
Abstract
Severe sepsis is consistently associated with systemic activation of the hemostatic system. There is abundant proof of a wide-ranging bidirectional cross talk between coagulation and inflammation, which is probably implicated in the pathogenesis of organ dysfunction in patients with sepsis. Inflammation not only leads to initiation and propagation of coagulation activity, but coagulation also markedly influences inflammation. Molecular mechanisms that play a role in inflammation-induced effects on the hemostatic system have been identified in much detail. Pro-inflammatory cells and cyto- and chemokines can activate the coagulation system and downregulate crucial physiological anticoagulant mechanisms. Initiation of coagulation activation and consequent thrombin generation is caused by expression of tissue factor on activated monocytes and endothelial cells and is ineffectually offset by tissue factor pathway inhibitor. At the same time, endothelial-associated anticoagulant pathways, in particular the protein C system, are impaired by pro-inflammatory cytokines. Also, fibrin elimination is severely blocked by inactivation of the endogenous fibrinolytic system, mainly as a result of upregulation of its principal inhibitor, plasminogen activator inhibitor type 1 (PAI-1). Enhanced fibrin generation and insufficient breakdown lead to deposition of (micro)vascular clots, which may contribute to tissue ischemia and ensue organ dysfunction. The cornerstone of the management of coagulation in sepsis is the explicit and thorough treatment of the underlying disorder by appropriate antibiotic treatment and source control interventions. Adjunctive strategies focused at the control of coagulation, including anticoagulants and restoration of physiological anticoagulant mechanisms, may supposedly be indicated, and some of these interventions have been found advantageous in experimental and initial clinical trials, although a beneficial effect in 28-day mortality has not been show unequivocally.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Sepsis and Coagulation
Sepsis is a very serious and potentially life-threatening complication of infection. Sepsis occurs when host defense mediators released into the circulation to combat the infection elicit systemic inflammatory responses throughout the body [1]. Sepsis is a frequently occurring medical condition with an incidence of about 2.5 per 1000 in the Western world and an almost 10% annual rise over the last two decades [2]. About 20% of patients with sepsis die within the hospital, and severe sepsis leads to a mortality rate of approximately 40% [3, 4].
Sepsis is consistently associated with coagulation abnormalities [5]. These deviations range from delicate activation of coagulation that can only be identified by highly sensitive assays for hemostatic factor activation to somewhat more severe coagulation activation that may be noticeable by a subtle fall in platelet count and subclinical elongation of global clotting assays to fulminant disseminated intravascular coagulation (DIC), manifested by profuse microvascular thrombosis in small- and mid-size vessels and simultaneous widespread hemorrhage from various sites [5,6,7]. Patients with sepsis and extensive forms of DIC may develop overt thromboembolic complications or clinically less apparent microvascular clot formation that may contribute to multiple organ failures [7, 8]. In other cases, severe hemorrhage may be the dominant presentation [9], and frequently sepsis and DIC lead to simultaneous thrombosis and bleeding. Hemorrhage is due to consumption and subsequent depletion of coagulation factors and platelets, caused by ongoing activation of the hemostatic system [10]. In its most extreme manifestation, this combination may present as the Waterhouse-Friderichsen syndrome, typically observed during fulminant meningococcal septicemia, although many other microorganisms may cause this clinical situation as well [11].
2 Frequency of Clinically Relevant Coagulopathy in Sepsis
Clinically relevant hemostatic changes may occur in up to 70% of septic patients, and approximately 35% of patients with sepsis will meet the criteria for DIC [12, 13]. The majority of septic patients will develop thrombocytopenia (platelet count <150 × 109/l) [14, 15]. Commonly, platelet count drops in the first 4 days following admission to the hospital [16]. The severity of sepsis correlates markedly with the decrease in platelet count [17]. Critical factors that cause thrombocytopenia in sepsis are decreased platelet production, enhanced consumption, obliteration, or sequestration in the spleen. Decreased production of megakaryocytes in the bone marrow may seem incongruous with the high levels of platelet production-stimulating pro-inflammatory mediators, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, and elevated levels of thrombopoietin in patients with sepsis, which theoretically should stimulate megakaryopoiesis [18]. However, in a substantial number of patients with sepsis, significant hemophagocytosis occurs, consisting of active phagocytosis of platelet precursors and other hematopoietic cells by mononuclear cells, presumably caused by elevated concentrations of macrophage colony-stimulating factor (M-CSF) in sepsis [19]. Platelet consumption is presumably also significant in sepsis, due to platelet activation secondary to continuous formation of thrombin. Platelet activation, consumption, and destruction take place at the endothelial surface as a result of the widespread endothelial cell-platelet interaction in sepsis, although the extent may differ between various vascular beds of different organs [20]. Prolonged global coagulation assays (such as the prothrombin time (PT) or the activated partial thromboplastin time (aPTT)) are detectable in 15–30% of septic patients [21]. Other coagulation assay changes include high fibrin degradation products (in more than 95% of patients with sepsis) [22, 23] and low levels of physiological anticoagulants, such as antithrombin and protein C (90% of sepsis patients) [23, 24].
