
Abstract
Medulloblastomas are embryonal cerebellar tumors and the most common central nervous system malignancies in children. While progress in clinical management dramatically improved outcomes for what was once a uniformly fatal disease, the prognosis for children with metastatic or recurrent medulloblastoma remains poor despite intensive multimodal therapy. As we reach a limit of effect and toxicities with conventional therapies, a new understanding of medulloblastoma biology has emerged, paving the way for the next generation of clinical trials. Current consensus identifies four medulloblastoma entities – WNT, SHH, Group 3, and Group 4 – with distinct clinical presentations, genetic and molecular features, responses to therapy, and patterns of relapse. The first prospective studies designed to deliver subgroup-specific treatment are underway and hold the promise of better survival outcomes and fewer long-term toxicities.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Introduction
Medulloblastomas are highly malignant and invasive neuroepithelial embryonal tumors that arise in the cerebellum. Cushing and Bailey first introduced the term in 1926 (Bailey and Cushing 1926), describing undifferentiated cerebellar tumors that invariably relapsed and quickly progressed, with short survival after surgery. The evolution of treatment including the introduction of adjuvant radiotherapy and, subsequently, chemotherapy allowed for gradual improvement in outcome and long-term survival. Once an exceedingly dismal disease, medulloblastoma can now be cured in the majority of affected children using a combination of surgery, radiotherapy (for children older than 3 years of age), and chemotherapy. Notwithstanding the progress made, current therapy often fails for patients with metastatic medulloblastoma and at relapse the disease remains universally fatal.
Improved outcomes led to the recognition of a host of tumor- and treatment-related long-term effects, including endocrine dysfunction, hearing loss, secondary malignancies, and neurocognitive impairment, which preclude many adult survivors of childhood medulloblastoma from leading an independent life.
Over the last decade, insights into tumor biology revealed that, rather than a single disease, medulloblastoma comprises four entities with distinct clinical, histopathological, genetic, and outcome features: the subgroups WNT (wingless), SHH (sonic hedgehog), Group 3, and Group 4 (Taylor et al. 2012). Armed with the evidence that tumor biology largely dictates clinical presentation and outcome, there is an ongoing effort to establish a biology-based risk classification in order to allow proper allocation of therapy in clinical trials, avoid overtreatment of low-risk subgroups, identify high-risk patients that require intensification of treatment, and develop better therapies.
Epidemiology
The overall incidence of medulloblastoma is approximately 1.5 per million. Though it may present at any age, medulloblastoma is 10 times more likely to affect children than adults (incidence 6.0 per million children aged 1–9 years, compared to 0.6 per million adults), and is rare after the fourth decade (Smoll and Drummond 2012). There are two age peaks at 3–5 and 7–10 years, with 80% of the patients being diagnosed under the age of 15 years. Most studies report a male to female 1.5:1 preponderance. Up to one-third of the patients present with leptomeningeal metastatic disease at diagnosis (Terterov et al. 2010).
Clinical Presentation
Due to its location in the posterior fossa, medulloblastoma typically causes mass effect, obstruction to cerebrospinal fluid flow at the level of the fourth ventricle, and secondary hydrocephalus. As such, most children present with symptoms of raised intracranial pressure and cerebellar dysfunction, with headache, vomiting, and ataxia reported in 75% of the patients at diagnosis. Fatigue, lethargy, and systemic symptoms related to poor nutrition and dehydration may be present as well. Other symptoms include diplopia, blurred vision, irritability, behavioral changes, vertigo, and hearing loss. Diagnosis may be challenging in infants and young children, who often present with a history of developmental delay and loss of psychomotor milestones.
On physical exam, ataxia with impaired heel-to-toe and broad-based gait, and ocular signs, such as nystagmus and cranial nerve VI palsy, are common. Papilledema is typically seen in the presence of acute hydrocephalus, though often absent if short duration of symptoms. Patients with lateral, hemispheric tumors may present with dysmetria on finger-to-nose testing. In infants, other findings include bulging of the fontanelle and macrocephaly.
