
Abstract
Cyclodextrins are naturally occurring cyclic oligosaccharides, which are non-toxic and biodegradable. The main feature of cyclodextrins is the ability to encapsulate lipophilic compounds, and thus many applications have been developped in various disciplines. Although many cyclodextrin derivatives have become available in the market, their price is in the range of fine chemicals, and thus they are still often synthesised in laboratories. The actual number of cyclodextrin derivatives exceeds 11,000, but new cyclodextrin derivatives are still needed for more advanced applications. Therefore, many newcomers or beginners in cyclodextrin chemistry struggle with a reliable choice of a synthetic route.
This chapter reviews the synthesis cyclodextrin derivatives that are able to be subsequently modified. Indeed, the modification of a cyclodextrin already substituted with a suitable functional group is much easier than the optimisation of the substitution for every new cyclodextrin derivative desired. This chapter describes the synthesis of different types of cyclodextrine derivatives: persubstituted, randomly substituted, persubstituted at selected positions, selectively substituted and monosubstituted.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Cyclodextrin
- Derivative
- Synthesis
- Reactivity
- Persubstitution
- Random substitution
- Selective substitution
- Monosubstitution
- Deprotection
- Protection
2.1 Introduction
The history of cyclodextrin derivatives goes back to the beginning of the twentieth century when cyclodextrin properties were examined and the first derivatives were prepared (Crini 2014). Since then, cyclodextrin derivatives have come a long way, and they can currently find their use in many kinds of human activities: in pharmaceutical and biomedical applications (Jambhekar and Breen 2016; Sharma and Baldi 2016; Oliveri and Vecchio 2016; Coisne et al. 2016; Leclercq 2016; di Cagno 2017; Saokham and Loftsson 2017); nanotherapeutics (Bonnet et al. 2015; Swaminathan et al. 2016; Antoniuk and Amiel 2016; Mejia-Ariza et al. 2017; Venuti et al. 2017); cosmetics, toiletries and personal care (Sharma and Baldi 2016); nutrition industry (Astray et al. 2009; Fenyvesi et al. 2016; Sharma and Baldi 2016); textile and packing industry (Radu et al. 2016; Sharma and Baldi 2016); separation techniques (Řezanka et al. 2014; Zhou et al. 2015; Adly et al. 2016; Saz and Marina 2016; Zhu and Scriba 2016); and as artificial enzymes or catalysts (Kryjewski et al. 2015; Macaev and Boldescu 2015; Aghahosseini and Ramazani 2016; Letort et al. 2016).
Probably the most known application of cyclodextrin derivatives in daily life is the use of octakis[6-(2-carboxyethylthio)-6-deoxy]-γ-cyclodextrin sodium salt (sugammadex) in Bridion® (Donati 2008). When an individual undergoes surgery, anaesthesia must be provided. Drugs like rocuronium bromide are administered as they block transmission at the cholinergic nicotinic receptor at the neuromuscular junction. After the surgery the patient is transited from the anaesthetised to the fully recovered state. Therefore, sugammadex is administered. It is designed to bind the neuromuscular blocking agent instead of relying on rocuronium pharmacokinetic properties or on the inhibition of acetylcholine breakdown with a reversal agent.
2.1.1 Inclusion Complexes of Cyclodextrin Derivatives
As it was illustrated in the previous paragraph, the key property of cyclodextrin derivatives lies in their ability to complex compounds in their cavity. These compounds are usually denoted as “guests”. There are several driving forces that could lead to the formation of cyclodextrin-guest complexes: van der Waals interactions, electrostatic interactions, hydrogen bonding, hydrophobic interactions, release of conformational strain, exclusion of “high-energy” water from the cavity, and charge-transfer interactions. Liu and Guo (Liu and Guo 2002) have shown that with the exception of the release of conformation strain and exclusion of water from the cyclodextrin cavity, the other interactions contribute to the complex formation. They concluded that hydrophobic and van der Waals interactions are the main driving forces, whereas hydrogen bonding and electrostatic interaction can significantly affect the stability of the inclusion complex.
The shape and size of guests forming complexes with cyclodextrin or its derivatives are variable and therefore the strength of a complex depends on the cavity size (Szente and Fenyvesi 2017). In the case of cyclodextrin derivatives, the substituents present on a cyclodextrin skeleton should not be overlooked – as they could significantly affect the formation of the inclusion complex. For example, an association constant of the abovementioned sugammadex with rocuronium bromide is 107 M−1 (Bom et al. 2002), while native γ-cyclodextrin has the constant only in the order of 104 M−1 (Cameron et al. 2002). The association constants for complexes of cyclodextrin derivatives with a wide variety of guests can be found in the review by Rekharsky and Inoue (Rekharsky and Inoue 1998).
Cyclodextrin derivative-guest complexes are prepared by several methods: co-precipitation, freeze-drying, kneading, melting, neutralization, grinding, sealed-heating and others (Iacovino et al. 2017). Cyclodextrin complexes usually have a cyclodextrin derivative:guest ratio of 1:1. However many other cyclodextrin derivative:guest ratios are known (Song et al. 2009b; Lima et al. 2016), and cyclodextrins can even form a wide variety of other supramolecular structures including catenanes, rotaxanes (Harada et al. 2009; Garcia-Rio et al. 2014) and supramolecular polymers (Wang et al. 2016).
2.1.2 Cyclodextrin Derivatives Properties
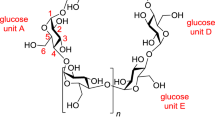
Nomenclature usually used by researchers for the description of cyclodextrin derivatives is depicted in Fig. 2.1. A glucose unit possesses substitution sites at positions 2, 3 and 6. Glucose units are named A, B, C etc. respectively in superscript. Sometimes the glucose units are numbered with roman numerals in superscript – i. e. I, II, III etc. Simplicity also often wins over precision. For example, per-O-methyl- 2A-O-allyl-β-cyclodextrin refers to β-cyclodextrin, where allyl substituent is at position 2 at one glucose unit and all 20 the other hydroxyl groups are protected with methyl groups.

Structure and numbering of α-, β-, and γ-cyclodextrin. Glucose units are named A, B, C etc. respectively and carbon atoms in each unit are numbered as usual
As it was already shown in the previous section, substituents present on a cyclodextrin skeleton play a key role in its properties. For example cyclodextrins with hydrophobic chains could form micelles or vesicles in water environment (Sallas and Darcy 2008), other cyclodextrins are capable of catalysing decomposition of organophosphorus compounds (Letort et al. 2016), and positively charged cyclodextrin derivatives are used in chiral separations in capillary electrophoresis as they are able to interact with carboxylic acids (Tang and Ng 2008a).
The dependence of solubility on substituents could be demonstrated in the following example. Only 1.85 g of β-cyclodextrin is soluble in 100 ml water at room temperature (Szejtli 1998), while almost 24 g of per-O-methyl-β-cyclodextrin could be dissolved in the same amount of water (Szente and Szejtli 1999). Moreover, when heated, the solubility of the β-cyclodextrin increases, while in the case of the latter its solubility decreases. Permethylated cyclodextrins are also well soluble in most organic solvents.
Cyclodextrin derivatives toxicity depends on the substituents present as well. The vast majority of toxicity tests are performed only on cyclodextrins randomly substituted, persubstituted or persubstituted at 2-, 3-, or 6-positions – as they are of interest of the pharmaceutical industry. Cytotoxic and hemolytic properties of these derivatives were compared by Kiss et al. on Caco-2 cells. Cell toxicity of several methylated β-cyclodextrin was the highest, while ionic derivatives were observed to be less toxic. The authors concluded cholesterol-solubilising properties could be a predictive factor for cyclodextrin derivatives cell toxicity (Kiss et al. 2010).
Randomly substituted sulfobutyl-β-cyclodextrin sodium salt with a degree of substitution 6.0–7.1 is well tolerated in male volunteers when administered intravenously at doses up to 200 mg/kg. Only a mild toxicity in the kidneys and liver was observed in rats at the dose of 3 g/kg, which is approximately 50-fold greater than the dose usually administered to men (Luke et al. 2010).
Randomly substituted 2-hydroxypropyl-β-cyclodextrin is well tolerated in rats, mice and dogs, particularly when administered orally. This cyclodextrin derivative is also well tolerated in humans, with the main adverse effect being diarrhoea. Moreover, intraperitoneal single dose of 10 g/kg in mice was neither lethal nor produced any toxicity (Gould and Scott 2005).
When speaking about randomly substituted derivatives, it should be taken into account there could be differences between derivatives with different degree of substitution (Li et al. 2016). Three randomly substituted 2-hydroxypropyl-β-cyclodextrin with degree of substitution 4.55, 6.16 and 7.76 were compared when administered intravenously to rats once daily at a dose of 500 mg/kg for 7 days. It was concluded cyclodextrin derivative with lower degree of substitution resulted in more changes in hematological and biochemical parameters. However, the effects were reversible at the end of recovery.
2.2 Cyclodextrin Derivatives
When native cyclodextrins are not suitable for a given application, their derivatives come into play. What type of a reaction should be used depends on the purpose of the product. For example, if a more soluble cyclodextrin derivative is required for the pharmaceutical application, then a random conversion of hydroxyl groups e.g. to sulphate (or another hydrophilic) group is carried out. However, such a product is a mixture of single compounds – usually characterised by degree of substitution. Moreover, the mixture differs in the detailed representation of individual isomers from batch to batch, even when degree of substitution remains constant (Estrada III and Vigh 2012).
When a single isomer cyclodextrin derivative is desired, the most straightforward way is to synthesise a persubstituted derivative. The direct synthesis of other single isomer derivatives is more challenging due to the number of theoretically possible isomers (Wenz 1994). The number of possible isomers is three for monosubstituted, dozens for disubstituted, and more than one hundred for trisubstituted derivatives.
The number of known cyclodextrin derivatives is huge. A search in SciFinder® for any substituted cyclodextrin skeleton revealed there are more than 2000 derivatives for α-cyclodextrin, almost 8000 for β-cyclodextrin, and more than 1000 for γ-cyclodextrin. Large ring cyclodextrins, i.e. having more than 8 glucose units (Endo 2011) or pre-α-cyclodextrin (Nakagawa et al. 1994) derivatives are not covered herein as their use is scarce. However, the reactivity of these cyclodextrins is expected to be similar to the three basic ones, with the exception of reactions where the reagent interferes with the cavity – see below. The large ring cyclodextrins’ cavity is more flexible as well as more spacious, while pre-α-cyclodextrin has the smallest one.
Listing of more than 11,000 α-, β-, and γ-cyclodextrin derivatives would exceed the possibilities of this chapter. Thus the aim of this chapter is to provide a comprehensive view on the synthesis of favourite or interesting cyclodextrin derivatives, especially on those which are suitable for further modifications. The reason for this approach is obvious – the modification of a cyclodextrin already substituted with one or more suitable functional groups is much easier than the optimisation of substitution for every new cyclodextrin derivative desired. The chapter is divided into several sections, each focused on one type of cyclodextrin derivatives: persubstituted cyclodextrins, randomly substituted cyclodextrins, cyclodextrins persubstituted at selected positions, selectively substituted cyclodextrins, and monosubstituted cyclodextrins (Fig. 2.2). A mini-summary could be found at the end of these sections as a help for busy readers.

Schematic representation of the cyclodextrin derivatives types
For the purpose of this review, the term monosubstituted cyclodextrins or selectively substituted cyclodextrins also refers to corresponding cyclodextrin derivatives with all the remaining hydroxyl groups protected – methylated, acetylated, benzylated etc.
Preparation of other cyclodextrin derivatives not described herein could be found in the previous reviews (Table 2.1).
2.2.1 Reactivity of Cyclodextrins
The modification reactions on cyclodextrins take place at the hydroxyl groups. As the hydroxyl groups are nucleophiles, the reaction proceeds via an electrophilic attack. However, a selective substitution of cyclodextrins is a great challenge for chemists as there are three types of hydroxyl groups present in one glucose unit (at position 2, 3 or 6). Moreover, several glucose units of which cyclodextrins are composed make the process rather difficult. Hydroxyl groups at positions 2, 3 and 6 compete against each other during the reaction. Fortunately, there are at least some differences among them. Hydroxyl groups at positions 6 are primary and at positions 2 and 3 are secondary. Hydroxyl groups at positions 6 are the least acidic and the most accessible, at positions 2 are the most acidic, and at positions 3 are the least accessible (Khan et al. 1998).
When all the hydroxyl groups are deprotonated with an excess of a base, an electrophilic reagent reacts at position 6, because it is the most accessible. The more reactive agents attack the hydroxyl groups at position 6 less selectively, and therefore they react also with the hydroxyl groups on the secondary rim. The less reactive reagents react at position 6 selectively. The best known examples of this feature are syntheses of 6A–O-tosyl-cyclodextrin where multisubstituted derivatives are the only by-products (Řezanka 2016).
Bases first deprotonate the hydroxyl groups at position 2, because they are the most acidic, having pKa = 12.2 (Sallas and Darcy 2008). The oxyanion formed is more nucleophilic than other non-deprotonated hydroxyl groups. Thus, the use of a corresponding amount of a strong base, e.g. NaH or LiH, often leads to the substitution at position 2 predominantly (Řezanka 2016).
Substitution at position 3 is the most difficult one for the abovementioned reasons. Fortunately, some reagents interfere with the cavity of cyclodextrins, making this process much easier. For example, cinnamyl bromide forms a complex with β-cyclodextrin and allows the substitution to be selective at position 3 with multisubstituted derivatives as the only by-products (Jindřich and Tišlerová 2005). On the other hand, such interference of a reagent with the cavity could be a complication in other modifications of cyclodextrins and should be always taken into account.
Solvents play another important role during the modification of a cyclodextrin. They can affect both nucleophilicity of oxyanions, as well as the strength of a complex with a substitution agent. If the complex is strong, the predominant product will be driven by the orientation of the substitution agent in the interior of cyclodextrin. Considering the abovementioned facts, the commonly achieved yields of substituted cyclodextrin derivatives are very low. Exceptions to this rule will be of interest in the next sections.
2.2.2 Persubstituted Cyclodextrin Derivatives
A great variety of persubstituted cyclodextrins, i. e. cyclodextrin derivatives, where every hydroxyl group is substituted by the same functional group, are available from common commercial sources. However, they are still synthesised by researches because of their simple synthesis and lower overall cost when compared to the commercial ones. The modification usually aims to increase the solubility of cyclodextrins – either in organic solvents or water – or to use them in deprotection reactions – see below.
As it was stated above, the persubstitutions proceed smoothly and they are usually carried out by the reaction of a cyclodextrin with an excess of the reagent, e.g. alkyl halogenide, in the presence of a base. Per-O-methylated cyclodextrins are obtained by the reaction of a corresponding cyclodextrin with NaH and methyl iodide in N,N′-dimethylformamide (Nakazono et al. 2010; Stefanache et al. 2014) or dimethyl sulfoxide (Szejtli et al. 1980). Similarly, per-O-benzylated cyclodextrins are prepared by the reaction of a cyclodextrin with benzyl halogenide (Bjerre et al. 2007; Normand et al. 2012); per-O-trimethylsilylated cyclodextrins with trimethylsilyl chloride (Kurochkina et al. 2014) or imidazole (Harabagiu et al. 2004) and per-O-(2-hydroxypropyl) cyclodextrins with propylene carbonate (Trotta et al. 2002).
Per-O-acetylated cyclodextrins are usually prepared by the reaction of a cyclodextrin with acetanhydride instead of acetyl halogenide – as the latter one is more expensive and less easy to handle. The reaction is promoted by acids (Zhang et al. 2011; Jicsinszky et al. 2015) or bases (Ghanem and Schurig 2001; Lian et al. 2014).
When a reagent for cyclodextrin persubstitution is chosen wisely, it allows a subsequent modification, which leads to other derivatives as it is illustrated by the example from Kraus et al. (Scheme 2.1) (Kraus et al. 2001). Firstly, per-O-allyl-β-cyclodextrin is prepared. The subsequent oxidation steps led to an unstable formylmethyl derivative and it was thus directly transformed into a hydroxyethyl derivative. Finally, the oxidation and methylation reactions were carried out to obtain per-O-methoxycarbonylmethyl-β-cyclodextrin.

