
Abstract
Massive amounts of methane are produced on Earth. Methane is useful as an energy source and as an energy storage material for H2. However, there is increasing concern about methane concentrations in the atmosphere because it is a potent greenhouse gas. Methane is biologically produced primarily by methanogenic archaea, most of which produce methane hydrogenotrophically from H2 and CO2. Many enzymes involved in the hydrogenotrophic methanogenic pathway are shared in the methanogenic pathway from C1 compounds or acetate. The methanogenic pathways contain unique enzymes and their prosthetic groups using unique electron and C1 carriers. Here, we describe an overview of the hydrogenotrophic methanogenic pathway, including the energy conservation and energy-coupling systems. The catalytic functions and mechanisms of the methanogenic enzymes are discussed based on their crystal structures.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


1 Introduction
Hydrogenotrophic methanogens are archaea that can grow on H2 and CO2 with the production of methane, an important intermediate in the global carbon cycle. They have a unique biochemistry that has been unraveled over the last 40 years. Methane is produced mainly by the anaerobic decomposition of plant biomass in anoxic environments, where the concentrations of sulfate, Fe(III), Mn(IV), and nitrate are low, such as in freshwater sediments, wetlands, and the intestinal tract of animals (Thauer et al. 2008). In anoxic environments, methane is generated by methanogenic archaea, and via this action, approximately 1 Gt of methane is formed globally every year, which is approximately 1% of the net carbon fixed from CO2 into plant biomass every year via photosynthesis. Most methane diffuses into oxic environments, where approximately 60% is oxidized to CO2 with O2 by methanotrophic bacteria. The remaining 40% escapes into the atmosphere, where most of it is photochemically converted to CO2. The concentration of methane in the atmosphere has more than doubled in the last 100 years, indicating that the rate of methane release (from all sources) into the atmosphere has increased relative to the rate of methane oxidation (Thauer et al. 2008). This is of concern, since methane is a potent greenhouse gas considered to contribute significantly to global warming. Methanogenesis is also of biotechnological interest in sewage treatment plants and in biogas production plants. The microbial formation of methane from H2 and CO2 has also been discussed as a means of H2 storage (Thauer et al. 2010).
Phylogenetic analysis indicated that methanogenic organisms are exclusively classified into archaea (Boone et al. 1993). Most methanogenic archaea are found in the lineage of Euryarchaeota, but recent metagenomic analysis has shed light on the presence of two lineages – Bathyarchaeota and Verstraetearchaeota – that are phylogenetically distant from Euryarchaeota (Fig. 1). Most orders of methanogenic archaea produce methane from H2 and CO2, from formate, or from H2 and methanol; these are referred to as hydrogenotrophic methanogens. Only one order, the Methanosarcinales, can also produce methane from acetate and from the disproportionation of C1 compounds such as methanol, methylamines, and methylthiols; these are referred to as acetoclastic methanogens and methylotrophic methanogens, respectively. The hydrogenotrophic methanogens differ from the archaea in the order of Methanosarcinales, as they are devoid of cytochromes and methanophenazine (Fig. 2) and use only sodium ions rather than protons for chemiosmotic energy conservation (Thauer et al. 2008).

Phylogeny of methanogenic archaea. Maximum likelihood tree was constructed from 16S rRNA sequences aligned by ClustalW. There were a total of 1396 comparable positions. Bootstrap values >50% based on 100 resamplings are indicated at the nodes. Crenarchaeota was used as the outgroup. Bar indicates 0.05 substitutions per nucleotide position. The tree consists of all known methanogenic families, orders, and phyla including Candidatus taxon

2 Energy Metabolism on H2 and CO2
The standard free energy change of methane formation from 4H2 and CO2 (ΔG°′) is −131 kJ/mol. Under physiological conditions where the partial pressure of H2 is only approximately 10 Pa, the free energy change is only approximately −30 kJ/mol of methane formed. The biosynthesis of ATP from ADP and inorganic phosphate in vivo is estimated to be between −60 and −70 kJ/mol, although under energy limitations, the value might be considerably lower. It is likely that less than 1 mol ATP is formed per mol methane. The exact ATP gain (mol ATP/mol CH4) is of general interest because it is an open question how close to thermodynamic equilibrium the energy metabolism of strict anaerobes can operate and how small the minimal free energy change increment must be to sustain life in anaerobic environments such as the deep biosphere (Thauer et al. 2008).
3 Reactions Involved in Methanogenesis from H2 and CO2
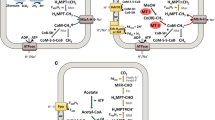
From mainly the works of Wolfe (Dimarco et al. 1990; Wolfe 1991), Gottschalk (Gottschalk and Blaut 1990; Deppenmeier et al. 1996), and Thauer (Thauer et al. 2008) and their collaborators, methanogenesis from H2 and CO2 is known to involve five coenzymes (Fig. 2) and ten reactions (Fig. 3). The structure of methanopterin was elucidated by Keltjens and Vogels (van Beelen et al. 1984). The pathway begins with the reduction of CO2 on methanofuran (MFR) with reduced ferredoxin (Fdred) to formyl-MFR catalyzed by formylmethanofuran dehydrogenase; in most methanogens there are two isoenzymes, one containing molybdenum (Fmd) and the other containing tungsten (Fwd). Subsequently, the formyl group of formyl-MFR is transferred to tetrahydromethanopterin (H4MPT) by formyltransferase (Ftr). N5-Formyl-H4MPT is subsequently converted in three steps to methyl-H4MPT via methenyl- and methylene-H4MPT as intermediates using methenyl-H4MPT+ cyclohydrolase (Mch), F420-dependent methylene-H4MPT dehydrogenase (Mtd), and F420-dependent methylene-H4MPT reductase (Mer). An alternate reaction to Mtd is catalyzed by H2-forming methylene-H4MPT dehydrogenase (Hmd or [Fe]-hydrogenase), which catalyzes the conversion of methenyl-H4MPT+ to methylene-H4MPT using H2 as an electron donor. F420 is a 5-deazaflavin that is converted to the reduced form (F420H2, Fig. 2) by H2 catalyzed by F420-reducing [NiFe]-hydrogenase (Frh). F420H2 is used as an electron donor for the two reduction steps in hydrogenotrophic methanogenesis and for other anabolic reduction reactions in the methanogenic archaea. Under nickel-limiting conditions, the [NiFe]-hydrogenase Frh is substituted by Hmd; Hmd is coupled with Mtd to reduce F420 with electrons from H2. After methyl-H4MPT is formed, its methyl group is transferred to coenzyme M (CoM-SH), yielding methyl-S-CoM in an exergonic reaction catalyzed by a membrane-associated methyltransferase complex (MtrA-H). The exergonic methyl-transfer reaction is coupled to endergonic sodium-ion translocation (Gottschalk and Thauer 2001). The sodium ion motive force thus generated is utilized by an A1AO-type ATP synthase to drive the phosphorylation of ADP (Vonck et al. 2009). In the next step, methyl-S-CoM is reduced with coenzyme B (CoB-SH) to methane and a heterodisulfide (CoM-S-S-CoB); this reaction is catalyzed by methyl-S-CoM reductase (Ermler et al. 1997a). CoM-S-S-CoB is reduced with H2 to CoM-SH and HS-CoB, catalyzed by the electron-bifurcating [NiFe]-hydrogenase/heterodisulfide reductase complex (MvhADG-HdrABC). This complex couples the exergonic reduction of CoM-S-S-CoB with H2 to the endergonic reduction of ferredoxin with H2. The reduced ferredoxin thus generated is used in the first step of the hydrogenotrophic methanogenesis, the reduction of CO2 to formyl-MFR.

