense [CHIRP HU0001–14–2-0041 to M.S. and R.T.C.].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Saoji, M., Cox, R.T. (2018). Mitochondrial RNase P Complex in Animals: Mitochondrial tRNA Processing and Links to Disease. In: Cruz-Reyes, J., Gray, M. (eds) RNA Metabolism in Mitochondria. Nucleic Acids and Molecular Biology, vol 34. Springer, Cham. https://doi.org/10.1007/978-3-319-78190-7_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-78190-7_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78189-1
Online ISBN: 978-3-319-78190-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)