Synthesis of persubstituted cyclodextrin derivatives
Mini-Summary
Persubstituted cyclodextrin derivatives are prepared by the reaction of native cyclodextrin with an excess of the reagent.
2.2.3 Randomly Substituted Cyclodextrin Derivatives
Randomly substituted cyclodextrin derivatives are modified at various positions and they are usually characterised by degree of substitution. The exact structure and ratio of single derivatives forming the mixture of randomly substituted cyclodextrins is unknown. As well as in the case of persubstituted derivatives, randomly substituted cyclodextrin derivatives are largely available from common commercial sources and their synthesis is thus often claimed in patents (Wimmer 1995; Yanli 2005).
The purchase of randomly substituted cyclodextrin derivatives conceals many pitfalls. It was shown there could be a significant difference in the relative abundances of the isomers with the same degree of substitution between two batches (Estrada III and Vigh 2012). Moreover, the authors also found the information about degree of substitution from the supplier may be affected by an error.
Researches should be careful when preparing randomly substituted cyclodextrin derivatives as they do not know the exact composition of the single isomers in the product. As it has been shown before (Řezanka et al. 2016), diverse single cyclodextrin isomers have different properties and their ratio could therefore affect the properties of randomly substituted cyclodextrin derivatives. They could be synthesised using conditions similar to preparation of persubstituted derivatives. The only need is to add a lesser amount of a reagent that is needed for a fully substituted derivative. Syntheses of the most common randomly substituted cyclodextrin derivatives are summarised in Table 2.2.
2.2.3.1 Random Cyclodextrin Polymers
The randomly substituted cyclodextrin derivatives also include a group of random cyclodextrin polymers, where cyclodextrins are interconnected to the other ones in a random way. Such polymers could be synthesised via three approaches: i) cyclodextrins or their derivatives are directly cross-linked by a suitable agent; ii) cyclodextrins are first randomly modified with reactive groups, which are subsequently used for the attachment onto a polymer backbone; iii) cyclodextrin is substituted with a functional group that is available for polymerisation, e.g. double bond. The first approach often gives branched polymers, whereas the second and the third result in linear polymers substituted with cyclodextrins. Prepared random cyclodextrin polymers could have various properties depending on the amount and nature of their cross-linking agent or the type of connection and the polymer used. However in the vast majority of cases, the cyclodextrin polymers retain their key property – the ability to include guests in their cavity.
The best known example of the first method is the direct reaction of epichlorohydrin – 2-(chloromethyl)oxirane – with a cyclodextrin (van de Manakker et al. 2009; Morin-Crini and Crini 2013; Concheiro and Alvarez-Lorenzo 2013; Gidwani and Vyas 2014). If the degree of cross-linking is sufficiently high, the resulting polymer becomes insoluble in water. Epichlorohydrin could be used for cross-linking of randomly substituted cyclodextrin derivatives as well (Zhang et al. 2012).
Epichlorohydrin-cyclodextrin polymers are used in various applications (Morin-Crini and Crini 2013; Folch-Cano et al. 2014), for example in lithium rechargeable batteries, where cyclodextrin cross-linked hyperbranched network structure covers the electrode, which results in a great improvement in both capacity and cycle life (Jeong et al. 2014).
Another example of the first approach is the use of N,N-carbonyldiimidazole as a reagent allowing cyclodextrin to be reacted with a cross-linker. For example, N,N- carbonyldiimidazole was first reacted with β-cyclodextrin forming a reactive derivative, which was subsequently used for the cross-linking with tris(2-aminoethyl)amine (Scheme 2.2) (Wang et al. 2013).

Cross-linking of β-cyclodextrin using N,N-carbonyldiimidazole and tris(2-aminoethyl)amine
Recently a review on cyclodextrin-polyurethane polymers for the removal of pollutants from waste water has been issued (Taka et al. 2017). Commonly used cross-linking agents for the synthesis of random cyclodextrin polymers are summarised in Fig. 2.3 (Mocanu et al. 2001; Concheiro and Alvarez-Lorenzo 2013; Karoyo and Wilson 2015).

Commonly used cross-linking agents for preparation of random cyclodextrin polymers
Direct cross-linking of cyclodextrin has also recently been published in Nature (Alsbaiee et al. 2016). Cross-linking of β-cyclodextrin with tetrafluoroterephthalonitrile resulted in a high-surface-area mesoporous polymer capable of adsorption of organic micropollutants. Moreover, only a mild washing procedure was required for the polymer regeneration and the polymer exhibited no loss in performance after recycling.
An example of the second approach, where cyclodextrins are first randomly modified by reactive groups and subsequently attached onto a polymer backbone, is the use of randomly carboxymethylated β-cyclodextrin for the attachment onto chitosan (Krauland and Alonso 2007; Prabaharan and Gong 2008). Opening of anhydride in poly[(methyl vinyl ether)-alt-(maleic anhydride)] by deprotonated β-cyclodextrin fits this approach as well (Renard et al. 2005).
The third approach requires the introduction of functional groups with the ability to be polymerised. This requirement is usually fulfilled by the synthesis of cyclodextrin ester of acrylic acid – e.g. by the reaction of cyclodextrin with m-nitrophenyl acrylate. Polymerisation of randomly acryloylated cyclodextrins is then initialised by potassium persulfate (Mocanu et al. 2001; Zhang et al. 2009).
Mini-Summary
Randomly substituted cyclodextrin derivatives are prepared by the reaction of native cyclodextrins with fewer equivalents of the reagent than the number of hydroxyl groups is. Random cyclodextrin polymers are most often synthesised by cross-linking with epichlorohydrin.
2.2.4 Cyclodextrins Persubstituted at Selected Positions
Cyclodextrins persubstituted at selected positions include cyclodextrins persubstituted either at 2 or 3 or 6 positions or any combination of thereof (Fig. 2.2). Synthesis of these derivatives is based on the different reactivity of hydroxyl groups described above and often employs the protection/deprotection methodology to achieve a desired derivative. The most favourite reactions are those carried out at position 6, because it possesses primary hydroxyl groups. Cyclodextrins could be easily substituted at this position with tert-butyldimethylsilyl or halogens. Such derivatives are useful precursors for amphiphilic cyclodextrin derivatives (Sallas and Darcy 2008). Otherwise very popular p-toluenesulfonyl, also so-called “tosyl”, group is not employed very often as it could spontaneously undergo the reaction to 3,6-anhydro form (Khan et al. 1998).
2.2.4.1 Syntheses Based on per-6-O-(tert-Butyldimethylsilyl) Cyclodextrins
Synthesis of per-6-O-(tert-butyldimethylsilyl)-α-, β- and γ-cyclodextrins is carried out by the reaction of native cyclodextrin with tert-butyldimethylsilyl chloride and BaO (Takeo et al. 1988; Takeo et al. 1989), pyridine (Fugedi 1989; Ashton et al. 1996) or imidazole (Vincent et al. 1997; Maynard and Vigh 2000) (Scheme 2.3). Among the bases, pyridine gave the best yields (Ashton et al. 1996).

Synthesis of per-6-O-(tert-butyldimethylsilyl)-α-, β- and γ-cyclodextrins
Hexakis(6-O-tert-butyldimethylsilyl)-α-cyclodextrin could be protected at positions 2 and 3 by acetyl, methyl or benzyl groups. Subsequent deprotection of silyl groups by BF3 with tetrahydrofurane, sodium methanolate or tetrabutylammonium fluoride results in useful derivatives, where hydroxyl groups at positions 6 are ready for any modification desired (Scheme 2.4) (Takeo et al. 1988). The reactions proceed similarly with β- (Jullien et al. 1994; Ashton et al. 1996; Vincent et al. 1997; Kirschner and Green 2005) or γ-cyclodextrin derivatives (Jullien et al. 1994). TBDMS group in heptakis(2,3-O-dimethyl-6-O-tert-butyldimethylsilyl)-β-cyclodextrin was also transformed by triphenylphosphine and bromine to heptakis(2,3-O-dimethyl-6-O-bromo)-β-cyclodextrin (Ashton et al. 1996).

Synthesis of α-cyclodextrins protected at positions 2 and 3
Heptakis(6-O-tert-butyldimethylsilyl)-β-cyclodextrin was also used for the synthesis of carboxymethyl β-cyclodextrin derivatives (Scheme 2.5). Firstly, positions 2 and 3 were allylated and the silyl group deprotected by tetrabutylammonium fluoride (Baer et al. 1992b). Subsequently, hydroxyl groups at position 6 were methylated and allyl groups oxidised in a similar way as described above (Kraus et al. 2001).

Synthesis of carboxymethyl β-cyclodextrin derivatives
The β-cyclodextrin derivatives similar to those depicted in Scheme 2.4 were also used for syntheses of other useful precursors (Scheme 2.6 and 2.7). Heptakis(6-O- tert-butyldimethylsilyl)-β-cyclodextrin was tosylated and the subsequent desilylation yielded heptakis(2-O-tosyl)-β-cyclodextrin (Coleman et al. 1991). Although this derivative could also be obtained by the direct substitution reaction of β-cyclodextrin with tosylimidazole in the presence of Cs2CO3, the yield is only 5% (Yu et al. 2006). It is noteworthy the subsequent substitution of tosyl group leads (according to Walden inversion) to non-cyclodextrin derivatives with aldohexoses in manno configuration.

Synthesis of useful β-cyclodextrin precursors

Synthesis of β-cyclodextrins persubstituted at position 6
β-cyclodextrins protected at positions 2 and 3 with methyl or acetyl group could undergo oxidation by (2,2,6,6-tetramethylpiperidin-1-yl)oxyl forming 5-carboxy-5- dehydroxymethyl derivatives (Scheme 2.7). Moreover, the acetylated derivative enables deprotection at positions 2 and 3 (Kraus et al. 2000). Uccello-Barretta et al. also synthesised other derivatives persubstituted at position 6 by common reactions (Scheme 2.7) (Uccello-Barretta et al. 2005).
Vigh and his colleagues devoted a great effort to synthesis of cyclodextrin derivatives persubstituted at selected positions (Vincent et al. 1997; Maynard and Vigh 2000; Busby and Vigh 2005; Tutu and Vigh 2011). Their methodology is based on modifications of per-6-O-(tert-butyldimethylsilyl)-α-, β- and γ-cyclodextrins (Scheme 2.8, 2.9 and 2.10). Synthesis of heptakis(2-O-methyl)-β-cyclodextrin includes a crucial step that involves the migration of silyl groups to position 3 and the simultaneous methylation of the hydroxyl group at position 2 (Scheme 2.8). The migration mechanism was suggested on the basis of NMR spectroscopy and subsequently confirmed by X-ray crystallography (Maynard and Vigh 2000).