Overview of the hydrogenotrophic methanogenic pathway (Bai et al. 2017)
4 Methanogenic Enzymes
4.1 Formylmethanofuran Dehydrogenase (Fmd and Fwd)
Hydrogenotrophic methanogenesis begins with the reductive bonding of CO2 to the amino group of the C1 carrier methanofuran to form formylmethanofuran. This reversible reaction is catalyzed by Fmd or Fwd. The redox potential of the formylmethanofuran/methanofuran couple is very low (E°′ = −530 mV) (Bertram and Thauer 1994); therefore, the reduction requires high-energy electrons from reduced ferredoxin (E′ = ~ −500 mV) (Kaster et al. 2011).
Crystal structure analysis of Fwd from Methanothermobacter wolfeii revealed a Fwd(ABCDFG)4 organization (Fig. 4) (Wagner et al. 2016a). FwdA is similar to the amidohydrolases, i.e., urease, phosphotriesterase, and dihydroorotase/hydantoinase. The metal ligands, including the posttranslationally modified N6-carboxylysine and a catalytically crucial aspartate, are strictly conserved in FwdA. Crystals soaked with methanofuran led to the identification of the binding site: a cavity extending from the dinuclear metal center to bulk solvent (Fig. 5a, c). FwdB and FwdD form a formate dehydrogenase-like catalytic unit, which is a member of the molybdo-/tungstopterin-dependent DMSO reductase superfamily. The redox-active tungsten of FwdBD is coordinated to four dithiolene thiolates of two tungstopterin guanine dinucleotide molecules (Fig. 5b). The [4Fe-4S]-cluster, tungstopterin-binding, the active site residues, and tungsten ligation mode are essentially conserved between FwdBD and formate dehydrogenases. FwdC shares the highest structural similarities to a C-terminal glutamate synthase domain (Binda et al. 2000), which has an architectural function. FwdF is the first polyferredoxin to be structurally analyzed; it is composed of four fused similar ferredoxin modules, each carrying two [4Fe-4S] clusters that are arranged in a “T”-shaped conformation (Fig. 4).

Structure of tungsten-containing formylmethanofuran dehydrogenase (Fwd) from Methanothermobacter wolfeii (Wagner et al. 2016a). (a) The Fwd(ABCDFG)4 complex. Four FwdABCDFG heterohexamers are shown in white, black, sand, and multiple colors of subunits. (b) The Fwd(ABCDFG)4 complex in 90° rotated angle from the orientation in panel a

Active site structures of Fwd. (a) FwdABD contains two metal active sites responsible for CO2 reduction and condensation of formate on MFR. FwdBD harbors the tungstopterin (light blue stick model for the organic part and black ball model for tungsten), and FwdA has a dinuclear zinc site (gray ball model). Iron-sulfur clusters in FwdG and FwdB are depicted as brown and yellow ball. CO2 entrance channel (blue surface) and formate transport channel (red surface) are shown, connecting the tungstopterin active site (b) and dinuclear zinc amidohydrolase active site (c)
The crystal structure of the FwdABCDFG complex provided evidence of the catalytic mechanism. The Fwd(ABCDFG)4 complex can be subdivided into an electron-supplying core (FwdF and FwdG) flanked by four catalytic units formed by FwdABCD (Fig. 5a). Each catalytic unit hosts two spatially separated active sites for the dual reactions. First, CO2 is funneled through a narrow 35-Å-long hydrophobic channel to the FwdBD tungstopterin center, namely, the formate dehydrogenase core (Fig. 5b). Previous biochemical studies indicated weak formate dehydrogenase activity for formylmethanofuran dehydrogenases (Bertram et al. 1994). The deeply buried redox-active tungsten center is connected to the [4Fe-4S] chains to efficiently transfer low-potential electrons to reduce CO2 to formate. The produced formate is transferred via an internal, 27-Å-long, hydrophilic tunnel and reacts with the amino group of methanofuran to form formylmethanofuran at the binuclear metal center of FwdA (Fig. 5c). The Fwd(ABCDFG)4 complex harbors 46 [4Fe-4S] clusters in the electron-supplying unit (Fig. 4), which is composed of iron-sulfur cluster chain links with short edge-to-edge distances for efficient electron transfer. The electron wires connect the redox-active tungsten sites of the 12-mer Fwd(ABCDFG)2 and the 24-mer Fwd(ABCDFG)4 over distances of ca. 188 Å and 206 Å, respectively.
4.2 Formyltransferase (Ftr)
The formyl group bound to methanofuran is transferred to H4MPT to form N5-formyl-H4MPT. This formyl transfer reaction is catalyzed by Ftr. The crystal structures of Ftr from Methanopyrus kandleri, Methanosarcina barkeri, and Archaeoglobus fulgidus have been solved. Methanopyrus kandleri is a hyperthermophilic methanogen (optimum growth temperature, 98 °C), and its Ftr contains a homotetramer in the crystal structure (Fig. 6a, c) (Ermler et al. 1997b). Biophysical experiments using analytical ultracentrifugation indicated that Ftr from M. kandleri is in equilibrium of monomer/dimer/tetramer, which is dependent on the concentration of lyotropic salts (i.e., potassium phosphate and ammonium sulfate) in the enzyme solution (Shima et al. 1998). The larger oligomeric forms appear with increasing salt concentration. At low salt concentration, this enzyme is inactive (as a monomer). Ftr is activated at higher salt concentrations when it forms dimer or tetramer. This finding suggests that the active form is a homodimer. This hypothesis was supported by the X-ray crystal structure analysis of Ftr in complex with the substrates formylmethanofuran and H4MPT (Acharya et al. 2006). The structure shows that each substrate is bound to different subunits, as shown in Fig. 6a, b, which indicate localization of the active site at the dimeric interface. For M. kandleri, the major dimer/dimer interaction of Ftr is the salt bridges between Glu64 and Arg261 (Fig. 6c). The tetrameric form of Ftr from M. kandleri stabilizes this protein against heat rather than catalytic activity (Shima et al. 2000a).

Structure and function of formyl-MFR:H4MPT formyltransferase (Ftr). (a) Cartoon model of the dimer of Ftr from M. kandleri in complex with its substrates formyl-MFR and H4MPT (Acharya et al. 2006). The bulky coenzymes MFR (orange) and H4MPT (green) are embedded into large surface clefts located between two monomers (purple and sand). (b) Surface model of the formyl-MFR and the H4MPT binding site shows the substrate-binding cleft. Two monomers are colored in purple (H4MPT binding) and sand (formyl-MFR binding). (c) In the tetrameric form, the contacts between two dimers (light blue and purple) involve salt bridges between Arg261 and Glu64. (d) A tetrahedral oxyanion intermediate proposed in the catalytic reaction
A catalytic mechanism for Ftr was proposed based on the ternary complex of Ftr with formyl-MFR and H4MPT (Acharya et al. 2006). Hydrogen bonds between Ser209 and formamide-N of formyl-MFR and between the formamide-O and the protonated carboxy-group of Glu245 increase the electrophilicity of the formamide-C. Nucleophilic attack of N5 of H4MPT produces a tetrahedral oxyanion intermediate (Fig. 6d), which is then stabilized by protonation from the protonated carboxy of Glu245. A proton is transferred to the nitrogen of MFR and formyl-H4MPT is finally formed.
4.3 Cyclohydrolase (Mch)
Mch reversibly catalyzes the condensation reaction of formyl-H4MPT to methenyl-H4MPT+. The first crystal structure of Mch was solved using heterologously produced enzyme from M. kandleri. The catalytic reaction of Mch was studied based on the heterologously produced enzyme from the sulfate-reducing archaeon Archaeoglobus fulgidus, which has a C1 pathway containing H4MPT (Klein et al. 1993). Mch is a homotrimeric enzyme (Fig. 7a), and the substrate N5-formyl-H4MPT binds to the cleft between domain A and B of each monomer (Fig. 7b, c) as observed in the catalytically inactive mutant E186Q. In the proposed catalytic mechanism, from methenyl-H4MPT+ to formyl-H4MPT, the substrate water molecule trapped between Arg183 and Glu186 nucleophilically attacks the C14a of methenyl-H4MPT+ to form a tetrahedral imidazolidin-2-ol intermediate (Fig. 7d) (Upadhyay et al. 2012). A proton of the intermediate is transferred to N10 of H4MPT via the carboxy group of Glu186, which preferentially selects N5 as the leaving group. A proton on the intermediate is finally transferred to Glu186, which forms N5-formyl-H4MPT.