Children are usually diagnosed within 2–3 months after onset of symptoms, and longer time to diagnosis does not negatively impact outcome. In fact, shorter time to diagnosis correlates with worse survival (Halperin et al. 2001; Gerber et al. 2012), reflecting subgroup-specific differences, rapid disease progression, and clinical deterioration in more aggressive tumors (Ramaswamy et al. 2014).
Association with Cancer Predisposition Syndromes
Though most cases are sporadic, approximately 5% of medulloblastomas – particularly of the SHH subgroup – may manifest in the context of cancer predisposition syndromes. Every child with medulloblastoma should be carefully evaluated for potential underlying genetic syndromes. The study of these rare genetic diseases has provided important insights into the etiology of medulloblastoma.
Gorlin Syndrome
Also known as nevoid basal cell carcinoma syndrome, Gorlin syndrome is an autosomal dominant disease caused by activating mutations in the PTCH1 tumor suppressor gene on chromosome 9, which encodes the Shh receptor Patched (Amlashi et al. 2003). It is characterized by multiple congenital malformations – fused ribs, vertebrae anomalies, early calcification of the falx cerebri, macrocephaly, frontal bossing, hypertelorism, syndactyly, odontogenic cysts – and predisposition to early onset cancer, including basal cell carcinoma, ovarian carcinoma, and medulloblastoma. Less than 5% of patients with Gorlin syndrome will develop medulloblastoma, accounting for less than 2% of all medulloblastomas. Typical onset occurs within the first 2 years of life; these tumors display desmoplastic histology and have a good overall prognosis. The clinical features may be subtle at this young age but early diagnosis is crucial; due to the high risk of basal cell carcinoma after radiation, these patients should not be irradiated.
Germline mutations of SUFU on chromosome 10 – encoding the suppressor of fused of the Shh pathway – predispose to SHH medulloblastoma of desmoplastic histology and the affected children have a Gorlin-like phenotype (Taylor et al. 2002). Though the prognostic implications are still being elucidated, patients with SUFU-associated Gorlin syndrome have a 20× higher risk of developing medulloblastoma when compared to PTCH1-associated Gorlin syndrome (Smith et al. 2014).
Turcot Syndrome
Turcot syndrome refers to two types of clinical entities characterized by an association between brain tumors and colonic polyposis. Type II – familial adenomatous polyposis (FAP) – is characterized by germline mutations in the adenomatous polyposis coli gene (APC) on chromosome 5 and activation of the Wnt pathway; medulloblastoma is the predominant brain tumor affecting these families and carries a good prognosis (Hamilton et al. 1995). Type I – hereditary nonpolyposis colorectal cancer – is caused by germline mutations in DNA mismatch repair genes and is associated with gliomas.
Li-Fraumeni Syndrome
Li-Fraumeni syndrome is an autosomal dominant cancer predisposition syndrome caused by germline mutations in the tumor suppressor gene TP53, leading to early onset sarcomas, breast cancer, adrenocortical carcinoma and leukemia, as well as brain tumors. Though gliomas are more common, it is also associated with medulloblastomas.
Rubinstein-Taybi Syndrome
Medulloblastomas have been described in patients with Rubinstein-Taybi syndrome, associated with an interstitial 16p13.3 deletion. This region often includes CREBBP – a tumor suppressor gene mutated in a subset of medulloblastomas – and its loss is likely the cause of medulloblastoma in these patients (Bourdeaut et al. 2014).