Synthesis of heptakis(2-O-methyl)-β-cyclodextrin

Synthesis of heptakis(2-O-methyl-3-O-acetyl)-β-cyclodextrin. Yields are not stated

Synthesis of heptakis(3-O-methyl-6-O-acetyl)-β-cyclodextrin. Yields are not stated
The migration also allows synthesis of heptakis(2-O-methyl-3-O-acetyl)-β- cyclodextrin, where substituents at every position are different from each other (Scheme 2.9). However, in this case, protection and migration of triethylsilyl instead of tert-butyldimethylsilyl groups is taken into advantage. Triethylsilyl is subsequently selectively hydrolysed by imidazole hydrochloride and free hydroxyl groups at position 3 are acetylated. The final product is obtained by the removal of tert-butyldimethylsilyl groups (Busby and Vigh 2005).
Finally, Tutu and Vigh synthesised heptakis(3-O-methyl-6-O-acetyl)-β-cyclodextrin (Scheme 2.10). The product was prepared by the standard protection/deprotection methodology with the selective benzylation of per-6-O-(tert-butyldimethylsilyl)-β-cyclodextrin at position 2 as the key step (Tutu and Vigh 2011).
2.2.4.2 Syntheses Based on per-6-Halogeno-per-6-Deoxy Cyclodextrins
Another group of favourite starting materials for synthesis of cyclodextrins persubstituted at selected positions are cyclodextrins perhalogenated at position 6. They are synthesised from native cyclodextrins by the reaction with triphenylphosphine and bromine (Takeo et al. 1974) or iodine (Gadelle and Defaye 1991; Fernandez et al. 1995; Ashton et al. 1996; Benkhaled et al. 2008) in N,N′-dimethylformamide (Scheme 2.11). However, the synthesis of bromo derivatives has been declining due to more convenient handling of iodine.

Synthesis of per-6-halogeno-per-6-deoxy-α-, β- and γ-cyclodextrins
These halogen derivatives are very useful precursors and could be easily transformed for example into azides and amines (Ashton et al. 1996; Gorin et al. 1996) or thiols (Rojas et al. 1995; Gorin et al. 1996) by standard procedures (Scheme 2.12). The remaining hydroxyl groups of these derivatives could be peracetylated (Boger et al. 1978; Baer et al. 1992a), methylated (Boger et al. 1978) or benzylated (Jullien et al. 1994) by the same methods described above.

Synthesis of azido, amino and thio derivatives of β-cyclodextrin
Jicsinszky et al. have recently described the use of per-6-iodo-per-6-deoxy-β- and γ-cyclodextrins for the synthesis of azido or thio derivatives in a planetary ball mill under solvent-free conditions. The authors found out the mechanochemical synthesis not only simplified the isolation and purification processes, but also allowed easy scale-up (Jicsinszky et al. 2016a).
Per-6-azido-per-6-deoxy cyclodextrins are the perfect starting materials for nowadays favourite copper-catalysed azide-alkyne cycloaddition reactions (Faugeras et al. 2012; Letort et al. 2016) or, as described by Kraus et al., for modifications at positions 2 and 3 (Kraus et al. 2002) (Scheme 2.13). The key synthetic step is the use of BaO/Ba(OH)2 for the introduction allyl groups at positon 2. The property of this base is not unique for azido derivatives and it is used as well for other syntheses, where the introduction of a substituent at position 2 is needed – see the beginning of the next section.

Modifications of hexakis(6-azido-6-deoxy)-α-cyclodextrin
2.2.4.3 Other Syntheses
As it was described above, BaO/Ba(OH)2 was successfully applied for the introduction of a substituent at position 2. Moreover, when the conditions are applied to a native cyclodextrin, the reaction proceeds to per-2,6-dialkyl cyclodextrin derivatives. This can be used e. g. in the synthesis of heptakis(2,6-O-dimethyl)-β-cyclodextrin (Szejtli et al. 1980) or heptakis(2,6-O-diallyl)-β-cyclodextrin (Bergeron et al. 1976) (Scheme 2.14). Allyl groups in heptakis(2-O-allyl-3-O-methyl-6-O-allyl)-β-cyclodextrin could be oxidised similarly to Scheme 2.5 to carboxymethyl derivatives (Kraus et al. 2001).

Synthesis of heptakis(2,6-O-dialkyl) β-cyclodextrin derivatives
Boger et al. used the selective deprotection strategy as the key step for syntheses of various cyclodextrin derivatives (Boger et al. 1978). Firstly, per-O-benzoyl-α-cyclodextrin is prepared. Secondly, the deprotection step by potassium isopropylalcoholate is carried out. The base is selective due to its steric hindrance and thus is able to deprotect only the primary hydroxyl groups (Scheme 2.15). The resulting product was transformed to various 6-azido or 6-amino derivatives.

Selective deprotection of per-O-benzoyl-α-cyclodextrin
The selective deprotection strategy was also used for acetolysis of perbenzylated α-cyclodextrin (Angibeaud and Utille 1991). The reaction yielded hexakis(2,3-O-dibenzyl-6-O-acetyl)-α-cyclodextrin, in which either acetyl or benzyl groups could be selectively deprotected (Scheme 2.16). Moreover, perbenzylated α-cyclodextrin was also used for deprotection by triethylsilane and iodine (Guitet et al. 2012). The method was originally developed for debenzylation of multiple-O-benzylated mono- and disaccharides (Pastore et al. 2011) and in the case of α-cyclodextrin proceeds at position 3, which is normally the least accessible (Scheme 2.16).

Selective deprotection of perbenzylated α-cyclodextrin
Mini-Summary
Synthesis of cyclodextrins persubstituted at selected positions utilises the different reactivity of hydroxyl groups and employs protection/deprotection methodology to achieve a desired derivative. Most of the syntheses begin with the substitution at position 6 – either by tert-butyldimethylsilyl or a halogen. BaO/Ba(OH)2 direct the substitution to positions 2 and 6. When position 6 is blocked, the substitution proceeds selectively at position 2. The selective substitution at otherwise the least accessible position 3 could be achieved by deprotection of perbenzylated cyclodextrin by triethylsilane with iodine.
2.2.5 Selectively Substituted Cyclodextrins
Selectively substituted cyclodextrins fill the gap between all the above discussed cyclodextrin derivatives and monosubstituted cyclodextrins, i.e., they are single isomer compounds with a known structure, where two or more substituents are attached to the cyclodextrin skeleton. Their synthesis is the most challenging among all cyclodextrin derivatives, as the number of possible isomers starts at dozens and ends at millions for different substituents at different positions. For the purposes of this chapter, the term of selectively substituted cyclodextrins also refers to the derivatives that contain several free hydroxyl groups with the rest being protected by e. g. methyl, acetyl, benzyl or other groups.
2.2.5.1 Syntheses Based on Direct Substitution
One of the first attempts to synthesise selectively substituted cyclodextrins was the use of disulfonates for the selective modification of selected glucose units at position 6 (Scheme 2.17). 6A,6B derivative is formed when β-cyclodextrin is reacted with 4,6-dimethoxybenzene-1,3-disulfonyl chloride (Breslow et al. 1990). The use of benzophenone-3,3′-disulfonyl chloride led to 6A,6C derivative, while trans-stilbene- 4,4′-disulfonyl chloride led to 6A,6D derivative (Tabushi et al. 1981). The disulfonates could be transformed into diiodo (Breslow et al. 1990), diazido, diamino (Tabushi et al. 1977; Di Blasio et al. 1996) or dithio derivatives (Tabushi et al. 1977) by common reactions.

Regioselective synthesis of disulfonate cyclodextrin derivatives
Similarly, 2A,2B disulfonates were synthesised reacting benzophenone-3,3′-disulfonyl imidazole with α- or β-cyclodextrin in 30% yields (Teranishi 2000). However, a further substitution of these derivatives leads to non-glucose cyclodextrin derivatives due to the stereogenic centre inversion. This problem could be avoided using non-sulfonate derivatives.
α,α-Dimethoxytoluene is able to protect two hydroxyl groups on a cyclodextrin skeleton, but the reaction is non-selective. Heptakis(6-O-pivaloyl)-β-cyclodextrin was therefore prepared to overcome this disadvantage (Scheme 2.18). Pivaloyl derivative is, unlike a native cyclodextrin, capable of furnishing 2A,3B derivative in a good yield (Sakairi and Kuzuhara 1993). The subsequent benzylation of the remaining hydroxyl groups together with the exchange of pivaloyl residues to benzyl groups and deprotection of benzylidene led to per-O-benzyl-2A,3B–dihydroxy-β-cyclodextrin. This approach was also applied for α- and γ-cyclodextrin as well (Sakairi et al. 1996b). Such derivatives are very useful precursors for further syntheses and are similar to the permethylated ones discussed below.

Regioselective protection by α,α-dimethoxytoluene
It is also possible to disubstitute just one glucose unit to obtain 2A,3A derivative. Such regioselectivity was achieved for the reaction of β-cyclodextrin with α,α’-dibromo-o-xylene (Balbuena et al. 2007). The reaction proceeds with lithium diisopropylamide as a base in a 30% yield (Scheme 2.19). The subsequent methylation of all remaining hydroxyl groups together with hydrogenolysis of the xylene group gave per-O-methyl-2A,3A–dihydroxy-β-cyclodextrin. This approach was also successfully applied on α- and γ-cyclodextrin (Balbuena et al. 2013).

Synthesis of per-O-methyl-2A,3A–dihydroxy-β-cyclodextrin
Similarly, methallyl dichloride could be used instead of α,α’-dibromo-o-xylene (Fenger et al. 2011). The reaction proceeds on α- and β-cyclodextrin and in similar yields.
When a trisubstituted cyclodextrin derivative is needed, the reaction of α-cyclodextrin with triphenylmethyl chloride, so-called “trityl chloride”, comes into play. The reaction yields symmetrically trisubstituted cyclodextrins due to sterical hindrance (Scheme 2.20). Protection of all the remaining hydroxyl groups by methyl iodide and hydrolysis of trityl groups proceeded to a useful trisubstituted precursor – per-O-methyl-6A,6C,6E–trihydroxy-α-cyclodextrin (Boger et al. 1979). The quantitative analysis of α-cyclodextrin tritylation was studied later by ultra-fast liquid chromatography (Yoshikiyo et al. 2015).

Synthesis of symmetrically trisubstituted α-cyclodextrin
2.2.5.2 Syntheses Based on Selective Debenzylation
Protection/deprotection strategies are vastly used for syntheses of selectively substituted cyclodextrins, where sterical properties of a reagent together with cyclodextrin reactivity play the key role. Selective deprotection of perbenzylated cyclodextrins by diisobutylaluminium hydride represents the most favourite method for synthesis of selectively substituted cyclodextrins, as well as monosubstituted cyclodextrins – see below. It was first described by Pearce and Sinaÿ (Pearce and Sinaÿ 2000). The method involves a selective deprotection of two opposite benzyl groups in high yields and is applicable to α-, β-, and γ-cyclodextrin (Scheme 2.21). Benzyl groups are removed from glucose units A and D at position 6, but in the case of γ-cyclodextrin also from glucose units A and E. Free hydroxyl groups could be subsequently used for organic chemistry transformations leading to the desired cyclodextrin derivative (Petrillo et al. 2009; Volkov et al. 2015).

Selective bis-O-debenzylation
It has been proposed the reaction involves at least two molecules of diisobutylaluminium hydride and the mechanism occurs by a stepwise process (Sollogoub 2013). This was proved by the synthesis of per-O-benzyl-6A-hydroxy derivatives, see below. This derivative could be used for the synthesis of per-O- benzyl-6A-deoxy-6A-azido derivatives and for the second debenzylation (Scheme 2.22). Azido groups provide steric hindrance to direct the second deprotection step towards the opposed glucose unit (Guieu and Sollogoub 2008a) allowing to introduce two different functional groups.

Azide promoted debenzylation
Such step by step debenzylation and substitution reactions could lead even to far more complicated structures – cyclodextrins trisubstituted (Guieu and Sollogoub 2008b; Rawal et al. 2010), tetrasubstituted (Rawal et al. 2010; Sollogoub 2013), pentasubstituted (Guieu and Sollogoub 2008b) or heaxasubstituted at position 6, even with all the substituents different from each other (Wang et al. 2014). Moreover, together with Et3SiH/I2 hexakis-O-debenzylation at position 3 (Guitet et al. 2012) (see above) they allow a simultaneous selective deprotection on both the primary and secondary rim.
2.2.5.3 Syntheses Based on Other Selective Deprotections
Shortly after the selective bis-O-debenzylation was discovered, another selective double diisobutylaluminium hydride deprotection from Sinaÿ saw the light of day (du Roizel et al. 2002). It was found out both per-O-methyl α- and β-cyclodextrin were able to undergo a regioselective bis-O-demethylation, but on the secondary rim (Scheme 2.23). This opened the way for a direct access to 2A,3B derivatives (Letort et al. 2015).

Selective bis-O-demethylation
As it was shown by Xiao et al. the reaction requires 2A methoxy group and an oxygen atom present at position 3B (Xiao et al. 2013) to proceed. The authors also showed it was possible to carry out two or even three bis-O-demethylations on permethylated α- or β-cyclodextrins (Scheme 2.24). However, in contrast to the multiple bis-O-debenzylations described above, these bis-O-demethylations are carried out in one step.

Multiple bis-O-demethylations
The last from frequently used deprotections is selective bis-O-de(tert-butyldimethyl)silylation (Ghosh et al. 2012). It proceeds at 6A,6D positions in 70% yields and is carried out on hexakis(2,3-O-dimethyl-6-O-tert-butyldimethylsilyl)-α-cyclodextrin or heptakis(2,3-O-dimethyl-6-O-tert-butyldimethylsilyl)-β-cyclodextrin (Scheme 2.25). The desilylation proceeds in even better yields on substrates where secondary hydroxyl groups are protected by benzyl instead of methyl groups. The methylated precursors were used in further transformations (Ghosh et al. 2012; Tatar et al. 2017).