Structure and function of methenyl-H4MPT+ cyclohydrolase (Mch) (Upadhyay et al. 2012). (a) Homotrimer of Mch from M. kandleri. (b) The substrate-binding site of the monomer built up between domains A (pink) and B (blue) from the Mch of Archaeoglobus fulgidus. (c) Location of the catalytic R183 and Q186 and formyl-H4MPT binding site of the E186Q mutant. (d) A tetrahedral imidazolidin-2-ol intermediate proposed in the catalytic reactions
4.4 F420-Dependent Methylene-Tetrahydromethanopterin Dehydrogenases (Mtd)
Mtd catalyzes reversible hydride transfer from F420H2 to methenyl-H4MPT+ to form methylene-H4MPT. The crystal structure of Mtd from M. kandleri was reported (Hagemeier et al. 2003) and indicated that Mtd is a homohexameric protein composed of a trimer of dimers (Fig. 8a). Mtd has no structural similarity to known proteins, including those binding F420 and the H4MPT derivatives. Based on the ternary Mtd complex structure with F420H2 and methenyl-H4MPT+, a catalytic mechanism was proposed (Ceh et al. 2009). The substrates bind to the active site formed in the cleft on a subunit at the interface of two domains (Fig. 8b, c), in which both substrates face each other (Fig. 8b, c, d). This substrate arrangement indicated the direct hydride transfer between C5 of F420H2 and C14a of methenyl-H4MPT+, which allows stereospecific hydride transfer (Fig. 8d).

Structure of F420-dependent methylene-H4MPT dehydrogenases (Mtd). (a) Structure of the homohexameric enzyme complex from M. kandleri in complex with methenyl-H4MPT+ (green stick) and F420 (yellow stick). (b) The binding site of methenyl-H4MPT+ and F420H2 is at the interface of the two domains (in pink and blue) from one monomer shown with transparent surface model. (c) Active site cleft (surface model) binding the substrates with the same color code than shown in panel b. (d) Stereospecific hydride transfer of the proS hydride bound to C5 of F420H2 to the proR side of the C14a atom of methenyl-H4MPT+
4.5 H2-Forming Methylene-Tetrahydromethanopterin Dehydrogenase (Hmd)
Hmd ([Fe]-hydrogenase) catalyzes reversible hydride transfer from H2 to methenyl-H4MPT+ (Shima and Ermler 2011). The products of this reaction are methylene-H4MPT and a proton. This enzyme is found in the majority of hydrogenotrophic methanogenic archaea. Hmd contains a unique iron guanylylpyridinol (FeGP) cofactor (Fig. 9a, b). Crystallographic, spectroscopic, and chemical analyses of [Fe]-hydrogenase revealed that its iron center is ligated by Cys176-sulfur, two CO, one solvent molecule, an sp2-hybridized pyridinol-nitrogen, and an acyl carbon in the substituent of the pyridinol ring. The FeGP cofactor is extractable from [Fe]-hydrogenase, and the active holoenzyme can be reconstituted from the isolated cofactor and the apoenzyme that is heterologously produced in Escherichia coli (Shima and Ermler 2011).

H2-forming methylene-H4MPT dehydrogenase (Hmd) . (a) C176A-mutated holoenzyme from M. jannaschii in complex with methylene-H4MPT. The FeGP cofactor and substrate are indicated in stick models. (b) Chemical structure of the FeGP cofactor. The solvent-binding site was proposed as the H2-binding site (Shima et al. 2015). (c) The closed and open conformations of the Hmd homodimer. Two monomers are shown in black and green (open)/light blue (closed) cartoon models. (d) An iron-hydride intermediate representation from the proposed catalytic mechanism
In the crystal structures, the apoenzyme (without the FeGP cofactor) and holoenzyme (with the FeGP cofactor) of [Fe]-hydrogenase have closed and open conformations in the active site cleft, respectively. The crystal structure of the C176A-mutated holoenzyme-substrate binary complex of [Fe]-hydrogenase was reported and revealed an open cleft with a distance of 9.3 Å between the iron and the C14a atom of the substrate (Fig. 9a). This distance is obviously too long for the direct transfer of hydride ions. To model a catalytically productive conformation, the closed conformation of the apoenzyme was used. The movement of the peripheral unit from the open to the closed form essentially corresponds to a rotation of 35° (Fig. 9c). A structure-based mechanism of [Fe]-hydrogenase has been proposed based on biochemical and biophysical studies (Vogt et al. 2008; Hiromoto et al. 2009; Yang and Hall 2009; Hedegard et al. 2015; Shima et al. 2015). The catalytic cycle is initiated by the binding of methenyl-H4MPT+ to the open form, which triggers the closure of the cleft. Subsequently, H2 is supplied to the active site in the closed form and is captured in the “open coordination” site (Fig. 9b) of the iron center. The H2 molecule likely binds to the iron. The base of the reaction may be the deprotonated form of the pyridinol hydroxy group. Semisynthetic Hmd enzymes built up with heterologously produced apoenzyme and chemically synthesized mimics reveal that the deprotonated 2-hydroxy group is crucial for enzyme activity, which supports the base function of the 2-pyridinol hydroxyl group (Shima et al. 2015). Density functional theory (DFT) calculations support the catalytic mechanism including the iron-hydride intermediate (Fig. 9d). However, experimental evidence of the iron-hydride intermediate has not been reported.
4.6 Methylenetetrahydromethanopterin Reductase (Mer)
Mer catalyzes the reversible reduction of methylene-H4MPT to form methyl-H4MPT. The crystal structure of Mer was obtained using the purified enzymes from M. marburgensis, M. kandleri, and M. barkeri (Fig. 10) (Shima et al. 2000b; Aufhammer et al. 2005). Heterologous expression of Mer in E. coli was unsuccessful, likely because of the presence of a non-prolyl cis-peptide bond (Fig. 10c). Mer is organized as a TIM-barrel fold that forms a homodimer (for M. marburgensis) or homotetramer (M. barkeri and M. kandleri). The enzyme is homologous to F420-dependent secondary alcohol dehydrogenase (Aufhammer et al. 2004) and bacterial luciferase family proteins (Fig. 10d) (Baldwin et al. 1995; Aufhammer et al. 2005). The crystal structure of Mer from M. barkeri was solved in the complex structure with F420 (Fig. 10a, b), but the crystal structure of the complex with methylene-H4MPT or methyl-H4MPT has not yet been reported.