Imaging
General Imaging Characteristics
Medulloblastomas show specific neuroradiological characteristics. On noncontrast computer tomography (CT) scan, medulloblastomas typically appear as a hyperdense posterior fossa mass that homogeneously enhances after contrast administration and is surrounded by peritumoral edema. Due to an involvement of the fourth ventricle, an obstructive hydrocephalus is present at the time of diagnosis in up to 95% of patients. Magnetic resonance imaging (MRI) is superior to CT in terms of detecting atypical imaging features and tumor dissemination and is the preferred imaging modality to plan neurosurgical interventions as well as for follow-up examinations. For pediatric posterior fossa tumors, the standard MRI protocol includes T1- and T2-weighted, T1-weighted post-contrast, FLAIR (fluid-attenuated inversion recovery), and diffusion sequences. On T1-weighted images, medulloblastomas are usually iso- to hypointense, well-defined masses. The extent of contrast enhancement and the signal on T2-weighted images are variable and often heterogenous (Eran et al. 2010) (Fig. 1). FLAIR images typically show hyperintensity compared to the surrounding brain tissue. Due to the high cellularity, medulloblastomas commonly present restricted diffusion of water on diffusion-weighted sequences (bright signal on DWI sequence, dark signal on ADC image) (Raybaud et al. 2015).

MRI characteristics of a medulloblastoma in the area of the fourth ventricle: (a) On the sagittal T1-weighted image, the mass is hypointense compared to the normal cerebellum tissue (arrow). (b) The sagittal T1-weighted post-gadolinium image shows a nearly homogeneous enhancement of the tumor (arrow). (c) On the sagittal T2-weighted image the mass appears mildly hyperintense compared to the surrounding normal cerebellum tissue (arrow). (d) The coronal T2-weighted image shows a hydrocephalic enlargement of the cerebral ventricles (arrows). (e, f) Postcontrast sagittal T1-weighted images show leptomeningeal dissemination along the surface of the spinal cord (arrows)
The diagnosis of medulloblastoma should be considered in any child presenting with a posterior fossa tumor, even in the presence of atypical neuroradiological findings (Eran et al. 2010). Tumor extension through the foramina of Magendie and Luschka and evidence of calcification – often detected in patients with ependymoma – are uncommon but may be present in medulloblastoma. The presence of necrosis or a large cyst in a posterior fossa mass often suggests the diagnosis of a pilocytic astrocytoma but cysts, often multiple and smaller, may also be detected in medulloblastoma.
Preoperative MRI imaging of the whole neuroaxis is critical to assess disease dissemination prior to treatment because leptomeningeal tumor dissemination is seen in about 30% of cases at time of diagnosis (Terterov et al. 2010).
Imaging Correlates with Molecular Subgroups
The molecular subgroups of medulloblastoma have characteristic radiologic presentations, which are helpful to presurgically assess the tumor type and aggressiveness (Raybaud et al. 2015).
Tumors located along the cerebellar peduncle/cerebellopontine angle (CP/CPA) most likely belong to the WNT molecular subgroup. These tumors arise from the lower rhombic lip, are usually well-defined, compact, and homogenous masses filling the fourth ventricle; they are often associated with obstructive hydrocephalus. The tumors are mildly hyperdense on CT and are highly restricted on diffusion-weighted sequences, typically enhancing after contrast administration. Leptomeningeal dissemination at the time of initial diagnosis is uncommon.
A cerebellar hemispheric location is characteristic of the SHH subgroup, in contrast with the other three subgroups that are typically localized in the midline. These tumors appear well-circumscribed and highly restricted on diffusion-weighted images, and show extensive enhancement with a multinodular pattern. Midline tumors in the immediate proximity of the fourth ventricle are usually associated with a high-pressure hydrocephalus, they are radiologically heterogeneous and belong predominantly to the Group 3 and 4 molecular subgroups. An enhancing midline mass with ill-defined tumor margins is most likely a Group 3 medulloblastoma. Minimal or absent enhancement after contrast administration is characteristic of Group 4 medulloblastomas. Metastatic dissemination is also quite common in both Group 3 and Group 4 tumors (Perreault et al. 2014; Raybaud et al. 2015).
