
Abstract
Currently, approximately 95% of pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC), which is the most aggressive form and the fourth leading cause of cancer death with extremely poor prognosis [1]. Poor prognosis is primarily attributed to the late diagnosis of the disease when patients are no longer candidates for surgical resection [2]. Cancer cells are dependent on the oncogenes that allow them to proliferate limitlessly. Thus, targeting the expression of known oncogenes in pancreatic cancer has been shown to lead to more effective treatment [3]. This chapter will discuss the complexity of metabolic features in pancreatic cancers. To be able to fully comprehend the heterogeneous nature of cancer metabolism, we need to take into account the close relationship between cancer metabolism and genetics. Gene expression varies tremendously, not only among different types of cancers, but also within the same type of cancer among different patients. Cancer metabolism heterogeneity is often prompted and perpetuated not only by genetic mutations in oncogenes and tumor suppressor genes but also by the innate diversity of the tumor microenvironment. Much effort has been focused on elucidating the genetic alterations that correlate with disease progression and treatment response [4]. However, the precise mechanism by which tumor metabolism contributes to cancer growth, survival, mobility, and aggressiveness represents a functional readout of tumor progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pancreatic ductal adenocarcinoma
- KRAS mutation
- Glucose metabolism
- Glutamine metabolism
- Combined therapy
-
Oncogenic KRAS regulates glucose and glutamine metabolism in pancreatic cancer cells.
-
MUC1 overexpression leads to increased glucose metabolism.
-
p53 functions predict the sensitivity of pancreatic cancer tumors to glycolytic inhibition.
-
Targeting alpha-ketoglutarate dehydrogenase function by CPI-613 to slow mitochondrial metabolism.
-
The antidiabetic drug, metformin, targets pancreatic cancer stem cells.
-
Combined therapy is used to target pancreatic metabolism heterogeneity.
Currently, approximately 95% of pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC), which is the most aggressive form and the fourth leading cause of cancer death with extremely poor prognosis [1]. Poor prognosis is primarily attributed to the late diagnosis of the disease when patients are no longer candidates for surgical resection [2]. Cancer cells are dependent on the oncogenes that allow them to proliferate limitlessly. Thus, targeting the expression of known oncogenes in pancreatic cancer has been shown to lead to more effective treatment [3]. This chapter will discuss the complexity of metabolic features in pancreatic cancers. To be able to fully comprehend the heterogeneous nature of cancer metabolism, we need to take into account the close relationship between cancer metabolism and genetics. Gene expression varies tremendously, not only among different types of cancers, but also within the same type of cancer among different patients. Cancer metabolism heterogeneity is often prompted and perpetuated not only by genetic mutations in oncogenes and tumor suppressor genes but also by the innate diversity of the tumor microenvironment. Much effort has been focused on elucidating the genetic alterations that correlate with disease progression and treatment response [4]. However, the precise mechanism by which tumor metabolism contributes to cancer growth, survival, mobility, and aggressiveness represents a functional readout of tumor progression.
1 Oncogenic KRAS Regulates Metabolism in Pancreatic Cancer Cells (Fig. 1)
1.1 Oncogenic KRAS Regulates Glutamine Metabolism
A cancer cell’s specific metabolic adaptations in nutrient uptake and biosynthesis have been linked to a particular genetic mutation. The KRAS (Kirsten rat sarcoma) oncogene homolog is a known regulator of glutamine metabolism among other intermediary metabolic pathways that renders cancer cells addicted to glutamine [5,6,7]. A range of mutations in the KRAS oncogene occurs in over 90% of PDAC [8, 9].


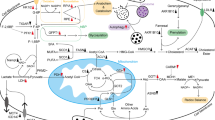
Oncogenic KRAS regulates glutamine and glucose metabolism in PDAC
Normally, glutamate feeds into the TCA cycle after being converted to alpha-ketoglutarate in the mitochondrion via glutamate dehydrogenase 1 (GLUD1). A study by Son et al. showed that KRAS regulated the reprogramming of glutamine metabolism through transcriptional regulation of key metabolic enzymes of transaminase reactions which, in turn, determine PDAC tumor growth. Notably, they concluded that PDAC cells greatly depend on these reactions for redox homeostasis. Given that this pathway is nonessential in normal cells, the unique importance of this pathway in PDAC suggests novel approaches to therapy in treating PDAC [6]. KRAS mutation led to the reprogramming of glutamine metabolism, which was partially due to increased aspartate aminotransferase or Glutamic-oxaloacetic transaminase 1 (GOT1) expression and decreased GLUD1 expression. The change in the ratio of expression of GOT1 and GLUD1 shunts glutamine flux through the aspartate aminotransferase pathway. Furthermore, they demonstrated that GOT knockdown failed to impair growth in several normal cell lines. According to Lyssiotis et al., the observation that the glutamine metabolism pathway is downstream of mutant KRAS serves as an explanation for the distinct glutamine dependency of pancreatic cancer. Not only do their results yield novel targets for pancreatic cancer therapy, but they also suggest that inhibiting glutamine metabolism in pancreatic cancer therapies may synergize with therapies that increase ROS [7].