Structure and function of F420-dependent methylene-H4MPT reductase (Mer) (Shima et al. 2000b; Aufhammer et al. 2004). (a) The tetrameric form of Mer from M. barkeri. Each monomer is shown in a different color. (b) F420 binding site. (c) A protein segment of Mer from M. kandleri, which contains cis-peptide bond between Gly64 and Ile65, indicated by a black arrow. (d) Superposition of Mer from M. barkeri (purple) F420 complex and bacterial luciferase (LuxA) from Vibrio harveyi with bound FMN (green). Spheres in cyan/blue indicate the N-terminal positions, and red/orange indicate the C-terminal positions
4.7 Integral Membrane Methyltransferase (MtrA-H)
The membrane-associated MtrA-H complex catalyzes an exergonic cobalamin-dependent methyltransferase reaction and couples it to the electrogenic translocation of two sodium ions, as demonstrated by experiments using reconstituted ether lipid liposomes (Gottschalk and Thauer 2001). From the primary structure, it is predicted that MtrH is a peripheral protein without a membrane anchor; that MtrA, MtrB, MtrF, and MtrG are peripheral proteins with one transmembrane helix anchor; and that the three other subunits, MtrC, MtrD, and MtrE, are integral membrane proteins with at least six transmembrane helices (Fig. 11). MtrH has been shown to catalyze the methyl-transfer reaction from methyl-H4MPT to the corrinoid prosthetic group of MtrA. From the methylated corrinoid, the methyl group is transferred to CoM-SH. The latter methyl-transfer reaction is dependent on the presence of sodium ion, which suggests its involvement in sodium-ion translocation.

Reaction and model of the membrane-associated MtrA-H complex catalyzing methyl transfer from methyl-H4MPT to coenzyme M (HS-CoM). MtrH has been shown to catalyze methyl transfer from methyl-H4MPT to Co(I) of the corrinoid bound to MtrA. MtrE is proposed to catalyze methyl transfer from CH3-CoIII-MtrA to CoM-SH and to couple this reaction with the translocation of two sodium ions. Demethylation rather than methylation has been shown to be dependent on sodium ions (Gottschalk and Thauer 2001)
The crystal structure of the MtrA soluble domain indicated that MtrA has a unique cobalamin-binding site (Fig. 12) (Wagner et al. 2016b). The cobalt coordination in the crystal structure is hexa-coordinated including an external histidine residue from another monomer. From the coordination chemistry of B12, previous site-directed mutagenesis studies, and the crystal structure, it was predicted that in the reduced non-methylated Co(I) form, the histidine ligand will be decoupled from cobalt (tetra-coordination), and in the methylated Co(III) form, the histidine will bind to cobalt (hexa-coordination). The switch to cobalt coordination upon demethylation would drive sodium-ion translocation using the conformational change in a protein segment (Gottschalk and Thauer 2001).

Crystal structure of the cytoplasmic MtrA with cobalamin in the Co(III) oxidation state (Wagner et al. 2016b). (a) Structure of MtrA was shown by cartoon and surface models. Cobalamin is depicted as ball and stick model (carbons in magenta). (b) The lower axial ligand H84, E54, and I111 and cobalamin are shown as stick model
4.8 Methyl-coenzyme M Reductase (Mcr)
The common final step of all methanogenic pathways is the Methyl-coenzyme M reductase (Mcr) reaction (Thauer 1998). This enzyme catalyzes the reduction of methyl-coenzyme M (methyl-S-CoM) with coenzyme B (CoB-SH) to methane and heterodisulfide (CoB-S-S-CoM) (see Figs. 2 and 3). It is known that Mcr also catalyzes the reverse reaction, consisting of the anaerobic oxidation of methane, the first reaction in the metabolism involved in anaerobic methanotrophic archaea, coupled to the reduction of sulfate (Shima et al. 2012) and nitrate (Haroon et al. 2013).
Mcr is composed of α-, β-, and γ-subunits in an (αβγ)2 configuration. The crystal structures of Mcr from M. marburgensis (Fig. 13a), M. kandleri, M. barkeri, and M. wolfeii were solved in several inactive states (Ermler et al. 1997a; Grabarse et al. 2000, 2001; Wagner et al. 2016c). The active site of Mcr contains a nickel porphinoid F430 as a prosthetic group (Fig. 13b). The Ni(I), Ni(II), and Ni(III) states of F430 are involved in the catalytic reactions (Thauer and Shima 2007). Two F430 molecules are embedded in the protein core composed of the α-, α′-, β-, and γ-subunits. The catalytic core is connected to bulk solvent via a channel occupied by coenzyme B. The active site is mainly constructed with α-, β-, and γ-subunits, but the reverse side of F430 is ligated with glutamine oxygen from another α-subunit. This structural feature is of interest because biochemical experiments suggest that the active site of Mcr is coupled with the other active site to couple endergonic and exergonic catalytic reaction steps (Thauer and Shima 2007). The crystal structures of Mcr in complex with coenzyme B and coenzyme M (MCRox1-silent form) (Fig. 14a) and with heterodisulfide (MCRsilent form) (Fig. 14b) were reported. The coenzyme B moiety of heterodisulfide in the MCRsilent structure and coenzyme B in the MCRox1-silent structure are bound to the same site of the substrate entrance channel. By contrast, coenzyme M binding sites are different. In the MCRsilent form, the coenzyme M moiety is bound to the nickel site of F430 through its sulfonate oxygen. In the MCRox1-silent form, coenzyme M is bound to the nickel site of F430 with its sulfur (Ermler et al. 1997a).

Structure of methyl-coenzyme M reductase (Mcr). (a) Whole MCR structure from M. marburgensis. F430, CoM-SH, and CoB-SH are represented in ball and stick. (b) Chemical structure of F430

