
Abstract
As very powerful anti-inflammatory and immunosuppressive drugs, glucocorticoids have gained attention over the past decades as a first line treatment for chronic inflammatory and autoimmune diseases, in addition to its wide use in the field of oncology. An extensive body of research has accumulated with regards to the molecular anti-inflammatory mechanisms by which glucocorticoids exert their effects on the cells of the immune system. Moreover, some pro-inflammatory properties have recently emerged, based on the analysis of glucocorticoid-regulated genes. Understanding the physiology and the pharmacology of these hormones and drugs in the context of inflammation and the immune system will allow for the comprehension of their still incomplete function in homeostasis and of their practical clinical applications.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
When discussing the most potent anti-inflammatory drugs, glucocorticoids (GCs) represent the first class of drugs that is typically considered. Since the discovery of their importance for the life of a human being in the half of the nineteenth century, GCs have been studied for decades; however, the molecular mechanisms of GCs have been incompletely understood to date. Thomas Addison was the first to describe adrenal insufficiency syndrome in 1849, and Harvey Cushing reported hypercortisolism syndrome later in 1932; however, it was not until 1949 when the use of corticosteroids started being introduced into the clinics for patients affected by rheumatoid arthritis, by Hench and Kendall. The discovery of the extraordinary anti-inflammatory properties of GCs was new and unexpected at that time, and it paved the way for the subsequent extensive clinical use of GCs for a multitude of inflammatory diseases that were incurable. The impact of treatment with GCs was so revolutionary that Philip Hench and Edward Kendall received the Nobel prize in 1950 together with Tadeus Reichstein, who first isolated and identified the steroid hormones of the adrenal cortex. Subsequently, approximately 30 synthetic GCs have been synthetized by the pharmaceutical industry and are employed to treat a variety of inflammatory and autoimmune diseases. Despite their therapeutic potency, considerable adverse effects occur during long-term treatment with GCs. The continuous and probing research into the molecular mechanisms of action of these compounds is helping to determine which synthetic GCs will exert only beneficial effects.
2 GCs in the Stress-Related Immunomodulation
Stress is defined as an actual or anticipated threat of the homeostasis of an organism, by which external or internal stimuli (e.g. social threats, maternal deprivation, fear, physiological challenge and surgery) termed stressors, activate the so-called “stress system” [1]. The stress system consists of: (1) the locus caeruleus/norepinephrine autonomic nervous system that acts within seconds; and (2) the hypothalamic–pituitary–adrenal (HPA) axis, comprising the hypothalamus, pituitary and adrenal glands, whose response starts more slowly, from hours to days. GCs are the final products of the HPA axis, and are released by the zona fasciculata of the adrenal cortex as a response to psychogenic (e.g., fear) and physical (e.g., cellular lesion or pathogen invasion) stressors. They act through the GC receptor (GR) via genomic and non-genomic pathways in virtually any tissues, thereby controlling the basic and physiological functions of the cardiovascular and immune systems, the metabolism of carbohydrates, lipids, and proteins, bone and muscles, as well as behavioral processes (e.g. emotion) (Table 1).
During the acute phase of stress, GCs at low concentrations can bind to both the membrane-bound GR and the mineral corticoid receptor (MR) . This, in turn, activates multiple signal transduction pathways that ultimately lead to a series of physical and behavioral changes that aim to increase the survival of the organism [2, 3]. While the cytoplasmic GR receptor is ubiquitously expressed in the brain, with high levels of expression in the neurons and microglia, MR is expressed in a few regions (e.g. the hippocampus, amygdala and neurons within the paraventricular nucleus of the hypothalamus) [4]. The cytoplasmic GR is responsible for the genomic effects of GCs, the membrane GR and MR mediate the rapid non-genomic effects of GCs in the brain in response to a stress stimulus, with important consequences to the adaptive changes in the organism. However, how these receptors function still requires further investigation. Following a stress stimulus, GCs restore their own hypersecretion via a negative feedback mechanism on the HPA axis, but if the stressor persists, GC hypersecretion may not be properly controlled and may become a potential threat for the organism. There is evidence that the prolonged exposure to stressful conditions can induce the structural remodeling of neurons and alterations in glial functions, which are frequently maladaptive, thus contributing to the development of acute or chronic diseases [5].
Both acute and chronic stress affects the immune response by activating both the sympathetic nervous system and the HPA axis, from which the release of cortisol is required to re-establish homeostasis. The interplay between these systems are complex and remain only partially understood, occurring in both the peripheral tissues and the central nervous system (CNS). As an example, GCs are known to control the expression of α1B and β adrenergic receptors, with important consequences in the clinical use of GCs [6, 7]. Immune responses are affected by disturbances in HPA axis activity. The hypersecretion of cortisol (e.g. during stress) can increase susceptibility to infections and neoplastic diseases and, simultaneously, enhance resistance to autoimmune or aseptic inflammatory diseases. Conversely, a deficiency in cortisol production reduces the susceptibility to infectious agents while also diminishing the susceptibility to autoimmune or aseptic inflammatory diseases. Therefore, the level of circulating cortisol influences immune cell activity due to the potent effects of GCs on cells of the innate and adaptive immune system (as described later). As an example, surgery-induced stress reduces the number of T cells and shifts the Th1/Th2 balance to a Th2 response in humans, in conjunction with an increase of T regulatory (Treg) cells that contribute to Th1 suppression [8]. Similarly, other studies have demonstrated that corticosteroids, both alone or combined with catecholamines, can increase the expression of Th2 cytokines in the peripheral blood cells of human subjects and shift the Th1/Th2 balance towards a Th2 phenotype during long-term GC exposure. Simultaneously, this shift was accompanied by an altered ratio of IFNγR/IL-4R, with a dominant expression of IL-4R in Th2 cells [9, 10]. A Th1/Th2 unbalance predominantly impacts diseases such as asthma, Th2 cytokine-mediated allergic airway inflammation of the lungs. For example, psychological stress exacerbates allergic symptoms due to endogenously released GCs and, simultaneously, induces a reduction in GR expression, causing an insensitivity to the anti-inflammatory effects of exogenously administered GCs for treatment of asthma [11, 12]. Such reduced GR expression is not the only mechanism responsible for the failure to elicit a response to GCs in this pathology. GC resistance also involves other mechanisms, including defects in caspase-induced apoptosis in eosinophils, the major pro-inflammatory effector cells in asthma. Furthermore, GCs inhibit IL-12 release by monocytes and macrophages, thus contributing to the shift towards a Th2 phenotype [13]. This effect is further exacerbated by the reduction of T regulatory (Treg) cells caused by GCs; stress-derived GCs suppress the proportion of FoxP3+ Treg cells in subjects experiencing mental stress, thus worsening the symptoms of asthma. In addition, a decrease in Treg numbers and activity was found in different rodent asthma models, likely due to the reduction of T cell production in the thymus [14, 15].