Selective bis-O-tert-butyldimethylsilylation
Mini-Summary
The synthesis of selectively substituted cyclodextrins is quite a challenge due to the high number of theoretically possible isomers. However, selective bis-O-debenzylation (forming 6A,6D derivatives) and bis-O-demethylation (forming 2A,3B derivatives) make this process much easier. Such an approach allows to satisfy a sweet tooth of almost the majority of chemists. The deprotections could be used multiple times or combined with the other methods. Moreover, when chemists are still picky, selective 6A,6B-, 6A,6C-, 6A,6D–sulfonations, 2A,3A–xylylation or 6A,6C,6E–tritylation could be offered.
2.2.6 Monosubstituted Cyclodextrins
Monosubstituted cyclodextrins could be either substituted at positions 2, 3 or 6. Nowadays, there are two main methods for their synthesis: direct and indirect (Řezanka 2016). When using the first method, a cyclodextrin is directly reacted with a substitution agent. A desired monosubstituted product is then purified from other isomers, unreacted cyclodextrins and multiple-substituted by-products. The location of a substituent on a glucose unit is driven by the cyclodextrin reactivity, which is described in Sect. 2.1. Yields usually strongly depend on solvent, base, type of cyclodextrin, and structure of the substitution agent and vary from units of percent up to a few dozens of percent. This dependency has been studied by several authors (Masurier et al. 2006; Martina et al. 2010; Řezanka 2016).
When reproducing synthesis according to the described procedure, please have in mind not all the authors use satisfactory purification steps and characterise the product sufficiently. Precipitation of a reaction mixture (e.g. by acetone) as the only purification method is inadequate. Without a proof of purity such as chromatogram from high-performance liquid chromatography, copy of 1H nuclear magnetic resonance spectra etc. it is expected the product contains several impurities – e.g. unreacted cyclodextrin and multiple-substituted cyclodextrin derivatives.
The second – indirect – method is based on a high-yielding deprotection step of a persubstituted cyclodextrin, such as permethylated, perbenzylated etc. Persubstituted cyclodextrins are easily obtained in high yields and thus the selective deprotection furnishes a monodeprotected derivative, where the free hydroxyl group is ready for further transformations. This method is more universal than the first one, as the substitution of the free hydroxyl group usually results in high yields regardless the nature of the substitution agent used.
There are also other methods available for cyclodextrin monosubstitution. It is possible to synthesise a monosubstituted derivative de novo. However, the synthesis requires many steps and the overall yield is very low (Sakairi et al. 1991; Sakairi et al. 1995; Chaise et al. 2008). An interesting approach of monosubstituted cyclodextrin derivative synthesis is the use of proteases, which are able to direct the substituents regioselectively (Xiao et al. 2004; Wang et al. 2005). Unfortunately, this method has not been examined in detail yet. The last from the methods for monosubstituted cyclodextrin derivative synthesis is the use of several protection/deprotection steps to achieve a desired product. However, this method requires a lot of steps and its use is now being superseded with direct and indirect strategies.
The syntheses of monosubstituted allyl, cinnamyl, propargyl, formylmethyl, carboxymethyl, azido and amino cyclodextrin derivatives have recently been studied in detail (Řezanka 2016). The following sections thus summarise the main findings and add information about other derivatives, which could be used as precursors for further synthesis.
2.2.6.1 Monosubstitution at Position 2
The direct method takes advantage of the above mentioned fact the hydroxyl group at position 2 is the most acidic. The use of a strong base thus mostly leads to substitution predominantly at this position and the yields reach up to 40%. However, sometimes the desired 2A-O substituted derivative is hard to separate from its isomer and the purification step is done after peracetylation (Scheme 2.26) (Řezanka and Jindřich 2011). Peracetylation has also other advantages such as easy distinguishing between 2, 3 and 6 isomers directly from 1H nuclear magnetic resonance spectrum (Řezanka 2016) and protecting the rest of hydroxyl groups against side reactions, for example, when oxidation of double bond is needed. Peracetylated derivatives could be easily deprotected by Zemplén deacetylation (Řezanka et al. 2010).

Synthesis of allyl derivatives
The best results for alkylation are generally achieved in dimethyl sulfoxide with a strong base like LiH together with a catalytic amount of LiI (Hanessian et al. 1995; Řezanka and Jindřich 2011); sodium ethoxide (Masurier et al. 2006, 2009); lithium diisopropylamide (Masurier et al. 2006); or NaH (Kalakuntla et al. 2013).
It is also possible to use the indirect method for the synthesis of cyclodextrin monosubstituted at position 2. As it has been mentioned above, regioselective bis-O-demethylation yields permethylated 2A,3B-dihydroxy derivatives. The diol could be selectively alkylated at position 2 and subsequently methylated on the remaining hydroxyl group yielding permethylated 2-O derivative (Scheme 2.27) (Guan et al. 2009; Řezanka et al. 2015). A great variety of permethylated α-cyclodextrin derivatives was prepared using this methodology (Xiao et al. 2013).

Synthesis of permethylated cyclodextrin derivatives monosubstituted at position 2
The other option of a selective deprotection, which yields 2-O substituted derivative, is selective debenzoylation (Sakairi et al. 1996a). The deprotection is performed on perbenzoylated β-cyclodextrin by hydrazine (Scheme 2.28). Free benzoyl group is then ready for further transformations. However, be aware of Walden inversion at this position, e.g. when substituting tosyl group in this position. The product is a non-cyclodextrin derivative with one unit in manno configuration.

Selective debenzoylation
2.2.6.2 Monosubstitution at Position 3
As it has been mentioned above, position 3 is the least accessible, which results in difficulties during the synthesis of such derivatives. However, there are several methods allowing substitution at position 3. Jindřich and Tišlerová found the alkylation of β-cyclodextrin with cinnamyl bromide result selectively in 3A-O-cinnamyl-β-cyclodextrin in a good yield (Jindřich and Tišlerová 2005). This behaviour is caused by the inclusion of cinnamyl bromide in the cavity. The resulting complex has the reactive centre of the alkylation agent oriented towards position 3. Cinnamyl group could be then oxidised after peracetylation and used for further synthesis (Scheme 2.29).

Synthesis of 3A-O-cinnamyl-β-cyclodextrin
Another possibility to introduce a substituent at position 3 selectively is the use of copper(II) sulfate in water together with NaOH as a base (Masurier et al. 2009). Alkylation using these conditions for several allylic or benzylic reagents resulted in 3-substituted derivatives in 40% yields. Other alkylation methods using NaOH in water or water/acetonitrile result only in 10% yields for cyclodextrin derivatives monosubstituted at position 3. Moreover, they have to be separated from their 2A-O and 6A-O isomers formed as by-products during the reaction (Řezanka and Jindřich 2011; Zhou et al. 2012a, 2012b; Bláhová et al. 2013).
The abovementioned regioselective bis-O-demethylation is also useful for the synthesis of permethylated cyclodextrin derivatives monosubstituted at position 3. The 2A,3B-diol could be selectively methylated at position 2 (Scheme 2.30) and the free hydroxyl group at position 3 serves as the reaction centre for further modifications (Xiao et al. 2013). Alternatively, the same derivative was obtained by selective demethylation of permethylated α-cyclodextrin by phenylthiotrimethylsilane in the presence of zinc(II) iodide (Scheme 2.30) (Chaise et al. 2004).

Synthesis of per-O-methyl-3A-hydroxy-α-cyclodextrin
2.2.6.3 Monosubstitution at Position 6
Cyclodextrins monosubstituted at position 6 represent unique precursors for the attachment to another molecule or to a surface. Connection through position 6 leaves the wider rim of a cyclodextrin open for interactions of guests with the cavity. Moreover, cyclodextrins monosubstituted at position 6 are easy to synthesise compared to the other derivatives. The reason has already been mentioned above – the hydroxyl group at position 6 is primary and the least acidic. The deprotonation of all hydroxyl groups thus predominantly leads to the substitution at this position, as there is the lowest steric hindrance. Although a lot of cyclodextrins monosubstituted at position 6 are commercially available, they are still synthesised by researchers due to their high price.
The most important cyclodextrins monosubstituted at position 6 are 6A-deoxy-6A-tosyl-α-, β- and γ-cyclodextrin. To synthesise them, cyclodextrin is reacted with tosyl chloride (Petter et al. 1990; Brown et al. 1993; Hamasaki et al. 1993; Gao et al. 1995; Liu et al. 1998; Tang and Ng 2008b; Trellenkamp and Ritter 2010; Xu et al. 2010), tosyl anhydride (Zhong et al. 1998) or 1-tosylimidazole (Tang and Ng 2007; Cao et al. 2009; Nielsen et al. 2010; Kulkarni et al. 2012). While some authors do not use purification methods, it is recommended to treat the product by chromatography (Brown et al. 1993; Hamasaki et al. 1993; Kulkarni et al. 2012) or to recrystallise it from water (Gao et al. 1995; Tang and Ng 2008b; Nielsen et al. 2010; Xu et al. 2010) or 50% MeOH in H2O (Popr et al. 2014; Bednářová et al. 2016).
Tosyl group is a useful precursor for azide, amino and thio groups (Scheme 2.31). Tosylated cyclodextrins could react with sodium azide in water (Hamasaki et al. 1993; Tang and Ng 2008b; Trellenkamp and Ritter 2010; Kulkarni et al. 2012) or N,N′-dimethylformamide (Petter et al. 1990; Nielsen et al. 2010; Xu et al. 2010). It is also possible to synthesise azide under solvent-free conditions in a ball mill (Jicsinszky et al. 2016b). The azide group could be subsequently transformed into amine by the reduction with triphenylphosphine and ammonia (Hamasaki et al. 1993; Xu et al. 2010; Kulkarni et al. 2012) or water (Tang and Ng 2008b). Both azido and amino derivatives are the favourite compounds for the synthesis of defined cyclodextrin polymers (Pun et al. 2004; Zhou and Ritter 2010).

Synthesis of cyclodextrins monosubstituted at position 6
In order to synthesise a thiol, tosyl group is treated with thiourea and subsequently with sodium hydroxide (Fujita et al. 1982; Fujita et al. 1985; Martinelli et al. 2014). Tosyl derivatives also serve as precursors for N-alkyl (Tang and Ng 2008a; Popr et al. 2014) or S-alkyl compounds (Bednářová et al. 2016).
When O-alkyl derivatives are needed, cyclodextrin could be directly alkylated in an excess of NaOH in water (Řezanka et al. 2010). It was found the method could be used for various alkylation agents on α-, β-, γ-cyclodextrins (Řezanka 2016). The yields typically reach values above 10% and no other monosubstituted isomers (2A-O and 3A-O) are formed.
The best known indirect method for the synthesis of cyclodextrin derivatives monosubstituted at position 6 is selective debenzylation of perbenzylated α-, β-, and γ-cyclodextrins (Scheme 2.32) (Pearce and Sinaÿ 2000). The reactions proceed in very good yields, unusual for monosubstituted cyclodextrins. The free hydroxyl group could be subsequently modified and benzyl groups removed by H2 on Pd (Lindbäck et al. 2012).

Selective debenzylation of cyclodextrins
The second indirect method is the abovementioned regioselective bis-O-demethylation. It provides per-O-methyl-6A-hydroxy-α- or β-cyclodextrin as by-products in 20% yield (Scheme 2.33) (du Roizel et al. 2002). It is also possible to use the protection/deprotection methodology for the synthesis of the latter one (Chen et al. 1996; Lupescu et al. 1999).