1.2 Oncogenic KRAS Regulates Glucose Metabolism
The KRAS oncogene is also known to contribute to the glucose metabolism in pancreatic cancer cells via upregulation of glucose uptake and diversion of glucose into the hexosamine biosynthesis pathways [10]. Oncogenic KRAS controls the diversion of glycolytic intermediates into ribose biosynthesis pathways via upregulation of the non-oxidative pentose phosphate pathway (PPP), a pathway that is fundamental to nucleic acid synthesis and thus cancer cell proliferation [10]. Expression of GLUT1 (glucose transporter-1), hexokinase-II (HK2), and LDHA that catalyzes the reaction of pyruvate to lactate is greatly enhanced by KRAS in pancreatic tumor cells [10]. Subsequently, glycolytic flux, the production of lactate from glucose, was high in KRAS-mutant tumors. It is of note that these alterations are not nearly as pronounced in the stromal cells of these tumors which are able to uptake the lactate generated by tumor cells and use pyruvate dehydrogenase (PDH) to convert the lactate back to pyruvate in order to fuel the TCA cycle [11, 12]. Yun et al. found that cells with mutated KRAS undergo the Warburg effect and survive in low-glucose environments compared to cells with wild-type KRAS due to the fact that KRAS upregulated GLUT1 [13]. This suggests that KRAS mutation is involved in the altering of a cancer cell’s bioenergetics that is seen in most PDAC tumor cells, which take advantage of altered metabolic pathways to successfully proliferate and grow.
2 Other Alternative Metabolisms in Pancreatic Cancer
2.1 MUC1 Overexpression Leads to Increased Glucose Metabolism
A study by Chaika et al. revealed that the overexpression of transmembrane protein MUC1 led to elevated glucose metabolism and related activities, such as increased glucose uptake and lactate production resulting from increases in GLUT1 expression and LDHA expression, respectively. These metabolic effects are particularly pronounced under hypoxic conditions, which are associated with the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α), a transcription factor for many genes involved in regulating glucose uptake, through the overexpression of MUC1 [14]. Pancreatic cancer cells that do not overexpress MUC1 have a reduction in lactate and glycolytic intermediates. Overall, the overexpression of MUC1 is capable of influencing glucose metabolism, the elevation of amino acid metabolism, and the TCA cycle, all of which are important in the biosynthesis of cellular building blocks, and thus tumorigenesis. The signaling pathway associated between MUC1 and HIF-1α plays an important role in the facilitation of tumor growth and metastasis, serving as a potential target for manipulation in the treatment of diseases reliant upon these proteins [14].
2.2 p53 Functions Predict the Sensitivity of Pancreatic Cancer Tumors to Glycolytic Inhibition
The heterogeneity of metabolic alterations within the same cancer types is best illustrated by a recent study by Rajeshkumar et al. They showed that PDAC’s sensitivity to the same metabolic inhibition could vary drastically from one tumor to another, depending on the specific tumor’s genetic status and unique metabolic phenotype [15]. More specifically, they uncovered that responses to LDHA inhibition by the small-molecule FX11 were determined by a tumor’s p53 status, a tumor suppressor gene that is largely inactivated in many cancers [16]. Within the same PDAC type, tumors with wild-type TP53 demonstrated resistance to FX11, while those with mutant TP53 exhibited sensitivity in the form of increased apoptosis, reduced proliferation, and attenuated tumor growth. Their data show that FX11 specifically reduces pyruvate to lactate conversion by LDHA only in the TP53-mutant tumor , suggesting LDHA inhibition as a possible therapeutic target to reduce TP53-mutant tumor growth. Resistance in TP53-WT tumors is thought to result from reduced dependence on glucose, as corroborated by their data showing higher levels of TIGAR, a p53-inducible protein that lowers glycolytic flux [17]. This study supports not only growing evidence for variable metabolic phenotypes across cancer types but also within cancers of the same type. From a clinical perspective, this insight emphasizes the importance of metabolic phenotypes in pancreatic cancer sub-characterization in order to pair drug therapies according to phenotypic sensitivity for a more selective and personalized treatment.