In the CNS , the primary cell types exhibiting immune functionality is microglia cells. Microglia cells are associated with the HPA axis, respond to infections by secreting pro-inflammatory cytokines, and help mobilize other immune cells to restore homeostasis. They express high levels of GR and represent the first immune target of GCs in the brain [16]. When a stressor activates the HPA axis, the released GCs stimulate the production of anti-inflammatory cytokines (e.g. IL-4 and IL-10), which function to counteract the inflammatory signals generated by the microglia. Moreover, stress-induced GCs stimulate the proliferation of microglia cells [17]. This cross-talk between GCs and microglia cells is included in the signaling network between the immune, endocrine and nervous systems. Such cross-talk is necessary to restore homeostasis following an inflammatory insult to prevent an inappropriate reaction that can result in detrimental effects to neurons; however, GCs both promote anti-inflammatory effects but also induce pro-inflammatory effects in the CNS following a stress stimulus, depending on the timing of exposure followed by an immunogenic challenge. For example, GCs can potentiate or suppress the same pro-inflammatory cytokines depending on whether the cells have been treated with GCs either before or after LPS administration, respectively [18, 19]. Thus, stress-released GCs via the HPA axis can prime neuroinflammatory processes to subsequent immune events, shifting the surveillance state of microglia cells to a primed one [20]. Since exaggerated immune and inflammatory responses in the brain are currently considered to be pathogenic in the context of a number of psychiatric disorders (e.g. depression or bipolar disorder), stressors that lead to neuroinflammation by means of released GCs are thought to contribute to the development of psychiatric disorders [21].
The immune system is also a target of the stress-related effects in the periphery. Cathecolamines released by the sympathetic nervous system (SNS) are directly released into the lymphoid organs. Increased norepinephrine in the bone marrow causes a release in the circulation of myeloid cells, (e.g. monocytes and granulocytes). These cells are immature and therefore, naturally “inflammatory”, producing pro-inflammatory cytokines in the tissues which they easily infiltrate [22]. Furthermore, myeloid cells are insensitive to GCs, thus accounting for the GC-resistance that develops during chronic stress. This functional insensitivity is primarily associated with the reduction of the transcriptional activity mediated by GR. In addition, inflammatory peripherally derived macrophages traffic to the brain, thus contributing to the neuroinflammation of the brain with consequences in neurobiology and behavior [23].
In several stress-related long-term diseases , (e.g. acute coronary syndrome or major depression), the percentage of Tregs is decreased, which may serve as a means to facilitate an immune response [15]. When stress becomes chronic, it can lead an increased percentage of Tregs to counteract the inflammatory status and the increased proliferation of effector T cells [24]. However, heightened levels of Tregs have also been found in acutely-stressed healthy individuals, which may represent an altered response to acute stress that may predispose individuals to future disease development [25].
Immunomodulation by stress can influence tumor growth, progression, and metastasis. Cytokines (e.g. IL-6) can be produced at high levels by immature, inflammatory myeloid derived-cells that are GC insensitive and contribute to enhanced inflammatory responses to infectious agents [26, 27]. Therefore, chronic inflammation can lead to tumor development via the mediation of cells of the immune system [28]. As an example, Tregs can favor tumor progression by locally suppressing the anti-tumor immunity [29,30,31]. In addition, stress-released GCs induce immunosuppression , thereby decreasing immune defense against cancer and facilitating tumor growth. Furthermore, epidemiologic studies have linked stress to tumor progression through the activation of the SNS [32]. Molecular studies have also revealed a role for the Glucocorticoid-Induced leucine Zipper (GILZ) protein and its isoform long-GILZ as novel anti-proliferative GC-mediators. GILZ can bind to Ras, thereby inhibiting Ras- and Raf-dependent cell proliferation, whereas L-GILZ can bind to p53, thereby exerting an anti-proliferative effect [33, 34]. However, the mechanism by which GCs influence tumor cell growth, either under conditions of stress or during a pharmacological treatment, requires further study.
3 Local Production of Glucocorticoids
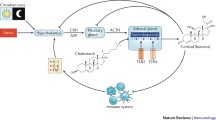
Circulating GCs are released by the adrenal cortex of the adrenal glands following a circadian local rhythm and this mechanism is independent of systemic influences (e.g. stress or peripheral constant CRH infusion). This regulation protects the organism from threats (e.g. infections) by avoiding possible serious consequences derived from the long-term activation of the immune system, which communicates with the HPA axis via the secretion of cytokines, including TNF-α, IL-1 and IL-6 [35]. GCs are derived from cholesterol via a series of enzymatic conversions (Fig. 1) thus leading to corticosterone production in species, such as reptiles, birds, or mice, and to cortisol release in species such as fish, primates or humans. Both corticosterone and cortisol bind to the GC and mineralocorticoid receptors, thereby regulating the activity of several organs.

Glucocorticoid synthesis in different species. A series of enzymatic reactions lead to corticosterone and cortisol production in rodents and humans, respectively
In addition to the systemic production of GCs, extra-adrenal synthesis can be found in organs such as primary lymphoid organs, skin, intestine, brain, heart, and vasculature. This local production does not affect serum levels of GCs, since the removal of adrenals does not lead to a detectable amount of circulating GCs; however, it rather seems to exert control over eventual inflammatory conditions [36]. The first organ in which the local production of GCs was demonstrated was the thymus, approximately 20 years ago [37]. Although the level of enzymes required for the de novo synthesis of GCs is up to 10,000-fold lower than that in the adrenals, the level of GCs produced by thymic epithelial cells (TECs) is sufficient to activate GC-responsive genes in the surrounding cells. TECs are the main cells that produce GCs in the thymus, which is high at birth but decreases with aging. In contrast, corticosterone produced by double positive (DP) CD4+ CD8+ thymocytes increases with age. Once thymocytes have passed positive selection, they upregulate the expression of CD69 and cease producing GCs. Immature thymocytes are the primary target of the effects of both adrenal-released and locally produced GCs. In fact, GCs produced by immature thymocytes induce apoptosis in these cells in an autocrine manner [38]. In contrast, ACTH inhibits GC synthesis in thymocytes while inducing their synthesis in adrenal and TECs. This opposing effect has not been fully characterized, but it is believed to function as a limiting factor for the control of excessive apoptosis in the thymus, thus protecting this organ from a strong activation of the HPA axis that can result in detrimental effects [39, 40]. However GCs can also prevent apoptosis in thymocytes when the TCR is triggered, in so called “mutual antagonism ”, in which the pathways activated by the TCR and the GR interact. This mechanism helps protect the thymocytes that would be negatively selected if they had received only one signal (from the GR or TCR). In particular, local GCs protect thymocytes with an intermediate affinity for the TCR from pro-apoptotic signals, thus undergoing to positive selection and survival, while low and strong affinity for TCR results in thymocyte apoptosis. Overall, positive selection can occur as a result of GC-mediated TCR antagonism [41]. At the molecular level, GCs induce the upregulation of Bcl2 expression and GR interactions with AP1, NFAT, and NF-κB transcription factors, thus contributing to prevention of apoptosis [40]. The dual production of GCs, one by the adrenal glands and the other by both the local thymocytes and TECs, appears to serve two purposes: (1) peripheral production is required to respond to and regulate strong systemic immune responses; and (2) local production controls homeostasis and the development of thymocytes and possibly other cell types, including dendritic cells, fibroblasts and macrophages.