3 Pathways Leading to Coagulation Abnormalities in Sepsis
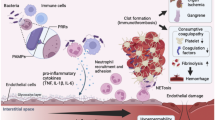
In the last three decades, the pathways involved in the coagulopathy of sepsis have been elucidated for an important part [6]. It is clear that various mechanisms in the coagulation system act simultaneously toward a prohemostatic state. Apparently the most important factors that mediate this derangement of the coagulation system during sepsis are cytokines. Ample evidence indicates an extensive cross talk between inflammation and coagulation, where besides inflammation-induced coagulation activation, coagulation also markedly influences inflammatory activity (Fig. 4.1) [25]. Of note, systemic activation of coagulation and inflammation in sepsis may manifest with organ-specific presentations that are relevant for the specific organ failure resulting from severe sepsis [26].

Interaction between inflammation and coagulation in sepsis. Expression of tissue factor in mononuclear cells and subsequent exposure to blood results in thrombin generation followed by fibrinogen to fibrin conversion. Simultaneously, platelet vessel wall interaction and activation of platelets contribute to (micro)vascular clot formation. Platelet-derived P-selectin further enhances tissue factor expression. Binding of tissue factor, thrombin, and other activated coagulation proteases to specific protease-activated receptors (PARs) and binding of fibrin to Toll-like receptor (TLR) 4 on inflammatory cells affect inflammation through the consequent release of pro-inflammatory cytokines and chemokines, which further modulate coagulation and fibrinolysis [6]
The most important initiator of thrombin formation in sepsis is tissue factor. Studies of experimental or human endotoxemia or cytokinemia have demonstrated a central role of the tissue factor/factor VIIa system in the initiation of thrombin generation [27]. Abrogation of the tissue factor/factor VII(a) pathway by specific interventions aimed at tissue factor or factor VIIa activity resulted in a complete abrogation of thrombin generation in experimental settings [28, 29]. Also, in severe Gram-negative sepsis, ex vivo tissue factor expression on monocytes of patients was demonstrated [30]. Experimental low-dose endotoxemia in healthy humans resulted in a 125-fold increase in tissue factor mRNA levels in blood monocytes [31]. An alternative source of tissue factor may be its localization on other blood cells [32], although it is not likely that these cells themselves produce tissue factor in substantial quantities [33]. Based on the assessment of transfer of tissue factor from mononuclear cells to activated platelets in an ex vivo perfusion setting, it was postulated that this “blood-borne” tissue factor is shuttled between cells through microparticles [34].
Platelets have a central role in the development of coagulation abnormalities in sepsis. Platelets can be triggered directly by pro-inflammatory mediators, such as platelet-activating factor [35]. Generated thrombin will further activate platelets. Activation of platelets may also stimulate fibrin formation by alternative mechanism. The expression of P-selectin on the platelet membrane not only mediates the adherence of platelets to leukocytes and endothelial cells but also enhances the expression of tissue factor on monocytes [36]. The underlying molecular pathway relies on nuclear factor kappa-B (NFκB) expression, induced by binding of activated platelets to neutrophils and monocytes. P-selectin can be shed from the surface of platelet membrane, and soluble P-selectin levels are indeed increased during systemic inflammation [36].
In normal circumstances activation of coagulation is controlled by three important physiological anticoagulant pathways: the antithrombin system, the activated protein C system, and the tissue factor pathway inhibitor (TFPI). In sepsis all three pathways are importantly deranged [37]. Due to a combination of impaired synthesis, ongoing consumption and proteolytic degradation (e.g., by neutrophilic elastase) levels of all three coagulation inhibitors are low. Also, significant downregulation of thrombomodulin and endothelial protein C receptor (EPCR) in inflammatory conditions will cause impaired conversion of protein C to activated protein C. In addition, at the time of the greatest activation of coagulation in sepsis, endogenous fibrinolysis is largely turned off. After the acute release of plasminogen activators (i.e., tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA)) from storage sites in vascular endothelial cells during inflammatory conditions, the increase in plasminogen activation and subsequent plasmin generation is annihilated by a sustained increase in plasminogen activator inhibitor type 1 (PAI-1) [38]. Of interest, studies have shown that a functional mutation in the PAI-1 gene, the 4G/5G polymorphism, not only affected the plasma levels of PAI-1 but was also linked to the clinical outcome of Gram-negative sepsis. Patients with the 4G/4G genotype had significantly higher PAI-1 concentrations and an increased mortality [39]. Other studies showed that the PAI-1 polymorphism increased the risk of developing septic shock from meningococcal infection [40].
4 Inflammation and the Coagulopathy of Sepsis
Like virtually all systemic inflammatory effects of infection, the derangement of the hemostatic system in sepsis is orchestrated by several cytokines. Most pro-inflammatory cytokines have been demonstrated to initiate coagulation activation in vitro. In sepsis high levels of cytokines can be found in the circulation of affected patients and investigational infection or experimental endotoxemia results in the transient increase in plasma levels of these cytokines [27]. The cytokine tumor necrosis factor (TNF) is the first mediator that becomes detectable, followed by an increase in serum levels of several interleukins (IL), of which IL-6 and IL-1 are prominent. Simultaneously, anti-inflammatory cytokines (such as IL-10) may have an inhibitory role in the activation of coagulation.
As TNF is the principal cytokine to become detectable in the circulation upon bacteremia and this cytokine has potent procoagulant effects, it was initially believed that hemostatic activation in sepsis was mediated by TNF. However, in several trials using different strategies to block TNF activity, it was shown that endotoxin induction of TNF could be completely abrogated, whereas activation of coagulation was not affected, albeit that the effects on coagulation inhibitors and fibrinolysis seemed to be regulated by TNF [27]. Also, in animals infused with a lethal dose of E. coli, an anti-TNF antibody had little or no effect on fibrinogen consumption or clinical outcome [41]. In line with this, clinical trials with an anti-TNF monoclonal antibody in septic patients did not show any advantage [42]. Interestingly, it was demonstrated in subsequent studies that strategies that blocked IL-6 caused a complete inhibition of endotoxin-induced activation of coagulation [43]. Also, studies in cancer patients with recombinant IL-6 showed that following the infusion of this cytokine, marked thrombin generation occurred [44]. Hence, these results suggest that IL-6 rather than TNF is important as a mediator for cytokine-induced coagulation activation. Whereas IL-1 is a potent agonist of tissue factor expression in vitro, its role has not been fully elucidated in vivo. Administration of an IL-1 receptor antagonist partly blocked the procoagulant response in experimental sepsis models and inhibited thrombin generation in patients [45]. However, most of the alterations in coagulation occur well before IL-1 becomes detectable in the circulation, leaving a potential role of IL-1 in the coagulopathy of sepsis an unsettled issue.
Coagulation factors and anticoagulant proteins do not only play a role in hemostatic activation but also interact with specific cell receptors leading to activation of signaling pathways (Fig. 4.1). Specifically, protease interactions that modulate inflammatory processes may be important in sepsis. The most significant pathway by which coagulation factors regulate inflammation is by binding to protease-activated receptors (PARs). PARs are transmembrane G-protein-coupled receptors, and four different types (PAR 1–4) have been recognized [46]. A typical property of PARs is that they serve as their own ligand. Proteolytic cleavage by an activated coagulation factor leads to exposure of a neo-amino terminus that is capable of activating the same receptor (and presumably adjacent receptors), leading to transmembrane signaling. PAR-1, PAR-3, and PAR4 are receptors that are activated by thrombin, while PAR-2 is triggered by the tissue factor/factor VIIa complex, factor Xa, and trypsin. PAR-1 is also a receptor for the tissue factor/factor VIIa complex and factor Xa.
It has become apparent that there is a significant cross talk between coagulation inhibitors and inflammatory mediators as well. Antithrombin can serve as a regulator of inflammation, e.g., by direct binding to inflammatory cells, thereby reducing cytokine and chemokine receptor expression [47]. Also, there is ample evidence that the protein C system importantly regulates inflammatory activity [48]. Activated protein C has been demonstrated to attenuate endotoxin-induced production of TNF-α, IL-1β, IL-6, and IL-8 by monocytes/macrophages [49]. In addition, activated protein C blocks cytokine release and leukocyte activation in experimental bacteremia in vivo [50]. Inhibition of the protein C pathway by a monoclonal antibody aggravates the inflammatory response, as shown by enhanced levels of pro-inflammatory cytokines and increased leukocyte activation and tissue damage [51]. Mice with a heterozygous protein C deficiency due to targeted disruption of the protein C gene have not only a stronger hemostatic response to experimental endotoxemia but also show marked differences in inflammatory responses (e.g., higher levels of circulating pro-inflammatory cytokines) [52].
5 Diagnosis of the Coagulopathy in Sepsis
There are several other causes for coagulation changes in septic patients. A low platelet count is almost invariably present in patients with severe sepsis, but thrombocytopenia may also be due to other (potentially concurrently present) conditions, such as immune thrombocytopenia, heparin-induced thrombocytopenia, thrombotic microangiopathies, or medication-induced bone marrow depression [53]. It is crucial to adequately diagnose these differential causes of thrombocytopenia, as they may necessitate specific management strategies [20]. Laboratory assays can be useful in differentiating the coagulopathy in sepsis from various other hemostatic conditions, such as vitamin K deficit or liver insufficiency. As these disorders may be present at the same time with sepsis-associated coagulopathy, this differentiation is not always easy [54, 55].
According to the contemporary thinking about sepsis-associated coagulopathy, the assessment of soluble fibrin in plasma appears to be important [56]. Generally, the sensitivity of assays for soluble fibrin for sepsis-associated coagulopathy is better than the specificity. Some clinical investigations have shown that at certain concentrations of soluble fibrin sepsis-associated coagulopathy is highly probable [22]. Most of the clinical trials show a sensitivity of 90–100% but simultaneously a rather low specificity [57]. Fibrin degradation products (FDPs) may be assayed by specific ELISAs or by latex agglutination assays, enabling quick and bedside determination in urgent cases. None of the available tests for fibrin degradation products distinguishes degradation products of cross-linked fibrin or fibrinogen degradation, which may cause falsely abnormal results [58]. The specificity of high levels of fibrin split products is therefore modest, and a series of other clinical situations, such as trauma, recent surgery, inflammation, or venous thromboembolism, may cause elevated FDPs. More modern tests are specifically targeted at the determination of neo-antigens on degraded cross-linked fibrin. Typically these assays react with an epitope related to plasmin-degraded cross-linked γ-chain, resulting in fragment D-dimer. These tests better distinguish degradation of cross-linked fibrin from fibrinogen or fibrinogen degradation products [59].
Ongoing coagulation activation results in depletion of coagulation factors in septic patients. Also, reduced synthesis, e.g., caused by impaired liver function or vitamin K deficiency, and loss of coagulation factors, due to massive bleeding, may be important. Measurement of fibrinogen levels has been widely promoted as a helpful tool for the diagnosis of coagulation abnormalities in sepsis, but in fact this is not very helpful in most cases [10, 60]. Fibrinogen acts as an acute-phase reactant, and despite considerable turnover, plasma concentrations can be well within the normal range. In a consecutive series of patients, the sensitivity of a low fibrinogen level for the diagnosis of DIC was less than 30%, and hypofibrinogenemia was established in extreme cases of Gram-negative septicemia only. Sequential assessment of fibrinogen might be more helpful and yield diagnostic insight.
Thrombelastography is increasingly employed in critically ill patients with a hypercoagulable state, including those with DIC [61, 62]. Procoagulant as well as anticoagulant states in DIC as indicated with thrombelastography was demonstrated to have a good correlation with clinically important organ dysfunction and survival although its advantage over usual coagulation assays has not yet been confirmed [63,64,65]. In a systematic review of 2 randomized controlled trials and 16 observational studies in patients with sepsis, it was demonstrated that thrombelastography was helpful in correctly identifying the endogenous fibrinolytic state [66]. The authors also found a correlation between hypocoagulability in sepsis and increased mortality. The accurate use of thrombelastography for the diagnosis of DIC has not been rigorously evaluated, although supporters believe that the assay may be helpful for appraising the state of coagulation in patients with critical illness [67, 68].
Based on retrospective analyses of databases from critically ill patients, composite scores for the diagnosis of sepsis-associated coagulopathy have been devised by the International Society on Thrombosis and Hemostasis (ISTH) [69]. The system is based on readily available laboratory tests, i.e., platelet count, prothrombin time, D-dimer, and fibrinogen levels. A diagnosis of DIC is compatible with a score of 5 or more points. The prothrombin time expressed in seconds in the scoring system may be replaced by the INR, making consistency between centers and standardization easier [70]. Validation analyses have shown a high diagnostic accuracy of the scoring system [71, 72]. The intensity of the coagulopathy as judged by this composite score is strongly associated with survival rates in critically ill patients [73]. Combining predictive intensive care measurement systems such as Acute Physiology and Chronic Health Evaluation (APACHE-II) with the DIC score seems to be a potent method to predict the prognosis in septic patients. Similar composite scores have been designed and studied in Japan [74]. The most relevant discrepancies between the ISTH and Japanese scores are a higher sensitivity and a higher proportion of patients with hemato-oncological diseases that are diagnosed with DIC by the Japanese systems [75, 76].
6 Supportive Treatment of Coagulation Abnormalities in Sepsis
The foundation of the treatment of septic coagulopathy is adequate management of the sepsis, e.g., by appropriate antibiotics and source control. However, in many situations, adjunctive supportive treatment, aimed at the replacement of organ function, is necessary. Likewise, coagulation may need supportive measures as the coagulopathy may proceed even after adequate sepsis treatment has been initiated. Some studies show that adjunctive interventions aimed at the derangement of coagulation may positively influence morbidity and mortality. The increase in the understanding of the various pathways that are important in coagulopathy of sepsis has indeed been helpful in the development of such adjunctive management strategies.
Low levels of platelets and coagulation factors may increase the risk of bleeding. However, plasma or platelet substitution therapy should not be instituted on the basis of laboratory results alone; it is indicated only in patients with active bleeding and in those requiring an invasive procedure or otherwise at risk for bleeding complications [77]. The presumed efficacy of treatment with plasma, fibrinogen, cryoprecipitate, or platelets is not based on randomized controlled trials but appears to be rational therapy in bleeding patients or in patients at risk for bleeding with a significant depletion of these hemostatic factors [37]. It may be required to use large volumes of plasma to restore normal concentrations of coagulation factors. Coagulation factor concentrates, such as prothrombin complex concentrate, may overcome this impediment, but these agents may lack important factors (e.g., factor V). Moreover, in older literature, caution is advocated with the use of prothrombin complex concentrates in systemic coagulation activation, as it may aggravate the coagulopathy due to small traces of activated factors in the concentrate. It is, however, less likely that this is still the case for the concentrates that are currently in use. Specific deficiencies in coagulation factors, such as fibrinogen, may be corrected by administration of purified coagulation factor concentrates [37].
Experimental studies have shown that heparin can at least partly inhibit the activation of coagulation in sepsis [78]. Uncontrolled case series in patients with sepsis and DIC have claimed to be successful. However, an advantageous effect of heparin on clinically important outcome events in patients with DIC has never been clearly demonstrated in controlled clinical trials [79], although there is cumulating evidence that heparin might be beneficial [80, 81]. In addition, there are several studies showing that critically ill patients with sepsis need adequate prophylaxis for venous thromboembolism, usually with (low molecular weight) heparin [82, 83]. Therapeutic doses of heparin are indicated in patients with clinically overt thromboembolism or extensive fibrin deposition, like purpura fulminans or acral ischemia. Patients with sepsis may benefit from prophylaxis to prevent venous thromboembolism, which may not be achieved with standard low-dose subcutaneous heparin [84].
Restoration of the levels of physiological anticoagulants in sepsis may be a rational approach [85]. Based on successful preclinical studies, the use of antithrombin concentrates has been examined mainly in randomized controlled trials in patients with severe sepsis. All trials have shown some beneficial effect in terms of improvement of laboratory parameters, shortening of the duration of the coagulopathy, or even improvement in organ function. In several small clinical trials, the use of very high doses of antithrombin concentrate showed even a modest reduction in mortality, however, without being statistically significant. A large-scale, multicenter, randomized controlled trial also showed no significant reduction in mortality of patients with sepsis [86]. Interestingly, post hoc subgroup analyses of this study indicated some benefit in patients who did not receive concomitant heparin and in those with the most severe coagulopathy [87]. Recent propensity-adjusted retrospective data from Japan demonstrated a significant benefit of antithrombin-treated patients with severe infection and sepsis [88, 89]. However these observations still need prospective validation.
Adjunctive therapy with activated protein C (APC) has also been widely studied. A phase III trial of APC concentrate in patients with sepsis was prematurely stopped because of efficacy in reducing mortality in these patients [23]. All-cause mortality at 28 days after inclusion was 24.7% in the APC group versus 30.8% in the control group (a 19.4% relative risk reduction). And there was also an improvement of coagulation abnormalities and reduced organ failure in APC-treated patients. Of note, patients with the most severe coagulopathy benefited most from this treatment [73]. However, a series of negative trials in specific populations of patients with severe sepsis led to scepticism regarding the use of APC in sepsis, and meta-analyses of published literature concluded that the basis for treatment with APC, even in patients with a high disease severity, was not very strong or even insufficient [90]. On top of that, there was uncertainty regarding the bleeding risk of APC in patients with severe sepsis. The last large placebo-controlled trial in patients with severe sepsis and septic shock was prematurely stopped due to the lack of any significant benefit of APC [91]. Subsequently, the manufacturer of APC has decided to withdraw the product from the market, which has resulted in a revision of current guidelines for treatment of DIC [92].
The most promising intervention at this moment is recombinant soluble thrombomodulin. Several preclinical studies in experimental sepsis models have shown that soluble thrombomodulin is capable of improving the derangement of coagulation and may restore organ dysfunction [93]. In phases I–II clinical studies, the pharmacokinetic profile of recombinant soluble thrombomodulin was determined [94]. In a subsequent phase III randomized double-blind clinical trial in patients with DIC, administration of the soluble thrombomodulin had a significantly better effect on bleeding manifestations and coagulation parameters than heparin, but the mortality rate at 28 days was similar in the two study groups [95]. When limiting these results to patients with severe infection and sepsis, DIC resolution rates were 67.5% in thrombomodulin-treated patients and 55.6% in the control group, and 28-day mortality rates were 21.4% and 31.6%, respectively. Subsequently, soluble thrombomodulin was evaluated in a phase II/III clinical study in 750 patients with sepsis and disseminated intravascular coagulation [96]. Twenty-eight-day mortality was 17.8% in the thrombomodulin group and 21.6% in the placebo group. Markers of coagulation activation were lower in the thrombomodulin group than in the placebo group. There were no differences between groups in bleeding or thrombotic events. The promising results with recombinant soluble thrombomodulin are supported by retrospective data in large series of Japanese patients and are currently being evaluated in a large international multicenter trial [97, 98].
7 New Pathways and Targets in the Management of DIC
In view of the overwhelming evidence for the central role of impaired natural anticoagulant pathways in the pathogenesis of DIC, much attention has been focused on the restoration of physiological anticoagulation as (adjunctive) treatment of DIC [6]. However, despite the fact that these interventions (such as recombinant human-activated protein C or antithrombin concentrate) have shown efficacy in reversing the coagulopathy, they have not resulted in an improvement on clinically relevant outcomes, such as survival or improvement of organ dysfunction [99]. One of the factors responsible for this may be that all these anticoagulants are clearly limited by the potential risk of major hemorrhage in critically ill patients. Therefore, it has been hypothesized that molecules that have less anticoagulant properties but have retained their anti-inflammatory effects may be promising new agents for the management of DIC. For example, non-anticoagulant heparin inhibits the expression and function of adhesion molecules, such as P-selectin and L-selectin. Moreover, this compound directly affects pro-inflammatory mediators, such as nuclear factor (NF)-κβ and cytokines, and attenuates endothelial cell dysfunction through the nitric oxide system. Non-anticoagulant heparin has a strong affinity for extracellular histones that result from cellular destruction during severe inflammation and that are robustly associated with endothelial dysfunction, organ failure, and death during sepsis [100]. Binding of this non-anticoagulant heparin to histones strongly inhibited cytotoxic activity in vitro and translated to impaired inflammation and improved survival in animal models of systemic infection and inflammation. Similarly, recent experiments indicate a beneficial effect of activated protein C variants that have lost their anticoagulant properties [101].
Another interesting new target may the glycocalyx covering the endothelial surface of the vascular bed [102]. The endothelium of the capillary bed is the most important interface in which the interaction between inflammation and coagulation takes place. All physiologic anticoagulant systems and various adhesion molecules that may modulate both inflammation and coagulation are connected to the endothelium. In sepsis, endothelial glycosaminoglycans present in the glycocalyx are downregulated by pro-inflammatory cytokines, thereby impairing the functions of antithrombin (AT), tissue factor pathway inhibitor (TFPI), leukocyte adhesion, and leukocyte transmigration. Because the glycocalyx also plays a role in other endothelial functions, including maintenance of the vascular barrier function, nitric oxide-mediated vasodilation, and antioxidant activity, all these processes can be impaired in DIC. Moreover, specific disruption of the glycocalyx results in thrombin generation and platelet adhesion within a few minutes. Novel interventions aimed at restoration of the glycocalyx may potentially maintain adequate physiological anticoagulation to balance activated coagulation in DIC [103].
References
Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(9):840–51.
Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–54.
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–10.
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–20.
Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017;149:38–44.
Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev. 2016;2:16037.
Levi M. The coagulant response in sepsis. Clin Chest Med. 2008;29(4):627–42.
Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341(8):586–92.
Miller DL, Welty-Wolf K, Carraway MS, Ezban M, Ghio A, Suliman H, et al. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002;26(6):650–8.
Levi M, ten Cate H, van der Poll T. Disseminated intravascular coagulation: state of the art. Thromb Haemost. 1999;82:695–705.
Ratnoff OD, Nebehay WG. Multiple coagulative defects in a patient with the Waterhouse-Friderichsen syndrome. Ann Intern Med. 1962;56:627.
Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340(3):207–14.
Levi M, de Jonge E, van der Poll T. Sepsis and disseminated intravascular coagulation. J Thromb Thrombolysis. 2003;16(1–2):43–7.
Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, et al. Thrombocytopenia and prognosis in intensive care. Crit Care Med. 2000;28(6):1871–6.
Strauss R, Wehler M, Mehler K, Kreutzer D, Koebnick C, Hahn EG. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements, and outcome. Crit Care Med. 2002;30(8):1765–71.
Akca S, Haji Michael P, de Medonca A, Suter PM, Levi M, Vincent JL. The time course of platelet counts in critically ill patients. Crit Care Med. 2002;30:753–6.
Mavrommatis AC, Theodoridis T, Orfanidou A, Roussos C, Christopoulou-Kokkinou V, Zakynthinos S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med. 2000;28(2):451–7.
Folman CC, Linthorst GE, van Mourik J, van Willigen G, de Jonge E, Levi M, et al. Platelets release thrombopoietin (Tpo) upon activation: another regulatory loop in thrombocytopoiesis? Thromb Haemost. 2000;83(6):923–30.
Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med. 1997;103(2):114–20.
Warkentin TE, Aird WC, Rand JH. Platelet-endothelial interactions: sepsis, HIT, and antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program. 2003;1:497–519.
MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55(1):39–44.
Shorr AF, Thomas SJ, Alkins SA, Fitzpatrick TM, Ling GS. D-dimer correlates with proinflammatory cytokine levels and outcomes in critically ill patients. Chest. 2002;121(4):1262–8.
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709.
Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79(6):1111–5.
Levi M, van der Poll T, Buller HR. The bidirectional relationship between coagulation and inflammation. Circulation. 2004;109:2698–704.
Aird WC. Vascular bed-specific hemostasis: role of endothelium in sepsis pathogenesis. Crit Care Med. 2001;29(7 Suppl):S28–34.
Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38(2 Suppl):S26–34.
Taylor FB Jr, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, et al. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33(3):127–34.
Levi M, ten Cate H, Bauer KA, van der Poll T, Edgington TS, Buller HR, et al. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Investig. 1994;93(1):114–20.
Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983;49(1):5–7.
Franco RF, de Jonge E, Dekkers PE, Timmerman JJ, Spek CA, van Deventer SJ, et al. The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood. 2000;96(2):554–9.
Giesen PL, Rauch U, Bohrmann B, Kling D, Roque M, Fallon JT, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96(5):2311–5.
Osterud B, Rao LV, Olsen JO. Induction of tissue factor expression in whole blood – lack of evidence for the presence of tissue factor expression on granulocytes. Thromb Haemost. 2000;83:861–7.
Rauch U, Bonderman D, Bohrmann B, Badimon JJ, Himber J, Riederer MA, et al. Transfer of tissue factor from leukocytes to platelets is mediated by CD15 and tissue factor. Blood. 2000;96(1):170–5.
Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. 2002;30(5 Suppl):S294–301.
Shebuski RJ, Kilgore KS. Role of inflammatory mediators in thrombogenesis. J Pharmacol Exp Ther. 2002;300(3):729–35.
Levi M, van der Poll T. Coagulation in patients with severe sepsis. Semin Thromb Hemost. 2015;41(1):9–15.
Biemond BJ, Levi M, ten CH, van dP, Buller HR, Hack CE, et al. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci. 1995;88(5):587–94.
Hermans PW, Hibberd ML, Booy R, Daramola O, Hazelzet JA, de GR, et al. 4G/5G promoter polymorphism in the plasminogen-activator-inhibitor-1 gene and outcome of meningococcal disease. Meningococcal Research Group. Lancet. 1999;354(9178):556–60.
Westendorp RG, Hottenga JJ, Slagboom PE. Variation in plasminogen-activator-inhibitor-1 gene and risk of meningococcal septic shock. Lancet. 1999;354(9178):561–3.
Hinshaw LB, Tekamp-Olson P, Chang AC, Lee PA, Taylor FB Jr, Murray CK, et al. Survival of primates in LD100 septic shock following therapy with antibody to tumor necrosis factor (TNF alpha). Circ Shock. 1990;30(3):279–92.
Abraham E, Wunderink R, Silverman H, Perl TM, Nasraway S, Levy H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA. 1995;273(12):934–41.
van der Poll T, Levi M, Hack CE, ten Cate H, van Deventer S, Eerenberg AJ, et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med. 1994;179(4):1253–9.
Stouthard JM, Levi M, Hack CE, Veenhof CH, Romijn HA, Sauerwein HP, et al. Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost. 1996;76(5):738–42.
Boermeester MA, van Leeuwen P, Coyle SM, Wolbink GJ, Hack CE, Lowry SF, et al. Interleukin-1 blockade attenuates mediator release and dysregulation of the hemostatic mechanism during human sepsis. Arch Surg. 1995;130(7):739–48.
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64.
Kaneider NC, Forster E, Mosheimer B, Sturn DH, Wiedermann CJ. Syndecan-4-dependent signaling in the inhibition of endotoxin-induced endothelial adherence of neutrophils by antithrombin. Thromb Haemost. 2003;90(6):1150–7.
Esmon CT. New mechanisms for vascular control of inflammation mediated by natural anticoagulant proteins. J Exp Med. 2002;196(5):561–4.
Yuksel M, Okajima K, Uchiba M, Horiuchi S, Okabe H. Activated protein C inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappa B and activator protein-1 in human monocytes. Thromb Haemost. 2002;88(2):267–73.
Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, et al. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87(2):642–7.
Taylor FB Jr, Stearns-Kurosawa DJ, Kurosawa S, Ferrell G, Chang AC, Laszik Z, et al. The endothelial cell protein C receptor aids in host defense against Escherichia coli sepsis. Blood. 2000;95(5):1680–6.
Levi M, Dorffler-Melly J, Reitsma PH, Buller HR, Florquin S, van der Poll T, et al. Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein C deficient mice. Blood. 2003;101:4823–7.
Baughman RP, Lower EE, Flessa HC, Tollerud DJ. Thrombocytopenia in the intensive care unit. Chest. 1993;104(4):1243–7.
Levi M, de Jonge E, Meijers J. The diagnosis of disseminated intravascular coagulation. Blood Rev. 2002;16(4):217–23.
Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. 2009;145(1):24–33.
Dempfle CE, Pfitzner SA, Dollman M, Huck K, Stehle G, Heene DL. Comparison of immunological and functional assays for measurement of soluble fibrin. Thromb Haemost. 1995;74(2):673–9.
Horan JT, Francis CW. Fibrin degradation products, fibrin monomer and soluble fibrin in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27(6):657–66.
Prisco D, Paniccia R, Bonechi F, Francalanci I, Abbate R, Gensini GF. Evaluation of new methods for the selective measurement of fibrin and fibrinogen degradation products. Thromb Res. 1989;56(4):547–51.
Shorr AF, Trotta RF, Alkins SA, Hanzel GS, Diehl LF. D-dimer assay predicts mortality in critically ill patients without disseminated intravascular coagulation or venous thromboembolic disease. Intensive Care Med. 1999;25(2):207–10.
Levi M, Meijers JC. DIC: which laboratory tests are most useful. Blood Rev. 2011;25(1):33–7.
Dempfle CE, Borggrefe M. Point of care coagulation tests in critically ill patients. Semin Thromb Hemost. 2008;34(5):445–50.
Levi M, Hunt BJ. A critical appraisal of point-of-care coagulation testing in critically ill patients. J Thromb Haemost. 2015;13(11):1960–7.
Johansson PI, Stensballe J, Vindelov N, Perner A, Espersen K. Hypocoagulability, as evaluated by thrombelastography, at admission to the ICU is associated with increased 30-day mortality. Blood Coagul Fibrinolysis. 2010;21(2):168–74.
Park MS, Martini WZ, Dubick MA, Salinas J, Butenas S, Kheirabadi BS, et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J Trauma. 2009;67(2):266–75.
Collins PW, Macchiavello LI, Lewis SJ, Macartney NJ, Saayman AG, Luddington R, et al. Global tests of haemostasis in critically ill patients with severe sepsis syndrome compared to controls. Br J Haematol. 2006;135(2):220–7.
Muller MC, Meijers JC, Vroom MB, Juffermans NP. Utility of thromboelastography and/or thromboelastometry in adults with sepsis: a systematic review. Crit Care. 2014;18(1):R30.
Brenner T, Schmidt K, Delang M, Mehrabi A, Bruckner T, Lichtenstern C, et al. Viscoelastic and aggregometric point-of-care testing in patients with septic shock – cross-links between inflammation and haemostasis. Acta Anaesthesiol Scand. 2012;56(10):1277–90.
Daudel F, Kessler U, Folly H, Lienert JS, Takala J, Jakob SM. Thromboelastometry for the assessment of coagulation abnormalities in early and established adult sepsis: a prospective cohort study. Crit Care. 2009;13(2):R42.
Taylor FB Jr, Toh CH, Hoots WK, Wada H, Levi M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86(5):1327–30.
Kim HK, Hong KH, Toh CH. Application of the international normalized ratio in the scoring system for disseminated intravascular coagulation. J Thromb Haemost. 2010;8(5):1116–8.
Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective validation of the international society of thrombosis and haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004;32:2416–21.
Toh CH, Hoots WK. The scoring system of the scientific and standardisation committee on disseminated intravascular coagulation of the International Society on thrombosis and haemostasis: a five year overview. J Thromb Haemost. 2007;5:604–6.
Dhainaut JF, Yan SB, Joyce DE, Pettila V, Basson BR, Brandt JT, et al. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004;2:1924–33.
Wada H, Gabazza EC, Asakura H, Koike K, Okamoto K, Maruyama I, et al. Comparison of diagnostic criteria for disseminated intravascular coagulation (DIC): diagnostic criteria of the International Society of Thrombosis and Hemostasis and of the Japanese Ministry of Health and Welfare for overt DIC. Am J Hematol. 2003;74(1):17–22.
Wada H, Thachil J, Di NM, Mathew P, Kurosawa S, Gando S, et al. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013;11:761–7.
Levi M. Settling the score for disseminated intravascular coagulation. Crit Care Med. 2005;33(10):2417–8.
Levi M. Platelets in critical illness. Semin Thromb Hemost. 2016;42(3):252–7.
du Toit HJ, Coetzee AR, Chalton DO. Heparin treatment in thrombin-induced disseminated intravascular coagulation in the baboon. Crit Care Med. 1991;19(9):1195–200.
Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60(2):284–7.
Zarychanski R, Abou-Setta AM, Kanji S, Turgeon AF, Kumar A, Houston DS, et al. The efficacy and safety of heparin in patients with sepsis: a systematic review and metaanalysis. Crit Care Med. 2015;43(3):511–8.
Levi M. The dual face of heparin in severe infection. Blood. 2014;123(7):947–8.
Boonyawat K, Crowther M. Venous thromboembolism prophylaxis in critically ill patients. Semin Thromb Hemost. 2015;41(1):68–74.
Levi M, Levy M, Williams MD, Douglas I, Artigas A, Antonelli M, et al. Prophylactic heparin in patients with severe sepsis treated with drotrecogin alfa (activated). Am J Respir Crit Care Med. 2007;176(5):483–90.
Dorffler-Melly J, de Jonge E, Pont AC, Meijers J, Vroom MB, Buller HR, et al. Bioavailability of subcutaneous low-molecular-weight heparin to patients on vasopressors. Lancet. 2002;359(9309):849–50.
Levy JH, Sniecinski RM, Welsby IJ, Levi M. Antithrombin: anti-inflammatory properties and clinical applications. Thromb Haemost. 2016;115(4):712–28.
Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286(15):1869–78.
Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, et al. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost. 2006;4(1):90–7.
Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: an observational nationwide study. J Thromb Haemost. 2014;12(9):1470–9.
Iba T, Saitoh D, Wada H, Asakura H. Efficacy and bleeding risk of antithrombin supplementation in septic disseminated intravascular coagulation: a secondary survey. Crit Care. 2014;18(5):497.
Levi M. Activated protein C in sepsis: a critical review. Curr Opin Hematol. 2008;15(5):481–6.
Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin Alfa (activated) in adults with septic shock. N Engl J Med. 2012;366(22):2055–64.
Thachil J, Toh CH, Levi M, Watson HG. The withdrawal of activated protein C from the use in patients with severe sepsis and DIC [amendment to the BCSH guideline on disseminated intravascular coagulation]. Br J Haematol. 2012;157(4):493–4.
Levi M, van der Poll T. Thrombomodulin in sepsis. Minerva Anestesiol. 2013;79(3):294–8.
Tsuruta K, Yamada Y, Serada M, Tanigawara Y. Model-based analysis of covariate effects on population pharmacokinetics of thrombomodulin alfa in patients with disseminated intravascular coagulation and normal subjects. J Clin Pharmacol. 2011;51(9):1276–85.
Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. 2007;5(1):31–41.
Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, et al. A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41(9):2069–79.
Yamakawa K, Aihara M, Ogura H, Yuhara H, Hamasaki T, Shimazu T. Recombinant human soluble thrombomodulin in severe sepsis: a systematic review and meta-analysis. J Thromb Haemost. 2015;13(4):508–19.
Levi M. Recombinant soluble thrombomodulin: coagulation takes another chance to reduce sepsis mortality. J Thromb Haemost. 2015;13(4):505–7.
Levi M, Schultz M, van der Poll T. Sepsis and thrombosis. Semin Thromb Hemost. 2013;39(5):559–66.
Wildhagen KC, Garcia de FP, Reutelingsperger CP, Schrijver R, Areste C, Ortega-Gomez A, et al. Non-anticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014;123(7):1098–101.
Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204(10):2439–48.
Schouten M, Wiersinga WJ, Levi M, van Der PT. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83(3):536–45.
Nieuwdorp M, Meuwese MC, Mooij HL, van Lieshout MH, Hayden A, Levi M, et al. Tumor necrosis factor-alpha inhibition protects against endotoxin-induced endothelial glycocalyx perturbation. Atherosclerosis. 2009;202(1):296–303.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Levi, M. (2018). The Coagulation System in Sepsis. In: Wiersinga, W., Seymour, C. (eds) Handbook of Sepsis. Springer, Cham. https://doi.org/10.1007/978-3-319-73506-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-73506-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-73505-4
Online ISBN: 978-3-319-73506-1
eBook Packages: MedicineMedicine (R0)