The active site structure of methyl-coenzyme M reductase (Shima 2016). The active site structure of Mcr isoenzyme I from M. marburgensis; the active site of the Mcrox1-silent form contains CoM-S-Ni(F430) and CoB-SH (a) and that of Mcrsilent form contains the heterodisulfide (b). (c) The modified amino acid residues found near the active site. (d) Key intermediates of two proposed Mcr catalytic mechanisms. In mechanism 2, the methyl radical is shown in brackets as it is only transiently present
One of the intriguing features of Mcr is the posttranslationally modified amino acid residues near the active site (Fig. 14c) (Kahnt et al. 2007; Wagner et al. 2016c). In Mcr from M. marburgensis, four methylated amino acids, one thioglycine, and a didehydroaspartate were identified. In addition, in Mcr from the ANME-1 methanotrophic archaeon, a 7-hydroxy-tryptophan was found in the crystal structure (Shima et al. 2012), and most recently a 6-hydroxy-tryptophan was identified in Methanotorris formicicus (Wagner et al. 2017). Didehydroaspartate, methylcysteine, and hydroxytryptophan were not conserved in other Mcr (Wagner et al. 2016c), which suggests that these modifications are not necessary for catalytic activity but improve catalytic activity and/or stability.
Based on the crystal structure of the MCRox1-silent form, the first catalytic mechanism was proposed (Ermler et al. 1997a; Grabarse et al. 2001), in which the Ni(I) of F430 attacks methyl-coenzyme M to make methyl-Ni(III) and CoM anion (Fig. 14d). After electron transfer from coenzyme M anion to methyl-Ni(III) forming methyl-Ni(II) and CoM thiyl radical, a hydrogen atom is transferred from CoB-SH to methyl-Ni(II) to produce methane (Ermler et al. 1997a). This mechanism is analogous to Co(I) chemistry in cobalamin-dependent enzymes, in which a methyl-cobalt intermediate is formed. The presence of Ni-H and Ni-S bonds is revealed by electron paramagnetic resonance (EPR) spectroscopic data (Harmer et al. 2005, 2008).
The second radical-based catalytic mechanism was proposed using a density function theory (DFT) calculation, again based on the crystal structure of MCRox1-silent. In the second mechanism, Ni(I) attacks methyl-S-CoM to produce methyl radicals and CoM-S-Ni(II) (Fig. 14d) (Pelmenschikov et al. 2002). Subsequently, the methyl radical accepts hydrogen atoms from CoB-SH to produce methane. To avoid rapid racemization of methyl radicals, C–S bond cleavage and C–H bond formation proceed in one step (Pelmenschikov et al. 2002; Scheller et al. 2017). Recently, Ragsdale and his colleagues have identified the CoM-S-Ni(II) intermediate of the reaction of Mcr using spectroscopic methods. Ultraviolet-visible spectroscopy, electron magnetic resonance spectroscopy, and magnetic circular dichroism spectroscopy were used to detect the intermediates trapped with a stopped-flow system using an analogue of CoB-SH containing a hexanoyl, instead of a heptanoyl side chain, which slows the reaction rate (Wongnate et al. 2016).
4.9 Heterodisulfide-Reductase/[NiFe]-Hydrogenase Complex (Hdr-Mvh)
The reduction of the heterodisulfide of coenzyme M and coenzyme B (CoM-S-S-CoB) with H2 is an exergonic reaction (ΔG°′ = −49 kJ/mol) (Thauer et al. 2010). Methanogens with cytochromes contain a membrane-associated heterodisulfide reductase (HdrDE) and a membrane-associated [NiFe]-hydrogenase (VhtAGC), which couple the exergonic reduction of the heterodisulfide with H2 to the endergonic translocation of protons through the cytoplasmic membrane (Peinemann et al. 1990; Deppenmeier et al. 1992; Abken et al. 1998). By contrast, in methanogens without cytochromes, a cytoplasmic electron-bifurcating heterodisulfide reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD) couples the reduction of CoM-S-S-CoB with H2 to the endergonic reduction of ferredoxin (Hedderich et al. 1989; Setzke et al. 1994; Kaster et al. 2011). Under physiological conditions, the redox potential E′ of ferredoxin is near −500 mV and that of the 2H+/H2 couple is near −400 mV. Therefore, ferredoxin can only be fully reduced by H2 when it is coupled to an exergonic reaction (Buckel and Thauer 2013). Experimental observations indicated that the HdrABC-MvhAGD complex catalyzes the complete reduction of ferredoxin with H2, but only in the presence of CoM-S-S-CoB (Kaster et al. 2011). The stoichiometry was found to be: 2H2 + CoM-S-S-CoB + Fdox = CoM-SH + CoB-SH + Fdred2− + 2H+. A model of the HdrABC-MvhAGD complex that considers these findings is illustrated in Fig. 15.

Composition and reaction of the heterodisulfide-reductase/[NiFe]-hydrogenase complex (HdrABC-MvhAGD) (Buckel and Thauer 2013)
The HdrABC-MvhAGD complex is composed of the hydrogenase module (MvhAGD) and the heterodisulfide reductase module (HdrABC). MvhA (53 kDa) is the large subunit of [NiFe]-hydrogenase, which contains a [NiFe] catalytic center. MvhG (34 kDa) is the small subunit of the hydrogenase, which contains three iron-sulfur clusters. MvhD (16 kDa) contains one [2Fe-2S] cluster and is predicted to provide the electronic connection to HdrABC. HdrA (72 kDa) contains one flavin binding site, four [4Fe-4S] clusters, and four characteristically spaced conserved cysteines. HdrB (33 kDa) harbors a zinc-binding motif at the N-terminal domain and two copies of a cysteine-rich sequence, CX31-39CCX35-36CXXC, which is proposed to be involved in binding of an unusual [4Fe-4S] cluster. HdrB harbors the active site for heterodisulfide reduction, and HdrC (21 kDa) serves as an electron connector between HdrA and HdrB, which provides two [4Fe-4S] clusters.
In the complex, the six subunits are present in a 1:1:1:1:1:1 stoichiometry. The apparent molecular mass of the HdrABC-MvhAGD complex was found to be approximately 500 kDa, indicating that the heterohexamer forms a dimer. The dimer is in equilibrium with the heterohexamer and is composed of the subcomplexes MvhAGD (103 kDa) and HdrABC (126 kDa) (Setzke et al. 1994). The purified complex contained 0.6 mol nickel, 0.9 mol FAD, 26 mol non-heme iron, and 22 mol acid-labile sulfur per mol of heterohexamer. FAD is only loosely bound; therefore, FAD must be added to the buffers used for purification. In most methanogens, the genes encoding these proteins are organized into three transcription units, mvhDGAB, hdrA, and hdrBC. The gene mvhB encodes a polyferredoxin with 12 [4Fe-4S] clusters.
In the model shown in Fig. 15, one FAD of HdrA is assumed to be the site of electron bifurcation. The FAD is reduced by 2 × 2 electrons from H2 and is oxidized by 2 × 1 electrons bifurcated to CoM-S-S-CoB and ferredoxin. However, the mechanism of flavin-based electron bifurcation requires that FAD is reduced with H2 in a 2e− reduction step (a hydride transfer). How this is achieved by only one FAD is difficult to envisage on the basis of the model, since iron-sulfur proteins generally transfer only one electron at a time.
4.10 [NiFe]-Hydrogenases
In the hydrogenotrophic methanogenic pathway of methanogens without cytochromes, three types of [NiFe]-hydrogenases are involved: F420-reducing hydrogenase (Frh), heterodisulfide-reductase-associating hydrogenase (Mvh), and integral membrane energy-conserving hydrogenase (Eha and Ehb). In methanogens with cytochromes, integral membrane energy-conserving hydrogenase (Ech) homologous to Eha and Ehb, methanophenazine-reducing [NiFe]-hydrogenase (VhtAGC) is additionally involved (Thauer et al. 2010).
Frh is found in most methanogenic archaea. In the hydrogenotrophic methanogenic pathway, Frh uses electrons from H2 to produce F420H2, which is used as hydride donor for the reactions catalyzed by Mtd and Mer and other reactions. In the methanogenic pathway from C1 compounds, F420H2 is generated from the oxidation of methyl-H4MPT to CO2 in the reverse reactions of those shown in Fig. 3. In methanogenesis, using formate, F420 can be reduced to F420H2 by F420-dependent formate dehydrogenase, and F420H2 is used for the formation of H2, which is catalyzed by Frh. However, Leigh et al. reported that F420-dependent formate dehydrogenase forms a complex with heterodisulfide reductase, which suggests that electrons from formate could be directly transferred to the heterodisulfide reductase system (Costa et al. 2010, 2013). Thus, Frh is used for both, the oxidation and reduction of H2 under physiological conditions. The catalytic unit of Frh appears to be the FrhAGB heterotrimer (Mills et al. 2013; Vitt et al. 2014) (Fig. 16a). The FrhA and FrhG subunits correspond to the large and small subunits of [NiFe]-hydrogenase, respectively. FrhA contains the [NiFe] dinuclear catalytic center, similar to that of other [NiFe]-hydrogenases, in which one CO and two CN ligands are coordinated to the iron site. FrhG contains three [4Fe-4S] clusters, which are slightly different from other [NiFe]-hydrogenase because the medial iron-sulfur cluster of other [NiFe]-hydrogenases is a [3Fe-4S] cluster rather than a [4Fe-4S] cluster; in addition, one of the ligands of the proximal [4Fe-4S] cluster is substituted to aspartate (instead of cysteine). In addition, one of the ligands of the distal [4Fe-4S] cluster was substituted to cysteine instead of histidine, which is found in the standard [NiFe]-hydrogenases. The redox potentials of the three [4Fe-4S] clusters in FrhG are lower than −400 mV, which is close to the redox potential of the H2/2H+ couple (E°′ = −414 mV) and the F420/F420H2 couple (E°′ = −360 mV). The redox potential of the iron-sulfur clusters is substantially lower than those in other [NiFe]-hydrogenases, which might be responsible for the reversibility of Frh.