The thymus is not the only lymphoid organ that produces GCs. The bone marrow and spleen are also lymphoid organs particularly devoted to the synthesis of GCs in early life when the adrenal contribution is low. This production increases with age, suggesting that lymphoid GC synthesis is required for lymphocyte development throughout life. In this context, in addition to T lymphocytes extensively studied in the thymus, B cells may also be the target of locally released GCs because of their high GR expression, and their maturation can be influenced by the effect of local GCs. However, additional studies are necessary to assess the role of lymphoid GCs in extra-thymic lymphoid organs and their effects on poorly studied cells other than T lymphocytes. One interesting feature of locally produced GCs is their synthesis via GC regeneration, as demonstrated by the high expression of the enzyme, Hsd11b1, that converts the inactive 11-keto metabolite DHC and cortisone into active GCs (Fig. 1) [42, 43]. While GC synthesis from cholesterol is independent of serum (adrenal) steroids, GC regeneration is dependent on the availability of circulating synthesized GC metabolites (DHC in mice or cortisone in humans), which can vary acutely in response to stressors. Therefore, stressors, chronic stress, or diseases that affect the availability of circulating GC metabolites can control T cell development via regenerated GCs in the peripheral lymphoid organs.
Another important organ with the ability to synthesize GCs is the intestine . The proliferating cells of the intestinal crypts are devoted to the production of GCs, which have a controlling task in the maintenance of local immune homeostasis. Any stimulus that activates the immune local system triggers the production of GCs that control and limit the immune response to avoid an exaggerated response and consequent tissue damage. Such function of locally produced GCs occurs in the mucosal tissues, in which the strict contact between immune cells and microorganism does exist. In addition to this sentinel role, GCs are also responsible for maintaining the integrity and permeability of the epithelial barrier by antagonizing the destruction of tight junctions caused by TNFα during inflammation (e.g. Crohn’s disease) [44]. TNFα is also the most important cytokine able to induce local GC synthesis: in mice lacking TNFα or its receptor, GC synthesis in the gut is either reduced or absent [45]. In line with this fact, GCs produced in the intestine were found to reduce the damage in inflammatory bowel disease, both in experimental colitis in rodents and in human disease, confirmed by the decreased expression of GC synthetic-enzymes in Crohn’s patients [46].
Tumors derived from transformed epithelial cells of the adrenal cortex produce GCs; similarly, transformed cells from intestinal crypts (i.e., colorectal tumor cells) constitutively synthetise GCs. Although the reason these tumor cells release GCs has not yet been elucidated, it is believed that local GC synthesis may exert immune suppression on immune cells infiltrating the tumor microenvironment, as a mechanism of immune escape [47].
Lungs, like the intestine , are lined with a single epithelial layer that allows for the exchange of gases with the external environment; however, this permits access to potential pathogen agents. For this reason, the mucosa is rich in resident immune cells involved in the surveillance against invading microorganisms. Similar to the intestine, the lungs are the site of extra-adrenal GC synthesis that serve to prevent tissue damage caused by an exaggerated immune response. However, the metabolic pathway of GC synthesis in the lungs differs from that in the intestine. The lungs appear to reactivate circulating non-active dehydrocortisone to corticosterone, whereas in the intestine, GCs are synthesized from cholesterol or cholesterol metabolites. Unlike the intestine, steroidogenesis is not triggered by TNFα or other pro-inflammatory cytokines, but rather is dependent on the serum dehydrocorticosterone released by the adrenal glands, as the surgical removal of these glands abolishes local GC synthesis in the lungs [48, 49]. This differential regulation of local steroidogenesis could be due to different regulation of local inflammation in the intestine and lungs. This may be related to the need to restrict the inflammatory response to the lamina propria to prevent bacteria spreading while maintaining hormonal regulation by the adrenal glands in the lungs, since local inflammation could easily turn into systemic inflammation due to the high vascularization of lungs.
Another barrier between the organism and the external environment is the skin. Steroidogenic enzymes have been found in melanocytes, fibroblasts and keratinocytes. Moreover, cortisol is the major steroid produced in the skin . Local production of GCs is induced by inflammation (i.e. TNFα and IL-1β cytokines), UV radiation, and tissue damage, which trigger a reproducible HPA axis response with local ACTH production. GCs produced in the skin play an immunosuppressive and anti-inflammatory role, in contrast to serum GCs, which promote cell migration from the blood to the skin in response to infections or tissue damage in the short-term. Therefore, their function is to control an excessive immune response, similar to other extra-adrenal organs [39].
Rat and human brains express enzymes for glucocorticoid and mineralocorticoid synthesis and the mouse brain can synthesize corticosterone in both hippocampal pyramidal neurons and granule neurons. In developing neurons, it is believed that GC levels are maintained at low levels due to their potential toxic effects, together with low levels of serum GCs; however this hypothesis requires validation [39]. As described earlier in this chapter, GCs exerts extremely important functions in the brain, but dissecting the effects caused by circulating GCs from those triggered by locally synthesized GCs is extremely difficult.
Overall, the local production of GCs appears to control an excessive immune reaction, since it takes place in organs in which the immune surveillance is physiologically relevant (e.g. thymus, intestine and lungs). The concentration of locally synthesized GCs is much higher than the serum levels. Furthermore, circulating GCs are poorly available when needed due to their ability to bind to the serum corticosteroid-binding globulin. This local high concentration allows for a higher number of GC receptors to be activated and initiate the genomic and non-genomic responses with typical anti-inflammatory and immunosuppressive outcomes at specific sites. In addition, the extra-adrenal and adrenal GC syntheses are occasionally in opposition, such as in the lymphoid organs, where local GC production is high during development and decreases over time, compared to the systemic GC synthesis that is low during development and increases over time. Since local GC production has been found in animal species other than rodents and humans, it is believed that this mechanism represents an evolutionary adaptation that is useful to obtain a focused control of the immune reactions thereby avoiding harmful effects in all other tissues eventually exposed to high levels of circulating GCs. Finally, this local production raises the question whether therapeutic use of synthetic GCs should be adapted to reconstitute local concentration levels of endogenous GCs that have been eventually decreased in diseases.