3 Suggested Therapy (Fig. 2)
The KRAS gene may be a solution to this type of disease since KRAS appears to have a prominent role in the metabolic rewiring of PDAC tumors and plays critical roles in PDAC pathogenesis [9]. While oncogenic KRAS alters the PDAC cell’s metabolism, it requires the cancer cell to become dependent on the oncogenic KRAS to continue proliferation [18]. This is known as oncogene addiction, in which the cancer cell becomes dependent on the activity of the oncogene for survival and proliferation [3]. Since KRAS mutations are found in a majority of PDAC cancer cells and KRAS regulates cancer cell’s metabolism, targeting these regulations for cancer therapy is an approach that researchers are taking [18].


Overview of therapeutic options targeting pancreatic cancer metabolism
3.1 Alpha-Ketoglutarate Dehydrogenase Function by CPI-613 to Slow Mitochondrial Metabolism
Drugs have been developed to target mitochondrial metabolism in cancers [18]. One of these drugs is CPI-613, an inhibitor of cancer-specific mitochondrial energy metabolism. The drug causes tumor cell apoptosis, necrosis, and autophagy by selectively targeting alterations in mitochondrial enzyme activities and redox status [19]. CPI-613 is a small molecule that attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process [20]. The drug is known to simultaneously attack multiple essential components of tumor cell regulation [20]. However, the exact mechanism is not well understood. CPI-613 has been recognized to be effective against various types of cancers [21], including patients with metastatic pancreatic cancer [19]. CPI-613 used in combination with modified FOLFIRINOX (mFOLFIRINOX) in patients with metastatic pancreatic cancer demonstrated better survival, but since this phase I study was not designed to determine the efficacy of adding CPI-613 to mFOLFIRINOX, the results should be interpreted with caution. Nevertheless, Alistar et al. have obtained encouraging results from the phase I studies and are currently performing a randomized phase II trial to compare FOLFIRINOX against mFOLFIRINOX with CPI-613. These results suggest that targeting mitochondrial metabolism holds enormous potential in combating pancreatic cancer.
3.2 Antidiabetic Drug, Metformin, Targets Pancreatic Cancer Stem Cells
Recent studies have shown that tumorigenic cancer stem cells (CSCs), a highly chemoresistant subclass of PDAC, are strongly dependent on oxidative metabolism [22, 23]. Retrospective analysis showed that oral administration of metformin in patients with type 2 diabetes was associated with reduced risk of developing PDAC [24] along with a better outcome for patients that had established PDAC [25]. More recently, it has been discovered that metformin targets pancreatic CSCs but not the differentiated progenies (non-CSCs) [22]. KRAS targeting has resulted in tumor shrinkage but fails to kill all the CSCs [26]. Viale et al. established that dormant tumor cells that survived oncogene ablation were shown to have high sensitivity to oxidative phosphorylation inhibitors [26]. Lonardo et al. uncovered that metformin uniformly reduced ATP levels in adherent cells and sphere cells from CSCs, but not in non-CSCs [23]. Although the mechanism for metformin in CSCs is largely unknown, what is known is that metformin slowly accumulates in the mitochondria and directly inhibits complex 1 (NADH dehydrogenase) in the electron transport chain, affecting oxidative phosphorylation [23]. Therefore, a potentially strong therapeutic strategy to manage pancreatic cancer is the combined targeting of the KRAS pathway and mitochondrial respiration [26].
3.3 Combined Therapy to Target Pancreatic Metabolism Heterogeneity
Combination therapy to target multiple metabolic pathways in pancreatic cancer has been demonstrated as a favorable therapeutic solution. Elgogary et al. found that targeting glutamine metabolism using the glutaminase inhibitor bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) encapsulated in nanoparticles effectively shrinks pancreatic cancer tumor size and slows proliferation [27]. They also found, using metabolomics technologies, that the tumor cells remaining after glutaminase inhibition were dependent on glycolysis and glycogen synthesis. Elgogary et al. continued the study by adding both BPTES nanoparticles and metformin to target both glutamine and glucose metabolisms in pancreatic cancer cells. They discovered that the combined therapy provided enhanced efficacy that inhibited tumor growth significantly more compared to the single treatment of BPTES or metformin alone. This highlights the fact that there is great heterogeneity in pancreatic cell metabolism since targeting only glutamine metabolism did not kill all the pancreatic cancer cells, but targeting both glutamine and glucose metabolisms reduced the tumor growth of the cells with considerably larger efficacy than targeting either glutamine or glucose metabolism alone. This has been observed in pancreatic cancer cells, but more clinical trials must be done in order to see if combination therapy can assist in pancreatic cancer patient survival.