Structure of F420-reducing hydrogenase (Frh) (Vitt et al. 2014). (a) A FrhAGB heterotrimer; the FrhA, FrhB, and FrhG subunits are shown in cyan, magenta, and green, respectively. The Ni, Fe, and S of the [NiFe] center and the [4Fe-4S] cluster are shown as green, brown, and yellow spheres, and FAD is shown as stick model. (b) Dimer of the FrhAGB. The three subunits of one trimer are colored as panel a. (c) Cartoon model of “nanoball” structure of hexameric FrhAGB dimer. The three subunits of one trimer are colored as panel a
The plausible catalytic unit of FrhAGB forms a homodimer of heterotrimers (Fig. 16b), and the six molecules of the homodimer form a cubic hexamer (Fig. 16c). In the hexameric “nanoball,” the entrances of the substrates H2 and F420 are located on the surface of the nanoball, which indicates that the internal space of the nanoball appears to be not involved in catalytic reactions. Shielding of the electron chains and the [NiFe]-active site from bulk solvent and stabilization of the protein in the physiological cytoplasmic environments are discussed (Vitt et al. 2014).
In the hydrogenotrophic methanogenic pathway, reduced ferredoxin is regenerated by the electron-bifurcating heterodisulfide-reductase/[NiFe]-hydrogenase complex. However, a part of reduced ferredoxin is used for the other anabolic reactions, and an intermediate of the methanogenic pathway, methyl-H4MPT, is consumed for anabolic metabolism. Therefore, the ferredoxin used for the anabolic reactions must be compensated by the other enzyme system. The integral membrane [NiFe]-hydrogenase complexes Eha and Ehb catalyze reduction of ferredoxin (E′ = ~ −500 mV) with oxidation of H2 (E°′ = ~ −414 mV). This endergonic reaction is driven by a sodium ion potential created by the integral membrane MtrA-H complex. EhaA-T and EhbA-Q are homologues of the energy-converting [NiFe]-hydrogenase EchA-F identified in methanogens with cytochromes. EchE is the large subunit containing a [NiFe] site, and EchC is the small subunit but contains only one [4Fe-4S] cluster. EchF contains two [4Fe-4S] clusters. EchD has no prosthetic groups, and its function is unknown. EchA and EchB are integral membrane proteins, which can mediate ion translocation. EhaA-T and EhbA-Q contain homologous subunits to EchA-G, and similar catalytic functions are predicted. However, 14 and 11 subunits with unknown function are additionally found in the gene cluster of Eha and Ehb, respectively (Tersteegen and Hedderich 1999).
5 Research Needs
Over the last two decades, the catalytic mechanism of methanogenic enzymes has been studied based on the crystal structures of the enzymes and enzyme complex with substrates and/or inhibitors. However, the crystal structures of some methanogenic enzymes have not yet been solved, and their catalytic mechanisms are not fully understood, as described below.
For instance, the catalytic mechanism of the heterodisulfide reductase/[NiFe]-hydrogenase complex (HdrABC/MvhAGD) using an electron bifurcation mechanism is unknown. The HdrB subunit, which is proposed to catalyze heterodisulfide reduction, contains a unique CCG motif, which might bind a new iron-sulfur cluster. This plausible iron-sulfur cluster-binding motif is conserved in more than 2,000 proteins involved in the three domains of life (Pereira et al. 2011). To unravel the catalytic mechanism of the enzyme complex, a high-resolution crystal structure of the HdrABC-MvhAGD complex is necessary. The unique energy conservation reaction of the MtrA-H complex using the methyl-transfer reaction to translocate sodium ions through the membrane must be elucidated. The structure of these two complexes will open the way to understand these unique machineries.
The methyl-coenzyme M reductase reaction is still the major target of interest. The findings for the CoM-S-Ni(II) intermediate, together with the previous finding that secondary deuterium isotope effects are consistent with the formation of methyl radical (Scheller et al. 2013a, b, 2017; Wongnate et al. 2016), support the methyl radical catalytic mechanism (mechanism 2) (Fig. 14d). However, to unravel the reaction mechanism of MCR, further experiments are required. All proposed mechanisms are based on crystal structures of the inactive forms of MCR; therefore, a crystal structure of the active form must be solved. 19F-ENDOR data for the active MCR in the presence of HS-CoM and CF3-S-CoB indicated a shift in the 7-thioheptanoyl chain toward nickel by more than 2 Å (Ebner et al. 2010), which reflects the potential of MCR to undergo a major conformational change in the active enzyme states during catalysis. Furthermore, MCR contains many modified amino acids near the active site. Investigations to analyze the function of posttranslational modifications and their biosynthesis are of interest.
References
Abken HJ, Tietze M, Brodersen J, Baumer S, Beifuss U, Deppenmeier U (1998) Isolation and characterization of methanophenazine and function of phenazines in membrane-bound electron transport of Methanosarcina mazei Gö1. J Bacteriol 180:2027–2032
Acharya P, Warkentin E, Ermler U, Thauer RK, Shima S (2006) The structure of formylmethanofuran: tetrahydromethanopterin formyltransferase in complex with its coenzymes. J Mol Biol 357:870–879. https://doi.org/10.1016/j.jmb.2006.01.015
Aufhammer SW, Warkentin E, Berk H, Shima S, Thauer RK, Ermler U (2004) Coenzyme binding in F420-dependent secondary alcohol dehydrogenase, a member of the bacterial luciferase family. Structure 12:361–370. https://doi.org/10.1016/j.str.2004.02.010
Aufhammer SW, Warkentin E, Ermler U, Hagemeier CH, Thauer RK, Shima S (2005) Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci 14:1840–1849. https://doi.org/10.1110/ps.041289805
Bai L, Fujishiro T, Huang G, Koch J, Takabayashi A, Yokono M, Tanaka A, Xu T, Hu X, Ermler U, Shima S (2017) Towards artificial methanogenesis: biosynthesis of the [Fe]-hydrogenase cofactor and characterization of the semi-synthetic hydrogenase. Faraday Discuss 198:37. https://doi.org/10.1039/c6fd00209a
Baldwin TO, Christopher JA, Raushel FM, Sinclair JF, Ziegler MM, Fisher AJ, Rayment I (1995) Structure of bacterial luciferase. Curr Opin Struct Biol 5:798–809. https://doi.org/10.1016/0959-440x(95)80014-X
Beifuss U, Tietze M, Baumer S, Deppenmeier U (2000) Methanophenazine: structure, total synthesis, and function of a new cofactor from methanogenic archaea. Angew Chem Int Ed 39:2470–2472. https://doi.org/10.1002/1521-3773(20000717)39:14<2470::AID-ANIE2470>3.0.CO;2-R
Bertram PA, Thauer RK (1994) Thermodynamics of the formylmethanofuran dehydrogenase reaction in Methanobacterium thermoautotrophicum. Eur J Biochem 226:811–818