4 GC Effects on Cells of the Immune System
Among the most sensitive GR expressing cells in an organism, immune cells have been largely studied for being exquisitely responsive to the effects of GC . The anti-inflammatory and immunosuppressive effects of GCs occur in all cells of the immune system , with either overlapping or different mechanisms linked to various cell types.
4.1 T Cells
T lymphocytes are the most studied immune cells under the effects of GCs. Starting from their maturation and development in the thymus, T cells are sensitive to GC effects throughout their lifespan. T cells are either positively or negatively selected at the stage of CD4+ CD8+ cells, in which a strong TCR signal leads to apoptosis as well as an absent TCR signal. At this stage, T lymphocytes are very sensitive to GC-induced apoptosis, which is antagonized by TCR engagement when both stimuli are present, as described earlier in this chapter [50]. The intensity of TCR signaling determines the fate of the cell: only TCRs with an intermediate signal intensity and avidity for self-antigens will allow the survival of the thymocyte due to the “mutual exclusion” effect of GCs, through which the TCR and GR signals oppose each other, allowing the cell to escape apoptosis. Thymic selection of mature T cells has also been explored in studies that have used transgenic mice with a mutated GR. Mice expressing reduced GR levels due to the presence of an antisense transgene exhibit an impaired transition from CD4- CD8- precursors to CD4+ CD8+ and increased apoptosis, supporting a role for endogenous GC in balancing TCR-mediated signals during thymic selection and agreeing with in vitro obtained results [51, 52]. Conversely, other studies that have used mice with GR-deficient thymocytes demonstrated that GR signaling is not essential for T cell development or selection in the thymus [53, 54]. To dissect the specific roles of GCs in the thymus, further studies using in vivo mouse models and targeted mutations in the GR will be able to assess the actual role of GC in thymic cell development.
GCs also exert their effects on mature T cells. T-cell-specific inactivation of the GR, as well as mice with a selective functional mutation in the GR are useful for demonstrating that GCs can suppress activation-induced cell death by inhibiting the expression of FasL, which induces cell death by binding to its cognate receptor, Fas, on T lymphocytes , thereby limiting excessive activation [55]. In addition to this genomic pathway, other non-genomic pathways mediate the effect of GCs on T lymphocytes. Soon after GC administration, kinases (i.e. LCK and Fyn) are downregulated with a consequent dissociation from the TCR complex [56]. Although less sensitive to GC-induced apoptosis due to a strong CD28 signal, mature lymphocytes are therefore influenced by GCs with regards to their activation, survival, and cell death through multiple mechanisms.
An important GC function is the ability to drive T cell subtype differentiation. Upon an antigenic or inflammatory stimulus, T lymphocytes can differentiate into distinct subtypes such as Th1, Th2, Th17, Tregs and, more recently, Th9 cells. Each subtype has a specific role, either physiological or even pathogenic, expresses specific transcription factors, and secretes a distinct pattern of cytokines. Each subset is differently sensitive to GC-induced apoptosis; for example, Th1 cells undergo apoptosis when subjected to GC effects while Th2 and Th17 cells are resistant. Moreover, Tregs have been demonstrated to be both sensitive and resistant in distinct experimental settings. Furthermore, GCs can induce cytokine suppression distinctly in different subtypes, one of the mechanisms of their anti-inflammatory properties. Cytokines from Th1 and Th2 cells, including IFN-γ, IL-4, IL-5, and IL-13, can be suppressed by GCs. In contrast, IL-17A and IL-17F from Th17 cells are resistant to GC suppression in primary Th17 cells, despite being sensitive in multiple sclerosis or severe asthma [57]. Moreover, Th17 cells and their released cytokine IL-17 appear to be partially responsible for the resistance to GC treatment in one third of patients with inflammatory disorders that do not respond to steroid therapy [58].
GCs can cause a shift from Th1 to Th2 immunity at physiological doses by suppressing Th1 cytokines. More specifically, GCs can suppress the production of interleukin (IL)-12, interferon IFN-γ, IFN-α, and TNF-α by antigen-presenting cells (APCs) and Th1 cells, but can upregulate the production of IL-4, IL-10, and IL-13 by Th2 cells, thereby promoting their differentiation [59]. One consequence of this unbalance is the exacerbation of pathologies (e.g. asthma) as mentioned above, in which the Th2 phenotype contributes to disease pathogenesis.
The role of GCs in Tregs remains still not completely defined: data obtained in vitro in human PBMC revealed a rapid decrease of FoxP3 expression within 24 h of exposure to Dexamethasone (Dex) , not due to apoptosis. In support of these in vitro data, in transplanted patients with a normal Treg number, Tregs can be suppressed by GC treatment. However, an increased number of Treg cells was reported in patients with allergic rhinitis, asthma or other autoimmune disease, treated with high doses of corticosteroids, in which the number of Tregs could be modified by the disease. Additionally, in an experimental model of EAE, short-term simultaneous administration of Dex and IL-2 markedly expanded functional suppressive Foxp3+ CD4+ CD25+ T cells in murine peripheral lymphoid tissues [14, 60, 61]. These opposing observations may vary according to the diseases in which Tregs are studied or differential experimental settings. Recently, the mechanism through which GCs increase the number of Treg cells in mice has been discovered; GILZ, an anti-inflammatory protein rapidly induced by GCs, increases the number of Treg cells by virtue of its ability to cooperate with TGF-β in the induction of FoxP3 [62]. The Treg field still remains an incompletely explored field and additional studies are needed to unravel the relationship between GCs and Tregs.
4.2 B Cells
If considered with respect to T lymphocytes , less is known about GC function in B lymphocytes. Only recent work has partially clarified the role of GCs in B cells. GCs are used to treat B cell malignancies due to their ability to suppress B-cell checkpoint genes across multiple developmental stages. A very recent study reported that supraphysiological levels of GCs can either push immature cells to the next stage of development, with consequent apoptosis, or they may arrest cells by removing a positive growth signal [63]. An in-depth analysis in murine B cells derived from the spleen and bone marrow demonstrated that Dex stimulated apoptosis in all B-cell developmental subsets, suggesting that GC signaling plays a pivotal role in B-cell life-or-death decisions [64]. To further underline this important role in the life of a B cell, a GC-induced protein, GILZ, was found to be the mediator of the effects of GCs on B cell lifespan. In mice with a GILZ deletion, an accumulation of B lymphocytes in the bone marrow, blood, and lymphoid tissues was found, as well as decreased B-cell apoptosis. This supports an important role of GCs, through GC-induced genes, in the regulation of B-cell survival [65, 66]. Immunoglobulin synthesis is also regulated by GCs; low levels of GCs do not exhibit any effects on immunoglobulin synthesis, whereas high doses decrease immunoglobulin levels in the blood due to an increase of their catabolism at the beginning, followed by a reduction in synthesis [67, 68].