Conclusion
Pancreatic ductal adenocarcinoma is the fourth leading cause of cancer death in the United States and is expected to be the second largest by 2030 [28, 29]. The deadliness of this disease can be attributed to its metabolic heterogeneity which developed through cancerous evolution (Fig. 3). With that in mind, the investigation of PDAC within the past few years has been exponentially increasing with improved technology and research methods that allow us to understand these intricate mechanisms better. Exploration of more aspects of a pancreatic cell enables scientists and clinicians to better target multiple facets of a pancreatic cell, resulting in more effective therapeutic methods.


Overview of pancreatic cancer metabolic heterogeneity
Abbreviations
- ASP:
-
Aspartate
- EGFR:
-
Epidermal growth factor receptor
- GLS:
-
Glutaminase
- GLUD1:
-
Glutamate dehydrogenase 1
- GLUT:
-
Glucose transporter
- GOT1:
-
Glutamic-oxaloacetic transaminase 1
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
- HK2:
-
Hexokinase 2
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- LDH:
-
Lactate dehydrogenase
- MCT:
-
Monocarboxylate transporter
- OAA:
-
Oxaloacetate
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PFK1:
-
Phosphofructokinase 1
- TCA:
-
Tricarboxylic acid cycle
References
Hariharan, D., Saied, A., & Kocher, H. M. (2008). Analysis of mortality rates for pancreatic cancer across the world. HPB: The Official Journal of the International Hepato Pancreato Biliary Association, 10(1), 58–62.
Hidalgo, M. (2010). Pancreatic cancer. The New England Journal of Medicine, 362(17), 1605–1617.
Weinstein, I. B., & Joe, A. (2008). Oncogene addiction. Cancer Research, 68(9), 3077–3080. discussion 3080.
Verhaak, R. G., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17(1), 98–110.
Birnbaum, D. J., et al. (2011). Genome profiling of pancreatic adenocarcinoma. Genes, Chromosomes & Cancer, 50(6), 456–465.
Son, J., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature, 496(7443), 101–105.
Lyssiotis, C. A., et al. (2013). Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle, 12(13), 1987–1988.
di Magliano, M. P., & Logsdon, C. D. (2013). Roles for KRAS in pancreatic tumor development and progression. Gastroenterology, 144(6), 1220–1229.
Sousa, C. M., & Kimmelman, A. C. (2014). The complex landscape of pancreatic cancer metabolism. Carcinogenesis, 35(7), 1441–1450.
Ying, H., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell, 149(3), 656–670.
Chaika, N. V., et al. (2012). Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS One, 7(3), e32996.
Maher, J. C., et al. (2005). Differential sensitivity to 2-deoxy-D-glucose between two pancreatic cell lines correlates with GLUT-1 expression. Pancreas, 30(2), e34–e39.
Yun, J., et al. (2009). Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science, 325(5947), 1555–1559.
Chaika, N. V., et al. (2012). MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 109(34), 13787–13792.
Rajeshkumar, N. V., et al. (2015). Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Research, 75(16), 3355–3364.
Surget, S., Khoury, M. P., & Bourdon, J. C. (2013). Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco Targets and Therapy, 7, 57–68.
Bensaad, K., et al. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell, 126(1), 107–120.
Weinberg, S. E., & Chandel, N. S. (2015). Targeting mitochondria metabolism for cancer therapy. Nature Chemical Biology, 11(1), 9–15.
Alistar, A., et al. (2017). Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-Centre, open-label, dose-escalation, phase 1 trial. The Lancet Oncology, 18(6), 770–778.
Stuart, S. D., et al. (2014). A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer & Metabolism, 2(1), 4.
Pardee, T. S., et al. (2014). A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clinical Cancer Research, 20(20), 5255–5264.
Sancho, P., et al. (2015). MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metabolism, 22(4), 590–605.
Lonardo, E., et al. (2013). Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One, 8(10), e76518.
Evans, J. M., et al. (2005). Metformin and reduced risk of cancer in diabetic patients. BMJ, 330(7503), 1304–1305.
Sadeghi, N., et al. (2012). Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clinical Cancer Research, 18(10), 2905–2912.
Viale, A., et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature, 514(7524), 628–632.
Elgogary, A., et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(36), E5328–E5336.
Rahib, L., et al. (2014). Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Research, 74(11), 2913–2921.
Rossi, M. L., Rehman, A. A., & Gondi, C. S. (2014). Therapeutic options for the management of pancreatic cancer. World Journal of Gastroenterology, 20(32), 11142–11159.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter
Cite this chapter
Camelo, F., Le, A. (2018). The Intricate Metabolism of Pancreatic Cancers. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1063. Springer, Cham. https://doi.org/10.1007/978-3-319-77736-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-77736-8_5
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-77735-1
Online ISBN: 978-3-319-77736-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)