Synthesis of per-O-methyl-6A-hydroxy-α- and β-cyclodextrins
Mini-Summary
The direct syntheses of monosubstituted cyclodextrin derivatives benefit from the different reactivity of hydroxyl groups. 2A-O substituted derivatives are obtained using a strong base in dimethyl sulfoxide. 3A-O substituted derivatives are synthesised using either selective introduction of cinnamyl group or selected alkylation agents in the presence of copper(II) sulfate. Cyclodextrins monosubstituted at position 6 are the most favourite ones and 6A-deoxy-6A-tosyl-cyclodextrins overshadow all the other derivatives, as it is the most used precursor for further synthesis. Deprotection of methyl groups from permethylated cyclodextrins could lead selectively to either 2, 3, or 6 monosubstituted derivatives and debenzylation of perbenzylated cyclodextrins furnishes per-O-benzyl-6A-hydroxy-cyclodextrins selectively in high yields.
2.3 Conclusion
A lot of cyclodextrin derivatives have become available on the market over the years. However, their price is in the range of fine chemicals and thus they are still often synthesised in laboratories. Randomly substituted cyclodextrin derivatives are the only exception. Synthesis of persubstituted cyclodextrin derivatives remains more or less the same and the methods for cyclodextrins persubstituted at selected positions are now very well examined.
The synthesis of selectively substituted and monosubstituted cyclodextrin derivatives has changed much over the years. Originally used direct methods subside and the indirect methods are now on the rise. The only exceptions are 6A-deoxy-6A-tosyl-α-, β- and γ-cyclodextrins. They are the most favourite precursors for further syntheses.
The synthesis of tosyl derivatives is quick, high yielding, using cheap chemicals and the tosyl group is suitable for further reactions. Moreover, the synthesis requires only an easy purification process, i.e. recrystallisation, which is the key step. Cyclodextrin derivatives aspiring to be similarly successful should fulfil these conditions.
References
Adly FG, Antwi NY, Ghanem A (2016) Cyclodextrin-functionalized monolithic capillary columns: preparation and chiral applications. Chirality 28(2):97–109. https://doi.org/10.1002/chir.22550
Aghahosseini H, Ramazani A (2016) General overview on cyclodextrin-based artificial enzymes’ activity. Curr Org Chem 20(26):2817–2836. https://doi.org/10.2174/1385272820666160328201207
Alsbaiee A, Smith BJ, Xiao L, Ling Y, Helbling DE, Dichtel WR (2016) Rapid removal of organic micropollutants from water by a porous beta-cyclodextrin polymer. Nature 529(7585):190–194. https://doi.org/10.1038/nature16185
Angibeaud P, Utille J-P (1991) Cyclodextrin chemistry; Part I. Application of a regioselective acetolysis method for benzyl ethers. Synthesis-Stuttgart 1991(9):737–738. https://doi.org/10.1055/s-1991-26560
Antoniuk I, Amiel C (2016) Cyclodextrin-mediated hierarchical self-assembly and its potential in drug delivery applications. J Pharm Sci 105(9):2570–2588. https://doi.org/10.1016/j.xphs.2016.05.010
Ashton PR, Königer R, Stoddart JF, Alker D, Harding VD (1996) Amino acid derivatives of β-cyclodextrin. J Org Chem 61(3):903–908. https://doi.org/10.1021/jo951396d
Astray G, Gonzalez-Barreiro C, Mejuto JC, Rial-Otero R, Simal-Gandara J (2009) A review on the use of cyclodextrins in foods. Food Hydrocoll 23(7):1631–1640. https://doi.org/10.1016/j.foodhyd.2009.01.001
Baer H, Berenguel A, Shu Y, Defaye J, Gadelle A, Gonzalez F (1992a) Improved preparation of Hexakis(6-deoxy)cyclomaltohexaose and Heptakis(6-deoxy)cyclomaltoheptaose. Carbohydr Res 228(1):307–314. https://doi.org/10.1016/S0008-6215(00)90568-8
Baer H, Shen Y, Gonzalez F, Berenguel A, Garcia J (1992b) Synthesis of a cycloheptaose consisting of (1-4)-linked 7-Amino-6,7-Dideoxy-alpha-D-Gluco-Heptopyranosyl units: a new analog of cyclomaltoheptaose. Carbohydr Res 235:129–139. https://doi.org/10.1016/0008-6215(92)80083-D
Bakó P, Fenichel L, Töke L, Szente L, Szejtli J (1994) Methylation of cyclodextrins by phase-transfer catalysis. J Incl Phenom Mol Recognit Chem 18(3):307–314. https://doi.org/10.1007/BF00708737
Balbuena P, Lesur D, Alvarez MJG, Mendicuti F, Mellet CO, Fernandez JMG (2007) One-pot regioselective synthesis of 2(I),3(I)-O-(o-xylylene)-capped cyclomaltooligosaccharides: tailoring the topology and supramolecular properties of cyclodextrins. Chem Commun 2007(31):3270–3272. https://doi.org/10.1039/b705644c
Balbuena P, Goncalves-Pereira R, Jimenez Blanco JL, Isabel Garcia-Moreno M, Lesur D, Ortiz Mellet C, Garcia Fernandez JM (2013) o-Xylylene protecting group in carbohydrate chemistry: application to the regioselective protection of a single vic-diol segment in cyclodextrins. J Org Chem 78(4):1390–1403. https://doi.org/10.1021/jo302178f
Bednářová E, Hybelbauerová S, Jindřich J (2016) Optimized methods for preparation of 6(I)-(omega-sulfanyl-alkylene-sulfanyl)-beta-cyclodextrin derivatives. Beilstein J Org Chem 12:349–352. https://doi.org/10.3762/bjoc.12.38
Bellia F, Mendola DL, Pedone C, Rizzarelli E, Saviano M, Vecchio G (2009) Selectively functionalized cyclodextrins and their metal complexes. Chem Soc Rev 38(9):2756–2781. https://doi.org/10.1039/B718436K
Benkhaled A, Cheradame H, Fichet O, Teyssie D, Buchmann W, Guegan P (2008) Synthesis and characterization of amphiphilic per-(6-thio-2,3-trimethylsilyl)cyclodextrin: application to Langmuir film formation. Carbohydr Polym 73(3):482–489. https://doi.org/10.1016/j.carbpol.2007.12.031
Bergeron R, Meeley M, Machida Y (1976) Selective alkylation of cycloheptaamylose. Bioorg Chem 5(1):121–126. https://doi.org/10.1016/0045-2068(76)90018-3
Bjerre J, Fenger TH, Marinescu LG, Bols M (2007) Synthesis of some trifluoromethylated cyclodextrin derivatives and analysis of their properties as artificial glycosidases and oxidases. Eur J Org Chem 2007(4):704–710. https://doi.org/10.1002/ejoc.200600762
Bláhová M, Bednářová E, Řezanka M, Jindřich J (2013) Complete sets of monosubstituted γ-cyclodextrins as precursors for further synthesis. J Org Chem 78(2):697–701. https://doi.org/10.1021/jo301656p
Boger J, Corcoran R, Lehn J (1978) Cyclodextrin chemistry - selective modification of all primary hydroxyl-groups of alpha-cyclodextrins and beta-cyclodextrins. Helv Chim Acta 61(6):2190–2218. https://doi.org/10.1002/hlca.19780610622
Boger J, Brenner D, Knowles J (1979) Symmetrical triamino-per-O-methyl-alpha-cyclodextrin - preparation and characterization of primary trisubstituted alpha-cyclodextrins. J Am Chem Soc 101(25):7630–7631. https://doi.org/10.1021/ja00519a035
Bom A, Bradley M, Cameron K, Clark JK, van Egmond J, Feilden H, MacLean EJ, Muir AW, Palin R, Rees DC, Zhang M-Q (2002) A novel concept of reversing neuromuscular block: chemical encapsulation of rocuronium bromide by a cyclodextrin-based synthetic host. Angew Chem Int Ed 41(2):265–270. https://doi.org/10.1002/1521-3773(20020118)41:2<265::AID-ANIE265>3.0.CO;2-Q
Bonnet V, Gervaise C, Djédaïni-Pilard F, Furlan A, Sarazin C (2015) Cyclodextrin nanoassemblies: a promising tool for drug delivery. Drug Discov Today 20(9):1120–1126. https://doi.org/10.1016/j.drudis.2015.05.008
Breslow R, Canary JW, Varney M, Waddell ST, Yang D (1990) Artificial transaminases linking pyridoxamine to binding cavities: controlling the geometry. J Am Chem Soc 112(13):5212–5219. https://doi.org/10.1021/ja00169a032
Brown SE, Coates JH, Coghlan DR, Easton CJ, van Eyk SJ, Janowski W, Lepore A, Lincoln SF, Luo Y, May BL, Schiesser DS, Wang P, Williams ML (1993) Synthesis and properties of 6A-Amino-6A-deoxy-α and β-cyclodextrin. Aust J Chem 46(6):953–958. https://doi.org/10.1071/CH9930953
Busby MB, Vigh G (2005) Synthesis of heptakis(2-O-methyl-3-O-acetyl-6-O-sulfo)-cyclomaltoheptaose, a single-isomer, sulfated β-cyclodextrin carrying nonidentical substitutents at all the C2, C3, and C6 positions and its use for the capillary electrophoretic separation of enantiomers in acidic aqueous and methanolic background electrolytes. Electrophoresis 26(10):1978–1987. https://doi.org/10.1002/elps.200500057
di Cagno MP (2017) The potential of cyclodextrins as novel active pharmaceutical ingredients: a short overview. Molecules 22(1):1. https://doi.org/10.3390/molecules22010001
Cameron KS, Fletcher D, Fielding L (2002) An NMR study of cyclodextrin complexes of the steroidal neuromuscular blocker drug Rocuronium Bromide. Magn Reson Chem 40(4):251–260. https://doi.org/10.1002/mrc.1008
Cao H, He J, Deng L, Gao X (2009) Fabrication of cyclodextrin-functionalized superparamagnetic Fe3O4/amino-silane core–shell nanoparticles via layer-by-layer method. Appl Surf Sci 255(18):7974–7980. https://doi.org/10.1016/j.apsusc.2009.04.199
Chaise T, Bourgeaux E, Cardinael P, Combret JC (2004) Direct and convenient access to mono 3-hydroxy per-O-methylated alpha-cyclodextrin. Tetrahedron Lett 45(30):5853–5856. https://doi.org/10.1016/j.tetlet.2004.06.003
Chaise T, Cardinael P, Tisse S, Combret J-C, Bouillon J-P (2008) Indirect and direct approaches in the synthesis of a new mono-6-O-benzyl methylated gamma-cyclodextrin as chiral selector for enantioselective gas chromatography. Tetrahedron Asymmetry 19(3):348–357. https://doi.org/10.1016/j.tetasy.2008.01.015
Chen Z, Bradshaw JS, Lee ML (1996) A convenient synthesis of mono-6-hydroxy permethylated beta-cyclodextrin via tert-butyldimethylsilylation. Tetrahedron Lett 37(38):6831–6834. https://doi.org/10.1016/0040-4039(96)01545-6
Coisne C, Tilloy S, Monflier E, Wils D, Fenart L, Gosselet F (2016) Cyclodextrins as emerging therapeutic tools in the treatment of cholesterol-associated vascular and neurodegenerative diseases. Molecules 21(12):1748. https://doi.org/10.3390/molecules21121748
Coleman A, Zhang P, Parrotlopez H, Ling C, Miocque M, Mascrier L (1991) The first selective per-tosylation of the secondary OH-2 of beta-cyclodextrin. Tetrahedron Lett 32(32):3997–3998. https://doi.org/10.1016/0040-4039(91)80609-A
Concheiro A, Alvarez-Lorenzo C (2013) Chemically cross-linked and grafted cyclodextrin hydrogels: from nanostructures to drug-eluting medical devices. Adv Drug Deliv Rev 65(9):1188–1203. https://doi.org/10.1016/j.addr.2013.04.015
Crini G (2014) Review: a history of cyclodextrins. Chem Rev 114(21):10940–10975. https://doi.org/10.1021/cr500081p
Croft AP, Bartsch RA (1983) Synthesis of chemically modified cyclodextrins. Tetrahedron 39(9):1417–1474. https://doi.org/10.1016/S0040-4020(01)88551-3
Cui Y, Wang C, Mao J, Yu Y (2010) A facile and practical approach to randomly methylated beta-cyclodextrin. J Chem Technol Biotechnol 85(2):248–251. https://doi.org/10.1002/jctb.2295
Di Blasio B, Galdiero S, Saviano M, Pedone C, Benedetti E, Rizzarelli E, Pedotti S, Vecchio G, Gibbons WA (1996) Synthesis and structural characterization of 6(I),6(II)-diamino-6(I),6(II)-dideoxy-cyclomaltoheptaose, a difunctionalized beta-cyclodextrin. Carbohydr Res 282(1):41–52. https://doi.org/10.1016/0008-6215(95)00357-6
Donati F (2008) Sugammadex: a cyclodextrin to reverse neuromuscular blockade in anaesthesia. Expert Opin Pharmacother 9(8):1375–1386. https://doi.org/10.1517/14656560802046211
Endo T (2011) Large-ring cyclodextrins. Trends Glycosci Glycotechnol 23(130):79–92. https://doi.org/10.4052/tigg.23.79
Engeldinger E, Armspach D, Matt D (2003) Capped cyclodextrins. Chem Rev 103(11):4147–4174. https://doi.org/10.1021/cr030670y
Estrada R III, Vigh G (2012) Comparison of charge state distribution in commercially available sulfated cyclodextrins used as chiral resolving agents in capillary electrophoresis. J Chromatogr A 1226:24–30. https://doi.org/10.1016/j.chroma.2011.08.040
Faugeras P-A, Boens B, Elchinger P-H, Brouillette F, Montplaisir D, Zerrouki R, Lucas R (2012) When cyclodextrins meet click chemistry. Eur J Org Chem 2012(22):4087–4105. https://doi.org/10.1002/ejoc.201200013
Fenger TH, Marinescu LG, Bols M (2011) Cyclodextrin ketones with the catalytic group at the secondary rim and their effectiveness in enzyme-like epoxidation of stilbenes. Eur J Org Chem 2011(12):2339–2345. https://doi.org/10.1002/ejoc.201001696
Fenyvesi E, Vikmon M, Szente L (2016) Cyclodextrins in food technology and human nutrition: benefits and limitations. Crit Rev Food Sci Nutr 56(12):1981–2004. https://doi.org/10.1080/10408398.2013.809513
Fernandez J, Mellet C, Blanco J, Mota J, Gadelle A, Costesarguet A, Defaye J (1995) Isothiocyanoates and cyclic thiocarbamates of alpha,alpha’-trehalose sucrose, and cyclomaltooligosaccharides. Carbohydr Res 268(1):57–71. https://doi.org/10.1016/0008-6215(94)00312-4
Folch-Cano C, Yazdani-Pedram M, Olea-Azar C (2014) Inclusion and functionalization of polymers with cyclodextrins: current applications and future prospects. Molecules 19(9):14066–14079. https://doi.org/10.3390/molecules190914066
Fugedi P (1989) Synthesis of heptakis(6-O-tert-butyldimethylsilyl)cyclomaltoheptaose and octakis(6-O-tert-butyldimethylsilyl)cyclomalto-octaose. Carbohydr Res 192:366–369. https://doi.org/10.1016/0008-6215(89)85197-3
Fujita K, Ueda T, Imoto T, Tabushi I, Toh N, Koga T (1982) Guest-induced conformational change of beta-cyclodextrin capped with an environmentally sensitive chromophore. Bioorg Chem 11(1):72–84. https://doi.org/10.1016/0045-2068(82)90049-9
Fujita K, Ejima S, Imoto T (1985) 1:2 Host-guest binding by double gamma-cyclodextrin. Chem Lett 14(1):11–12. https://doi.org/10.1246/cl.1985.11
Gadelle A, Defaye J (1991) Selective halogenation at primary positions of cyclomaltooligosaccharides and a synthesis of per-3,6-anhydro cyclomaltooligosaccharides. Angew Chem Int Ed Engl 30(1):78–80. https://doi.org/10.1002/anie.199100781
Gan Y, Zhang Y, Xiao C, Zhou C, Zhao Y (2011) A novel preparation of methyl-beta-cyclodextrin from dimethyl carbonate and beta-cyclodextrin. Carbohydr Res 346(3):389–392. https://doi.org/10.1016/j.carres.2010.05.028
Gao X, Tong L, Inoue Y, Tai A (1995) Synthesis and characterization of novel multifunctional host compounds. 4. Cyclodextrin derivatives rearing chromophores. Synth Commun 25(5):703–710. https://doi.org/10.1080/00397919508011407
Garcia-Rio L, Otero-Espinar FJ, Luzardo-Alvarez A, Blanco-Mendez J (2014) Cyclodextrin based rotaxanes, polyrotaxanes and polypseudorotaxanes and their biomedical applications. Curr Top Med Chem 14(4):478–493. https://doi.org/10.2174/1568026613666131219123910
Ghanem A, Schurig V (2001) Peracetylated beta-cyclodextrin as additive in enzymatic reactions: enhanced reaction rate and enantiomeric ratio in lipase-catalyzed transesterifications in organic solvents. Tetrahedron-Asymmetry 12(19):2761–2766. https://doi.org/10.1016/S0957-4166(01)00482-7
Ghosh R, Zhang P, Wang A, Ling C-C (2012) Diisobutylaluminum hydride mediated regioselective o desilylations: access to multisubstituted cyclodextrins. Angew Chem Int Ed 51(7):1548–1552. https://doi.org/10.1002/anie.201105737
Gidwani B, Vyas A (2014) Synthesis, characterization and application of epichlorohydrin-beta-cyclodextrin polymer. Colloids Surf B Biointerfaces 114:130–137. https://doi.org/10.1016/j.colsurfb.2013.09.035
Gorin BI, Riopelle RJ, Thatcher GRJ (1996) Efficient perfacial derivatization of cyclodextrins at the primary face. Tetrahedron Lett 37(27):4647–4650. https://doi.org/10.1016/0040-4039(96)00916-1
Gould S, Scott RC (2005) 2-hydroxypropyl-beta-cyclodextrin (HP-beta-CD): a toxicology review. Food Chem Toxicol 43(10):1451–1459. https://doi.org/10.1016/j.fct.2005.03.007
Grachev MK (2013) Phosphorus-containing cyclodextrins. Characteristics of the synthesis and chemical behaviour. Russ Chem Rev 82(11):1034–1046. https://doi.org/10.1070/RC2013v082n11ABEH004381
Guan Z, Wang Y, Chen Y, Zhang L, Zhang Y (2009) Novel approach for synthesis of 2:1 permethylated β-cyclodextrin–C60 conjugate. Tetrahedron 65(6):1125–1129. https://doi.org/10.1016/j.tet.2008.12.001
Guieu S, Sollogoub M (2008a) Regiospecific tandem azide-reduction/deprotection to afford versatile amino alcohol-functionalized α- and β-cyclodextrins. Angew Chem Int Ed 47(37):7060–7063. https://doi.org/10.1002/anie.200801573
Guieu S, Sollogoub M (2008b) Multiple homo- and hetero-functionalizations of alpha-cyclodextrin through oriented deprotections. J Org Chem 73(7):2819–2828. https://doi.org/10.1021/jo7027085
Guitet M, de Beaumais SA, Bleriot Y, Vauzeilles B, Zhang Y, Menand M, Sollogoub M (2012) Cyclodextrins selectively modified on both rims using an O-3-debenzylative post-functionalisation, a consequence of the sorrento meeting. Carbohydr Res 356:278–281. https://doi.org/10.1016/j.carres.2011.12.002
Hamasaki K, Ikeda H, Nakamura A, Ueno A, Toda F, Suzuki I, Osa T (1993) Fluorescent sensors of molecular recognition. Modified cyclodextrins capable of exhibiting guest-responsive twisted intramolecular charge transfer fluorescence. J Am Chem Soc 115(12):5035–5040. https://doi.org/10.1021/ja00065a012
Hanessian S, Benalil A, Laferriere C (1995) The synthesis of functionalized cyclodextrins as scaffolds and templates for molecular diversity, catalysis, and inclusion phenomena. J Org Chem 60(15):4786–4797. https://doi.org/10.1021/jo00120a023
Harabagiu V, Simionescu BC, Pinteala M, Merrienne C, Mahuteau J, Guegan P, Cheradame H (2004) Synthesis and characterization of persilylated cyclodextrins. Carbohydr Polym 56(3):301–311. https://doi.org/10.1016/j.carbpol.2003.12.007
Harada A, Hashidzume A, Yamaguchi H, Takashima Y (2009) Polymeric rotaxanes. Chem Rev 109(11):5974–6023. https://doi.org/10.1021/cr9000622
Iacovino R, Caso JV, Di Donato C, Malgieri G, Palmieri M, Russo L, Isernia C (2017) Cyclodextrins as complexing agents: preparation and applications. Curr Org Chem 21(2):162–176. https://doi.org/10.2174/1385272820666160909111842
Ishiguro T, Morishita E, Iohara D, Hirayama F, Wada K, Motoyama K, Arima H, Uekama K (2011) Some pharmaceutical and inclusion properties of 2-hydroxybutyl-beta-cyclodextrin derivative. Int J Pharm 419(1–2):161–169. https://doi.org/10.1016/j.ijpharm.2011.07.044
Jambhekar SS, Breen P (2016) Cyclodextrins in pharmaceutical formulations I: structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov Today 21(2):356–362. https://doi.org/10.1016/j.drudis.2015.11.017
Jeong YK, Kwon T, Lee I, Kim T-S, Coskun A, Choi JW (2014) Hyperbranched beta-cyclodextrin polymer as an effective multidimensional binder for silicon anodes in lithium rechargeable batteries. Nano Lett 14(2):864–870. https://doi.org/10.1021/nl404237j
Jicsinszky L, Martina K, Caporaso M, Cintas P, Zanichelli A, Cravotto G (2015) Complexes of peracetylated cyclodextrin in a non-aqueous aprotic medium: the role of residual water. Phys Chem Chem Phys 17(26):17380–17390. https://doi.org/10.1039/c5cp02379c
Jicsinszky L, Caporaso M, Martina K, Gaudino EC, Cravotto G (2016a) Efficient mechanochemical synthesis of regioselective persubstituted cyclodextrins. Beilstein J Org Chem 12:2364–2371. https://doi.org/10.3762/bjoc.12.230
Jicsinszky L, Caporaso M, Tuza K, Martina K, Gaudino EC, Cravotto G (2016b) Nucleophilic substitutions of 6I-O-monotosyl-beta-cyclodextrin in a planetary ball mill. ACS Sustain Chem Eng 4(3):919–929. https://doi.org/10.1021/acssuschemeng.5b01006
Jindřich J, Tišlerová I (2005) Simple preparation of 3I-O-substituted β-cyclodextrin derivatives using cinnamyl bromide. J Org Chem 70(22):9054–9055. https://doi.org/10.1021/jo051339c
Jullien L, Canceill J, Lacombe L, Lehn J (1994) Analysis of the conformational behavior of perfunctionalized beta-cyclodextrins. Part 1. Evidence for insertion of one of the rim substituents into the cyclodextrin cavity in organic-solvents. J Chem Soc Perkin Trans 2(5):989–1002. https://doi.org/10.1039/p29940000989
Jung M, Francotte E (1996) Comparison of γ-cyclodextrin sulfobutyl ether and unmodified γ-cyclodextrin as chiral selectors in capillary electrophoresis. J Chromatogr A 755(1):81–88. https://doi.org/10.1016/S0021-9673(96)00589-4
Kalakuntla RK, Wille T, Le Provost R, Letort S, Reiter G, Mueller S, Thiermann H, Worek F, Gouhier G, Lafont O, Estour F (2013) New modified beta-cyclodextrin derivatives as detoxifying agents of chemical warfare agents (I). Synthesis and preliminary screening: evaluation of the detoxification using a half-quantitative enzymatic assay. Toxicol Lett 216(2–3):200–205. https://doi.org/10.1016/j.toxlet.2012.11.020
Karoyo AH, Wilson LD (2015) Nano-sized cyclodextrin-based molecularly imprinted polymer adsorbents for perfluorinated compounds-a mini-review. Nanomaterials 5(2):981–1003. https://doi.org/10.3390/nano5020981
Khan AR, Forgo P, Stine KJ, D’Souza VT (1998) Methods for selective modifications of cyclodextrins. Chem Rev 98(5):1977–1996. https://doi.org/10.1021/cr970012b
Kirschner DL, Green TK (2005) Nonaqueous synthesis of a selectively modified, highly anionic sulfopropyl ether derivative of cyclomaltoheptaose (beta-cyclodextrin) in the presence of 18-crown-6. Carbohydr Res 340(11):1773–1779. https://doi.org/10.1016/j.carres.2005.04.012
Kiss T, Fenyvesi F, Bacskay I, Varadi J, Fenyvesi E, Ivanyi R, Szente L, Tosaki A, Vecsernyes M (2010) Evaluation of the cytotoxicity of beta-cyclodextrin derivatives: evidence for the role of cholesterol extraction. Eur J Pharm Sci 40(4):376–380. https://doi.org/10.1016/j.ejps.2010.04.014
Krauland AH, Alonso MJ (2007) Chitosan/cyclodextrin nanoparticles as macromolecular drug delivery system. Int J Pharm 340(1–2):134–142. https://doi.org/10.1016/j.ijpharm.2007.03.005
Kraus T, Budesinsky M, Zavada J (2000) Synthesis of per(5-carboxy-5-dehydroxymethyl)-alpha-cyclodextrin and beta-cyclodextrin - self-assembly of the per(2,3-di-O-methyl)-protected homologues into highly stable dimers, driven by multiple hydrogen bonds. Eur J Org Chem 2000(18):3133–3137. https://doi.org/10.1002/1099-0690(200009)2000:18<3133::AID-EJOC3133>3.0.CO;2-P
Kraus T, Buděšínský M, Závada J (2001) General approach to the synthesis of persubstituted hydrophilic and amphiphilic β-cyclodextrin derivatives. J Org Chem 66(13):4595–4600. https://doi.org/10.1021/jo010046q
Kraus T, Budesinsky M, Cisarova I, Zavada J (2002) Per(6-amino-2-O-carboxymethyl-6-deoxy-3-O-methyl)-alpha-cyclodextrin: helical self-assembly of a polyionic amino acid into nanotubes. Angew Chem Int Ed 41(10):1715–1717. https://doi.org/10.1002/1521-3773(20020517)41:10<1715::AID-ANIE1715>3.0.CO;2-K
Kryjewski M, Goslinski T, Mielcarek J (2015) Functionality stored in the structures of cyclodextrin-porphyrinoid systems. Coord Chem Rev 300:101–120. https://doi.org/10.1016/j.ccr.2015.04.009
Kulkarni A, DeFrees K, Hyun S-H, Thompson DH (2012) Pendant polymer: amino-β-cyclodextrin:siRNA guest:host nanoparticles as efficient vectors for gene silencing. J Am Chem Soc 134(18):7596–7599. https://doi.org/10.1021/ja300690j
Kurochkina GI, Grachev MK, Levina II, Nifanťev EE (2014) (29)Si NMR spectroscopy as a reliable method for the determination of silyl protective groups positions at the cyclodextrin frame. Phosphorus Sulfur Silicon Relat Elem 189(1):33–39. https://doi.org/10.1080/10426507.2013.788006
Lammers JNJJ, Koole JL, Hurkmans J (1971) Properties of cyclodextrins. Part VI. Water-soluble cyclodextrin-derivatives. Preparation and analysis. Starch - Stärke 23(5):167–171. https://doi.org/10.1002/star.19710230504
Leclercq L (2016) Interactions between cyclodextrins and cellular components: towards greener medical applications? Beilstein J Org Chem 12:2644–2662. https://doi.org/10.3762/bjoc.12.261
Lee SA, Lim ST (1998) Preparation and solubility of phosphorylated beta-cyclodextrins. Cereal Chem 75(5):690–694. https://doi.org/10.1094/CCHEM.1998.75.5.690
Letort S, Mathiron D, Grel T, Albaret C, Daulon S, Djédaïni-Pilard F, Gouhier G, Estour F (2015) The first 2(IB), 3(IA)-heterodifunctionalized beta-cyclodextrin derivatives as artificial enzymes. Chem Commun 51(13):2601–2604. https://doi.org/10.1039/c4cc09189b
Letort S, Balieu S, Erb W, Gouhier G, Estour F (2016) Interactions of cyclodextrins and their derivatives with toxic organophosphorus compounds. Beilstein J Org Chem 12:204–228. https://doi.org/10.3762/bjoc.12.23
Li P, Song J, Ni X, Guo Q, Wen H, Zhou Q, Shen Y, Huang Y, Qiu P, Lin S, Hu H (2016) Comparison in toxicity and solubilizing capacity of hydroxypropyl-β-cyclodextrin with different degree of substitution. Int J Pharm 513(1–2):347–356. https://doi.org/10.1016/j.ijpharm.2016.09.036