4.3 Macrophages
Monocytes and macrophages are among the cells of the first line of defense in the immune system and thus targets of the actions of GCs. Suppressing the intracellular signaling cascade of MAP kinases is one of the mechanisms through which GCs exert their anti-inflammatory effects, by inhibiting the transcription of pro-inflammatory cytokines like IFNγ, IL-1α and IL-1β. Furthermore, the suppression of these pro-inflammatory cytokines can be achieved via the direct interaction of ligand-activated GR with transcription factors like AP-1 and NF-κB at the promoter of target genes [68]. GCs can even promote the survival of anti-inflammatory monocytes by exerting anti-apoptotic effects. It is important to protect anti-inflammatory monocytes from apoptosis and let them differentiate so that they can be efficient in the down-regulation of inflammation. The mechanism by which GCs exert this specific effect is via the regulation of the A3 adenosine receptor [69].
Physiologically, low concentrations of corticosterone exert stimulatory effects on naïve macrophage chemotactic and phagocytic activities, in the absence of immune stimuli, and GR is at least partially responsible for these effects. Conversely, supraphysiological GC concentrations do not have any effects on macrophage functionality. Thus, during the early phase of stress, corticosterone may prime innate cells and contribute to defense against an infectious agent [70]. Overall GCs exert distinct functions on monocytes/macrophages, depending on the dose and the presence or absence of an immune stimulus.
4.4 Neutrophils and Other Granulocytes
Different from other immune cell types , neutrophils are protected from apoptosis when treated with GCs , exhibiting a doubling of their half-lives compared to untreated cells. This is due to the expression of members of the Bcl-2 family of survival proteins and the suppression of pro-apoptotic genes. In this manner, GCs can contribute with neutrophils to help the organism fight against infections with a primary defense cell, when all other cells of the immune system succumb to their apoptotic action. In contrast, the persistence of neutrophils in inflamed tissues further increases inflammation and contributes to the resistance to any pharmacological treatments with GCs (e.g. severe neutrophilic asthma, inflammatory bowel disease, and rheumatoid arthritis). It remains to be elucidated whether GCs differentially influence the distinct circulating phenotypes of neutrophils under inflammatory conditions [71]. Another anti-inflammatory effect GCs exert on neutrophils is the prevention of granulocyte trans-migration into inflamed tissues. GCs can arrest the extravasation of neutrophils from blood circulation by multiple mechanisms, including the reduction of selectin expression and integrin receptors on neutrophils and endothelial cells, respectively. Furthermore, GCs have been recently found to reduce neutrophil migration by the upregulation of GILZ protein and consequently Annexin A1, an anti-inflammatory and anti-migration protein [72]. GCs are also able to increase bone-marrow derived neutrophils in the blood stream, so that they are therapeutically useful for the treatment of neutropenia in combination with G-CSF [73]. Although studies describing the effects of GCs on granulocytes began many years ago, we are still far from fully understanding the influence of GCs on neutrophils, under both the physiological and inflammatory conditions.
Eosinophils and basophils are sensitive to GC-induced apoptosis, and this mechanism is mediated by the Fas/FasL system and the increased generation of cell-damaging molecules (e.g. reactive oxygen species [ROS]) by eosinophils [74, 75]. In addition, GCs promote eosinophil clearance by inhibiting pro-survival signals induced by the cytokines IL-3, IL-5 and granulocyte–macrophage colony-stimulating factor (GM-CSF) [76]. These effects explain why GCs have been used for many years to treat eosinophilic disorders, although new pharmacological treatments have replaced them as a result of their undesired adverse effects. GCs do not only reduce the number of basophils but can even inhibit their migration in a concentration-dependent manner and prevent the release of histamine. Recently, GCs have also been found to inhibit basophil activation via membrane-bound GR interferences with the formation of lipid raft nanoclusters [77, 78].
4.5 Mast Cells
Mast cells are effector cells characteristic of allergic and inflammatory reactions. When allergen-mediated aggregation of the FcεRI takes place, a signaling cascade is initiated that leads to the production of cytokines, chemokines, arachidonic and eicosanoid production, and cellular degranulation. Long-term treatment with GCs can inhibit mast cell activation by downregulating Erk1/2 and inhibiting of the PI3K signaling cascade, with the subsequent prevention of degranulation and mast cell activation. Other signaling pathways are also reduced by GC treatment, including the phosphorylation of p38 and JNK1/2 [79]. One of the major factors involved in the molecular pathways of the anti-inflammatory actions of GCs on mast cells is the activation of protein tyrosine phosphatases (PTPs) by GCs. Within this family, DUSP1 and DUSP2 have been functionally characterized in mast cells and found to be upregulated by GCs, thus are available for the dephosphorylation of Erk1/2 and the subsequent inhibition of cellular activation. Another important feature of mast cells is their accumulation at the site of inflammation. GCs are known to reduce mast cell accumulation by downregulating the stem cell factor released by fibroblasts with kinetics that remains to be established [80, 81]. While all of these actions mediated by GCs are genomic-derived, other effects occur rapidly after GC administration (e.g. in the treatment of allergic reactions, degranulation is rapidly decreased). Despite the potential role of membrane bound based on studies in other cell types, no studies has still been conducted in mast cells.
4.6 Dendritic Cells
Dendritic cells have been largely studied as a target of GC action. Throughout their life cycle, dendritic cells are influenced by GCs, differently from the monocytes from which they are derived. Dendritic cells mature after encountering an antigen and they are sensitive to GC-induced apoptosis only before this stage. Furthermore, GCs stimulate antigen uptake before maturing, thus helping the organism fight against invading pathogens by keeping these cells in an immature state. More importantly, dendritic cells become tolerogenic once they are exposed to GCs, exhibiting low levels of expression of MHCII molecules, costimulatory molecules, and cytokines (e.g. IL-1, IL-6, and IL-12) [68]. Under this state they can neither prime nor induce the proliferation or activation of T cells; however they can promote the formation of Treg cells [82]. The migration towards the lymph nodes is also inhibited by GCs.
It has been recently shown that endogenous GCs suppress the dendritic cell response to LPS exposure by reducing IL-12 production during sepsis, thus explaining the role of GCs in the treatment of sepsis [83].