Bertram PA, Karrasch M, Schmitz RA, Bocher R, Albracht SP, Thauer RK (1994) Formylmethanofuran dehydrogenases from methanogenic archaea. Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. Eur J Biochem 220:477–484
Binda C, Bossi RT, Wakatsuki S, Arzt S, Coda A, Curti B, Vanoni MA, Mattevi A (2000) Cross-talk and ammonia channeling between active centers in the unexpected domain arrangement of glutamate synthase. Structure 8:1299–1308
Boone DR, Whitman WB, Rouvière P (1993) Diversity and taxonomy of methanogens. In: Ferry JG (ed) Methanogenesis ecology, physiology, biochemistry & genetics. Chapman & Hall, New York/London
Buckel W, Thauer RK (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. https://doi.org/10.1016/j.bbabio.2012.07.002
Ceh K, Demmer U, Warkentin E, Moll J, Thauer RK, Shima S, Ermler U (2009) Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry 48:10098–10105. https://doi.org/10.1021/bi901104d
Costa KC, Wong PM, Wang TS, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA (2010) Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci U S A 107:11050–11055. https://doi.org/10.1073/pnas.1003653107
Costa KC, Lie TJ, Xia Q, Leigh JA (2013) VhuD facilitates electron flow from H2 or formate to heterodisulfide reductase in Methanococcus maripaludis. J Bacteriol 195:5160–5165. https://doi.org/10.1128/Jb.00895-13
Deppenmeier U, Blaut M, Schmidt B, Gottschalk G (1992) Purification and properties of a F420 nonreactive, membrane-bound hydrogenase from Methanosarcina strain Go1. Arch Microbiol 157:505–511
Deppenmeier U, Muller V, Gottschalk G (1996) Pathways of energy conservation in methanogenic archaea. Arch Microbiol 165:149–163. https://doi.org/10.1007/Bf01692856
Dimarco AA, Bobik TA, Wolfe RS (1990) Unusual coenzymes of methanogenesis. Annu Rev Biochem 59:355–394. https://doi.org/10.1146/annurev.biochem.59.1.355
Ebner S, Jaun B, Goenrich M, Thauer RK, Harmer J (2010) Binding of coenzyme B induces a major conformational change in the active site of methyl-coenzyme M reductase. J Am Chem Soc 132:567–575. https://doi.org/10.1021/ja906367h
Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK (1997a) Crystal structure of methyl coenzyme M reductase: the key enzyme of biological methane formation. Science 278:1457–1462
Ermler U, Merckel MC, Thauer RK, Shima S (1997b) Formylmethanofuran:tetrahydromethanopterin formyltransferase from Methanopyrus kandleri – new insights into salt-dependence and thermostability. Structure 5:635–646. https://doi.org/10.1016/S0969-2126(97)00219-0
Gottschalk G, Blaut M (1990) Generation of proton and sodium motive forces in methanogenic bacteria. Biochim Biophys Acta 1018:263–266. https://doi.org/10.1016/0005-2728(90)90263-4
Gottschalk G, Thauer RK (2001) The Na+-translocating methyltransferase complex from methanogenic archaea. Biochim Biophys Acta 1505:28–36
Grabarse W, Mahlert F, Shima S, Thauer RK, Ermler U (2000) Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation. J Mol Biol 303:329–344. https://doi.org/10.1006/jmbi.2000.4136
Grabarse W, Mahlert F, Duin EC, Goubeaud M, Shima S, Thauer RK, Lamzin V, Ermler U (2001) On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding. J Mol Biol 309:315–330. https://doi.org/10.1006/jmbi.2001.4647
Hagemeier CH, Shima S, Thauer RK, Bourenkov G, Bartunik HD, Ermler U (2003) Coenzyme F420 dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) from Methanopyrus kandleri: a methanogenic enzyme with an unusual quarternary structure. J Mol Biol 332:1047–1057. https://doi.org/10.1016/S0022-2836(03)00949-5
Harmer J, Finazzo C, Piskorski R, Bauer C, Jaun B, Duin EC, Goenrich M, Thauer RK, Van Doorslaer S, Schweiger A (2005) Spin density and coenzyme M coordination geometry of the ox1 form of methyl-coenzyme M reductase: a pulse EPR study. J Am Chem Soc 127:17744–17755. https://doi.org/10.1021/ja053794w
Harmer J, Finazzo C, Piskorski R, Ebner S, Duin EC, Goenrich M, Thauer RK, Reiher M, Schweiger A, Hinderberger D, Jaun B (2008) A nickel hydride complex in the active site of methyl-coenzyme m reductase: implications for the catalytic cycle. J Am Chem Soc 130:10907–10920. https://doi.org/10.1021/ja710949e
Haroon MF, Hu SH, Shi Y, Imelfort M, Keller J, Hugenholtz P, Yuan ZG, Tyson GW (2013) Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500:567–570. https://doi.org/10.1038/Nature12375
Hedderich R, Berkessel A, Thauer RK (1989) Catalytic properties of the heterodisulfide reductase involved in the final step of methanogenesis. FEBS Lett 255:67–71. https://doi.org/10.1016/0014-5793(89)81062-2
Hedegard ED, Kongsted J, Ryde U (2015) Multiscale modeling of the active site of [Fe] hydrogenase: the H2 binding site in open and closed protein conformations. Angew Chem Int Ed 54:6246–6250. https://doi.org/10.1002/anie.201501737
Hiromoto T, Warkentin E, Moll J, Ermler U, Shima S (2009) The crystal structure of an [Fe]-hydrogenase-substrate complex reveals the framework for H2 activation. Angew Chem Int Ed 48:6457–6460. https://doi.org/10.1002/anie.200902695
Kahnt J, Buchenau B, Mahlert F, Kruger M, Shima S, Thauer RK (2007) Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS J 274:4913–4921. https://doi.org/10.1111/j.1742-4658.2007.06016.x
Kaster AK, Moll J, Parey K, Thauer RK (2011) Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci U S A 108:2981–2986. https://doi.org/10.1073/pnas.1016761108
Klein AR, Breitung J, Linder D, Stetter KO, Thauer RK (1993) N5, N10-Methenyltetrahydromethanopterin cyclohydrolase from the extremely thermophilic sulfate reducing Archaeoglobus fulgidus – comparison of its properties with those of the cyclohydrolase from the extremely thermophilic Methanopyrus kandleri. Arch Microbiol 159:213–219. https://doi.org/10.1007/Bf00248474
Mills DJ, Vitt S, Strauss M, Shima S, Vonck J (2013) De novo modeling of the F420-reducing [NiFe]-hydrogenase from a methanogenic archaeon by cryo-electron microscopy. elife 2. https://doi.org/10.7554/eLife.00218
Peinemann S, Hedderich R, Blaut M, Thauer RK, Gottschalk G (1990) ATP synthesis coupled to electron-transfer from H2 to the heterodisulfide of 2-mercaptoethanesulfonate and 7-mercaptoheptanoylthreonine phosphate in vesicle preparations of the methanogenic bacterium strain Gö1. FEBS Lett 263:57–60. https://doi.org/10.1016/0014-5793(90)80704-M
Pelmenschikov V, Blomberg MRA, Siegbahn PEM, Crabtree RH (2002) A mechanism from quantum chemical studies for methane formation in methanogenesis. J Am Chem Soc 124:4039–4049. https://doi.org/10.1021/Ja011664r
Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS (2011) A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol 2(69). https://doi.org/10.3389/fmicb.2011.00069