Lian C-M, Jiang L-P, Liu D-L (2014) Synthesis and characterization of acetylated sept-D-glucopyranose carbamate as an oligosaccharide donor. Chin Chem Lett 25(1):134–136. https://doi.org/10.1016/j.cclet.2013.10.025
Lima PSS, Lucchese AM, Araujo-Filho HG, Menezes PP, Araujo AAS, Quintans-Junior LJ, Quintans JSS (2016) Inclusion of terpenes in cyclodextrins: preparation, characterization and pharmacological approaches. Carbohydr Polym 151:965–987. https://doi.org/10.1016/j.carbpol.2016.06.040
Lindbäck E, Zhou Y, Pedersen CM, Bols M (2012) Two diastereomeric artificial enzymes with different catalytic activity. Eur J Org Chem 2012(27):5366–5372. https://doi.org/10.1002/ejoc.201200699
Liu L, Guo Q-X (2002) The driving forces in the inclusion complexation of cyclodextrins. J Incl Phenom Macrocycl Chem 42(1–2):1–14. https://doi.org/10.1023/A:1014520830813
Liu Y, Han B-H, Li B, Zhang Y-M, Zhao P, Chen Y-T, Wada T, Inoue Y (1998) Molecular recognition study on supramolecular system. 14. Synthesis of modified cyclodextrins and their inclusion complexation thermodynamics with l-tryptophan and some naphthalene derivatives. J Org Chem 63(5):1444–1454. https://doi.org/10.1021/jo971466b
Luke DR, Tomaszewski K, Damle B, Schlamm HT (2010) Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci 99(8):3291–3301. https://doi.org/10.1002/jps.22109
Lupescu N, Ho CKY, Jia GC, Krepinsky JJ (1999) A convenient synthesis of per-O-methylated 6-O-monosubstituted beta-cyclodextrins. J Carbohydr Chem 18(1):99–104. https://doi.org/10.1080/07328309908543982
Ma D-Y, Zhang Y-M, Xu J-N (2016) The synthesis and process optimization of sulfobutyl ether beta-cyclodextrin derivatives. Tetrahedron 72(22):3105–3112. https://doi.org/10.1016/j.tet.2016.04.039
Macaev F, Boldescu V (2015) Cyclodextrins in asymmetric and stereospecific synthesis. Symmetry-Basel 7(4):1699–1720. https://doi.org/10.3390/sym7041699
van de Manakker F, Vermonden T, van Nostrum CF, Hennink WE (2009) Cyclodextrin-based polymeric materials: synthesis, properties, and pharmaceutical/biomedical applications. Biomacromolecules 10(12):3157–3175. https://doi.org/10.1021/bm901065f
Martina K, Puntambekar DS, Barge A, Gallarate M, Chirio D, Cravotto G (2010) Synthesis, characterization and potential application of monoacyl-cyclodextrins. Carbohydr Res 345(2):191–198. https://doi.org/10.1016/j.carres.2009.11.009
Martinelli J, Thangavel K, Tei L, Botta M (2014) Dendrimeric beta-cyclodextrin/Gd-III chelate supramolecular host-guest adducts as high-relaxivity MRI probes. Chem Eur J 20(35):10944–10952. https://doi.org/10.1002/chem.201402418
Masurier N, Estour F, Lefèvre B, Brasme B, Masson P, Lafont O (2006) Improved access to 2-O-monobenzyl ethers of beta-cyclodextrin as precursors of catalysts for organophosphoryl esters hydrolysis. Carbohydr Res 341(7):935–940. https://doi.org/10.1016/j.carres.2006.02.012
Masurier N, Lafont O, Le Provost R, Lesur D, Masson P, Djédaïni-Pilard F, Estour F (2009) Regioselective access to 3(I)-O-substituted-beta-cyclodextrin derivatives. Chem Commun 5:589–591. https://doi.org/10.1039/b812325j
Maynard DK, Vigh G (2000) Synthesis and analytical characterization of the sodium salt of heptakis(2-O-methyl-3,6-di-O-sulfo)cyclomalto-heptaose, a chiral resolving agent candidate for capillary electrophoresis. Carbohydr Res 328(3):277–285. https://doi.org/10.1016/S0008-6215(00)00114-2
Mejia-Ariza R, Grana-Suarez L, Verboom W, Huskens J (2017) Cyclodextrin-based supramolecular nanoparticles for biomedical applications. J Mater Chem B 5(1):36–52. https://doi.org/10.1039/c6tb02776h
Mocanu G, Vizitiu D, Carpov A (2001) Cyclodextrin polymers. J Bioact Compat Polym 16(4):315–342. https://doi.org/10.1106/JJUV-8F2K-JGYF-HNGF
Morin-Crini N, Crini G (2013) Environmental applications of water-insoluble beta-cyclodextrin-epichlorohydrin polymers. Prog Polym Sci 38(2):344–368. https://doi.org/10.1016/j.progpolymsci.2012.06.005
Nakagawa T, Ueno K, Kashiwa M, Watanabe J (1994) The stereoselective synthesis of cyclomaltopentaose. A novel cyclodextrin homologue with D.P. five. Tetrahedron Lett 35(12):1921–1924. https://doi.org/10.1016/S0040-4039(00)73196-0
Nakazono K, Takashima T, Arai T, Koyama Y, Takata T (2010) High-yield one-pot synthesis of permethylated alpha-cyclodextrin-based polyrotaxane in hydrocarbon solvent through an efficient heterogeneous reaction. Macromolecules 43(2):691–696. https://doi.org/10.1021/ma902161d
Nielsen TT, Wintgens V, Amiel C, Wimmer R, Larsen KL (2010) Facile synthesis of β-cyclodextrin-dextran polymers by “Click” chemistry. Biomacromolecules 11(7):1710–1715. https://doi.org/10.1021/bm9013233
Normand M, Kirillov E, Carpentier J-F, Guillaume SM (2012) Cyclodextrin-centered polyesters: controlled ring-opening polymerization of cyclic esters from beta-cyclodextrin-diol. Macromolecules 45(2):1122–1130. https://doi.org/10.1021/ma202400e
Oliveri V, Vecchio G (2016) Cyclodextrins as protective agents of protein aggregation: an overview. Chem Asian J 11(11):1648–1657. https://doi.org/10.1002/asia.201600259
Pastore A, Valerio S, Adinolfi M, Iadonisi A (2011) An easy and versatile approach for the regioselective de-O-benzylation of protected sugars based on the I2/Et3SiH combined system. Chem- Eur J 17(21):5881–5889. https://doi.org/10.1002/chem.201003332
Pearce AJ, Sinaÿ P (2000) Diisobutylaluminum-promoted regioselective de-O-benzylation of perbenzylated cyclodextrins: a powerful new strategy for the preparation of selectively modified cyclodextrins. Angew Chem Int Ed 39(20):3610–3612. https://doi.org/10.1002/1521-3773(20001016)39:20<3610::AID-ANIE3610>3.0.CO;2-V
Petrillo M, Marinescu L, Rousseau C, Bols M (2009) Selective discrimination of cyclodextrin diols using cyclic sulfates. Org Lett 11(9):1983–1985. https://doi.org/10.1021/ol900413s
Petter RC, Salek JS, Sikorski CT, Kumaravel G, Lin FT (1990) Cooperative binding by aggregated mono-6-(alkylamino)-beta-cyclodextrins. J Am Chem Soc 112(10):3860–3868. https://doi.org/10.1021/ja00166a021
Pitha J, Milecki J, Fales H, Pannell L, Uekama K (1986) Hydroxypropyl-beta-cyclodextrin - preparation and characterization - effects on solubility of drugs. Int J Pharm 29(1):73–82. https://doi.org/10.1016/0378-5173(86)90201-2
Popr M, Hybelbauerova S, Jindrich J (2014) A complete series of 6-deoxy-monosubstituted tetraalkylammonium derivatives of alpha-, beta-, and gamma-cyclodextrin with 1, 2, and 3 permanent positive charges. Beilstein J Org Chem 10:1390–1396. https://doi.org/10.3762/bjoc.10.142
Prabaharan M, Gong S (2008) Novel thiolated carboxymethyl chitosan-g-beta-cyclodextrin as mucoadhesive hydrophobic drug delivery carriers. Carbohydr Polym 73(1):117–125. https://doi.org/10.1016/j.carbpol.2007.11.005
Pun SH, Bellocq NC, Liu AJ, Jensen G, Machemer T, Quijano E, Schluep T, Wen SF, Engler H, Heidel J, Davis ME (2004) Cyclodextrin-modified polyethylenimine polymers for gene delivery. Bioconjug Chem 15(4):831–840. https://doi.org/10.1021/bc049891g
Qu QI, Tucker E, Christian SD (2002) Sulfoalkyl ether β-cyclodextrin derivatives: synthesis and characterizations. J Incl Phenom Macrocycl Chem 43(3–4):213–222. https://doi.org/10.1023/A:1021255314835
Radu C-D, Parteni O, Ochiuz L (2016) Applications of cyclodextrins in medical textiles - review. J Control Release 224:146–157. https://doi.org/10.1016/j.jconrel.2015.12.046
Rao CT, Pitha J, Lindberg B, Lindberg J (1992) Distribution of substituents in O-(2-hydroxypropyl) derivatives of cyclomalto-oligosaccharides (cyclodextrins): influence of increasing substitution, of the base used in the preparation, and of macrocyclic size. Carbohydr Res 223:99–107. https://doi.org/10.1016/0008-6215(92)80009-P
Rawal GK, Rani S, Ward S, Ling C-C (2010) DIBAL-H mediated triple and quadruple debenzylations of perbenzylated cyclodextrins. Org Biomol Chem 8(1):171–180. https://doi.org/10.1039/b915450g
Rekharsky MV, Inoue Y (1998) Complexation thermodynamics of cyclodextrins. Chem Rev 98(5):1875–1918. https://doi.org/10.1021/cr970015o
Renard E, Volet G, Amiel C (2005) Synthesis of a novel linear water-soluble beta-cyclodextrin polymer. Polym Int 54(3):594–599. https://doi.org/10.1002/pi.1742
Reuben J, Trinadha Rao C, Pitha J (1994) Distribution of substituents in carboxymethyl ethers of cyclomaltoheptaose. Carbohydr Res 258:281–285. https://doi.org/10.1016/0008-6215(94)84094-6
Řezanka M (2016) Monosubstituted cyclodextrins as precursors for further use. Eur J Org Chem 2016(32):5322–5334. https://doi.org/10.1002/ejoc.201600693
Řezanka M, Jindřich J (2011) Complete sets of monosubstituted cyclomaltohexaoses (alpha- cyclodextrins) as precursors for further synthesis. Carbohydr Res 346(15):2374–2379. https://doi.org/10.1016/j.carres.2011.08.011
Řezanka M, Eignerová B, Jindřich J, Kotora M (2010) Synthesis of mono(perfluoroalkyl) cyclodextrins via cross metathesis. Eur J Org Chem 2010(32):6256–6262. https://doi.org/10.1002/ejoc.201000807
Řezanka P, Navrátilová K, Řezanka M, Král V, Sýkora D (2014) Application of cyclodextrins in chiral capillary electrophoresis. Electrophoresis 35(19):2701–2721. https://doi.org/10.1002/elps.201400145
Řezanka M, Langton MJ, Beer PD (2015) Anion recognition in water by a rotaxane containing a secondary rim functionalised cyclodextrin stoppered axle. Chem Commun 51(21):4499–4502. https://doi.org/10.1039/C5CC00171D
Řezanka P, Řezanková K, Sedláčková H, Mašek J, Rokosová L, Bláhová M, Řezanka M, Jindřich J, Sýkora D, Král V (2016) Influence of substituent position and cavity size of the regioisomers of monocarboxymethyl-α-, β-, and γ-cyclodextrins on the apparent stability constants of their complexes with both enantiomers of Tröger’s base. J Sep Sci 39(5):980–985. https://doi.org/10.1002/jssc.201500845
du Roizel B, Baltaze J-P, Sinaÿ P (2002) Diisobutylaluminum-promoted secondary rim selective de-O-methylation of permethylated cyclodextrins. Tetrahedron Lett 43(13):2371–2373. https://doi.org/10.1016/S0040-4039(02)00274-5
Rojas MT, Koeniger R, Stoddart JF, Kaifer AE (1995) Supported monolayers containing preformed binding sites. Synthesis and interfacial binding properties of a thiolated beta-cyclodextrin derivative. J Am Chem Soc 117(1):336–343. https://doi.org/10.1021/ja00106a036
Sakairi N, Kuzuhara H (1993) Efficient and regioselective preparation of an eight-membered interglycosidic benzylidene derivative of β-cyclodextrin. Chem Lett 22(12):2077–2080. https://doi.org/10.1246/cl.1993.2077
Sakairi N, Wang L-X, Kuzuhara H (1991) Insertion of a D-glucosamine residue into the α-cyclodextrin skeleton; a model synthesis of ‘chimera cyclodextrins’. J Chem Soc Chem Commun (5):289–290. https://doi.org/10.1039/C39910000289
Sakairi N, Wang L-X, Kuzuhara H (1995) Modification of cyclodextrins by insertion of a heterogeneous sugar unit into their skeletons. Synthesis of 2-amino-2-deoxy-β-cyclodextrin from α-cyclodextrin. J Chem Soc Perkin 1(4):437–443. https://doi.org/10.1039/P19950000437
Sakairi N, Kuzuhara H, Okamoto T, Yajima M (1996a) Synthesis and biological evaluation of 2-amino-2-deoxy- and 6-amino-6-deoxy-cyclomaltoheptaose polysulfates as synergists for angiogenesis inhibition. Bioorg Med Chem 4(12):2187–2192. https://doi.org/10.1016/S0968-0896(96)00222-2
Sakairi N, Nishi N, Tokura S, Kuzuhara H (1996b) Interglycosidic acetals. Part 3. Synthesis and structure determination of cyclic monobenzylidene acetals of cyclodextrin derivatives bridging between two contiguous D-glucopyranosyl residues. Carbohydr Res 291:53–62. https://doi.org/10.1016/S0008-6215(96)00192-9
Sallas F, Darcy R (2008) Amphiphilic cyclodextrins – advances in synthesis and supramolecular chemistry. Eur J Org Chem 2008(6):957–969. https://doi.org/10.1002/ejoc.200700933
Saokham P, Loftsson T (2017) Gamma-cyclodextrin. Int J Pharm 516(1–2):278–292. https://doi.org/10.1016/j.ijpharm.2016.10.062
Saz JM, Marina ML (2016) Recent advances on the use of cyclodextrins in the chiral analysis of drugs by capillary electrophoresis. J Chromatogr A 1467:79–94. https://doi.org/10.1016/j.chroma.2016.08.029
Sharma N, Baldi A (2016) Exploring versatile applications of cyclodextrins: an overview. Drug Deliv 23(3):739–757. https://doi.org/10.3109/10717544.2014.938839
Shirin S, Buncel E, vanLoon GW (2003) The use of β-cyclodextrins to enhance the aqueous solubility of trichloroethylene and perchloroethylene and their removal from soil organic matter: effect of substituents. Can J Chem 81(1):45–52. https://doi.org/10.1139/v02-205
Sollogoub M (2013) Site-selective heterofunctionalization of cyclodextrins: discovery, development, and use in catalysis. Synlett 24(20):2629–2640. https://doi.org/10.1055/s-0033-1339877
Song A, Wang J, Liu C, Deng L (2009a) Sulfoalkyl ether β-cyclodextrin derivatives synthesized by a single step method as pharmaceutical biomaterials. Chin Sci Bull 54(18):3187–3199. https://doi.org/10.1007/s11434-009-0262-8
Song LX, Bai L, Xu XM, He J, Pan SZ (2009b) Inclusion complexation, encapsulation interaction and inclusion number in cyclodextrin chemistry. Coord Chem Rev 253(9–10):1276–1284. https://doi.org/10.1016/j.ccr.2008.08.011
Stefanache A, Silion M, Stoica I, Fifere A, Harabagiu V, Farcas A (2014) Poly[2,7-(9,9- dioctylfluorene)-alt-(5,5′-bithiophene/permethylated beta-cyclodextrin)] main-chain polyrotaxane: synthesis, characterization and surface morphology. Eur Polym J 50:223–234. https://doi.org/10.1016/j.eurpolymj.2013.11.001
Swaminathan S, Cavalli R, Trotta F (2016) Cyclodextrin-based nanosponges: a versatile platform for cancer nanotherapeutics development. Wiley Interdiscip Rev Nanomed Nanobiotechnol 8(4):579–601. https://doi.org/10.1002/wnan.1384
Szejtli J (1998) Introduction and general overview of cyclodextrin chemistry. Chem Rev 98(5):1743–1754. https://doi.org/10.1021/cr970022c
Szejtli J, Lipták A, Jodál I, Fügedi P, Nánási P, Neszmélyi A (1980) Synthesis and 13C-NMR spectroscopy of methylated beta-cyclodextrins. Starch - Stärke 32(5):165–169. https://doi.org/10.1002/star.19800320506
Szente L, Fenyvesi E (2017) Cyclodextrin-lipid complexes: cavity size matters. Struct Chem 28(2):479–492. https://doi.org/10.1007/s11224-016-0884-9
Szente L, Szejtli J (1999) Highly soluble cyclodextrin derivatives: chemistry, properties, and trends in development. Adv Drug Deliv Rev 36(1):17–28. https://doi.org/10.1016/S0169-409X(98)00092-1
Tabushi I, Shimokawa K, Fujita K (1977) Specific bifunctionalization on cyclodextrin. Tetrahedron Lett 18(18):1527–1530. https://doi.org/10.1016/S0040-4039(01)93093-X
Tabushi I, Kuroda Y, Yokota K, Yuan L (1981) Regiospecific A,C- and A,D-disulfonate capping of beta-cyclodextrin. J Am Chem Soc 103(3):711–712. https://doi.org/10.1021/ja00393a056
Taka AL, Pillay K, Mbianda XY (2017) Nanosponge cyclodextrin polyurethanes and their modification with nanomaterials for the removal of pollutants from waste water: a review. Carbohydr Polym 159:94–107. https://doi.org/10.1016/j.carbpol.2016.12.027
Takeo K, Sumimoto T, Kuge T (1974) An improved synthesis of 6-deoxy-analogues of cyclodextrins and amylose - further interpretations of the proton magnetic resonance spectra of the peracetates of cyclodextrins and amylose. Starch 26(4):111–118. https://doi.org/10.1002/star.19740260403
Takeo K, Uemura K, Mitoh H (1988) Derivatives of alpha-cyclodextrin and the synthesis of 6-O-alpha-D-glucopyranosyl-alpha-cyclodextrin. J Carbohydr Chem 7(2):293–308. https://doi.org/10.1080/07328308808058926
Takeo K, Mitoh H, Uemura K (1989) Selective chemical modification of cyclomalto- oligosaccharides via tert-butyldimethylsilylation. Carbohydr Res 187(2):203–221. https://doi.org/10.1016/0008-6215(89)80004-7
Tang W, Ng S-C (2007) Synthesis of cationic single-isomer cyclodextrins for the chiral separation of amino acids and anionic pharmaceuticals. Nat Protoc 2(12):3195–3200. https://doi.org/10.1038/nprot.2007.479
Tang W, Ng SC (2008a) Monosubstituted positively charged cyclodextrins: synthesis and applications in chiral separation. J Sep Sci 31(18):3246–3256. https://doi.org/10.1002/jssc.200800357
Tang W, Ng S-C (2008b) Facile synthesis of mono-6-amino-6-deoxy-α-, β-, γ-cyclodextrin hydrochlorides for molecular recognition, chiral separation and drug delivery. Nat Protoc 3(4):691–697. https://doi.org/10.1038/nprot.2008.37
Tatar A, Grishina A, Budesinsky M, Kraus T (2017) Per-2,3-O-alkylated beta-cyclodextrin duplexes connected with disulfide bonds. Supramol Chem 29(1):40–48. https://doi.org/10.1080/10610278.2016.1164860
Teranishi K (2000) Practicable regiospecific bifunctionalization on the secondary face of alpha- and beta-cyclodextrins. Chem Commun (14):1255–1256. https://doi.org/10.1039/b002445g
Tongiani S, Velde DV, Ozeki T, Stella VJ (2005) Sulfoalkyl ether-alkyl ether cyclodextrin derivatives, their synthesis, NMR characterization, and binding of 6 alpha-methylprednisolone. J Pharm Sci 94(11):2380–2392. https://doi.org/10.1002/jps.20367
Trellenkamp T, Ritter H (2010) Poly(N-vinylpyrrolidone) bearing covalently attached cyclodextrin via click-chemistry: synthesis, characterization, and complexation behavior with phenolphthalein. Macromolecules 43(13):5538–5543. https://doi.org/10.1021/ma100812q
Trotta F, Costa L, Giancarlo C (2002) A new easy access to enantiomerically pure 2,2′-dihydroxy- 1,1′-binaphthyl. J Incl Phenom Macrocycl Chem 44(1–4):341–344. https://doi.org/10.1023/A:1023069324382
Tutu E, Vigh G (2011) Synthesis, analytical characterization and initial capillary electrophoretic use in an acidic background electrolyte of a new, single-isomer chiral resolving agent: heptakis(2- O- sulfo-3-O-methyl-6-O-acetyl)-β-cyclodextrin. Electrophoresis 32(19):2655–2662. https://doi.org/10.1002/elps.201100104
Uccello-Barretta G, Sicoll G, Balzano F, Salvadori P (2005) NMR spectroscopy: a powerful tool for detecting the conformational features of symmetrical persubstituted mixed cyclomaltoheptaoses (beta-cyclodextrins). Carbohydr Res 340(2):271–281. https://doi.org/10.1016/j.carres.2004.11.022
Venuti V, Rossi B, Mele A, Melone L, Punta C, Majolino D, Masciovecchio C, Caldera F, Trotta F (2017) Tuning structural parameters for the optimization of drug delivery performance of cyclodextrin-based nanosponges. Expert Opin Drug Deliv 14(3):331–340. https://doi.org/10.1080/17425247.2016.1215301
Vincent JB, Kirby DM, Nguyen TV, Vigh G (1997) A family of single-isomer chiral resolving agents for capillary electrophoresis. 2. Hepta-6-sulfato-β-cyclodextrin. Anal Chem 69(21):4419–4428. https://doi.org/10.1021/ac970418o
Volkov S, Kumprecht L, Budesinsky M, Lepsik M, Dusek M, Kraus T (2015) A gamma- cyclodextrin duplex connected with two disulfide bonds: synthesis, structure and inclusion complexes. Org Biomol Chem 13(10):2980–2985. https://doi.org/10.1039/c4ob02464h
Wang N, Wu Q, Xiao YM, Chen CX, Lin MF (2005) Regioselective synthesis of cyclodextrin mono-substituted conjugates of non-steroidal anti-inflammatory drugs at C-2 secondary hydroxyl by protease in non-aqueous media. Bioorg Med Chem 13(11):3667–3671. https://doi.org/10.1016/j.bmc.2005.03.031
Wang Y, Zhao Y, Han B (2013) Preparation of tris(2-aminoethyl)amine-cross-linked cyclodextrin-based porous nanospheres and their application as drug delivery systems. Chin J Chem 31(5):657–662. https://doi.org/10.1002/cjoc.201300296
Wang B, Zaborova E, Guieu S, Petrillo M, Guitet M, Blériot Y, Ménand M, Zhang Y, Sollogoub M (2014) Site-selective hexa-hetero-functionalization of α-cyclodextrin an archetypical C6-symmetric concave cycle. Nat Commun 5:5354. https://doi.org/10.1038/ncomms6354
Wang J, Qiu Z, Wang Y, Li L, Guo X, Pham D-T, Lincoln SF, Prud’homme RK (2016) Supramolecular polymer assembly in aqueous solution arising from cyclodextrin host-guest complexation. Beilstein J Org Chem 12:50–72. https://doi.org/10.3762/bjoc.12.7
Wenz G (1994) Cyclodextrins as building blocks for supramolecular structures and functional units. Angew Chem Int Ed 33(8):803–822. https://doi.org/10.1002/anie.199408031
Wimmer T (1995) Process for the preparation of alkylated cyclodextrin derivatives, the methylated cyclodextrin derivatives obtainable from this process and the use of the products, Patent DE4333598 (A1)
Xiao Y, Wu Q, Wang N, Lin X (2004) Regioselective monoacylation of cyclomaltoheptaose at the C-2 secondary hydroxyl groups by the alkaline protease from Bacillus subtilis in nonaqueous media. Carbohydr Res 339(7):1279–1283. https://doi.org/10.1016/j.carres.2004.02.024
Xiao S, Yang M, Yu F, Zhang L, Zhou D, Sinaÿ P, Zhang Y (2013) Synthesis of four mono-functionalized α-cyclodextrin derivatives for further confirming DIBAL-H-promoted bis- de-O-methylation mechanism. Tetrahedron 69(20):4053–4060. https://doi.org/10.1016/j.tet.2013.03.070
Xu M, Wu S, Zeng F, Yu C (2010) Cyclodextrin supramolecular complex as a water-soluble ratiometric sensor for ferric ion sensing. Langmuir 26(6):4529–4534. https://doi.org/10.1021/la9033244
Yanli M (2005) Process for synthesizing random methylated beta-cyclodextrin, Patent CN1709918 (A)
Yoshikiyo K, Matsui Y, Yamamoto T (2015) Qualitative evaluation of regioselectivity in the formation of di- and tri-6-O-tritylates of alpha-cyclodextrin. Beilstein J Org Chem 11:1530–1540. https://doi.org/10.3762/bjoc.11.168
Yu H, Teramoto A, Fukudome M, Xie R-G, Yuan D-Q, Fujita K (2006) A facile sulfonylation method enabling direct syntheses of per(2-O-sulfonyl)-beta-cyclodextrins. Tetrahedron Lett 47(50):8837–8840. https://doi.org/10.1016/j.tetlet.2006.10.061
Yuan C, Liu B, Liu H (2015) Characterization of hydroxypropyl-beta-cyclodextrins with different substitution patterns via FTIR, GC-MS, and TG-DTA. Carbohydr Polym 118:36–40. https://doi.org/10.1016/j.carbpol.2014.10.070
Zhang W, Qin L, He X-W, Li W-Y, Zhang Y-K (2009) Novel surface modified molecularly imprinted polymer using acryloyl-beta-cyclodextrin and acrylamide as monomers for selective recognition of lysozyme in aqueous solution. J Chromatogr A 1216(21):4560–4567. https://doi.org/10.1016/j.chroma.2009.03.056
Zhang J, Zhang B, Zhou J, Li J, Shi C, Huang T, Wang Z, Tang J (2011) H2SO4-SiO2: highly efficient and reusable catalyst for per-O-acetylation of carbohydrates under solvent-free conditions. J Carbohydr Chem 30(3):165–177. https://doi.org/10.1080/07328303.2011.621042
Zhang XM, Peng CS, Xu GC (2012) Synthesis of modified beta-cyclodextrin polymers and characterization of their fuchsin adsorption. J Incl Phenom Macrocycl Chem 72(1–2):165–171. https://doi.org/10.1007/s10847-011-9956-z
Zhong N, Byun H-S, Bittman R (1998) An improved synthesis of 6-O-monotosyl-6-deoxy-beta-cyclodextrin. Tetrahedron Lett 39(19):2919–2920. https://doi.org/10.1016/S0040-4039(98)00417-1
Zhou J, Ritter H (2010) Cyclodextrin functionalized polymers as drug delivery systems. Polym Chem 1(10):1552–1559. https://doi.org/10.1039/c0py00219d
Zhou Y, Lindbäck E, Marinescu LG, Pedersen CM, Bols M (2012a) Synthesis of cyclodextrins with carboxylic acids at the secondary rim and an evaluation of their properties as chemzymes for glycoside hydrolysis. Eur J Org Chem 2012(21):4063–4070. https://doi.org/10.1002/ejoc.201200404
Zhou Y, Marinescu L, Pedersen CM, Bols M (2012b) Synthesis of tin-containing cyclodextrins as potential enzyme models. Eur J Org Chem 2012(32):6383–6389. https://doi.org/10.1002/ejoc.201200756
Zhou J, Tang J, Tang W (2015) Recent development of cationic cyclodextrins for chiral separation trac. Trends Anal Chem 65:22–29. https://doi.org/10.1016/j.trac.2014.10.009
Zhu Q, Scriba GKE (2016) Advances in the use of cyclodextrins as chiral selectors in capillary electrokinetic chromatography: fundamentals and applications. Chromatographia 79(21–22):1403–1435. https://doi.org/10.1007/s10337-016-3167-0
Acknowledgements
This work was supported by project LO1201 of the Ministry of Education, Youth and Sports in the framework of the targeted support of the “National Programme for Sustainability I”; and by the project 16-02316Y of the Czech Science Foundation (GA CR).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Řezanka, M. (2018). Synthesis of Cyclodextrin Derivatives. In: Fourmentin, S., Crini, G., Lichtfouse, E. (eds) Cyclodextrin Fundamentals, Reactivity and Analysis. Environmental Chemistry for a Sustainable World, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-319-76159-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-76159-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-76158-9
Online ISBN: 978-3-319-76159-6
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)