5 Anti-inflammatory Versus Pro-inflammatory Effects of GCs
As evidenced throughout this chapter, due to the specific effects of GCs on the cells of the immune system, GCs have historically gained attention as the most important anti-inflammatory and immunosuppressive drugs. Indeed GCs are currently used to treat pathologies such as asthma, rheumatoid arthritis, inflammatory bowel disease or even tumor pathologies such as acute lymphoblastic leukemia or to prevent the graft-vs-host disease in organ transplantations. The ability of GCs to suppress pro-inflammatory cytokines or other inflammatory mediators (listed in Table 2) in a variety of cells, guarantees its success in the treatment of inflammatory diseases. Nonetheless, GCs are not as anti-inflammatory as might be expected. It is generally accepted that in addition to the detrimental side effects described in Table 1 and gathered under the Cushingoid syndrome, the long term treatment with GCs may enhance inflammation and immunity depending on the dose, chronicity of treatment, and target organ [84, 85]. Genome-wide expression studies of GC-treated cells have revealed that GCs upregulate genes of innate immune cells that are involved in the recognition of pathogens (e.g. pattern recognition receptors –PRRs-), but inhibit the expression of pro-inflammatory cytokines in cells involved in the adaptive immune response [86, 87]. In other studies, the increased expression of cytokine receptors has been reported (e.g. TNFR, IL-1R, Il-6R, and receptors for IFNγ), as well as increased IL-1β production in response to LPS [88, 89]. The expression of pro-inflammatory genes, including iNOS and TNFα, together with a decreased expression of anti-inflammatory genes, including IL-1ra and IL-10, have also been shown in the frontal cortex of rats; these effects were shown to be GR-mediated and region-dependent [88]. Therefore, in some conditions, while the effects of GCs may be opposing, their functions respond to the specific needs of the organism and the mechanisms through which they occur remain incompletely understood. A model proposed by Cain and Cidlowski suggests that low levels of GCs in the absence of inflammation make cells sensitive to any harmful stimulus by promoting the expression of PRRs and other pro-inflammatory mediators. In contrast, during inflammatory conditions, high levels of GCs induced by stress shorten the duration of the inflammatory response by acting as anti-inflammatory agents [86].
6 Conclusions
There is no doubt regarding the clinical efficacy of GCs for the treatment of pathologies that cannot be treated with targeted drugs, even despite the occasional observance of resistance to GC treatment. However, basic knowledge regarding the functions of GCs on cells, not only of the immune system, but of the whole organism remains incomplete. Thus, gaining insights into the mechanism of GC action is required to both unravel their physiological role and to develop alternative drugs with the same anti-inflammatory properties as GCs, without the harmful adverse effects.
References
Nicolaides NC, Kyratzi E, Lamprokostopoulou A, Chrousos GP, Charmandari E (2015) Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 22(1–2):6–19
Nguyen ET, Streicher J, Berman S, Caldwell JL, Ghisays V, Estrada CM, Wulsin AC, Solomon MB (2017) A mixed glucocorticoid/mineralocorticoid receptor modulator dampens endocrine and hippocampal stress responsivity in male rats. Physiol Behav 178:82–92
Charmandari E, Tsigos C, Chrousos G (2005) Endocrinology of the stress response. Annu Rev Physiol 67:259–284
Vyas S, Rodrigues AJ, Silva JM, Tronche F, Almeida OF, Sousa N, Sotiropoulos I (2016) Chronic stress and glucocorticoids: from neuronal plasticity to neurodegeneration. Neural Plast 2016:6391686
Sousa N, Almeida OF (2012) Disconnection and reconnection: the morphological basis of (mal)adaptation to stress. Trends Neurosci 35(12):742–751
Mak JC, Nishikawa M, Shirasaki H, Miyayasu K, Barnes PJ (1995) Protective effects of a glucocorticoid on downregulation of pulmonary beta 2-adrenergic receptors in vivo. J Clin Invest 96(1):99–106
Sakaue M, Hoffman BB (1991) Glucocorticoids induce transcription and expression of the alpha 1B adrenergic receptor gene in DTT1 MF-2 smooth muscle cells. J Clin Invest 88(2):385–389
Ogawa K, Hirai M, Katsube T, Murayama M, Hamaguchi K, Shimakawa T, Naritake Y, Hosokawa T, Kajiwara T (2000) Suppression of cellular immunity by surgical stress. Surgery 127(3):329–336
Salicru AN, Sams CF, Marshall GD (2007) Cooperative effects of corticosteroids and catecholamines upon immune deviation of the type-1/type-2 cytokine balance in favor of type-2 expression in human peripheral blood mononuclear cells. Brain Behav Immun 21(7):913–920
Xiang L, Marshall GD Jr (2013) Immunomodulatory effects of dexamethasone on gene expression of cytokine and stress hormone receptors in peripheral blood mononuclear cells. Int Immunopharmacol 17(3):556–560
Chida Y, Sudo N, Sonoda J, Hiramoto T, Kubo C (2007) Early-life psychological stress exacerbates adult mouse asthma via the hypothalamus-pituitary-adrenal axis. Am J Respir Crit Care Med 175(4):316–322
Miyasaka T, Dobashi-Okuyama K, Takahashi T, Takayanagi M, Ohno I (2017) The interplay between neuroendocrine activity and psychological stress-induced exacerbation of allergic asthma. Allergol Int 67:32–42
DeKruyff RH, Fang Y, Umetsu DT (1998) Corticosteroids enhance the capacity of macrophages to induce Th2 cytokine synthesis in CD4+ lymphocytes by inhibiting IL-12 production. J Immunol 160(5):2231–2237
Olsen PC, Kitoko JZ, Ferreira TP, de- Azevedo CT, Arantes AC, Martins MA (2015) Glucocorticoids decrease Treg cell numbers in lungs of allergic mice. Eur J Pharmacol 747:52–58
Freier E, Weber CS, Nowottne U, Horn C, Bartels K, Meyer S, Hildebrandt Y, Luetkens T, Cao Y, Pabst C, Muzzulini J, Schnee B, Brunner-Weinzierl MC, Marangolo M, Bokemeyer C, Deter HC, Atanackovic D (2010) Decrease of CD4(+)FOXP3(+) T regulatory cells in the peripheral blood of human subjects undergoing a mental stressor. Psychoneuroendocrinology 35(5):663–673
Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K (2008) Steroid hormone receptor expression and function in microglia. Glia 56(6):659–674
Nair A, Bonneau RH (2006) Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol 171(1–2):72–85
Frank MG, Miguel ZD, Watkins LR, Maier SF (2010) Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun 24(1):19–30