Scheller S, Goenrich M, Thauer RK, Jaun B (2013a) Methyl-coenzyme M reductase from methanogenic archaea: isotope effects on label exchange and ethane formation with the homologous substrate ethyl-coenzyme M. J Am Chem Soc 135:14985–14995. https://doi.org/10.1021/Ja4064876
Scheller S, Goenrich M, Thauer RK, Jaun B (2013b) Methyl-coenzyme M reductase from methanogenic archaea: isotope effects on the formation and anaerobic oxidation of methane. J Am Chem Soc 135:14975–14984. https://doi.org/10.1021/Ja406485z
Scheller S, Ermler U, Shima S (2017) Catabolic pathways and enzymes involved in the anaerobic oxidation of methane. In: Boll M (ed) Handbook of hydrocarbon and lipid microbiology series anaerobic utilization of hydrocarbons, oils and lipids. Springer, Cham
Setzke E, Hedderich R, Heiden S, Thauer RK (1994) H2: heterodisulfide oxidoreductase complex from Methanobacterium thermoautotrophicum: composition and properties. Eur J Biochem 220:139–148. https://doi.org/10.1111/j.1432-1033.1994.tb18608.x
Shima S (2016) The biological methane-forming reaction: mechanism confirmed through spectroscopic characterization of a key intermediate. Angew Chem Int Ed 55:13648. https://doi.org/10.1002/anie.201606269
Shima S, Ermler U (2011) Structure and function of [Fe]-hydrogenase and its iron-guanylylpyridinol (FeGP) cofactor. Eur J Inorg Chem 2011:963–972. https://doi.org/10.1002/ejic.201000955
Shima S, Tziatzios C, Schubert D, Fukada H, Takahashi K, Ermler U, Thauer RK (1998) Lyotropic-salt-induced changes in monomer/dimer/tetramer association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri in relation to the activity and thermostability of the enzyme. Eur J Biochem 258:85–92. https://doi.org/10.1046/j.1432-1327.1998.2580085.x
Shima S, Thauer RK, Ermler U, Durchschlag H, Tziatzios C, Schubert D (2000a) A mutation affecting the association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri and its influence on the enzyme’s activity and thermostability. Eur J Biochem 267:6619–6623
Shima S, Warkentin E, Grabarse W, Sordel M, Wicke M, Thauer RK, Ermler U (2000b) Structure of coenzyme F420 dependent methylenetetrahydromethanopterin reductase from two methanogenic archaea. J Mol Biol 300:935–950
Shima S, Krueger M, Weinert T, Demmer U, Kahnt J, Thauer RK, Ermler U (2012) Structure of a methyl-coenzyme M reductase from Black Sea mats that oxidize methane anaerobically. Nature 481:98–101. https://doi.org/10.1038/Nature10663
Shima S, Chen DF, Xu T, Wodrich MD, Fujishiro T, Schultz KM, Kahnt J, Ataka K, Hu XL (2015) Reconstitution of [Fe]-hydrogenase using model complexes. Nat Chem 7:995–1002. https://doi.org/10.1038/Nchem.2382
Tersteegen A, Hedderich R (1999) Methanobacterium thermoautotrophicum encodes two multisubunit membrane-bound [NiFe] hydrogenases – transcription of the operons and sequence analysis of the deduced proteins. Eur J Biochem 264:930–943. https://doi.org/10.1046/j.1432-1327.1999.00692.x
Thauer RK (1998) Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 144:2377–2406
Thauer RK, Shima S (2007) Methyl-coenzyme M reductase in methanogens and methanotrophs. In: Garrett R, Klenk H-P (eds) Archaea, evolution, physiology and molecular biology. Blackwell Publishing, Malden, pp 275–283
Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R (2008) Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. https://doi.org/10.1038/nrmicro1931
Thauer RK, Kaster AK, Goenrich M, Schick M, Hiromoto T, Shima S (2010) Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem 79:507–536. https://doi.org/10.1146/annurev.biochem.030508.152103
Upadhyay V, Demmer U, Warkentin E, Moll J, Shima S, Ermler U (2012) Structure and catalytic mechanism of N5,N10-methenyltetrahydromethanopterin cyclohydrolase. Biochemistry 51:8435–8443. https://doi.org/10.1021/Bi300777k
van Beelen P, Labro JF, Keltjens JT, Geerts WJ, Vogels GD, Laarhoven WH, Guijt W, Haasnoot CA (1984) Derivatives of methanopterin, a coenzyme involved in methanogenesis. Eur J Biochem 139:359–365. https://doi.org/10.1111/j.1432-1033.1984.tb08014.x
Vitt S, Ma K, Warkentin E, Moll J, Pierik AJ, Shima S, Ermler U (2014) The F420-reducing [NiFe]-hydrogenase complex from Methanothermobacter marburgensis, the first X-ray structure of a group 3 family member. J Mol Biol 426:2813–2826. https://doi.org/10.1016/j.jmb.2014.05.024
Vogt S, Lyon EJ, Shima S, Thauer RK (2008) The exchange activities of [Fe] hydrogenase (iron-sulfur-cluster-free hydrogenase) from methanogenic archaea in comparison with the exchange activities of [FeFe] and [NiFe] hydrogenases. J Biol Inorg Chem 13:97–106. https://doi.org/10.1007/s00775-007-0302-2
Vonck J, Pisa KY, Morgner N, Brutschy B, Muller V (2009) Three-dimensional structure of A1A0 ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus by electron microscopy. J Biol Chem 284:10110–10119. https://doi.org/10.1074/jbc.M808498200
Wagner T, Ermler U, Shima S (2016a) The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 354:114–117. https://doi.org/10.1126/science.aaf9284
Wagner T, Ermler U, Shima S (2016b) MtrA of the sodium ion pumping methyltransferase binds cobalamin in a unique mode. Sci Rep 6. https://doi.org/10.1038/Srep28226
Wagner T, Kahnt J, Ermler U, Shima S (2016c) Didehydroaspartate modification in methyl-coenzyme M reductase catalyzing methane formation. Angew Chem Int Ed 55:10630–10633. https://doi.org/10.1002/anie.201603882
Wagner T, Wegner C-E, Kahnt J, Ermler U, Shima S (2017) Phylogenetic and structural comparisons of the three types of methyl-coenzyme M reductase from Methanococcales and Methanobacteriales. J Bacteriol 199:e00197
Wolfe RS (1991) My kind of biology. Annu Rev Microbiol 45:1–35
Wongnate T, Sliwa D, Ginovska B, Smith D, Wolf MW, Lehnert N, Raugei S, Ragsdale SW (2016) The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science 352:953–958. https://doi.org/10.1126/science.aaf0616
Yang XZ, Hall MB (2009) Monoiron hydrogenase catalysis: hydrogen activation with the formation of a dihydrogen, Fe-Hδ- – H deltaδ+-O, bond and methenyl-H4MPT+ triggered hydride transfer. J Am Chem Soc 131:10901–10908. https://doi.org/10.1021/Ja902689n
Acknowledgments
We thank Prof. Dr. Rolf Thauer for his helpful suggestions. This work was supported by a grant from the Max Planck Society, Deutsch Forschungsgemeinschaft Priority Programme “Iron-Sulfur for Life” (SH87/1-1) to S.S.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
Wagner, T., Watanabe, T., Shima, S. (2019). Hydrogenotrophic Methanogenesis. In: Stams, A., Sousa, D. (eds) Biogenesis of Hydrocarbons. Handbook of Hydrocarbon and Lipid Microbiology . Springer, Cham. https://doi.org/10.1007/978-3-319-78108-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-78108-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78107-5
Online ISBN: 978-3-319-78108-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences