
Abstract
The vitamin D receptor (VDR) is found in nearly every cell in the body. Moreover, the enzyme, CYP27B1, that produces the active form of the hormone, 1,25-dihydroxyvitamin D (1,25(OH)2D), is also widespread, although perhaps not as ubiquitous as VDR. These observations underlie the recent understanding that vitamin D does much more than regulate bone mineral metabolism through its actions on intestinal calcium and phosphate absorption, renal calcium and phosphate reabsorption, and bone development and remodeling. In this chapter, I will review the production of vitamin D in the skin, its subsequent metabolism to the major circulating metabolite 25-hydroxyvitamin D (25(OH)D) by the different 25-hydroxylases in the liver and elsewhere, and the metabolism of 25(OH)D to 1,25(OH)2D in the kidney and elsewhere. This last step is tightly regulated, but its regulation differs according to the cell involved. Transport of the vitamin D metabolites in blood by the vitamin D-binding protein (DBP) and albumin and its uptake by different tissues are then discussed emphasizing that some cells rely on diffusion of the metabolites across the membrane in free form, whereas others have a mechanism to take up the metabolite bound to DBP. The molecular mechanism of action of 1,25(OH)2D will then be reviewed, describing the very large number of cellular processes regulated by 1,25(OH)2D, emphasizing the cell specificity of this regulation. In the final section, I will provide examples of different physiologic functions of vitamin D signaling in its regulation of proliferation/differentiation, hormone regulation, and immune function.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Vitamin D
- Vitamin D receptor
- CYP27B1
- Calcitriol
- Calcium
- Vitamin D-binding protein
- Genomic regulation
- Cancer
- Immunity
- Hormone regulation
- Proliferation
- Differentiation
Introduction
With the findings that both the vitamin D receptor (VDR) and the enzyme (CYP27B1, the 1α-hydroxylase) required to convert the major circulating metabolite of vitamin D (25-hydroxyvitamin D (25(OH)D)) to the most biologically active metabolite, 1,25-dihydroxyvitamin D (1,25(OH)2D), are found in many if not all cells, interest in vitamin D metabolism and mechanisms of action has exploded. Much of this interest is attributed to the potential for vitamin D metabolites and analogs to affect not only the regulation by the classic tissues—the intestine, bone, and kidney—of bone and mineral metabolism but also that of most tissues and their functions not necessarily related to bone and mineral metabolism. This interest is further piqued by the observations from advanced genomic techniques such as RNA-seq and ChIP-seq that the VDR has thousands of binding sites throughout the genome affecting the transcription of hundreds of different genes, and the profile of the affected genes shows substantial diversity among the different cell types. In this chapter, I will first discuss vitamin D production and metabolism, then focus on the mechanism of action of 1,25(OH)2D, and conclude with a discussion of the impact of the vitamin D metabolites on a representative sampling of different tissues, both the classic and nonclassic.
Vitamin D Production
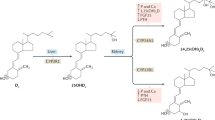
The skin contains substantial amounts of 7-dehydrocholesterol (7-DHC, provitamin D3), which when irradiated by UV light (UVB spectrum 280–320), typically from the sun, undergoes a two-step process to form vitamin D3 (D3) (cholecalciferol) (Fig. 1.1). In the first step, UVB breaks open the B ring of 7-DHC forming pre-D3 that isomerizes to D3 in a thermosensitive but noncatalytic process. Both UVB intensity and skin pigmentation level contribute to the extent of D3 formation [1]. Skin pigmentation and other chromogenic agents that can absorb UVB block D3 production , as do clothing and sunscreen. Moreover, both season and latitude affect the intensity of UVB from sunlight so that those living at higher latitudes have a shorter period of the year in which sunlight is capable of producing D3 [2]. Vitamin D can also be obtained from the diet . Most foods with the exception of fatty fish contain little vitamin D unless fortified, whereas fish contain only D3, which they obtain from plankton (or from the ingestion of other fish). Plants and fungi (e.g., mushrooms) contain ergosterol, which, similar to D3 production from 7-DHC, is converted to D2 (ergocalciferol) by UVB via the same two-step process. D2 differs from D3 in having a double bond between C22 and C23 and a methyl group at C24 in the side chain (Fig. 1.1). Many foods such as milk and orange juice are fortified by D2. Knowledge of which vitamin D is being consumed either as supplements or in fortified foods is important because their pharmacokinetics, metabolism, and measurement of their metabolites by various immunoassays differ. The structural differences between D2 and D3 in the side chain affect their affinity for DBP resulting in faster clearance of D2 from the circulation, limit the conversion of D2 to 25(OH)D2 by at least some of the 25-hydroxylases to be described subsequently, and alter its catabolism by the 24-hydroxyase (CYP24A1) [3,4,5]. As such, equivalent amounts of D2 do not give as high or as long-lasting level of 25(OH)D as does D3 [6]. That said, the active metabolites of D2 and D3, namely, 1,25(OH)2D2 and 1,25(OH)2D3, have comparable affinities for the VDR [4] and are thus expected to have comparable biologic activity. Therefore, in this review, if no subscript is employed, both D2 and D3 and their metabolites are being considered.

Vitamin D Production . Vitamin D3 is produced in the skin from 7-dehydrocholesterol (7-DHC). The B ring of 7-DHC is broken by ultraviolet light B (UVB) to form previtamin D3 which isomerizes in a temperature-dependent process to form vitamin D3. In the same way, ergosterol in plants and fungi when exposed to UVB forms previtamin D2 that isomerizes to vitamin D2. Vitamin D2 differs from vitamin D3 only in the side chain in having a double bond between C22 and C23 and a methyl group at C24
Vitamin D Metabolism
The three main steps in vitamin D metabolism, 25-hydroxylation, 1α-hydroxylation, and 24-hydroxylation, are all performed by cytochrome P450 mixed function oxidases (CYPs). These enzymes are located either in the endoplasmic reticulum (ER) (e.g., CYP2R1) or in the mitochondria (e.g., CYP27A1, CYP27B1, and CYP24A1). The ER enzymes utilize nicotinamide adenine dinucleotide phosphate (NADPH)-dependent P450 reductase as their electron donor, whereas the electron donor for mitochondrial enzymes is a complex of ferredoxin and ferredoxin reductase. Only the cytochrome P450 of the enzyme complex is specific for the substrate being hydroxylated. At this point, only CYP2R1 and CYP24A1 have been crystallized, but their homology with the other vitamin D-metabolizing enzymes suggests common structural features. These include 12 helices (A–L) and loops and a common prosthetic group, namely, the iron-containing protoporphyrin IX (heme) linked to the thiolate of cysteine. The I helix traverses the center of the enzyme above the heme where a threonine or serine and aspartic or glutamic acid pairing is essential for catalytic activity [7]. The ER enzyme CYP2R1 contains additional two helices thought to form a substrate channel in the bilayer of the ER [7] with the B′ helix serving as a gate closing on substrate. It is not known if such structures exist in the mitochondrial enzymes. With this overview, consideration of the individual enzymes follows.
25-Hydroxylase . The liver is the major but not the sole source of 25OHD production from vitamin D. A number of enzymes (all CYPs) have been shown to have 25-hydroxylase activity. CYP27A1 is the only known mitochondrial 25-hydroxylase, initially identified as a sterol 27-hydroxylase involved in bile acid synthesis. Its tissue distribution is wide, not limited to the liver. A number of studies have cast doubt on its being the main 25-hydroxyase. First of all, CYP27A1 does not 25-hydroxylate D2. Secondly, at least in the mouse, deletion of CYP27A1 actually increases 25OHD levels [8]. Finally, mutations of CYP27A1 in humans result in cerebrotendinous xanthomatosis with abnormal bile and cholesterol metabolism, but not rickets [9]. More recently, a microsomal 25-hydroxylase, CYP2R1, was identified in mouse liver [10]. CYP2R1 25-hydroxylates both D2 and D3 with comparable kinetics. Its expression is more tissue limited (primarily liver and testes), and this expression is increased in mice in which CYP27A1 is deleted, probably accounting for the rise in 25(OH)D in the CYP27A1-null mouse. In contrast to the CYP27A1 deletion, deletion of CYP2R1 does reduce 25(OH) (by 50%), but not to zero [8]. Even if both CYP2R1 and CYP27A1 are deleted, the blood level of 25OHD does not fall to zero. Moreover, these deletions do not significantly affect circulating levels of calcium and phosphate [8] indicating compensation by other enzymes with 25-hydroxylase activity. That said, a human mutation in CYP2R1 (leu99pro) was found in a Nigerian with severe bone disease associated with biochemical evidence of rickets [11], and in vitro testing determined that this mutation had a significant effect on CYP2R1 activity. Other enzymes including the drug-metabolizing enzyme CYP3A4 have 25-hydroxylase activity [12], but CYP2R1 appears to be the major 25-hydroxylase .
1α-Hydroxylase . The kidney is not the only tissue capable of producing 1,25(OH)2D, although it is the major source of circulating levels of 1,25(OH)2D. CYP27B1 is the only enzyme recognized to have 25-OHD 1α-hydroxylase activity as proven by its cloning by several laboratories from different tissues [13,14,15,16]. Mutations in CYP27B1 cause a hereditary form of rickets known as pseudo-vitamin D deficiency due to inadequate 1,25(OH)2D production. These individuals respond to 1,25(OH)2D but not to vitamin D itself [13,14,15,16]. CYP27B1 is highly homologous with the mitochondrial CYPs involved with vitamin D metabolism: CYP27A1 and CYP24A1. As mentioned above, the kidney is not the only tissue expressing CYP27B1, and regulation of this critical enzyme for vitamin D metabolism differs among the tissues in which it is expressed [17]. Examples of extrarenal CYP27B1 expression include the epithelial cells in the skin, lungs, breast, intestine, and prostate; endocrine glands including the parathyroid gland, pancreatic islets, thyroid, testes, ovary, and placenta; cells of the immune system including macrophages, T and B lymphocytes, and dendritic cells; osteoblasts and chondrocytes; and a variety of tumors derived from these cells. In the kidney, CYP27B1 is tightly regulated primarily by three hormones, PTH, FGF23, and 1,25(OH)2D itself (Fig. 1.2). PTH stimulates, whereas FGF23 inhibits CYP27B1. Increased levels of calcium and phosphate suppress CYP27B1 activity primarily by inhibiting PTH secretion (calcium) and stimulating FGF23 secretion from bone (phosphate), respectively, although these ions can have direct effects on renal CYP27B1 on their own [18, 19]. Whether 1,25(OH)2D has a direct inhibitory effect on CYP27B1 in the kidney or regulates 1,25(OH)2D levels indirectly remains unclear. 1,25(OH)2D has been reported to inhibit CYP27B1 expression directly through a complex mechanism involving VDR and a vitamin D inhibitory receptor (VDIR) that brings both histone deacetylases (HDACs) and DNA methyl transferases to the promoter of CYP27B1 inhibiting its transcription [20]. This observation has not been confirmed by other investigators. 1,25(OH)2D acts indirectly by inhibiting PTH and increasing FGF23 secretion. Moreover, 1,25(OH)2D induces CYP24A1 (see below) that metabolizes and thus reduces 1,25(OH)2D levels as well as its precursor 25(OH)D.

Vitamin D metabolism . The liver converts vitamin D to 25-OHD. The kidney converts 25-OHD to 1,25-(OH)2D and 24,25-(OH)2D. Control of metabolism is exerted primarily at the level of the kidney, where low levels of serum phosphorus, calcium, and fibroblast growth factor 23 (FGF23) and high levels of parathyroid hormone (PTH) favor production of 1,25-(OH)2D, whereas high serum levels of phosphorus, calcium, FGF23, and 1,25-(OH)2D and low levels of PTH favor 24,25-(OH)2D production
Regulation of extrarenal CYP27B1 differs from that in the kidney. This has been best studied in keratinocytes and macrophages. In keratinocytes, neither PTH nor FGF23 seem to play a role. Moreover, 1,25(OH)2D does not have a direct effect on CYP27B1 expression. Rather, 1,25(OH)2D regulates its own levels in the keratinocyte by inducing CYP24A1 [16]. However, CYP24A1 induction by 1,25(OH)2D and/or its function in macrophages is blunted [21]. The mechanism appears to involve the expression of a truncated form of CYP24, which includes the substrate-binding domain but not the mitochondrial targeting sequence [22]. Cytokines such as tumor necrosis factor-α (TNFα) [23] and interferon-γ (IGFγ) [24] appear to be the major regulators of CYP27B1 activity in the keratinocyte and macrophage [21, 25,26,27], although FGF23 has been shown to be inhibitory in monocytes [28]. In parathyroid cells, FGF23 is reported to stimulate CYP27B1 expression, opposite of its actions in the kidney [29].
24-Hydroxylase. Like CYP27B1, CYP24A1 is the only known 24-hydroxylase . This enzyme has both 24-hydroxylase and 23-hydroxylase activity, the ratio of which is species dependent [30]. The human enzyme has both, but the rat enzyme is primarily 24-hydroxylase [31]. Single base pair mutations can shift the ratio of 23- to 24-hydroxylase activity [32]. The 24-hydroxylase pathway leads to the production of calcitroic acid, a biologically inert end product, whereas the 23-hydroxylase pathway leads to the biologically active 1,25–26,23 lactone. All steps are performed by one enzyme [31]. 1,25(OH)2D and 25(OH)D are both substrates for CYP24A1. The initial product of CY24A1 metabolism of 1,25(OH)2D, 1,24,25(OH)3D, has approximately 1/10th the affinity of 1,25(OH)2D for the VDR and has biologic activity. 24,25(OH)2D may have biologic activity in the growth plate [33], although such a role is controversial. The biologic impact of deleting CYP24A1 results in defective mineralization of intramembranous (not endochondral) bone [34], but this appears to be due to large increases in 1,25(OH)2D and not to a deficiency of 24,25(OH)2D [34]. Inactivating mutations in CYP24A1 are one cause of idiopathic infantile hypercalcemia, which presents with severe hypercalcemia, hypercalciuria, and nephrocalcinosis, decreased PTH, low 24,25(OH)2D, and inappropriately normal to high 1,25(OH)2D [35]. In this syndrome, the failure of CYP24A1 to control 1,25(OH)2D levels appears to account for the phenotype .
Most tissues express CYP24A1, and increased expression is a nearly universal marker of 1,25(OH)2D action on that tissue. The promoter of CYP24A1 contains two vitamin D response elements (VDREs) upstream of the transcriptional start site to which VDR/RXR bind along with other transcription factors [36]. More distant sites downstream of the human CYP24A1 gene to which histone acetyl transferases and RNA polymerase II are recruited have been shown to play a role in CYP24A1 induction [37]. In the kidney, CYP24A1 regulation is the reciprocal of that of CYP27B1 in that PTH limits the induction of CYP24A1 by 1,25(OH)2D [38, 39], whereas FGF23 increases its expression [40]. FGF23 has a similar role in the uterus [41], but this has not been studied in other tissues. On the other hand, PTH enhances 1,25(OH)2D induction of CYP24A1 transcription in osteoblasts through the same apparent mechanism, namely, the cAMP/PKA pathway, by which it reduces CYP24A1 induction in the kidney [42]. Thus, like the regulation of CYP27B1, the regulation of CYP24A1 can be tissue specific .
3-Epimerase. The C-3 epimers of the vitamin D metabolites have recently gained widespread attention mainly as contaminants in LC-MS/MS assays of these metabolites. This issue is particularly important in assessing 25OHD levels in infants where levels of the C-3 epimer of 25OHD can equal or exceed that of 25OHD [43]. However, these epimers have been recognized for decades. 3-Epimerase activity was first identified in keratinocytes where it produces the 3-epi form of 1,25(OH)2D [44] but has also been found in colon cancer cells (Caco2), parathyroid cells, osteoblasts, and hepatocyte-derived cells (HepG2). Surprisingly, this epimer has not been found in renal preparations [43]. The enzyme has not yet been purified and so remains an activity that could be due to several enzymes. The 3-epimerase isomerizes the C-3 hydroxy group of the A ring from the alpha to beta orientation of all natural vitamin D metabolites. This does not affect subsequent metabolism but does reduce binding to DBP of the 3-epi form of 25(OH)D relative to 25OHD and binding to VDR of the 3-epi form of 1,25(OH)2D relative to 1,25(OH)2D [45]. Thus, the C-3 epimers have reduced biologic activity in general [45], but, surprisingly, the 3-epi form of 1,25(OH)2D appears to be equipotent to 1,25(OH)2D with respect to PTH suppression [46]. The extra effort required to measure the C-3 epimers separately from the classic metabolites may prove necessary especially in children to accurately determine vitamin D status.
CYP11A1. CYP11A1 , known also as the side chain cleavage enzyme, is a key enzyme essential for steroidogenesis. Recently, CYP11A1 has been shown to provide an alternative pathway for vitamin D activation converting vitamin D to 20(OH)D [47]. 20(OH)D, or its metabolite 20,23(OH)2D, appear to have activity similar to 1,25(OH)2D at least for some functions. It is unclear whether these metabolites require further metabolism by CYP27B1 to be active. The biologic significance of this pathway remains unclear, as it does not compensate for animals lacking CYP27B1.
Transport of Vitamin D Metabolites in the Blood and Their Cellular Uptake
The vitamin D metabolites are transported in blood bound primarily to vitamin D-binding protein (DBP) (85–88%) and albumin (12–15%) [48,49,50]. The normal range of DBP concentrations is 4-8 μM, such that DBP is only about 1–2% saturated by normal levels of the vitamin D metabolites. DBP has high affinity for these metabolites (Ka = 5 × 108M−1 for 25OHD and 24,25(OH)2D, 4 × 107M−1 for 1,25(OH)2D and vitamin D). Thus, under normal circumstances, only approximately 0.03% 25OHD and 24,25(OH)2D and 0.4% 1,25(OH)2D are free [49,50,51]. Conditions such as liver disease, nephrotic syndrome, and acute illness resulting in reduced DBP and albumin levels will lead to a reduction in total 25OHD and 1,25(OH)2D levels without necessarily affecting the free concentrations [52,53,54,55]. On the other hand, oral (not transdermal) estrogens and pregnancy [49] increase DBP levels and so may increase total levels of the vitamin D metabolites without increasing (and may even decrease) the free concentrations [49, 56]. High levels of 25(OH)D in cases of vitamin D intoxication can increase the degree of DBP saturation such that despite the normal levels of total 1,25(OH)2D, the free concentrations of 1,25(OH)2D are increased [57] contributing to the hypercalcemia/hypercalciuria observed in these cases. DBP is a 58 kDa protein with 458 amino acids that is homologous to albumin and α-fetoprotein (αFP) (40% homology at the nucleotide level, 23% at the amino acid level) [58]. DBP like albumin and αFP is made primarily but not exclusively in the liver. Other sites include the kidney, testes, and fat.
Direct measurement of the free levels of the vitamin D metabolites becomes important if most cells take up only the free concentration, a hypothesis known as the free hormone hypothesis . An early articulation of this hypothesis comes from observations that patients with nephrotic syndrome had low levels of circulating thyroid hormone (assessed as PBI) and increased urinary losses of PBI but yet appeared clinically euthyroid [59]. This suggested to the authors that the supply of hormone to the tissues in these patients was normal. Similar observations have recently been made in patients with nephrotic syndrome with regard to lack of changes in serum calcium, phosphate, PTH, and bone mineral density measurements despite lower vitamin D metabolite levels and increased urinary losses of DBP [53]. Similar conclusions regarding the importance of the free levels of vitamin D metabolites come from observations that the increase in 1,25(OH)2D levels with administration of oral contraceptives or during the third trimester of pregnancy is associated with a parallel increase in DBP but not with changes in calcium metabolism, at least until the latter stages of pregnancy when the measured free levels of 1,25(OH)2D increase despite the increase in DBP [49, 60]. The concept that the major role of DBP is as a blood transporter of the vitamin D metabolites is further demonstrated in mice in which the DBP gene was deleted. Although these mice lost substantial amounts of the vitamin D metabolites in the urine and their circulating levels of 25(OH)D were very low, they did not develop evidence of rickets until put on a low-vitamin D diet [61].
However, the free hormone hypothesis does not apply to all tissues. The renal tubule differs from most other tissues in its mechanism for at least 25(OH)D uptake and likely for all vitamin D metabolites. DBP and its bound 25(OH)D are filtered in the glomerulus and reabsorbed in the proximal tubule through endocytosis mediated by the megalin/cubilin complex. This provides 25(OH)D for further metabolism in the kidney tubule [62, 63]. The megalin/cubilin complex is not specific for DBP, but when megalin is deleted, the major protein lost in the urine is DBP, bone growth is slowed, and the skeleton is osteopenic [62]. Similar if less severe results were obtained with cubilin deletion [63]. The parathyroid gland and placenta also express megalin/cubilin [64], but at this point, experiments to determine the impact of either megalin or cubilin deletion from these tissues have not been reported.
Vitamin D Mechanism of Action
The best-known and most widely studied actions of vitamin D involve genomic actions regulated by 1,25(OH)2D interacting with its receptor VDR. However, a growing body of literature is concerned also with the nongenomic actions of 1,25(OH)2D, some mediated also by VDR and others by a nonnuclear receptor variously named membrane-associated rapid response steroid (MARRS)-binding protein, ERp57/GRp58/ERp60, and protein disulfide isomerase family A member 3 (Pdia3). In this section, the genomic and nongenomic actions will be described separately.
Genomic actions. All genomic actions of 1,25(OH)2D are mediated by the VDR. VDR is a transcription factor and member of the steroid hormone nuclear receptor family with which it has substantial homology especially in the DNA-binding domain. Based on the original cloning of the estrogen and glucocorticoid receptors, these nuclear hormone receptors were recognized to have six distinct domains: A–F. The A/B domain is the N-terminal region, known in other receptors as the activation domain 1. In VDR, this domain is quite short (24 amino acids), and in the f allele of the FokI polymorphism, it is further shortened by 3 amino acids [65]. The C domain is the DNA-binding domain with 65 amino acids containing 2 zinc fingers that bind to the grooves of the DNA at discrete sites called vitamin D response elements (VDREs). The highly flexible hinge region (domain D) with 143 amino acids separates the DNA-binding domain from the E/F domain (195 amino acids) that contains the ligand-binding domain and terminal activation domain (AF2). This domain also serves the function of dimerization with VDR partners (e.g., RXR) and binding of corepressors as well as coactivators (in AF2). These domains are illustrated in Fig. 1.3. The structure of the ligand-binding domain has been solved by X-ray crystallography [66]. It is comprised of 12 helices. Helix 12 serves as a gating mechanism closing around the incorporated ligand (i.e., 1,25(OH)2D) and forming an interface for coactivators and the nuclear hormone dimerization partners such as RXR. As mentioned above, the VDR binds to select regions in the genome called VDREs. The sequence of VDREs is highly variable, but those with the highest affinity for VDR are direct repeats of hexanucleotides with a spacing of three nucleotides between the half sites. This motif is called a DR3. VDR binding to its VDRE then recruits coregulatory complexes required for its genomic activity. These coregulatory complexes are required to remodel the chromatin, altering the condensation state by histone modifications to create binding sites for additional coregulatory complexes and facilitating the link to the RNA polymerase II to initiate transcription. The complexes that participate in these functions are the ATPase-containing SWI/SNF complex involved with remodeling the chromosome [67], complexes with activities that modify the histones via histone acetyl transferases (HATs) such as the coactivator CBP/P300 complex containing the steroid receptor coactivator family (SRC 1–3), histone methyl transferases (HMTs), and histone deacetylases (HDACs), which are part of the corepressor complexes of SMRT and NCoR, histone demethylases (DMTs) [68], and the mediator complex that is thought to link the RNA polymerase to the transcription start site [69]. The SRC and mediator complexes include a subunit that directly binds to the VDR generally through an LXXLL motif. Corepressors such as SMRT and NCoR, on the other hand, bind through a LXXXIXXX(I/L) motif. These complexes can be both gene and cell specific, enabling the selectivity of 1,25(OH)2D action among cell types on which it acts.

Domains of the VDR . The N terminus of VDR, domains A/B, forms the short AF1 domain. The C domain contains the DNA domain containing the two zinc fingers. The D domain includes the hinge region. The E/F domains include the ligand-binding domain and the C terminal AF2 domain to which coactivators bind following ligand binding
The newer techniques of RNA-seq and ChIP-seq [70, 71] have markedly expanded our understanding of vitamin D mechanism of action at the genomic level. Moreover, the development of CRISPR/Cas9 to specifically and relatively quickly delete regions of the genome has enabled testing of the various putative regulatory regions of the genome for functional significance [72]. For example, in the mouse osteoblast, 1200 VDR binding sites were found under basal (i.e., no 1,25(OH)2D) conditions, whereas 8000 sites were observed following 1,25(OH)2D administration [73]. In a separate study with human lymphoblastoid cell lines treated with 1,25(OH)2D, 2776 VDR binding sites were found altering the expression of 229 genes [74]. Although there is some overlap among different cell types, the profile of VDR binding sites and genes activated varies substantially from cell to cell as well as at different times after exposure to 1,25(OH)2D in the same cell [75]. These VDR binding sites can be anywhere in the genome, often quite distant from the coding region of the gene being regulated, and just because VDR binds to a site does not mean that the site is functional with respect to regulation of the expression of that gene in that cell. Other transcription factors and their binding sites are generally found in association with VDR at its binding site. In osteoblasts, for example, these include RUNX2 and C/EBP α and β, among others [76, 77]. These sites often demonstrate a distinct epigenetic histone signature involving methylation and/or acetylation of lysines within H3 and H4 [78]. In general, a gene is regulated by more than one enhancer element [71], and the adjacent transcription factors may vary altering the regulation of that gene. An interesting example of this is the gene, Tnfsf11, that encodes RANKL. This gene is regulated by parathyroid hormone (PTH), a number of cytokines in addition to 1,25(OH)2D. It plays a role not only in osteoclast activation but in immune regulation and other cellular functions. Five strong VDR binding sites (D1–D5) were identified by ChIP-seq up to 75 kb upstream of the transcription start site [79]. PTH-induced CREB binding was found at some of these sites, and IL6-induced STAT3 binding was found at another. These sites in combination with additional sites even further upstream seem to regulate which cell (e.g., osteoblast or hematopoietic cell) and/or which hormone (PTH, cytokine, 1,25(OH)2D) regulates the expression of Tnfsf1 [80, 81] in that cell. Thus, the key aspects of genomic regulation by VDR and its ligand 1,25(OH)2D can be summed up as follows:
-
1.
The profile of VDR binding sites in the genome varies from cell to cell and with time after 1,25(OH)2D administration.
-
2.
Most but not all binding sites require 1,25(OH)2D for VDR binding.
-
3.
The VDR binding sites are generally DR3 in which VDR binds in combination with RXR.
-
4.
VDR binding sites can be located nearly anywhere in the gene and may be close to or thousands of base pairs away from the transcription start site.
-
5.
The VDR binding sites are generally part of a cluster containing binding sites for a number of other transcription factors, which like the profile of the VDR binding sites themselves are cell specific.
Nongenomic actions . 1,25(OH)2D also exerts effects that are too rapid to involve a genomic action. The earliest description of this nongenomic action involved rapid stimulation of intestinal calcium transport in a vitamin D replete chick, called transcaltachia [82]. Of interest is that this phenomenon did not occur in a vitamin D-deficient chick indicating that the vitamin D-induced mechanisms for calcium transport need to be in place. Analogs of 1,25(OH)2D that had no apparent genomic activity were as effective as 1,25(OH)2D itself. Other examples emerged including effects on the chondrocytes in the growth plate [83] and keratinocytes in the skin [84]. Two receptors have been identified. One is the VDR itself albeit in a different configuration to enable binding by nongenomic VDR agonists [85]. The second is a novel receptor for 1,25(OH)2D variably known as membrane-associated rapid response steroid (MARRS)-binding protein, ERp57/GRp58/ERp60, and protein disulfide isomerase family A member 3 (Pdia3) as mentioned earlier [86]. These receptors are located in the membrane within caveolae/lipid rafts [87] where they are poised to activate kinases, phosphatases, and ion channels. This latter receptor has not been crystallized so the binding of 1,25(OH)2D to it is not known. On the other hand, the VDR has been crystallized, and the structure deduced indicated that the binding pocket in VDR would accommodate only agonists with a 6s-trans configuration. However, analogs with the 6s-cis configuration are active in inducing these nongenomic actions. Mizwicki and Norman [85] proposed an alternative model in which the 6s-cis analogs could fit into an alternative pocket in the VDR, although crystallographic evidence for this configuration has not been obtained. At this point, the physiologic significance of the nongenomic actions of 1,25(OH)2D remains unclear, although deletion of the MARRS (Pdia3) gene from the intestine in vivo [88], from osteoblasts in vitro [89], or in heterozygotes (global knockouts are embryonic lethal) [90] does disrupt the rapid actions of 1,25(OH)2D in those cells with altered intestinal calcium transport and bone and cartilage abnormalities in the relevant in vivo models.
Vitamin D Regulation of Cellular Function
In this section, I will first discuss the “classic” target tissues of vitamin D involved with bone mineral homeostasis, following which I will discuss the “nonclassic” tissues which although influenced by calcium regulation are not in themselves central to the regulation of calcium homeostasis.
Classic Vitamin D Target Tissues
Intestine . Intestinal calcium absorption , in particular the active component of transcellular calcium absorption, is one of the oldest and best-known actions of vitamin D. Absorption of calcium from the luminal contents of the intestine involves both transcellular and paracellular pathways. The transcellular pathway dominates in the duodenum, and this is the pathway primarily regulated by 1,25(OH)2D [91, 92]. Calcium entry across the brush border membrane (BBM) occurs down a steep electrical-chemical gradient and requires no input of energy. This is achieved by changes in membrane fluidity, a 1,25(OH)2D induced calcium channel TRPV6 in the BBM, and a 1,25(OH)2D induced translocation of calmodulin to the BBM. Calcium movement through the cell occurs with minimal elevation of the intracellular free calcium concentration [93] by packaging the calcium in calbindin-containing vesicles [94,95,96] that form in the terminal web following 1,25(OH)2D administration. Removal of calcium at the basolateral membrane must work against this gradient, and energy is required. This is achieved by the CaATPase (PMCA1b) and the sodium/calcium exchanger NCX. The first step, calcium entry across the BBM, is accompanied by changes in the lipid composition of the membrane including an increase in linoleic and arachidonic acid [97, 98] and an increase in the phosphatidylcholine/phosphatidylethanolamine ratio [99]. These changes are associated with increased membrane fluidity [98] and are rapid and nongenomic [99]. The epithelial-specific calcium channel, TRPV6, is homologous to TRPV5, a calcium channel originally identified in the kidney [100, 101]. Mice null for TRPV6 have a partial reduction in intestinal calcium transport [102], although the reduction is modest [103]. Calmodulin also participates in intestinal calcium transport. It is the major calcium-binding protein in the microvillus [104], and its concentration in the microvillus is increased by 1,25(OH)2D but not its overall levels in the cell and does not require new protein synthesis [105]. Inhibitors of calmodulin block 1,25(OH)2D-stimulated calcium uptake by BBMV [106]. Calmodulin has been shown to regulate TRPV6 activity [107]. Calmodulin is bound to myosin 1A (myo1A), binding that is increased by 1,25(OH)2D [105]. This complex increases with differentiation of the intestinal epithelial cell as does the capacity for calcium transport [108]. However, its exact role in calcium transport is unclear in that mice null for myo1A do not show reduced intestinal calcium transport (Bikle and Munson, unpublished observations). Calcium entering the cytoplasm across the BBM must then be moved into and through the cytoplasm without disrupting the function of the cell. In the vitamin D-deficient animal, calcium accumulates along the inner surface of the plasma membrane of the microvilli [109, 110], from which calcium is released following vitamin D or 1,25(OH)2D administration to enter the cytoplasm where it is found in mitochondria and calbindin-containing vesicles within the terminal web [94, 95, 109, 110]. The vesicles appear to shuttle the calcium to the lateral membrane, where it is pumped out of the cell. Calbindin is the dominant calcium-binding protein in the cytoplasm [104, 111]. However, the role of calbindin in intestinal calcium transport does not appear to be critical in that mice null for calbindin9k grow normally and have normal intestinal calcium transport and their serum calcium levels and bone mineral content are equivalent to wild-type mice regardless of the calcium content of the diet [112]. Moreover, even the double deletion of both TRPV6 and calbindin does not completely block 1,25(OH)2D-stimulated calcium transport [113]. The PMCA1b and NCX at the basolateral membrane are responsible for removing calcium from the cell against the same steep electrochemical gradient as it facilitates calcium entry at the BBM [114]. PMCA1b is induced by 1,25(OH)2D [115]. Calmodulin activates the pump, but calbindin may do likewise [116]. The effect of PMCA1b deletion on calcium transport has not been evaluated. The role of NCX is not considered to be as important as PMCA1b for intestinal calcium transport [117]. It is clear that both genomic and nongenomic actions of 1,25(OH)2D are involved in regulating intestinal calcium transport, but much remains to be learned regarding their relative importance .
Although less studied, intestinal phosphate transport is also under the control of vitamin D. Active phosphate transport is greatest in the jejunum, in contrast to active calcium transport that is greatest in the duodenum. NaPIIb, a sodium phosphate transporter in the small intestine homologous to the type IIa sodium phosphate transporter in the kidney, has been cloned and sequenced [118]. It is induced by 1,25(OH)2D [119], but the impact of deleting this transporter has not been reported. Moreover, it may not be the only or even the major phosphate transporter in the intestine [120]. Transport of phosphate through the cytoplasm is not well understood but, like calcium, may occur in vesicles [121].
Bone . Nutritional vitamin D deficiency, altered vitamin D responsiveness such as vitamin D receptor mutations (hereditary vitamin D-resistant rickets), and decreased 1,25(OH)2D production as in mutations in the CYP27B1 gene (pseudo-vitamin D deficiency) all have rickets as their main phenotype indicating the critical role of vitamin D and in particular 1,25(OH)2D in bone development and turnover. Like most other cells, VDR is found in bone cells [122, 123], and vitamin D metabolites have been shown to regulate many processes in bone. The VDR makes its first appearance in the fetal rat at day 13 of gestation with expression in osteoblasts and the proliferating and hypertrophic chondrocytes by day 17 [124]. However, fetal development is quite normal in vitamin D-deficient rats [125] and VDR knockout mice [126] suggesting that vitamin D and the VDR are not critical for skeletal formation. Rickets develops postnatally, becoming most manifest after weaning. Even at this point, the rickets resulting from vitamin D deficiency or VDR mutations (or knockouts) can be corrected by supplying adequate amounts of calcium and phosphate either by infusions or orally [127, 128]. Moreover, expressing the VDR in the intestine is sufficient to reverse the skeletal changes observed in the VDR-null mouse [129]. These observations suggest that the role of vitamin D on bone is primarily or totally indirect. However, arguing for a physiologically nonredundant direct action of vitamin D on bone is the development of osteoporosis and decreased bone formation in VDR- or CYP27B1-null mice that is not corrected by the high-calcium/high-phosphate diet [130]. In vivo studies of the impact of vitamin D on bone are complicated by the impact of vitamin D on systemic calcium homeostatic mechanisms such as PTH and FGF23. Furthermore, within bone, the vitamin D metabolites can alter the expression and/or secretion of a large number of skeletally derived factors including insulin-like growth factor-1 (IGF-I), transforming growth factor β (TGFβ) [131], vascular endothelial growth factor (VEGF) [132], and a number of cytokines all of which can exert effects on bone of their own as well as modulate the actions of the vitamin D metabolites on bone. Similarly, species differences, differences in responsiveness of bone and cartilage cells according to their states of differentiation, and differences in responsiveness in terms of the vitamin D metabolite being examined all contribute to the complexity and uncertainty in distinguishing the direct and indirect roles of the vitamin D metabolites on bone formation and turnover.
The impairment of endochondral bone formation observed in vitamin D deficiency is associated with decreased alkaline phosphatase activity of the hypertrophic chondrocytes [133], alterations in the lipid composition of the matrix [134] perhaps secondary to reduced phospholipase activity [135], and altered proteoglycan degradation [136] due to changes in metalloproteinase activity [136, 137]. Both 1,25(OH)2D and 24,25(OH)2D appear to be required for optimal endochondral bone formation [33]. Some of these actions of 1,25(OH)2D and 24,25(OH)2D on endochondral bone formation are nongenomic in that they take place with isolated matrix vesicles and membrane preparations from these cells [138]. On the other hand, deletion of the VDR or CYP27B1 specifically from chondrocytes does not have a direct impact on chondrocyte development and maturation but does affect bone through FGF23 regulation of phosphate [139, 140]. As mentioned above, osteoblasts at different stages of differentiation differ in their response to 1,25(OH)2D [141]. In the latter stages of differentiation, rat osteoblasts respond to 1,25(OH)2D with an increase in osteocalcin production [142], but do not respond to 1,25(OH)2D in the early stages. Mice, however, differ from rats in that 1,25(OH)2D inhibits osteocalcin expression [142]. Similar species differences are found for other proteins. Osteocalcin and osteopontin in human and rat cells have well-described VDREs in their promoters [143,144,145], but these genes in mouse cells do not [146]. These maturation-dependent effects of 1,25(OH)2D on bone cell function may explain the surprising ability of excess 1,25(OH)2D to block mineralization leading to hyperosteoidosis [147,148,149] as such doses may prevent the normal maturation of osteoblasts. That said, the phenotype of mice in which the VDR has been deleted in osteoblasts is modest and suggests more of an impact on bone resorption (decreased) than on bone formation [150].
1,25(OH)2D also promotes bone resorption by increasing the number and activity of osteoclasts [151]. It is unclear whether mature osteoclasts contain the VDR [152, 153], but the stimulation of osteoclastogenesis by 1,25(OH)2D is mediated by osteoblasts [154, 155]. 1,25(OH)2D induces a membrane-associated protein known as RANKL (receptor activator of nuclear factor (NF)-kB ligand) in osteoblasts that in combination with mCSF also induced by 1,25(OH)2D in osteoblasts activates RANK on osteoclasts and their hematopoietic precursors to stimulate the differentiation of osteoclast precursors and promote their activity [156]. As discussed earlier, the regulation of RANKL expression involves a number of different hormones working in conjunction with or independent of 1,25(OH)2D.
Kidney. The regulation of calcium and phosphate transport by vitamin D metabolites in the kidney has received less study than that in the intestine, but the two tissues have similar although not identical mechanisms. Most of the calcium in the glomerular filtrate is reabsorbed in the proximal tubule. This is a paracellular, sodium-dependent process with little or no regulation by PTH and 1,25(OH)2D. Regulation of calcium reabsorption by vitamin D takes place in the distal nephron where calcium moves against an electrochemical gradient (presumably transcellular) in a sodium-independent fashion [157]. The majority of phosphate reabsorption also takes place in the proximal tubule but in this case is closely regulated by PTH and FGF23 [158, 159]. In parathyroidectomized (PTX) animals, Puschett et al. [160,161,162] demonstrated acute effects of 25OHD and 1,25(OH)2D on calcium and phosphate reabsorption. Subsequent studies indicated that PTH could enhance or was required for the stimulation of calcium and phosphate reabsorption by vitamin D metabolites [163, 164].
The molecules critical for calcium reabsorption in the distal tubule include the VDR, calbindin, TRPV5, and BLM calcium pump (PMCA1b as in the intestine), a situation similar to the mechanism for calcium transport in the intestine [165]. The calbindin in the kidney in most species is 28 kDa, whereas the 9 kDa form is found in the intestine in most species. The kidney has mostly TRPV5, whereas the intestine is primarily TRPV6. Calmodulin and a brush border myosin I like protein are also found in the kidney brush border, but their role in renal calcium transport has not been explored. Not all distal tubules express these proteins [100, 101, 166, 167] suggesting that not all distal tubules are involved in calcium transport. 1,25(OH)2D upregulates the VDR [168], calbindin [169, 170], and TRPV5 expression [171]. Deletion studies of these proteins are limited.
Phosphate reabsorption in the proximal tubule is mediated at the brush border by sodium-dependent phosphate transporters (NaP2a and NaP2c) that rely on the basolateral membrane Na,K-ATPase to maintain the sodium gradient that drives the transport process [172]. It is not clear whether 1,25(OH)2D regulates the expression or activities of these transporters as it does the homologous NaP2b in the intestine.
Nonclassic Vitamin D Target Tissues
Vitamin D signaling in nonclassic target tissues can be categorized into three different not mutually exclusive actions:
-
1.
Regulation of proliferation and differentiation
-
2.
Regulation of hormone secretion
-
3.
Regulation of immune function
Examples of these mechanisms of action will be discussed in turn.
Regulation of Proliferation and Differentiation
In this section, I will discuss a normal tissue, the skin, as representing a good example of the regulation of proliferation and differentiation by VDR and 1,25(OH)2D, followed by cancer in which such regulation is lost.
Skin . Epidermal keratinocytes express the entire vitamin D metabolic pathway from the production of vitamin D3 from 7-DHC, its conversion to 25(OH)D by CYP27A1 [173] (expression of CYP2R1 has been described in fibroblasts [174] but not in keratinocytes), and its further conversion to 1,25(OH)2D by CYP27B1 [175]. Moreover, the skin also expresses CYP24A1, limiting the levels of 1,25(OH)2D in keratinocytes under vitamin D replete conditions [176, 177]. CYP27B1 is expressed primarily in the basal cells of the epidermis [178], as the cells differentiate the mRNA and protein levels of CYP27B1and its activity decline [179].
1,25(OH)2D regulates keratinocyte differentiation in partnership with calcium [180]. The keratinocytes express the calcium-sensing receptor (CaSR) critical for their response to calcium, and CaSR is induced by 1,25(OH)2D [181]. Keratinocytes grown at calcium concentrations below 0.07 mM continue to proliferate but fail to differentiate. Acutely increasing the extracellular calcium concentration (Cao) above 0.1 mM (calcium switch) initiates the differentiation process. Within hours of the calcium switch, keratinocytes switch from making the basal keratins K5 and K14 and begin making keratins K1 and K10 [182] followed, subsequently, by increased levels of profilaggrin (the precursor of filaggrin, an intermediate filament-associated protein), involucrin, and loricrin (precursors for the cornified envelope) [183, 184]. Loricrin, involucrin, and other proteins [185] are cross-linked into the insoluble cornified envelope (CE) by the calcium-sensitive, membrane-bound form of transglutaminase [186, 187], which like involucrin and loricrin increases within 24 h after the calcium switch [188]. 1,25(OH)2D increases the mRNA and protein levels for involucrin and transglutaminase and promotes CE formation at subnanomolar concentrations in preconfluent keratinocytes [189,190,191,192]. Deletion of either the VDR or CaSR from keratinocytes in vivo [193, 194] also blocks the formation of the lipids that are secreted into the cornified envelope by the lamellar bodies in the stratum granulosum to waterproof the permeability barrier. Moreover, deletion of CYP27B1 from keratinocytes in vitro blocks the induction of antimicrobial peptides that are likewise incorporated into the lamellar bodies and secreted into the cornified envelope as part of the barrier [195]. This will be discussed further in the section on innate immunity.
Calcium affects the ability of 1,25(OH)2D to stimulate keratinocyte differentiation and vice versa [196]. The calcium switch also leads to the rapid redistribution of a number of proteins from the cytosol to the membrane where they participate in the formation of intercellular contacts. These include the calcium-sensing receptor (CaSR), phospholipase C-γ1 (PLC-γ1), src kinases, and E-cadherin/catenin complex. This complex plays a critical role in calcium and vitamin D signaling in the keratinocyte. Besides E-cadherin, it contains phosphatidyl inositol 3 kinase (PI3K), phosphatidyl inositol 4-phosphate 5-kinase 1α (PIP5K1α), and the catenins Ctnna1, Ctnnb1, and Ctnnd1 (α- and β-catenin, p120). These all play important roles in calcium- and vitamin D-induced differentiation [197,198,199,200,201,202]. PI3K and PIP5K1α sequentially phosphorylate PIP and PIP2 to PIP3 that activates PLC-γ1. PLC-γ1 cleaves PIP2 to form IP3 and diacylglycerol. IP3 releases calcium from intracellular stores, important for the sustained increase in intracellular calcium (Cai) required for the differentiation process [203]. Diacylglycerol along with calcium activates protein kinase C alpha (PKCα) that also promotes differentiation [204]. 1,25(OH)2D is required for the formation of the E-cadherin/catenin complex and induces some of its constituents such as PLC-γ1 [205]. Deletion of the CaSR from keratinocytes reduces their stores of calcium and like the deletion of VDR blocks their response to extracellular calcium (Cao) including the formation of the E-cadherin/Ctnn complex and the permeability barrier [199, 206]. Thus, calcium and vitamin D signaling are essential partners for keratinocyte differentiation.
Cancer. The antiproliferative, prodifferentiating effects of vitamin D signaling on many, if not all, cell types have raised the hope that vitamin D, 1,25(OH)2D, or one or more of its analogs would prove useful in the prevention and/or treatment of cancer. This section will focus on the antiproliferative/prodifferentiating actions as shown in a number of cellular and animal studies, but a large number of other mechanisms have been invoked as recently reviewed [207].
Cellular mechanisms . Most tumors express the VDR and often express CYP27B1, but their expression is often lost as the tumor undergoes progressive dedifferentiation [208,209,210]. On the other hand, CYP24A1 expression is often increased in tumors and is associated with resistance to 1,25(OH)2D [210, 211]. These changes in vitamin D metabolism and responsiveness reduce the ability of 1,25(OH)2D to control the proliferation and differentiation of these tumors. Moreover, a number of miRNAs have been identified to be regulated by 1,25(OH)2D/VDR relevant to their antiproliferative actions [212]. These include increased expression of miR145, which blocks the expression of E2F3, a key regulator of proliferation [213] or miR-32 that blocks the proapoptotic protein Bim that somewhat paradoxically actually protects the cell (human myeloid leukemia) from AraC-induced apoptosis [214].
1,25(OH)2D typically causes arrest at the Go/G1 and/or G1/S transitions in the cell cycle associated with a decrease in cyclins and an increase in the inhibitors of the cyclin-dependent kinases (CDK) such as p21cip1 and p27kip1 [215, 216]. One class of transcription factors that have been shown to be involved in suppression of proliferation and increased apoptosis is the family of Forkhead box O (FoxO) proteins. 1,25(OH)2D promotes their interaction with VDR as well as their regulation by Sirt1 and protein phosphatase 1 maintaining these proteins in the transcriptionally active dephosphorylated state [217]. 1,25(OH)2D reduces the expression of proproliferative genes such as Myc, Fos, and Jun [77] while stimulating the expression of IGF-binding protein 3 (IGFBP3) in prostate and breast cancer cells, thus blocking the proproliferative actions of IGFs 1 and 2 [218, 219]. In epithelial cells, 1,25(OH)2D stimulates the expression of TGFβ2, which is antiproliferative in these cells [220,221,222], and suppresses components of the hedgehog pathway (HH), which when overexpressed result in basal cell carcinomas (BCC) [223, 224]. 1,25(OH)2D inhibits EGF stimulation of proliferation by inhibiting the expression of its receptor in breast cell lines [225]. Constitutive activation of the wnt/β-catenin pathway is the cause of most colorectal cancers (CRC). When activated, β-catenin enters the nucleus where it binds to TCF/LEF sites in genes promoting proliferation (e.g., cyclin D1). 1,25(OH)2D/VDR blocks this pathway both by binding to β-catenin, restricting its proproliferative actions in the nucleus, and by stimulating the formation of the E-cadherin/catenin complex in the cell membrane to which β-catenin binds restricting its translocation to the nucleus [226]. Moreover, 1,25(OH)2D can increase the expression of the wnt inhibitor dickkopf (DKK)-1 [227] while inhibiting that of the wnt activator DKK-4 [228] in colon cancer cells.
1,25(OH)2D promotes the apoptosis of a number of cell types [229, 230] by stimulating the expression of proapoptotic genes such as GOS2 (Go/G1 switch gene 2) [216], Bax [231], DAP (death-associated protein)-3, CFKAR (caspase 8 apoptosis-related cysteine peptidase), FADD (Fas-associated death domain), and caspases (e.g., caspase 3, 4, 6, and 8) [221] in a variety of cell lines, while suppressing the expression of proapoptotic genes such as Bcl2 and Bcl-XL in these and others [231,232,233].
Animal studies. Animal studies demonstrating the efficacy of 1,25(OH)2D in preventing or slowing the progression of different tumors are numerous with those of the colorectum (CRC), breast, prostate, and skin being most studied in both animal and human studies. A Western diet low in calcium and vitamin D fed to mice increases their risk of CRC, a risk that can be reversed with a diet supplemented with calcium and vitamin D [234]. Tumors induced by the combination of azoxymethane and dextran sulfate can be at least partially prevented with the administration of vitamin D metabolites [235]. Activation of the wnt/β-catenin pathway caused by mutations in adenomatous polyposis coli (APCmin) develops tumors much faster on a Western diet [236], on a vitamin D-deficient diet [237], or when bred with VDR-null mice [238]. As for CRC, the number of breast cancers induced in this case by dimethylbenzanthracene (DMBA) is increased when the rats are fed a Western diet [239] or when DMBA is given to VDR-null mice [240]. VDR agonists prevent the growth of breast cancer xenografts [241]. Vitamin D analogs can also inhibit the growth of prostate cancer regardless of androgen receptor status [242]. PC3 prostate cancer cells in bone grow more rapidly when the mice are fed a vitamin D-deficient diet [243]. Similarly, breeding the transgenic prostate tumor model, LPB-Tag with VDR-null mice, stimulates the growth of these tumors [244], whereas high doses of 1,25(OH)2D suppress the development of tumors in the TRAMP model of prostate cancer [245]. The most common skin cancers are squamous cell carcinomas (SCC) and basal cell carcinomas (BCC). In animals, these tumors are typically induced by DMBA topically or orally often followed by repeated topical application of phorbol esters or by chronic exposure to UVB. Nearly all VDR-null mice treated with DMBA or chronic UVB exposure develop skin tumors, but not their controls [224, 246]. Topical 1,25(OH)2D is protective at least of the early effects of UVB on markers of DNA damage such as cyclobutane pyrimidine dimers [247].
Clinical studies . Most of the evidence supporting a role for vitamin D in tumor prevention in humans is epidemiologic. The evidence for a link between vitamin D and CRC is reasonably strong [248, 249]. One such study found a risk reduction of 0.88 (CI 0.8–0.96) comparing the highest to lowest levels of vitamin D intake [248] and 0.67 (CI 0.54–0.80) comparing the highest to the lowest serum 25OHD levels [248]. In breast cancer, the largest cohort studies [250, 251] (Nurses’ Health Study with 88,891 participants and Women’s Health Study with 31,487 participants) showed a relative risk (RR) of 0.72 (CI 0.55–0.94) and 0.65 (CI 0.42–1.00), respectively, but only in premenopausal women. However, several meta-analyses did not make this distinction regarding menopause status. One such study demonstrated a risk reduction of 0.55 (CI 0.38–0.80) comparing the highest quintile of 25OHD levels to the lowest [252]. Another meta-analysis showed a RR of 0.89 (0.82–0.98) for a 10 ng/mL increase in 25OHD when all studies were included and 0.83 (0.79–0.87) when only case control studies were pooled [253]. In contrast to CRC and breast cancer, the role of vitamin D in prostate cancer is decidedly mixed. In a recent summary of 14 studies examining the association between 25OHD levels and the development of prostate cancer, 11 showed no association [254]. Similarly, studies examining the association of dietary vitamin D intake to prostate cancer did not show benefit [253, 255]. Studies examining the link between vitamin D and nonmelanoma skin cancer (NMSC) are difficult to interpret because UVB is the common etiologic agent for both cancer development and vitamin D production. Those studies that have been reported are mixed. NMSC incidence in the osteoporotic fractures in men (MrOS) study indicated that those with the highest baseline serum 25OHD levels (30 ng/mL) had a relative risk of 0.53 (CI 0.3–0.93) compared to those with the lowest baseline 25OHD levels [256]. However, other studies found that higher 25(OH)D levels were associated with an increased risk of BCC [257, 258].
Regulation of Hormone Secretion
Parathyroid hormone secretion. The promoter of the parathyroid hormone (PTH) gene contains a negative VDRE by which 1,25(OH)2D acting through its receptor is thought to control PTH synthesis [259]. More recent studies identified an E-box element in the PTH gene similar to that found in the CYP27B1 gene. VDR/RXR binds to this element but through the vitamin D inhibitory receptor complex similar to the inhibition by 1,25(OH)2D of CYP27B1 in the kidney [260, 261]. This leads to suppression of transcription via the same mechanisms (HDAC recruitment) as in the CYP27B1 gene. Of interest is that PTH levels are more highly correlated with circulating 25OHD levels than with circulating 1,25(OH)2D levels [262]. As noted earlier, the parathyroid gland (PTG) expresses the megalin/cubilin complex likely enabling uptake of the 25(OH)D/DBP complex into the gland providing the substrate for the CYP27B1 in the PTG to produce its own 1,25(OH)2D [263]. It was initially presumed that the locally produced 1,25(OH)2D utilized the PTG VDR to suppress PTH production, but when the VDR was specifically deleted in PTG, the effect on PTH secretion was modest, and hyperplasia of the gland was not observed [264]. However, deletion of CYP27B1 in the PTG had a much greater impact on PTH secretion (250% increase vs. 80% increase in the PTG VDR deletion) and a surprising drop in serum calcium and 1,25(OH)2D levels suggesting that the CYP27B1 in the PTG was also providing 1,25(OH)2D to the circulation [265]. The drop in serum calcium may have contributed to the greater increase in PTH secretion when CYP27B1 was deleted from the PTG than when the VDR was deleted. In addition to suppression of PTH secretion, 1,25(OH)2D inhibits the proliferation of parathyroid cells (PTC) in vivo [266] and in vitro [267]. In chronic kidney disease, epidermal growth factor receptor (EGFR) and its ligand transforming growth factor α (TGFα) are increased and thought to drive the PTG hyperplasia [268, 269]. 1,25(OH)2D decreases TGFα and EGFR expression [268] and increases the expression of the cell cycle inhibitors p21 and p27 to block the hyperplasia [270].
1,25(OH)2D interacts with other signaling mechanisms to enhance its regulation of PTH secretion and PTG proliferation. The promoter of the CaSR has two functional VDREs through which 1,25(OH)2D/VDR stimulates the expression of CaSR [271]. The CaSR may, in turn, increase VDR levels as suggested by the observation that low-calcium diets decrease the VDR in PTG but high-calcium diets increase the VDR in PTG [272]. 1,25(OH)2D also stimulates Klotho expression in PTC, which, along with FGF receptors, enables FGF23 responsiveness and in turn FGF23 stimulates CYP27B1 [29]. This effect of FGF23 on CYP27B1 in the PTC is opposite to the effect of FGF23 on CYP27B1 in the kidney for unclear reasons. The ability of 1,25(OH)2D to inhibit PTH production and secretion has been exploited clinically in that 1,25(OH)2D and several of its analogs are used to prevent and/or treat secondary hyperparathyroidism associated with renal failure.
Insulin secretion . 1,25(OH)2D stimulates insulin secretion , although the mechanism is not well defined [273, 274]. Moreover, insulin secretion is reduced in vitamin D deficiency [275] and in VDRKO mice [276]. However, calcium is important for insulin secretion, and low calcium levels can be suppressive [277]. Therefore, the early results with vitamin D deficiency may also have reflected the low calcium levels in this condition. When VDRKO mice were placed on a rescue diet to maintain normal calcium levels, insulin secretion was not different from wild type [278]. On the other hand, VDR, CYP27B1, and calbindin-D28k are found in pancreatic beta cells [279,280,281] suggesting a direct role of VDR and 1,25(OH)2D in insulin secretion. Moreover, studies using calbindin-D28k-null mice have suggested that calbindin-D28k, by regulating intracellular calcium, can modulate depolarization-stimulated insulin release [282]. Furthermore, calbindin-D28k, by buffering calcium, can protect against cytokine-mediated destruction of beta cells [283]. The renin/angiotensin system (RAS) may also play a role by impairing beta cell function and insulin sensitivity. 1,25(OH)2D suppresses the RAS in VDRKO mice, and this property in mouse islets may contribute to the ability of 1,25(OH)2D to stimulate insulin secretion [284].
Fibroblast growth factor (FGF23) . FGF23 is produced primarily by bone, and in particular by osteoblasts and osteocytes. 1,25(OH)2D3 stimulates this process, but the mechanism is not clear [285]. As noted earlier, FGF23 in turn inhibits 1,25(OH)2D production by the kidney resulting in a feedback loop similar to that for PTH secretion to maintain a balance in the levels of these hormones. Diseases in which FGF23 is overexpressed or not catabolized properly result in decreased 1,25(OH)2D levels, whereas the opposite is true when FGF23 fails to be secreted or in conditions such as Klotho gene deletion when its target tissues are unresponsive [286].
Renin . VDR- and CYP27B1-null mice have increased levels of renin [287, 288]. Renin converts angiotensinogen to angiotensin I, which is further converted to angiotensin II, a powerful vasoconstrictor as well as stimulator of aldosterone production. In mice lacking VDR or CYP27B1, blood pressure is increased, with increased cardiac hypertrophy, impaired systolic and diastolic function, and increased arterial stiffness [287,288,289]. In the global VDR knockout, renin expression is reduced both in the kidney and the heart as is the cardiac expression of atrial natriuretic factor (ANP) [290]. However, in the cardiomyocyte-specific VDRKO, the increased renin expression is found, but cardiomyopathy is not, suggesting that the effect of renin on the heart is indirect.
Regulation of the Immune System
The immune system is comprised of two interacting forms of immunity: adaptive and innate. Adaptive immunity refers to the process by which cells specialized in antigen presentation, dendritic cells (DC) primarily, and the cells responsible for antigen recognition, T and B lymphocytes, are activated by foreign antigens to initiate a series of functions such as cytokine production, antibody production, and cell killing. The major classes of T helper cells differentiating from the parent CD4 lymphocyte include Th1, Th2, Th9, Th17, and Treg. These responses adapt to the antigen presented. The innate immune response involves the activation of toll-like receptors (TLRs), of which there are ten in the human genome. These TLRs are established during cell development (innate) [291]. TLRs are found in a number of cells including polymorphonuclear cells (PMNs), monocytes, macrophages, and a wide variety of epithelial cells including keratinocytes of the skin, gingiva, intestine, vagina, bladder, and lungs. TLRs are pathogen-recognition receptors that recognize various products of infectious agents including bacteria and viruses and trigger the cell to produce various antimicrobial peptides (AMPs), the best studied of which is cathelicidin. In general, vitamin D signaling suppresses adaptive immunity but promotes innate immunity.
The VDR and CYP27B1 are expressed in most, if not all, cells of the immune system including the epithelial cells, at least when activated [292,293,294]. Moreover, several of these cells express CYP2R1 and so in combination with CYP27B1 can produce 1,25(OH)2D from circulating vitamin D as well as 25(OH)D [294]. As noted earlier, the regulation of CYP27B1 in these cells differs substantially from that in the kidney, being insensitive to hormonal regulators such as PTH and FGF23, its product 1,25(OH)2D, and calcium and phosphate levels. In these immune cells, CYP27B1 is stimulated by cytokines such as tumor necrosis-α (TNFα) and interferon-γ (IGFγ) [23,24,25,26]. Thus, activation of these immune cells in diseases such as sarcoidosis or lymphomas can lead to hypercalcemia with elevated 1,25(OH)2D levels.
Adaptive immunity. 1,25(OH)2D decreases the maturation of DC, thus decreasing their ability to present antigen [295]. Furthermore, 1,25(OH)2D suppresses the proliferation and differentiation of the T and B cells by suppressing IL-12 production, important for Th1 development, IL-23 and IL-6 production important for Th17 development, as well as their ability to secrete IGFγ and IL-2 (from Th1 cells) and IL-17 from Th17 cells [296,297,298]. The suppression of IL-12 also increases the development of Th2 cells and their production of IL-4, IL-5, and IL-13, which serve to further suppress Th1 while promoting Th2 cell number and function. 1,25(OH)2D reduces IL-9 production by Th9 cells [299], which, like the products from Th1 and Th17 cells, plays a role in inflammatory responses. Treg cells, on the other hand, are induced by 1,25(OH)2D [300]. Treg cells produce the regulatory cytokine IL-10 that suppresses the development of Th1 and Th17 leading to immune tolerance [301]. The regulation of a number of cytokines involved in the inflammatory process can be both direct and indirect. Inhibition of IL-2 expression involves blocking NFAT binding to the IL-2 promoter and sequestration of runx1 by VDR [297, 302]. Suppression of IFNα expression involves a negative VDRE in the promoter [303]. Suppression of IL-17 expression involves blocking NFAT binding to the IL-17 promoter and induction of Foxp3 [297]. 1,25(OH)2D blocks NFκB by inhibiting its nuclear translocation, its binding to the consensus sequences in the genes it regulates such as IL-8 and IL-12, and by degradation of IFκB (inhibitor of NFκB) [304]. 1,25(OH)2D3 has also been shown to bring an inhibitor complex containing histone deacetylase 3 (HDAC3) to the promoter of rel B, one of the members of the NFκB family, thus suppressing gene expression. The actions of 1,25(OH)2D3 on B cells have received less attention, but recent studies have demonstrated a reduction in proliferation, maturation to plasma cells, and immunoglobulin production [293].
Although overall myelopoiesis and composition of lymphoid tissue are normal in VDRKO mice, abnormalities in immune responses to stimuli have been observed. Moreover, a number of experimental models of autoimmune diseases including rheumatoid arthritis, psoriasis, type 1 diabetes mellitus (NOD mouse), systemic lupus erythematosus (SLE), experimental allergic encephalitis (EAE, model for multiple sclerosis), and inflammatory bowel disease (IBD) have been prevented/ameliorated with the use of 1,25(OH)2D or one of its analogs [305]. The severity in IBD is increased when IL-10 knockout mice are bred with VDRKO mice [306]. Rejection of transplanted tissues is reduced when the animals are treated with 1,25(OH)2D or one of its analogs [307]. On the other hand, the promotion of Th2 numbers and function may have adverse effects on allergic diseases such as asthma and atopic dermatitis. Calcipotriol, an analog of 1,25(OH)2D, stimulated thymic stromal lymphopoietin (TSLP) in keratinocytes leading to an increased expression of Th2 cytokines and increased inflammatory responses to allergen-induced atopic dermatitis and asthma [308]. However, 1,25(OH)2D was shown to be protective against experimentally induced asthma including a reduction in IL-4 production and eosinophilic infiltration in studies in normal mice [309] perhaps due to its suppression of IL-9, a potent part of the inflammatory response in the lungs [299]. Other studies have shown that mice lacking the VDR (VDRKO) are also protected from experimentally induced asthma [310]. The effects of 1,25(OH)2D on infections are also mixed. 1,25(OH)2D inhibition of IGFγ stimulation of reactive oxygen species and nitric oxide production [311] or suppression of IL-17 limiting its induction of AMPs and neutrophil recruitment [312] have been shown to reduce resistance to infectious organisms such as Leishmania [311], Toxoplasma [313], and Citrobacter [314].
Innate immunity . Stimulation of TLR 2/1 in macrophages [315] or TLR2 and its coreceptor CD14 in keratinocytes [195] leads to an increase in CYP27B1 and VDR expression enabling these cells to produce and respond their own 1,25(OH)2D. 1,25(OH)2D then induces antimicrobial peptides such as cathelicidin and defensins that kill intracellular organisms such as Mycobacterium tuberculosis. Cathelicidin also promotes the chemotaxis of neutrophils, monocytes, macrophages, and T cells into the skin thus linking the adaptive and immune responses in the skin and other tissues [316]. In this way, the innate immune function of these cells acts essentially as the first responder to invading organisms prior to the adaptive immune response. The murine cathelicidin gene lacks a VDRE and so is not responsive to 1,25(OH)2D. However, 1,25(OH)2D stimulates the inducible NOS pathway by which it induces M. tuberculosis killing in these macrophages [317]. Unfortunately, supplementation with vitamin D of patients with M. tuberculosis has not been universally successful [318,319,320,321]. In diseases such as atopic dermatitis, the production of cathelicidin and other antimicrobial peptides is reduced, predisposing these patients to microbial superinfections [322]. Th2 cytokines such as IL-4 and IL-13 suppress the induction of AMPs [323]. Since 1,25(OH)2D3 stimulates the differentiation of Th2 cells, its administration to patients with atopic dermatitis may not be useful in spite of its induction of cathelicidin in contrast to its proven value in patients with psoriasis in which suppression of Th1 and Th17 and their cytokines appears central to its therapeutic effect.
References
Holick MF, McLaughlin JA, Clark MB, Holick SA, PJ JT, Anderson RR, et al. Photosynthesis of previtamin D3 in human and the physiologic consequences. Science. 1980;210:203–5.
Webb AR, DeCosta BR, Holick MF. Sunlight regulates the cutaneous production of vitamin D3 by causing its photodegradation. J Clin Endocrinol Metab. 1989;68(5):882–7.
Houghton LA, Vieth R. The case against ergocalciferol (vitamin D2) as a vitamin supplement. Am J Clin Nutr. 2006;84(4):694–7.
Hollis BW. Comparison of equilibrium and disequilibrium assay conditions for ergocalciferol, cholecalciferol and their major metabolites. J Steroid Biochem. 1984;21(1):81–6.
Horst RL, Reinhardt TA, Ramberg CF, Koszewski NJ, Napoli JL. 24-Hydroxylation of 1,25-dihydroxyergocalciferol. An unambiguous deactivation process. J Biol Chem. 1986;261(20):9250–6.
Tripkovic L, Lambert H, Hart K, Smith CP, Bucca G, Penson S, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95(6):1357–64.
Sugimoto H, Shiro Y. Diversity and substrate specificity in the structures of steroidogenic cytochrome P450 enzymes. Biol Pharm Bull. 2012;35(6):818–23.
Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A. 2013;110(39):15650–5.
Moghadasian MH. Cerebrotendinous xanthomatosis: clinical course, genotypes and metabolic backgrounds. Clin Invest Med. 2004;27(1):42–50.
Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J Biol Chem. 2003;278(39):38084–93.
Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101(20):7711–5.
Gupta RP, Hollis BW, Patel SB, Patrick KS, Bell NH. CYP3A4 is a human microsomal vitamin D 25-hydroxylase. J Bone Miner Res. 2004;19(4):680–8.
Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, et al. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol (Baltimore, Md). 1997;11(13):1961–70.
Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, et al. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci U S A. 1997;94(24):12920–5.
Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277(5333):1827–30.
St-Arnaud R, Messerlian S, Moir JM, Omdahl JL, Glorieux FH. The 25-hydroxyvitamin D 1-alpha-hydroxylase gene maps to the pseudovitamin D-deficiency rickets (PDDR) disease locus. J Bone Miner Res. 1997;12(10):1552–9.
Bikle D. Extra renal synthesis of 1,25-dihydroxyvitamin D and its health implications. In: Holick M, editor. Vitamin D: physiology, molecular biology, and clinical applications. New York: Humana Press; 2010. p. 277–95.
Bikle DD, Rasmussen H. The ionic control of 1,25-dihydroxyvitamin D3 production in isolated chick renal tubules. J Clin Invest. 1975;55(2):292–8.
Bikle DD, Murphy EW, Rasmussen H. The ionic control of 1,25-dihydroxyvitamin D3 synthesis in isolated chick renal mitochondria. The role of calcium as influenced by inorganic phosphate and hydrogen-ion. J Clin Invest. 1975;55(2):299–304.
Kim MS, Fujiki R, Kitagawa H, Kato S. 1alpha,25(OH)2D3-induced DNA methylation suppresses the human CYP27B1 gene. Mol Cell Endocrinol. 2007;265–266:168–73.
Adams JS, Gacad MA. Characterization of 1 alpha-hydroxylation of vitamin D3 sterols by cultured alveolar macrophages from patients with sarcoidosis. J Exp Med. 1985;161(4):755–65.
Ren S, Nguyen L, Wu S, Encinas C, Adams JS, Hewison M. Alternative splicing of vitamin D-24-hydroxylase: a novel mechanism for the regulation of extrarenal 1,25-dihydroxyvitamin D synthesis. J Biol Chem. 2005;280(21):20604–11.
Bikle DD, Pillai S, Gee E, Hincenbergs M. Tumor necrosis factor-alpha regulation of 1,25-dihydroxyvitamin D production by human keratinocytes. Endocrinology. 1991;129(1):33–8.
Bikle DD, Pillai S, Gee E, Hincenbergs M. Regulation of 1,25-dihydroxyvitamin D production in human keratinocytes by interferon-gamma. Endocrinology. 1989;124(2):655–60.
Pryke AM, Duggan C, White CP, Posen S, Mason RS. Tumor necrosis factor-alpha induces vitamin D-1-hydroxylase activity in normal human alveolar macrophages. J Cell Physiol. 1990;142(3):652–6.
Gyetko MR, Hsu CH, Wilkinson CC, Patel S, Young E. Monocyte 1 alpha-hydroxylase regulation: induction by inflammatory cytokines and suppression by dexamethasone and uremia toxin. J Leukoc Biol. 1993;54(1):17–22.
Stoffels K, Overbergh L, Giulietti A, Verlinden L, Bouillon R, Mathieu C. Immune regulation of 25-hydroxyvitamin-D3-1alpha-hydroxylase in human monocytes. J Bone Miner Res. 2006;21(1):37–47.
Bacchetta J, Sea JL, Chun RF, Lisse TS, Wesseling-Perry K, Gales B, et al. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res. 2013;28(1):46–55.
Krajisnik T, Bjorklund P, Marsell R, Ljunggren O, Akerstrom G, Jonsson KB, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195(1):125–31.
Jones G, Prosser DE, Kaufmann M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin D. Arch Biochem Biophys. 2012;523(1):9–18.
Sakaki T, Sawada N, Komai K, Shiozawa S, Yamada S, Yamamoto K, et al. Dual metabolic pathway of 25-hydroxyvitamin D3 catalyzed by human CYP24. Eur J Biochem. 2000;267(20):6158–65.
Prosser DE, Kaufmann M, O’Leary B, Byford V, Jones G. Single A326G mutation converts human CYP24A1 from 25-OH-D3-24-hydroxylase into -23-hydroxylase, generating 1alpha,25-(OH)2D3-26,23-lactone. Proc Natl Acad Sci U S A. 2007;104(31):12673–8.
Plachot JJ, Du Bois MB, Halpern S, Cournot-Witmer G, Garabedian M, Balsan S. In vitro action of 1,25-dihydroxycholecalciferol and 24,25-dihydroxycholecalciferol on matrix organization and mineral distribution in rabbit growth plate. Metab Bone Dis Relat Res. 1982;4(2):135–42.
St-Arnaud R, Arabian A, Travers R, Barletta F, Raval-Pandya M, Chapin K, et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141(7):2658–66.
Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(5):410–21.
Zierold C, Darwish HM, DeLuca HF. Two vitamin D response elements function in the rat 1,25-dihydroxyvitamin D 24-hydroxylase promoter. J Biol Chem. 1995;270(4):1675–8.
Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. J Biol Chem. 2010;285(20):15599–610.
Zierold C, Reinholz GG, Mings JA, Prahl JM, DeLuca HF. Regulation of the procine 1,25-dihydroxyvitamin D3-24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 and parathyroid hormone in AOK-B50 cells. Arch Biochem Biophys. 2000;381(2):323–7.
Zierold C, Mings JA, DeLuca HF. Parathyroid hormone regulates 25-hydroxyvitamin D(3)-24-hydroxylase mRNA by altering its stability. Proc Natl Acad Sci U S A. 2001;98(24):13572–6.
Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–64.
Ohata Y, Yamazaki M, Kawai M, Tsugawa N, Tachikawa K, Koinuma T, et al. Elevated fibroblast growth factor 23 exerts its effects on placenta and regulates vitamin D metabolism in pregnancy of Hyp mice. J Bone Miner Res. 2014;29(7):1627–38.
Armbrecht HJ, Hodam TL, Boltz MA, Partridge NC, Brown AJ, Kumar VB. Induction of the vitamin D 24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 is regulated by parathyroid hormone in UMR106 osteoblastic cells. Endocrinology. 1998;139(8):3375–81.
Bailey D, Veljkovic K, Yazdanpanah M, Adeli K. Analytical measurement and clinical relevance of vitamin D(3) C3-epimer. Clin Biochem. 2013;46(3):190–6.
Reddy GS, Muralidharan KR, Okamura WH, Tserng KY, McLane JA. Metabolism of 1alpha,25-dihydroxyvitamin D(3) and its C-3 epimer 1alpha,25-dihydroxy-3-epi-vitamin D(3) in neonatal human keratinocytes. Steroids. 2001;66(3–5):441–50.
Kamao M, Tatematsu S, Hatakeyama S, Sakaki T, Sawada N, Inouye K, et al. C-3 epimerization of vitamin D3 metabolites and further metabolism of C-3 epimers: 25-hydroxyvitamin D3 is metabolized to 3-epi-25-hydroxyvitamin D3 and subsequently metabolized through C-1alpha or C-24 hydroxylation. J Biol Chem. 2004;279(16):15897–907.
Brown AJ, Ritter C, Slatopolsky E, Muralidharan KR, Okamura WH, Reddy GS. 1Alpha,25-dihydroxy-3-epi-vitamin D3, a natural metabolite of 1alpha,25-dihydroxyvitamin D3, is a potent suppressor of parathyroid hormone secretion. J Cell Biochem. 1999;73(1):106–13.
Slominski AT, Janjetovic Z, Fuller BE, Zmijewski MA, Tuckey RC, Nguyen MN, et al. Products of vitamin D3 or 7-dehydrocholesterol metabolism by cytochrome P450scc show anti-leukemia effects, having low or absent calcemic activity. PLoS One. 2010;5(3):e9907.
Cooke NE, Haddad JG. Vitamin D binding protein (Gc-globulin). Endocr Rev. 1989;10(3):294–307.
Bikle DD, Gee E, Halloran B, Haddad JG. Free 1,25-dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. J Clin Invest. 1984;74(6):1966–71.
Bikle DD, Siiteri PK, Ryzen E, Haddad JG. Serum protein binding of 1,25-dihydroxyvitamin D: a reevaluation by direct measurement of free metabolite levels. J Clin Endocrinol Metab. 1985;61(5):969–75.
Bikle DD, Gee E, Halloran B, Kowalski MA, Ryzen E, Haddad JG. Assessment of the free fraction of 25-hydroxyvitamin D in serum and its regulation by albumin and the vitamin D-binding protein. J Clin Endocrinol Metab. 1986;63(4):954–9.
Bikle DD, Halloran BP, Gee E, Ryzen E, Haddad JG. Free 25-hydroxyvitamin D levels are normal in subjects with liver disease and reduced total 25-hydroxyvitamin D levels. J Clin Invest. 1986;78(3):748–52.
Aggarwal A, Yadav AK, Ramachandran R, Kumar V, Kumar V, Sachdeva N, et al. Bioavailable vitamin D levels are reduced and correlate with bone mineral density and markers of mineral metabolism in adults with nephrotic syndrome. Nephrology (Carlton). 2016;21(6):483–9.
Kim HJ, Ji M, Song J, Moon HW, Hur M, Yun YM. Clinical utility of measurement of vitamin D-binding protein and calculation of bioavailable vitamin D in assessment of vitamin D status. Ann Lab Med. 2017;37(1):34–8.
Lai JC, Bikle DD, Lizaola B, Hayssen H, Terrault NA, Schwartz JB. Total 25(OH) vitamin D, free 25(OH) vitamin D and markers of bone turnover in cirrhotics with and without synthetic dysfunction. Liver Int. 2015;35(10):2294–300.
Schwartz JB, Lai J, Lizaola B, Kane L, Markova S, Weyland P, et al. A comparison of measured and calculated free 25(OH) vitamin D levels in clinical populations. J Clin Endocrinol Metab. 2014;99(5):1631–7.
Pettifor JM, Bikle DD, Cavaleros M, Zachen D, Kamdar MC, Ross FP. Serum levels of free 1,25-dihydroxyvitamin D in vitamin D toxicity. Ann Intern Med. 1995;122(7):511–3.
Cooke NE, David EV. Serum vitamin D-binding protein is a third member of the albumin and alpha fetoprotein gene family. J Clin Invest. 1985;76(6):2420–4.
Recant L, Riggs DS. Thyroid function in nephrosis. J Clin Invest. 1952;31(8):789–97.
Bouillon R, Van Assche FA, Van Baelen H, Heyns W, De Moor P. Influence of the vitamin D-binding protein on the serum concentration of 1,25-dihydroxyvitamin D3. Significance of the free 1,25-dihydroxyvitamin D3 concentration. J Clin Invest. 1981;67(3):589–96.
Safadi FF, Thornton P, Magiera H, Hollis BW, Gentile M, Haddad JG, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. 1999;103(2):239–51.
Nykjaer A, Dragun D, Walther D, Vorum H, Jacobsen C, Herz J, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96(4):507–15.
Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3). Proc Natl Acad Sci U S A. 2001;98(24):13895–900.
Lundgren S, Carling T, Hjalm G, Juhlin C, Rastad J, Pihlgren U, et al. Tissue distribution of human gp330/megalin, a putative Ca(2+)-sensing protein. J Histochem Cytochem. 1997;45(3):383–92.
Uitterlinden AG, Arp PP, Paeper BW, Charmley P, Proll S, Rivadeneira F, et al. Polymorphisms in the sclerosteosis/van Buchem disease gene (SOST) region are associated with bone-mineral density in elderly whites. Am J Hum Genet. 2004;75(6):1032–45.
Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173–9.
Carlson M, Laurent BC. The SNF/SWI family of global transcriptional activators. Curr Opin Cell Biol. 1994;6(3):396–402.
Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25(1):45–71.
Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene. 2000;246(1–2):9–21.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63.
Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111(17):6131–8.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23.
Meyer MB, Goetsch PD, Pike JW. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J Steroid Biochem Mol Biol. 2010;121(1–2):136–41.
Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20(10):1352–60.
Carlberg C, Seuter S, Heikkinen S. The first genome-wide view of vitamin D receptor locations and their mechanistic implications. Anticancer Res. 2012;32(1):271–82.
Zella LA, Meyer MB, Nerenz RD, Lee SM, Martowicz ML, Pike JW. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol Endocrinol (Baltimore, Md). 2010;24(1):128–47.
Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/beta-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol (Baltimore, Md). 2012;26(1):37–51.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–9.
Kim S, Yamazaki M, Zella LA, Shevde NK, Pike JW. Activation of receptor activator of NF-kappaB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol. 2006;26(17):6469–86.
Galli C, Zella LA, Fretz JA, Fu Q, Pike JW, Weinstein RS, et al. Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-kappaB ligand gene reduces bone remodeling and increases bone mass. Endocrinology. 2008;149(1):146–53.
Onal M, St John HC, Danielson AL, Markert JW, Riley EM, Pike JW. Unique distal enhancers linked to the mouse Tnfsf11 gene direct tissue-specific and inflammation-induced expression of RANKL. Endocrinology. 2016;157(2):482–96.
Norman AW, Okamura WH, Hammond MW, Bishop JE, Dormanen MC, Bouillon R, et al. Comparison of 6-s-cis- and 6-s-trans-locked analogs of 1alpha,25-dihydroxyvitamin D3 indicates that the 6-s-cis conformation is preferred for rapid nongenomic biological responses and that neither 6-s-cis- nor 6-s-trans-locked analogs are preferred for genomic biological responses. Mol Endocrinol (Baltimore, Md). 1997;11(10):1518–31.
Schwartz N, Verma A, Bivens CB, Schwartz Z, Boyan BD. Rapid steroid hormone actions via membrane receptors. Biochim Biophys Acta. 2016;1863(9):2289–98.
Sequeira VB, Rybchyn MS, Tongkao-On W, Gordon-Thomson C, Malloy PJ, Nemere I, et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1alpha,25-dihydroxyvitamin D3. Mol Endocrinol. 2012;26(4):574–82.
Mizwicki MT, Norman AW. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci Signal. 2009;2(75):re4.
Nemere I, Farach-Carson MC, Rohe B, Sterling TM, Norman AW, Boyan BD, et al. Ribozyme knockdown functionally links a 1,25(OH)2D3 membrane binding protein (1,25D3-MARRS) and phosphate uptake in intestinal cells. Proc Natl Acad Sci U S A. 2004;101(19):7392–7.
Huhtakangas JA, Olivera CJ, Bishop JE, Zanello LP, Norman AW. The vitamin D receptor is present in caveolae-enriched plasma membranes and binds 1 alpha,25(OH)2-vitamin D3 in vivo and in vitro. Mol Endocrinol (Baltimore, Md). 2004;18(11):2660–71.
Nemere I, Garbi N, Hammerling GJ, Khanal RC. Intestinal cell calcium uptake and the targeted knockout of the 1,25D3-MARRS (membrane-associated, rapid response steroid-binding) receptor/PDIA3/Erp57. J Biol Chem. 2010;285(41):31859–66.
Chen J, Olivares-Navarrete R, Wang Y, Herman TR, Boyan BD, Schwartz Z. Protein-disulfide isomerase-associated 3 (Pdia3) mediates the membrane response to 1,25-dihydroxyvitamin D3 in osteoblasts. J Biol Chem. 2010;285(47):37041–50.
Wang Y, Chen J, Lee CS, Nizkorodov A, Riemenschneider K, Martin D, et al. Disruption of Pdia3 gene results in bone abnormality and affects 1alpha,25-dihydroxy-vitamin D3-induced rapid activation of PKC. J Steroid Biochem Mol Biol. 2010;121(1–2):257–60.
Christakos S, Dhawan P, Porta A, Mady LJ, Seth T. Vitamin D and intestinal calcium absorption. Mol Cell Endocrinol. 2011;347(1–2):25–9.
Bikle DD, Morrissey RL, Zolock DT, Rasmussen H. The intestinal response to vitamin D. Rev Physiol Biochem Pharmacol. 1981;89:63–142.
Bikle DD, Shoback DM, Munson S. 1,25-dihydroxyvitamin D increases the intracellular free calcium concentration of duodenal epithelial cells. In: Schaefer K, Grigoleit HG, Herrath DV, editors. Vitamin D: chemical, biochemical and clinical update. New York: Walter de Gruyter; 1985. 416 p.
Morrissey RL, Zolock DT, Mellick PW, Bikle DD. Influence of cycloheximide and 1,25-dihydroxyvitamin D3 on mitochondrial and vesicle mineralization in the intestine. Cell Calcium. 1980;1:69–79.
Davis WL, Hagler HK, Jones RG, Farmer GR, Cooper OJ, Martin JH, et al. Cryofixation, ultracryomicrotomy, and X-ray microanalysis of enterocytes from chick duodenum: vitamin-D-induced formation of an apical tubulovesicular system. Anat Rec. 1991;229(2):227–39.
Nemere I, Leathers V, Norman AW. 1,25-Dihydroxyvitamin D3-mediated intestinal calcium transport. Biochemical identification of lysosomes containing calcium and calcium- binding protein (calbindin-D28K). J Biol Chem. 1986;261(34):16106–14.
Max EE, Goodman DB, Rasmussen H. Purification and characterization of chick intestine brush border membrane. Effects of 1alpha(OH) vitamin D3 treatment. Biochim Biophys Acta. 1978;511(2):224–39.
Brasitus TA, Dudeja PK, Eby B, Lau K. Correction by 1,25(OH)2D3 of the abnormal fluidity and lipid composition of enterocyte brush border membranes in vitamin D-deprived rats. J Biol Chem. 1981;256:3354–60.
Matsumoto T, Fontaine O, Rasmussen H. Effect of 1,25-dihydroxyvitamin D3 on phospholipid metabolism in chick duodenal mucosal cell. Relationship to its mechanism of action. J Biol Chem. 1981;256(7):3354–60.
Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, et al. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem. 1999;274(13):8375–8.
Peng JB, Chen XZ, Berger UV, Vassilev PM, Brown EM, Hediger MA. A rat kidney-specific calcium transporter in the distal nephron. J Biol Chem. 2000;275(36):28186–94.
Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, et al. Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res. 2007;22(2):274–85.
Kutuzova GD, Sundersingh F, Vaughan J, Tadi BP, Ansay SE, Christakos S, et al. TRPV6 is not required for 1alpha,25-dihydroxyvitamin D3-induced intestinal calcium absorption in vivo. Proc Natl Acad Sci U S A. 2008;105(50):19655–9.
Glenney JR Jr, Glenney P. Comparison of Ca++-regulated events in the intestinal brush border. J Cell Biol. 1985;100(3):754–63.
Bikle DD, Gee E. Free, and not total, 1,25-dihydroxyvitamin D regulates 25-hydroxyvitamin D metabolism by keratinocytes. Endocrinology. 1989;124(2):649–54.
Bikle DD, Munson S, Chafouleas J. Calmodulin may mediate 1,25-dihydroxyvitamin D-stimulated intestinal calcium transport. FEBS Lett. 1984;174(1):30–3.
Lambers TT, Weidema AF, Nilius B, Hoenderop JG, Bindels RJ. Regulation of the mouse epithelial Ca2(+) channel TRPV6 by the Ca(2+)-sensor calmodulin. J Biol Chem. 2004;279(28):28855–61.
Bikle DD, Munson S. The villus gradient of brush border membrane calmodulin and the calcium-independent calmodulin-binding protein parallels that of calcium-accumulating ability. Endocrinology. 1986;118(2):727–32.
Sampson HW, Matthews JL, Martin JH, Kunin AS. An electron microscopic localization of calcium in the small intestine of normal, rachitic, and vitamin-D-treated rats. Calcif Tissue Res. 1970;5(4):305–16.
Schaefer HJ. Ultrastructure and ion distribution of the intestinal cell during experimental vitamin D deficiency rickets in rats. Virchows Arch. 1973;359:111–23.
Wasserman RH, Taylor AN. Vitamin D-dependent calcium-binding protein. Response to some physiological and nutritional variables. J Biol Chem. 1968;243(14):3987–93.
Lee GS, Lee KY, Choi KC, Ryu YH, Paik SG, Oh GT, et al. Phenotype of a calbindin-D9k gene knockout is compensated for by the induction of other calcium transporter genes in a mouse model. J Bone Miner Res. 2007;22(12):1968–78.
Benn BS, Ajibade D, Porta A, Dhawan P, Hediger M, Peng JB, et al. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology. 2008;149(6):3196–205.
Ghijsen WE, De Jong MD, Van Os CH. ATP-dependent calcium transport and its correlation with Ca2+-ATPase activity in basolateral plasma membranes of rat duodenum. Biochim Biophys Acta. 1982;689(2):327–36.
Cai Q, Chandler JS, Wasserman RH, Kumar R, Penniston JT. Vitamin D and adaptation to dietary calcium and phosphate deficiencies increase intestinal plasma membrane calcium pump gene expression. Proc Natl Acad Sci U S A. 1993;90(4):1345–9.
Wasserman RH, Chandler JS, Meyer SA, Smith CA, Brindak ME, Fullmer CS, et al. Intestinal calcium transport and calcium extrusion processes at the basolateral membrane. J Nutr. 1992;122(3 Suppl):662–71.
Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85(1):373–422.
Xu H, Bai L, Collins JF, Ghishan FK. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics. 1999;62(2):281–4.
Xu H, Bai L, Collins JF, Ghishan FK. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). Am J Physiol Cell Physiol. 2002;282(3):C487–93.
Candeal E, Caldas YA, Guillen N, Levi M, Sorribas V. Intestinal phosphate absorption is mediated by multiple transport systems in rats. Am J Physiol Gastrointest Liver Physiol. 2017;312(4):G355–66.
Fuchs R, Peterlik M. Vitamin D-induced transepithelial phosphate and calcium transport by chick jejunum. Effect of microfilamentous and microtubular inhibitors. FEBS Lett. 1979;100(2):357–9.
Narbaitz R, Stumpf WE, Sar M, Huang S, DeLuca HF. Autoradiographic localization of target cells for 1 alpha, 25-dihydroxyvitamin D3 in bones from fetal rats. Calcif Tissue Int. 1983;35(2):177–82.
Boivin G, Mesguich P, Pike JW, Bouillon R, Meunier PJ, Haussler MR, et al. Ultrastructural immunocytochemical localization of endogenous 1,25-dihydroxyvitamin D3 and its receptors in osteoblasts and osteocytes from neonatal mouse and rat calvaria. Bone Miner. 1987;3(2):125–36.
Johnson JA, Grande JP, Roche PC, Kumar R. Ontogeny of the 1,25-dihydroxyvitamin D3 receptor in fetal rat bone. J Bone Miner Res. 1996;11(1):56–61.
Miller SC, Halloran BP, DeLuca HF, Lee WSS. Studies on the role of vitamin D in early skeletal development, mineralization, and growth in rats. Calcif Tissue Int. 1983;35:455–60.
Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94(18):9831–5.
Balsan S, Garabedian M, Larchet M, Gorski AM, Cournot G, Tau C, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77(5):1661–7.
Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139(10):4391–6.
Xue Y, Fleet JC. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology. 2009;136(4):1317–27, e1–2.
Panda DK, Miao D, Bolivar I, Li J, Huo R, Hendy GN, et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279(16):16754–66.
Sato T, Ono T, Tuan RS. 1,25-Dihydroxy vitamin D3 stimulation of TGF-beta expression in chick embryonic calvarial bone. Differentiation. 1993;52(2):139–50.
Wang DS, Yamazaki K, Nohtomi K, Shizume K, Ohsumi K, Shibuya M, et al. Increase of vascular endothelial growth factor mRNA expression by 1,25-dihydroxyvitamin D3 in human osteoblast-like cells. J Bone Miner Res. 1996;11(4):472–9.
Kyeyune-Nyombi E, Lau KH, Baylink DJ, Strong DD. 1,25-Dihydroxyvitamin D3 stimulates both alkaline phosphatase gene transcription and mRNA stability in human bone cells. Arch Biochem Biophys. 1991;291(2):316–25.
Irving JT, Wuthier RE. Histochemistry and biochemistry of calcification with special reference to the role of lipids. Clin Orthop. 1968;56:237–60.
Howell DS, Marquez JF, Pita JC. The nature of phospholipids in normal and rachitic costochondral plates. Arthritis Rheum. 1965;8(6):1039–46.
Dean DD, Boyan BD, Muniz OE, Howell DS, Schwartz Z. Vitamin D metabolites regulate matrix vesicle metalloproteinase content in a cell maturation-dependent manner. Calcif Tissue Int. 1996;59(2):109–16.
Roughley PJ, Dickson IR. A comparison of proteoglycan from chick cartilage of different types and a study of the effect of vitamin D on proteoglycan structure. Connect Tissue Res. 1986;14(3):187–97.
Boyan BD, Schwartz Z, Carnes DL Jr, Ramirez V. The effects of vitamin D metabolites on the plasma and matrix vesicle membranes of growth and resting cartilage cells in vitro. Endocrinology. 1988;122(6):2851–60.
Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116(12):3150–9.
Naja RP, Dardenne O, Arabian A, St Arnaud R. Chondrocyte-specific modulation of Cyp27b1 expression supports a role for local synthesis of 1,25-dihydroxyvitamin D3 in growth plate development. Endocrinology. 2009;150(9):4024–32.
Owen TA, Aronow MS, Barone LM, Bettencourt B, Stein GS, Lian JB. Pleiotropic effects of vitamin D on osteoblast gene expression are related to the proliferative and differentiated state of the bone cell phenotype: dependency upon basal levels of gene expression, duration of exposure, and bone matrix competency in normal rat osteoblast cultures. Endocrinology. 1991;128(3):1496–504.
Lian J, Stewart C, Puchacz E, Mackowiak S, Shalhoub V, Collart D, et al. Structure of the rat osteocalcin gene and regulation of vitamin D-dependent expression. Proc Natl Acad Sci U S A. 1989;86(4):1143–7.
Demay MB, Gerardi JM, DeLuca HF, Kronenberg HM. DNA sequences in the rat osteocalcin gene that bind the 1,25-dihydroxyvitamin D3 receptor and confer responsiveness to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1990;87(1):369–73.
Kerner SA, Scott RA, Pike JW. Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc Natl Acad Sci U S A. 1989;86(12):4455–9.
Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87(24):9995–9.
Zhang R, Ducy P, Karsenty G. 1,25-dihydroxyvitamin D3 inhibits Osteocalcin expression in mouse through an indirect mechanism. J Biol Chem. 1997;272(1):110–6.
Wronski TJ, Halloran BP, Bikle DD, Globus RK, Morey-Holton ER. Chronic administration of 1,25-dihydroxyvitamin D3: increased bone but impaired mineralization. Endocrinology. 1986;119(6):2580–5.
Hock JM, Gunness-Hey M, Poser J, Olson H, Bell NH, Raisz LG. Stimulation of undermineralized matrix formation by 1,25 dihydroxyvitamin D3 in long bones of rats. Calcif Tissue Int. 1986;38(2):79–86.
Suda T, Masuyama R, Bouillon R, Carmeliet G. Physiological functions of vitamin D: what we have learned from global and conditional VDR knockout mouse studies. Curr Opin Pharmacol. 2015;22:87–99.
Yamamoto Y, Yoshizawa T, Fukuda T, Shirode-Fukuda Y, Yu T, Sekine K, et al. Vitamin D receptor in osteoblasts is a negative regulator of bone mass control. Endocrinology. 2013;154(3):1008–20.
Suda T, Takahashi N, Abe E. Role of vitamin D in bone resorption. J Cell Biochem. 1992;49(1):53–8.
Merke J, Klaus G, Hugel U, Waldherr R, Ritz E. No 1,25-dihydroxyvitamin D3 receptors on osteoclasts of calcium-deficient chicken despite demonstrable receptors on circulating monocytes. J Clin Invest. 1986;77(1):312–4.
Mee AP, Hoyland JA, Braidman IP, Freemont AJ, Davies M, Mawer EB. Demonstration of vitamin D receptor transcripts in actively resorbing osteoclasts in bone sections. Bone. 1996;18(4):295–9.
Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption—a hypothesis. Calcif Tissue Int. 1981;33(4):349–51.
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–57.
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597–602.
Winaver J, Sylk DB, Robertson JS, Chen TC, Puschett JB. Micropuncture study of the acute renal tubular transport effects of 25-hydroxyvitamin D3 in the dog. Miner Electrolyte Metab. 1980;4:178–88.
Tenenhouse HS. Cellular and molecular mechanisms of renal phosphate transport. J Bone Miner Res. 1997;12(2):159–64.
Erben RG, Andrukhova O. FGF23-Klotho signaling axis in the kidney. Bone. 2017;100:62–8.
Puschett JB, Beck WS Jr, Jelonek A, Fernandez PC. Study of the renal tubular interactions of thyrocalcitonin, cyclic adenosine 3′,5′-monophosphate, 25-hydroxycholecalciferol, and calcium ion. J Clin Invest. 1974;53(3):756–67.
Puschett JB, Fernandez PC, Boyle IT, Gray RW, Omdahl JL, DeLuca HF. The acute renal tubular effects of 1,25-dihydroxycholecalciferol. Proc Soc Exp Biol Med. 1972;141(1):379–84.
Puschett JB, Moranz J, Kurnick WS. Evidence for a direct action of cholecalciferol and 25-hydroxycholecalciferol on the renal transport of phosphate, sodium, and calcium. J Clin Invest. 1972;51(2):373–85.
Popovtzer MM, Robinette JB, DeLuca HF, Holick MF. The acute effect of 25-hydroxycholecalciferol on renal handling of phosphorus. Evidence for a parathyroid hormone-dependent mechanism. J Clin Invest. 1974;53(3):913–21.
Yamamoto M, Kawanobe Y, Takahashi H, Shimazawa E, Kimura S, Ogata E. Vitamin D deficiency and renal calcium transport in the rat. J Clin Invest. 1984;74(2):507–13.
Lambers TT, Bindels RJ, Hoenderop JG. Coordinated control of renal Ca2+ handling. Kidney Int. 2006;69(4):650–4.
Kumar R, Schaefer J, Grande JP, Roche PC. Immunolocalization of calcitriol receptor, 24-hydroxylase cytochrome P-450, and calbindin D28k in human kidney. Am J Phys. 1994;266(3 Pt 2):F477–85.
Borke JL, Minami J, Verma AK, Penniston JT, Kumar R. Co-localization of erythrocyte Ca++-Mg++ ATPase and vitamin D-dependent 28-kDa-calcium binding protein. Kidney Int. 1988;34(2):262–7.
Peterlik M, Wasserman RH. Regulation by vitamin D of intestinal phosphate absorption. Horm Metab Res. 1980;12(5):216–9.
Christakos S, Brunette MG, Norman AW. Localization of immunoreactive vitamin D-dependent calcium binding protein in chick nephron. Endocrinology. 1981;109(1):322–4.
Roth J, Thorens B, Hunziker W, Norman AW, Orci L. Vitamin D--dependent calcium binding protein: immunocytochemical localization in chick kidney. Science. 1981;214(4517):197–200.
Song Y, Peng X, Porta A, Takanaga H, Peng JB, Hediger MA, et al. Calcium transporter 1 and epithelial calcium channel messenger ribonucleic acid are differentially regulated by 1,25 dihydroxyvitamin D3 in the intestine and kidney of mice. Endocrinology. 2003;144(9):3885–94.
Biber J, Hernando N, Forster I. Phosphate transporters and their function. Annu Rev Physiol. 2013;75:535–50.
Lehmann B, Tiebel O, Meurer M. Expression of vitamin D3 25-hydroxylase (CYP27) mRNA after induction by vitamin D3 or UVB radiation in keratinocytes of human skin equivalents—a preliminary study. Arch Dermatol Res. 1999;291(9):507–10.
Vantieghem K, De Haes P, Bouillon R, Segaert S. Dermal fibroblasts pretreated with a sterol Delta7-reductase inhibitor produce 25-hydroxyvitamin D3 upon UVB irradiation. J Photochem Photobiol B. 2006;85(1):72–8.
Bikle DD, Nemanic MK, Gee E, Elias P. 1,25-Dihydroxyvitamin D3 production by human keratinocytes. Kinetics and regulation. J Clin Invest. 1986;78(2):557–66.
Bikle DD, Halloran BP, Riviere JE. Production of 1,25 dihydroxyvitamin D3 by perfused pig skin. J Invest Dermatol. 1994;102(5):796–8.
Xie Z, Munson S, Huang N, Schuster I, Portale AA, Miller WL, et al. The mechanism of 1,25-dihydroxyvitamin D3 auto-regulation in keratinocytes. J Bone Min Res (Program & Abstracts). 2001;16(Suppl 1):S556.
Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86(2):888–94.
Pillai S, Bikle DD, Elias PM. 1,25-Dihydroxyvitamin D production and receptor binding in human keratinocytes varies with differentiation. J Biol Chem. 1988;263(11):5390–5.
Bikle DD. Vitamin D and the skin: physiology and pathophysiology. Rev Endocr Metab Disord. 2012;13(1):3–19.
Ratnam AV, Cho JK, Bikle DD. 1,25-dihydroxyvitamin D3 enhances the calcium response of keratinocytes. J Invest Dermatol. 1996;106:910.
Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol. 1989;109(3):1207–17.
Rice RH, Green H. Presence in human epidermal cells of a soluble protein precursor of the cross-linked envelope: activation of the cross-linking by calcium ions. Cell. 1979;18(3):681–94.
Hohl D, Lichti U, Breitkreutz D, Steinert PM, Roop DR. Transcription of the human loricrin gene in vitro is induced by calcium and cell density and suppressed by retinoic acid. J Invest Dermatol. 1991;96(4):414–8.
Simon M, Green H. Participation of membrane-associated proteins in the formation of the cross-linked envelope of the keratinocyte. Cell. 1984;36(4):827–34.
Hohl D. Cornified cell envelope. Dermatologica. 1990;180(4):201–11.
Thacher SM, Rice RH. Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell. 1985;40(3):685–95.
Hennings H, Steinert P, Buxman MM. Calcium induction of transglutaminase and the formation of epsilon(gamma-glutamyl) lysine cross-links in cultured mouse epidermal cells. Biochem Biophys Res Commun. 1981;102(2):739–45.
Su MJ, Bikle DD, Mancianti ML, Pillai S. 1,25-Dihydroxyvitamin D3 potentiates the keratinocyte response to calcium. J Biol Chem. 1994;269(20):14723–9.
Smith EL, Walworth NC, Holick MF. Effect of 1 alpha,25-dihydroxyvitamin D3 on the morphologic and biochemical differentiation of cultured human epidermal keratinocytes grown in serum-free conditions. J Invest Dermatol. 1986;86(6):709–14.
Pillai S, Bikle DD. Role of intracellular-free calcium in the cornified envelope formation of keratinocytes: differences in the mode of action of extracellular calcium and 1,25 dihydroxyvitamin D3. J Cell Physiol. 1991;146(1):94–100.
McLane JA, Katz M, Abdelkader N. Effect of 1,25-dihydroxyvitamin D3 on human keratinocytes grown under different culture conditions. In Vitro Cell Dev Biol. 1990;26(4):379–87.
Oda Y, Uchida Y, Moradian S, Crumrine D, Elias PM, Bikle DD. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol. 2009;129(6):1367–78.
Tu CL, Crumrine DA, Man MQ, Chang W, Elalieh H, You M, et al. Ablation of the calcium-sensing receptor in keratinocytes impairs epidermal differentiation and barrier function. J Invest Dermatol. 2012;132(10):2350–9.
Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117(3):803–11.
Oda Y, Sihlbom C, Huang L, Rachez C, Chang CP, Burlingame AL, et al. Sequential utilization of the VDR coactivators, DRIP/Mediator and SRC/p160, during keratinocyte differentiation. Mol Endocrinol. 2003;17:2329–39.
Tu CL, Chang W, Bikle DD. The extracellular calcium-sensing receptor is required for calcium-induced differentiation in human keratinocytes. J Biol Chem. 2001;276(44):41079–85.
Tu CL, Chang W, Bikle DD. Phospholipase cgamma1 is required for activation of store-operated channels in human keratinocytes. J Invest Dermatol. 2005;124(1):187–97.
Tu C, Chang W, Xie Z, Bikle DD. Inactivation of the calcium sensing receptor inhibits E-cadherin-mediated cell-cell adhesion and calcium-induced differentiation in human epidermal keratinocytes. J Biol Chem. 2008;283(6):3519–28.
Xie Z, Singleton PA, Bourguignon LY, Bikle DD. Calcium-induced human keratinocyte differentiation requires src- and fyn-mediated phosphatidylinositol 3-kinase-dependent activation of phospholipase C-gamma1. Mol Biol Cell. 2005;16(7):3236–46.
Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. J Biol Chem. 2007;282(12):8695–703.
Xie Z, Chang SM, Pennypacker SD, Liao EY, Bikle DD. Phosphatidylinositol-4-phosphate 5-kinase 1alpha mediates extracellular calcium-induced keratinocyte differentiation. Mol Biol Cell. 2009;20(6):1695–704.
Pillai S, Bikle DD. Adenosine triphosphate stimulates phosphoinositide metabolism, mobilizes intracellular calcium, and inhibits terminal differentiation of human epidermal keratinocytes. J Clin Invest. 1992;90(1):42–51.
Yang LC, Ng DC, Bikle DD. Role of protein kinase C alpha in calcium induced keratinocyte differentiation: defective regulation in squamous cell carcinoma. J Cell Physiol. 2003;195(2):249–59.
Xie Z, Bikle DD. Cloning of the human phospholipase C-gamma1 promoter and identification of a DR6-type vitamin D-responsive element. J Biol Chem. 1997;272(10):6573–7.
Tu CL, Chang W, Bikle DD. The role of the calcium sensing receptor in regulating intracellular calcium handling in human epidermal keratinocytes. J Invest Dermatol. 2007;127(5):1074–83.
Bikle DD. Extraskeletal actions of vitamin D. Ann N Y Acad Sci. 2016;1376(1):29–52.
Santagata S, Thakkar A, Ergonul A, Wang B, Woo T, Hu R, et al. Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J Clin Invest. 2014;124(2):859–70.
Matusiak D, Benya RV. CYP27A1 and CYP24 expression as a function of malignant transformation in the colon. J Histochem Cytochem. 2007;55(12):1257–64.
Brozek W, Manhardt T, Kallay E, Peterlik M, Cross HS. Relative expression of vitamin D hydroxylases, CYP27B1 and CYP24A1, and of cyclooxygenase-2 and heterogeneity of human colorectal cancer in relation to age, gender, tumor location, and malignancy: results from factor and cluster analysis. Cancers. 2012;4(3):763–76.
Tannour-Louet M, Lewis SK, Louet JF, Stewart J, Addai JB, Sahin A, et al. Increased expression of CYP24A1 correlates with advanced stages of prostate cancer and can cause resistance to vitamin D3-based therapies. FASEB J. 2014;28(1):364–72.
Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96(1):365–408.
Chang S, Gao L, Yang Y, Tong D, Guo B, Liu L, et al. miR-145 mediates the antiproliferative and gene regulatory effects of vitamin D3 by directly targeting E2F3 in gastric cancer cells. Oncotarget. 2015;6(10):7675–85.
Gocek E, Wang X, Liu X, Liu CG, Studzinski GP. MicroRNA-32 upregulation by 1,25-dihydroxyvitamin D3 in human myeloid leukemia cells leads to Bim targeting and inhibition of AraC-induced apoptosis. Cancer Res. 2011;71(19):6230–9.
Hager G, Formanek M, Gedlicka C, Thurnher D, Knerer B, Kornfehl J. 1,25(OH)2 vitamin D3 induces elevated expression of the cell cycle-regulating genes P21 and P27 in squamous carcinoma cell lines of the head and neck. Acta Otolaryngol. 2001;121(1):103–9.
Palmer HG, Sanchez-Carbayo M, Ordonez-Moran P, Larriba MJ, Cordon-Cardo C, Munoz A. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003;63(22):7799–806.
An BS, Tavera-Mendoza LE, Dimitrov V, Wang X, Calderon MR, Wang HJ, et al. Stimulation of Sirt1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol Cell Biol. 2010;30(20):4890–900.
Colston KW, Perks CM, Xie SP, Holly JM. Growth inhibition of both MCF-7 and Hs578T human breast cancer cell lines by vitamin D analogues is associated with increased expression of insulin-like growth factor binding protein-3. J Mol Endocrinol. 1998;20(1):157–62.
Huynh H, Pollak M, Zhang JC. Regulation of insulin-like growth factor (IGF) II and IGF binding protein 3 autocrine loop in human PC-3 prostate cancer cells by vitamin D metabolite 1,25(OH)2D3 and its analog EB1089. Int J Oncol. 1998;13(1):137–43.
Peehl DM, Shinghal R, Nonn L, Seto E, Krishnan AV, Brooks JD, et al. Molecular activity of 1,25-dihydroxyvitamin D3 in primary cultures of human prostatic epithelial cells revealed by cDNA microarray analysis. J Steroid Biochem Mol Biol. 2004;92(3):131–41.
Swami S, Raghavachari N, Muller UR, Bao YP, Feldman D. Vitamin D growth inhibition of breast cancer cells: gene expression patterns assessed by cDNA microarray. Breast Cancer Res Treat. 2003;80(1):49–62.
Yang L, Yang J, Venkateswarlu S, Ko T, Brattain MG. Autocrine TGFbeta signaling mediates vitamin D3 analog-induced growth inhibition in breast cells. J Cell Physiol. 2001;188(3):383–93.
Aszterbaum M, Rothman A, Johnson RL, Fisher M, Xie J, Bonifas JM, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998;110(6):885–8.
Teichert AE, Elalieh H, Elias PM, Welsh J, Bikle DD. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. J Invest Dermatol. 2011;131(11):2289–97.
McGaffin KR, Chrysogelos SA. Identification and characterization of a response element in the EGFR promoter that mediates transcriptional repression by 1,25-dihydroxyvitamin D3 in breast cancer cells. J Mol Endocrinol. 2005;35(1):117–33.
Byers SW, Rowlands T, Beildeck M, Bong YS. Mechanism of action of vitamin D and the vitamin D receptor in colorectal cancer prevention and treatment. Rev Endocr Metab Disord. 2011;13(1):31–8.
Aguilera O, Pena C, Garcia JM, Larriba MJ, Ordonez-Moran P, Navarro D, et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis. 2007;28(9):1877–84.
Pendas-Franco N, Garcia JM, Pena C, Valle N, Palmer HG, Heinaniemi M, et al. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene. 2008;27(32):4467–77.
Diaz GD, Paraskeva C, Thomas MG, Binderup L, Hague A. Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implications for prevention and therapy. Cancer Res. 2000;60(8):2304–12.
Pan L, Matloob AF, Du J, Pan H, Dong Z, Zhao J, et al. Vitamin D stimulates apoptosis in gastric cancer cells in synergy with trichostatin A/sodium butyrate-induced and 5-aza-2′-deoxycytidine-induced PTEN upregulation. FEBS J. 2010;277(4):989–99.
Kizildag S, Ates H, Kizildag S. Treatment of K562 cells with 1,25-dihydroxyvitamin D3 induces distinct alterations in the expression of apoptosis-related genes BCL2, BAX, BCLXL, and p21. Ann Hematol. 2009;89(1):1–7.
Weitsman GE, Ravid A, Liberman UA, Koren R. Vitamin D enhances caspase-dependent and independent TNF-induced breast cancer cell death: the role of reactive oxygen species. Ann N Y Acad Sci. 2003;1010:437–40.
Weitsman GE, Koren R, Zuck E, Rotem C, Liberman UA, Ravid A. Vitamin D sensitizes breast cancer cells to the action of H2O2: mitochondria as a convergence point in the death pathway. Free Radic Biol Med. 2005;39(2):266–78.
Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30(1):88–92.
Murillo G, Nagpal V, Tiwari N, Benya RV, Mehta RG. Actions of vitamin D are mediated by the TLR4 pathway in inflammation-induced colon cancer. J Steroid Biochem Mol Biol. 2003;121(1–2):403–7.
Yang K, Lamprecht SA, Shinozaki H, Fan K, Yang W, Newmark HL, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J Nutr. 2008;138(9):1658–63.
Xu H, Posner GH, Stevenson M, Campbell FC. Apc(MIN) modulation of vitamin D secosteroid growth control. Carcinogenesis. 2010;31(8):1434–41.
Zheng W, Wong KE, Zhang Z, Dougherty U, Mustafi R, Kong J, et al. Inactivation of the vitamin D receptor in APC(min/+) mice reveals a critical role for the vitamin D receptor in intestinal tumor growth. Int J Cancer. 2011;130(1):10–9.
Lipkin M, Newmark HL. Vitamin D, calcium and prevention of breast cancer: a review. J Am Coll Nutr. 1999;18(5 Suppl):392S–7S.
Zinser GM, Welsh J. Effect of Vitamin D3 receptor ablation on murine mammary gland development and tumorigenesis. J Steroid Biochem Mol Biol. 2004;89–90(1–5):433–6.
VanWeelden K, Flanagan L, Binderup L, Tenniswood M, Welsh J. Apoptotic regression of MCF-7 xenografts in nude mice treated with the vitamin D3 analog, EB1089. Endocrinology. 1998;139(4):2102–10.
Bhatia V, Saini MK, Shen X, Bi LX, Qiu S, Weigel NL, et al. EB1089 inhibits the parathyroid hormone-related protein-enhanced bone metastasis and xenograft growth of human prostate cancer cells. Mol Cancer Ther. 2009;8(7):1787–98.
Zheng Y, Zhou H, Ooi LL, Snir AD, Dunstan CR, Seibel MJ. Vitamin D deficiency promotes prostate cancer growth in bone. Prostate. 2011;71(9):1012–21.
Mordan-McCombs S, Brown T, Wang WL, Gaupel AC, Welsh J, Tenniswood M. Tumor progression in the LPB-Tag transgenic model of prostate cancer is altered by vitamin D receptor and serum testosterone status. J Steroid Biochem Mol Biol. 2010;121(1–2):368–71.
Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin North Am. 2010;39(2):401–18, table of contents.
Ellison TI, Smith MK, Gilliam AC, Macdonald PN. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J Invest Dermatol. 2008;128:2508–17.
Gupta R, Dixon KM, Deo SS, Holliday CJ, Slater M, Halliday GM, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127(3):707–15.
Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol. 2011;29(28):3775–82.
Yin L, Grandi N, Raum E, Haug U, Arndt V, Brenner H. Meta-analysis: serum vitamin D and colorectal adenoma risk. Prev Med. 2011;53(1–2):10–6.
Shin MH, Holmes MD, Hankinson SE, Wu K, Colditz GA, Willett WC. Intake of dairy products, calcium, and vitamin d and risk of breast cancer. J Natl Cancer Inst. 2002;94(17):1301–11.
Lin J, Manson JE, Lee IM, Cook NR, Buring JE, Zhang SM. Intakes of calcium and vitamin D and breast cancer risk in women. Arch Intern Med. 2007;167(10):1050–9.
Chen P, Hu P, Xie D, Qin Y, Wang F, Wang H. Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat. 2010;121(2):469–77.
Gandini S, Boniol M, Haukka J, Byrnes G, Cox B, Sneyd MJ, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer. 2011;128(6):1414–24.
van der Rhee HJ, de Vries E, Coebergh JW. Does sunlight prevent cancer? A systematic review. Eur J Cancer. 2006;42(14):2222–32.
Gilbert R, Martin RM, Beynon R, Harris R, Savovic J, Zuccolo L, et al. Associations of circulating and dietary vitamin D with prostate cancer risk: a systematic review and dose-response meta-analysis. Cancer Causes Control. 2011;22(3):319–40.
Tang JY, Parimi N, Wu A, Boscardin WJ, Shikany JM, Chren MM, et al. Inverse association between serum 25(OH) vitamin D levels and non-melanoma skin cancer in elderly men. Cancer Causes Control. 2010;21(3):387–91.
Asgari MM, Tang J, Warton ME, Chren MM, Quesenberry CP Jr, Bikle D, et al. Association of prediagnostic serum vitamin D levels with the development of basal cell carcinoma. J Invest Dermatol. 2010;130(5):1438–43.
Eide MJ, Johnson DA, Jacobsen GR, Krajenta RJ, Rao DS, Lim HW, et al. Vitamin D and nonmelanoma skin cancer in a health maintenance organization cohort. Arch Dermatol. 2011;147(12):1379–84.
Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM. Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1992;89(17):8097–101.
Kim MS, Fujiki R, Murayama A, Kitagawa H, Yamaoka K, Yamamoto Y, et al. 1Alpha,25(OH)2D3-induced transrepression by vitamin D receptor through E-box-type elements in the human parathyroid hormone gene promoter. Mol Endocrinol (Baltimore, Md). 2007;21(2):334–42.
Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, et al. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009;461(7266):1007–12.
Sai AJ, Walters RW, Fang X, Gallagher JC. Relationship between vitamin D, parathyroid hormone, and bone health. J Clin Endocrinol Metab. 2011;96(3):E436–46.
Ritter CS, Armbrecht HJ, Slatopolsky E, Brown AJ. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006;70(4):654–9.
Meir T, Levi R, Lieben L, Libutti S, Carmeliet G, Bouillon R, et al. Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am J Physiol Renal Physiol. 2009;297(5):F1192–8.
Cheng Z, Tu C, Li A, Santa-Maria C, Ho H, You M, et al. Endocrine actions of parathyroid Cyp27b1 in the Ca2+ and skeletal homeostasis: studies of parathyroid-specific knockout mice. ASBMR Abstract. 2012;1108:S36.
Szabo A, Merke J, Beier E, Mall G, Ritz E. 1,25(OH)2 vitamin D3 inhibits parathyroid cell proliferation in experimental uremia. Kidney Int. 1989;35(4):1049–56.
Liu W, Ridefelt P, Akerstrom G, Hellman P. Differentiation of human parathyroid cells in culture. J Endocrinol. 2001;168(3):417–25.
Dusso A, Cozzolino M, Lu Y, Sato T, Slatopolsky E. 1,25-Dihydroxyvitamin D downregulation of TGFalpha/EGFR expression and growth signaling: a mechanism for the antiproliferative actions of the sterol in parathyroid hyperplasia of renal failure. J Steroid Biochem Mol Biol. 2004;89–90(1–5):507–11.
Gogusev J, Duchambon P, Stoermann-Chopard C, Giovannini M, Sarfati E, Drueke TB. De novo expression of transforming growth factor-alpha in parathyroid gland tissue of patients with primary or secondary uraemic hyperparathyroidism. Nephrol Dial Transplant. 1996;11(11):2155–62.
Cozzolino M, Lu Y, Finch J, Slatopolsky E, Dusso AS. p21WAF1 and TGF-alpha mediate parathyroid growth arrest by vitamin D and high calcium. Kidney Int. 2001;60(6):2109–17.
Canaff L, Hendy GN. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J Biol Chem. 2002;277(33):30337–50.
Canadillas S, Canalejo R, Rodriguez-Ortiz ME, Martinez-Moreno JM, Estepa JC, Zafra R, et al. Upregulation of parathyroid VDR expression by extracellular calcium is mediated by ERK1/2-MAPK signaling pathway. Am J Physiol Renal Physiol. 2010;298(5):F1197–204.
Kadowaki S, Norman AW. Demonstration that the vitamin D metabolite 1,25(OH)2-vitamin D3 and not 24R,25(OH)2-vitamin D3 is essential for normal insulin secretion in the perfused rat pancreas. Diabetes. 1985;34(4):315–20.
Lee S, Clark SA, Gill RK, Christakos S. 1,25-Dihydroxyvitamin D3 and pancreatic beta-cell function: vitamin D receptors, gene expression, and insulin secretion. Endocrinology. 1994;134(4):1602–10.
Norman AW, Frankel JB, Heldt AM, Grodsky GM. Vitamin D deficiency inhibits pancreatic secretion of insulin. Science. 1980;209(4458):823–5.
Zeitz U, Weber K, Soegiarto DW, Wolf E, Balling R, Erben RG. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB J. 2003;17(3):509–11.
Hagstrom E, Hellman P, Lundgren E, Lind L, Arnlov J. Serum calcium is independently associated with insulin sensitivity measured with euglycaemic-hyperinsulinaemic clamp in a community-based cohort. Diabetologia. 2007;50(2):317–24.
Gysemans C, van Etten E, Overbergh L, Giulietti A, Eelen G, Waer M, et al. Unaltered diabetes presentation in NOD mice lacking the vitamin D receptor. Diabetes. 2008;57(1):269–75.
Clark SA, Stumpf WE, Sar M, DeLuca HF, Tanaka Y. Target cells for 1,25 dihydroxyvitamin D3 in the pancreas. Cell Tissue Res. 1980;209(3):515–20.
Morrissey RL, Bucci TJ, Richard B, Empson N, Lufkin EG. Calcium-binding protein: its cellular localization in jejunum, kidney and pancreas. Proc Soc Exp Biol Med. 1975;149(1):56–60.
Bland R, Markovic D, Hills CE, Hughes SV, Chan SL, Squires PE, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in pancreatic islets. J Steroid Biochem Mol Biol. 2004;89–90(1–5):121–5.
Sooy K, Schermerhorn T, Noda M, Surana M, Rhoten WB, Meyer M, et al. Calbindin-D(28k) controls [Ca(2+)](i) and insulin release. Evidence obtained from calbindin-d(28k) knockout mice and beta cell lines. J Biol Chem. 1999;274(48):34343–9.
Rabinovitch A, Suarez-Pinzon WL, Sooy K, Strynadka K, Christakos S. Expression of calbindin-D(28k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology. 2001;142(8):3649–55.
Cheng Q, Li YC, Boucher BJ, Leung PS. A novel role for vitamin D: modulation of expression and function of the local renin-angiotensin system in mouse pancreatic islets. Diabetologia. 2011;54(8):2077–81.
Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, et al. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289(6):G1036–42.
Fukumoto S, Yamashita T. FGF23 is a hormone-regulating phosphate metabolism—unique biological characteristics of FGF23. Bone. 2007;40(5):1190–5.
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–38.
Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D. Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int. 2008;74(2):170–9.
Andrukhova O, Slavic S, Zeitz U, Riesen SC, Heppelmann MS, Ambrisko TD, et al. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol Endocrinol (Baltimore, Md). 2014;28(1):53–64.
Xiang W, Kong J, Chen S, Cao LP, Qiao G, Zheng W, et al. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288(1):E125–32.
Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med. 2007;13(3):117–24.
Bikle DD. Vitamin D and immune function: understanding common pathways. Curr Osteoporos Rep. 2009;7(2):58–63.
Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. 2007;179(3):1634–47.
Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8(3):285–93.
van Etten E, Mathieu C. Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. J Steroid Biochem Mol Biol. 2005;97(1–2):93–101.
Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther. 2008;324(1):23–33.
Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol. 2011;31(17):3653–69.
Palmer MT, Lee YK, Maynard CL, Oliver JR, Bikle DD, Jetten AM, et al. Lineage-specific effects of 1,25-dihydroxyvitamin D(3) on the development of effector CD4 T cells. J Biol Chem. 2011;286(2):997–1004.
Keating P, Munim A, Hartmann JX. Effect of vitamin D on T-helper type 9 polarized human memory cells in chronic persistent asthma. Ann Allergy Asthma Immunol. 2014;112(2):154–62.
Gregori S, Casorati M, Amuchastegui S, Smiroldo S, Davalli AM, Adorini L. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J Immunol. 2001;167(4):1945–53.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–87.
Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol. 1995;15(10):5789–99.
Cippitelli M, Santoni A. Vitamin D3: a transcriptional modulator of the interferon-gamma gene. Eur J Immunol. 1998;28(10):3017–30.
Riis JL, Johansen C, Gesser B, Moller K, Larsen CG, Kragballe K, et al. 1alpha,25(OH)(2)D(3) regulates NF-kappaB DNA binding activity in cultured normal human keratinocytes through an increase in IkappaBalpha expression. Arch Dermatol Res. 2004;296(5):195–202.
Adorini L, Penna G. Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008;4(8):404–12.
Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol (Baltimore, Md). 2003;17(12):2386–92.
Amuchastegui S, Daniel KC, Adorini L. Inhibition of acute and chronic allograft rejection in mouse models by BXL-628, a nonhypercalcemic vitamin D receptor agonist. Transplantation. 2005;80(1):81–7.
Zhang Z, Hener P, Frossard N, Kato S, Metzger D, Li M, et al. Thymic stromal lymphopoietin overproduced by keratinocytes in mouse skin aggravates experimental asthma. Proc Natl Acad Sci U S A. 2009;106(5):1536–41.
Topilski I, Flaishon L, Naveh Y, Harmelin A, Levo Y, Shachar I. The anti-inflammatory effects of 1,25-dihydroxyvitamin D3 on Th2 cells in vivo are due in part to the control of integrin-mediated T lymphocyte homing. Eur J Immunol. 2004;34(4):1068–76.
Wittke A, Weaver V, Mahon BD, August A, Cantorna MT. Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. J Immunol. 2004;173(5):3432–6.
Ehrchen J, Helming L, Varga G, Pasche B, Loser K, Gunzer M, et al. Vitamin D receptor signaling contributes to susceptibility to infection with Leishmania major. FASEB J. 2007;21(12):3208–18.
Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2009;2(5):403–11.
Rajapakse R, Mousli M, Pfaff AW, Uring-Lambert B, Marcellin L, Bronner C, et al. 1,25-Dihydroxyvitamin D3 induces splenocyte apoptosis and enhances BALB/c mice sensitivity to toxoplasmosis. J Steroid Biochem Mol Biol. 2005;96(2):179–85.
Ryz NR, Patterson SJ, Zhang Y, Ma C, Huang T, Bhinder G, et al. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to Citrobacter rodentium by suppressing mucosal Th17 responses. Am J Physiol Gastrointest Liver Physiol. 2012;303(12):G1299–311.
Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–3.
Schauber J, Gallo RL. The vitamin D pathway: a new target for control of the skin’s immune response? Exp Dermatol. 2008;17(8):633–9.
Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285(5428):732–6.
Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, et al. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet. 2011;377(9761):242–50.
Salahuddin N, Ali F, Hasan Z, Rao N, Aqeel M, Mahmood F. Vitamin D accelerates clinical recovery from tuberculosis: results of the SUCCINCT Study [Supplementary Cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis’. BMC Infect Dis. 2013;13:22.
Daley P, Jagannathan V, John KR, Sarojini J, Latha A, Vieth R, et al. Adjunctive vitamin D for treatment of active tuberculosis in India: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2015;15(5):528–34.
Tukvadze N, Sanikidze E, Kipiani M, Hebbar G, Easley KA, Shenvi N, et al. High-dose vitamin D3 in adults with pulmonary tuberculosis: a double-blind randomized controlled trial. Am J Clin Nutr. 2015;102(5):1059–69.
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–60.
Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24(3):341–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Bikle, D.D. (2018). Vitamin D Biochemistry and Physiology. In: Liao, E. (eds) Extraskeletal Effects of Vitamin D. Contemporary Endocrinology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-73742-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-73742-3_1
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-73741-6
Online ISBN: 978-3-319-73742-3
eBook Packages: MedicineMedicine (R0)