Goujon E, Parnet P, Laye S, Combe C, Kelley KW, Dantzer R (1995) Stress downregulates lipopolysaccharide-induced expression of proinflammatory cytokines in the spleen, pituitary, and brain of mice. Brain Behav Immun 9(4):292–303
Frank MG, Weber MD, Watkins LR, Maier SF (2016) Stress-induced neuroinflammatory priming: a liability factor in the etiology of psychiatric disorders. Neurobiol Stress 4:62–70
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–741
Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2014) Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci 8:447
Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, Turner RB (2012) Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A 109(16):5995–5999
Dhabhar FS (2009) Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation 16(5):300–317
Ronaldson A, Gazali AM, Zalli A, Kaiser F, Thompson SJ, Henderson B, Steptoe A, Carvalho L (2016) Increased percentages of regulatory T cells are associated with inflammatory and neuroendocrine responses to acute psychological stress and poorer health status in older men and women. Psychopharmacology (Berl) 233(9):1661–1668
Mundy-Bosse BL, Thornton LM, Yang HC, Andersen BL, Carson WE (2011) Psychological stress is associated with altered levels of myeloid-derived suppressor cells in breast cancer patients. Cell Immunol 270(1):80–87
Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF (2002) Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. J Neuroimmunol 124(1–2):9–15
Eiro N, Vizoso FJ (2012) Inflammation and cancer. World J Gastrointest Surg 4(3):62–72
De Rosa V, Di Rella F, Di Giacomo A, Matarese G (2017) Regulatory T cells as suppressors of anti-tumor immunity: role of metabolism. Cytokine Growth Factor Rev 35:15–25
Chaudhary B, Elkord E (2016) Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel). 2016 Aug 6;4(3). pii: E28. https://doi.org/10.3390/vaccines4030028
Darrasse-Jeze G, Podsypanina K (2013) How numbers, nature, and immune status of foxp3(+) regulatory T-cells shape the early immunological events in tumor development. Front Immunol 4:292
Powell ND, Tarr AJ, Sheridan JF (2013) Psychosocial stress and inflammation in cancer. Brain Behav Immun 30(Suppl):S41–S47
Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C (2007) GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest 117(6):1605–1615
Ayroldi E, Petrillo MG, Bastianelli A, Marchetti MC, Ronchetti S, Nocentini G, Ricciotti L, Cannarile L, Riccardi C (2015) L-GILZ binds p53 and MDM2 and suppresses tumor growth through p53 activation in human cancer cells. Cell Death Differ 22(1):118–130
Dumbell R, Matveeva O, Oster H (2016) Circadian clocks, stress, and immunity. Front Endocrinol 7:37
Cima I, Corazza N, Dick B, Fuhrer A, Herren S, Jakob S, Ayuni E, Mueller C, Brunner T (2004) Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med 200(12):1635–1646
Vacchio MS, Papadopoulos V, Ashwell JD (1994) Steroid production in the thymus: implications for thymocyte selection. J Exp Med 179(6):1835–1846
Pazirandeh A, Jondal M, Okret S (2005) Conditional expression of a glucocorticoid receptor transgene in thymocytes reveals a role for thymic-derived glucocorticoids in thymopoiesis in vivo. Endocrinology 146(6):2501–2507
Taves MD, Gomez-Sanchez CE, Soma KK (2011) Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function. Am J Physiol Endocrinol Metab 301(1):E11–E24
Talaber G, Jondal M, Okret S (2015) Local glucocorticoid production in the thymus. Steroids 103:58–63
Vacchio MS, Ashwell JD (1997) Thymus-derived glucocorticoids regulate antigen-specific positive selection. J Exp Med 185(11):2033–2038
Nuotio-Antar AM, Hasty AH, Kovacs WJ (2006) Quantitation and cellular localization of 11beta-HSD1 expression in murine thymus. J Steroid Biochem Mol Biol 99(2–3):93–99
Taves MD, Plumb AW, Korol AM, Van Der Gugten JG, Holmes DT, Abraham N, Soma KK (2016) Lymphoid organs of neonatal and adult mice preferentially produce active glucocorticoids from metabolites, not precursors. Brain Behav Immun 57:271–281
Boivin MA, Ye D, Kennedy JC, Al-Sadi R, Shepela C, Ma TY (2007) Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am J Physiol Gastrointest Liver Physiol 292(2):G590–G598
Noti M, Corazza N, Mueller C, Berger B, Brunner T (2010) TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 207(5):1057–1066
Coste A, Dubuquoy L, Barnouin R, Annicotte JS, Magnier B, Notti M, Corazza N, Antal MC, Metzger D, Desreumaux P, Brunner T, Auwerx J, Schoonjans K (2007) LRH-1-mediated glucocorticoid synthesis in enterocytes protects against inflammatory bowel disease. Proc Natl Acad Sci U S A 104(32):13098–13103
Sidler D, Renzulli P, Schnoz C, Berger B, Schneider-Jakob S, Fluck C, Inderbitzin D, Corazza N, Candinas D, Brunner T (2012) Colon cancer cells produce immunoregulatory glucocorticoids. Oncoimmunology 1(4):529–530
Kostadinova F, Schwaderer J, Sebeo V, Brunner T (2014) Why does the gut synthesize glucocorticoids? Ann Med 46(7):490–497
Hostettler N, Bianchi P, Gennari-Moser C, Kassahn D, Schoonjans K, Corazza N, Brunner T (2012) Local glucocorticoid production in the mouse lung is induced by immune cell stimulation. Allergy 67(2):227–234
Herold MJ, McPherson KG, Reichardt HM (2006) Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci 63(1):60–72
Stephens GL, Ashwell JD, Ignatowicz L (2003) Mutually antagonistic signals regulate selection of the T cell repertoire. Int Immunol 15(5):623–632
Mittelstadt PR, Monteiro JP, Ashwell JD (2012) Thymocyte responsiveness to endogenous glucocorticoids is required for immunological fitness. J Clin Invest 122(7):2384–2394
Purton JF, Boyd RL, Cole TJ, Godfrey DI (2000) Intrathymic T cell development and selection proceeds normally in the absence of glucocorticoid receptor signaling. Immunity 13(2):179–186
Liddicoat DR, Purton JF, Cole TJ, Godfrey DI (2014) Glucocorticoid-mediated repression of T-cell receptor signalling is impaired in glucocorticoid receptor exon 2-disrupted mice. Immunol Cell Biol 92(2):148–155
Baumann S, Dostert A, Novac N, Bauer A, Schmid W, Fas SC, Krueger A, Heinzel T, Kirchhoff S, Schutz G, Krammer PH (2005) Glucocorticoids inhibit activation-induced cell death (AICD) via direct DNA-dependent repression of the CD95 ligand gene by a glucocorticoid receptor dimer. Blood 106(2):617–625
Lowenberg M, Tuynman J, Bilderbeek J, Gaber T, Buttgereit F, van Deventer S, Peppelenbosch M, Hommes D (2005) Rapid immunosuppressive effects of glucocorticoids mediated through Lck and Fyn. Blood 106(5):1703–1710
Banuelos J, Lu NZ (2016) A gradient of glucocorticoid sensitivity among helper T cell cytokines. Cytokine Growth Factor Rev 31:27–35
Schewitz-Bowers LP, Lait PJ, Copland DA, Chen P, Wu W, Dhanda AD, Vistica BP, Williams EL, Liu B, Jawad S, Li Z, Tucker W, Hirani S, Wakabayashi Y, Zhu J, Sen N, Conway-Campbell BL, Gery I, Dick AD, Wei L, Nussenblatt RB, Lee RW (2015) Glucocorticoid-resistant Th17 cells are selectively attenuated by cyclosporine A. Proc Natl Acad Sci U S A 112(13):4080–4085
Elenkov IJ (2004) Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci 1024:138–146
Chen X, Oppenheim JJ, Winkler-Pickett RT, Ortaldo JR, Howard OM (2006) Glucocorticoid amplifies IL-2-dependent expansion of functional FoxP3(+)CD4(+)CD25(+) T regulatory cells in vivo and enhances their capacity to suppress EAE. Eur J Immunol 36(8):2139–2149
Xiang L, Marshall GD Jr (2011) Immunomodulatory effects of in vitro stress hormones on FoxP3, Th1/Th2 cytokine and costimulatory molecule mRNA expression in human peripheral blood mononuclear cells. Neuroimmunomodulation 18(1):1–10
Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, Sorcini D, Venanzi A, Di Sante M, Riccardi C (2014) GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep 7(2):464–475
Kruth KA, Fang MM, Shelton DN, Abu-Halawa O, Mahling R, Yang HX, Weissman JS, Loh ML, Muschen M, Tasian SK, Bassik MC, Kampmann M, Pufall MA (2017) Suppression of B-cell development genes is key to glucocorticoid efficacy in treatment of acute lymphoblastic leukemia. Blood 129(22):3000–3008
Gruver-Yates AL, Quinn MA, Cidlowski JA (2014) Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology 155(2):463–474
Bruscoli S, Biagioli M, Sorcini D, Frammartino T, Cimino M, Sportoletti P, Mazzon E, Bereshchenko O, Riccardi C (2015) Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood 126(15):1790–1801
Alnemri ES, Fernandes TF, Haldar S, Croce CM, Litwack G (1992) Involvement of BCL-2 in glucocorticoid-induced apoptosis of human pre-B-leukemias. Cancer Res 52(2):491–495
Chalubinski M, Grzegorczyk J, Kowalski ML (2011) Glucocorticoid-induced immunoglobulin E synthesis by peripheral blood mononuclear cells from allergic and nonallergic subjects. Ann Allergy Asthma Immunol 107(3):251–257
Baschant U, Tuckermann J (2010) The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol 120(2–3):69–75
Barczyk K, Ehrchen J, Tenbrock K, Ahlmann M, Kneidl J, Viemann D, Roth J (2010) Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood 116(3):446–455
Zhong HJ, Wang HY, Yang C, Zhou JY, Jiang JX (2013) Low concentrations of corticosterone exert stimulatory effects on macrophage function in a manner dependent on glucocorticoid receptors. Int J Endocrinol 2013:405127
Wang J, Arase H (2014) Regulation of immune responses by neutrophils. Ann N Y Acad Sci 1319:66–81
Ricci E, Ronchetti S, Pericolini E, Gabrielli E, Cari L, Gentili M, Roselletti E, Migliorati G, Vecchiarelli A, Riccardi C (2017) Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J 31(7):3054–3065
Stroncek DF, Yau YY, Oblitas J, Leitman SF (2001) Administration of G--CSF plus dexamethasone produces greater granulocyte concentrate yields while causing no more donor toxicity than G--CSF alone. Transfusion 41(8):1037–1044
Meagher LC, Cousin JM, Seckl JR, Haslett C (1996) Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol 156(11):4422–4428
Druilhe A, Letuve S, Pretolani M (2003) Glucocorticoid-induced apoptosis in human eosinophils: mechanisms of action. Apoptosis 8(5):481–495
Fulkerson PC, Rothenberg ME (2013) Targeting eosinophils in allergy, inflammation and beyond. Nat Rev Drug Discov 12(2):117–129
Yamaguchi M, Hirai K, Nakajima K, Ohtoshi T, Takaishi T, Ohta K, Morita Y, Ito K (1994) Dexamethasone inhibits basophil migration. Allergy 49(5):371–375
Yamagata S, Tomita K, Sano H, Itoh Y, Fukai Y, Okimoto N, Watatani N, Inbe S, Miyajima H, Tsukamoto K, Santoh H, Ichihashi H, Sano A, Sato R, Tohda Y (2012) Non-genomic inhibitory effect of glucocorticoids on activated peripheral blood basophils through suppression of lipid raft formation. Clin Exp Immunol 170(1):86–93
Oppong E, Flink N, Cato AC (2013) Molecular mechanisms of glucocorticoid action in mast cells. Mol Cell Endocrinol 380(1–2):119–126
Nilsson G, Butterfield JH, Nilsson K, Siegbahn A (1994) Stem cell factor is a chemotactic factor for human mast cells. J Immunol 153(8):3717–3723
Kassel O, Schmidlin F, Duvernelle C, de Blay F, Frossard N (1998) Up- and down-regulation by glucocorticoids of the constitutive expression of the mast cell growth factor stem cell factor by human lung fibroblasts in culture. Mol Pharmacol 54(6):1073–1079
Bros M, Jahrling F, Renzing A, Wiechmann N, Dang NA, Sutter A, Ross R, Knop J, Sudowe S, Reske-Kunz AB (2007) A newly established murine immature dendritic cell line can be differentiated into a mature state, but exerts tolerogenic function upon maturation in the presence of glucocorticoid. Blood 109(9):3820–3829
Li CC, Munitic I, Mittelstadt PR, Castro E, Ashwell JD (2015) Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. PLoS Biol 13(10):e1002269
Duque Ede A, Munhoz CD (2016) The pro-inflammatory effects of glucocorticoids in the brain. Front Endocrinol 7:78
Cruz-Topete D, Cidlowski JA (2015) One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 22(1–2):20–32
Cain DW, Cidlowski JA (2017) Immune regulation by glucocorticoids. Nat Rev Immunol 17(4):233–247
Sorrells SF, Sapolsky RM (2007) An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun 21(3):259–272
Munhoz CD, Sorrells SF, Caso JR, Scavone C, Sapolsky RM (2010) Glucocorticoids exacerbate lipopolysaccharide-induced signaling in the frontal cortex and hippocampus in a dose-dependent manner. J Neurosci 30(41):13690–13698
Wiegers GJ, Reul JM (1998) Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol Sci 19(8):317–321
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Ronchetti, S., Riccardi, C. (2018). Glucocorticoids: Immunity and Inflammation. In: Riccardi, C., Levi-Schaffer, F., Tiligada, E. (eds) Immunopharmacology and Inflammation. Springer, Cham. https://doi.org/10.1007/978-3-319-77658-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-77658-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-77657-6
Online ISBN: 978-3-319-77658-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)