Classification and Molecular Subgrouping
Histopathology
Medulloblastomas are densely packed, small round blue-cell tumors, with high mitotic activity corresponding to WHO Grade IV. Homer-Wright rosettes are observed in up to 40% of cases and most medulloblastomas display immunohistochemical positivity for neuron-specific enolase and synaptophysin, consistent with neuronal differentiation. There are four histological variants: classic, large cell/anaplastic (LCA), desmoplastic/nodular, and medulloblastoma with extensive nodularity (MBEN). Classic histology is the most common, characterized by sheets of small round blue cells with a high nuclear/cytoplastic ratio and round nuclei. Marked nuclear pleomorphism and high mitotic activity with atypical forms are prominent features of anaplastic medulloblastoma, often containing cells with large nuclei, prominent nucleoli, and variable amounts of eosinophilic cytoplasm. Desmoplastic medulloblastoma is characterized by pale islands of reticulin-free tumor tissue, surrounded by highly proliferative cells with a dense intercellular reticulin fiber network. MBEN differ from desmoplastic medulloblastoma in that they have large elongated reticulin-free zones, which contain small round neurocytic cells in a fibrillary background.
Several studies found a correlation between anaplasia and adverse outcome (Kortmann et al. 2000; Gajjar et al. 2006; Giangaspero et al. 2006), whereas desmoplasia (Rutkowski et al. 2005) and extensive nodularity have a good prognosis (Giangaspero et al. 1999). However, when analyzed in the context of molecular subgroups, tumor subgroup affiliation combined with cytogenetic and clinical biomarkers provided a more accurate survival prediction than histopathological characteristics (Shih et al. 2014).
Molecular Subgroups
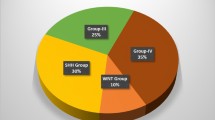
In order to transition from a histology-based to a molecular-based approach to diagnosis, the most recent 2016 World Health Organization classification of CNS tumors proposed an integrated classification, including “genetically defined” and “histologically defined” variants (Louis et al. 2016). Medulloblastoma genetic profiling studies have established that there are four molecular subgroups of medulloblastoma: WNT, SHH, Group 3, and Group 4 (Taylor et al. 2012), with distinct cell of origin, activated pathways, clinical and genetic characteristics (Fig. 2).

Molecular subgroups of medulloblastoma. (Reprinted by permission from: Springer publishers, Taylor et al. 2012)
WNT medulloblastoma arises from lower rhombic lip progenitor cells of the dorsal brainstem. Activation of the Wnt pathway is the hallmark of these tumors and is mostly due to CTNNB1 mutations, the gene encoding beta-catenin; these may be associated with TP53 mutations. In addition to a characteristic WNT-pathway signature, they are readily detectable by immunohistochemistry via beta-catenin staining in the nucleus rather than its normal pattern of expression at the cell membrane. Monosomy of chromosome 6 is a characteristic cytogenetic alteration that is almost never seen in other subgroups. WNT is the least common of the four subgroups, accounting for 10% of all medulloblastomas; they are often of classic histology and nonmetastatic. They have an equal gender distribution, and usually affect children older than 3 years and adults; they are associated with good prognosis.
SHH medulloblastoma derives from cerebellar granule precursor cells of the external granule layer and is characterized by activation of the SHH pathway, often through mutations in the tumor suppressor genes PTCH1 or SUFU or amplifications of GLI2 or MYCN. TP53 mutations are detected in 20% of SHH tumors, approximately 50% of them are somatic. Unlike WNT medulloblastoma – where TP53 mutations have no correlation with outcome – SHH tumors with TP53 mutations define a very high-risk group of patients (Ramaswamy et al. 2016a). The presence of chromotripsis (chromosome shredding), focal GLI2 or NMYC amplification and chromosome 14q loss correlate with poor outcome. Other common cytogenetic abnormalities are chromosome 9q and 10q loss. SHH tumors account for one-third of all medulloblastomas and have a bimodal age distribution, with age peaks in infancy and late childhood/adolescence. They are generally described as intermediate risk tumors; however, TP53-mutated SHH tumors have a very poor survival outcome. All histological variants may be seen; desmoplastic histology is restricted to SHH tumors and indicates a favorable outcome when compared to classical histology, which in turn has a better prognosis than LCA in SHH tumors (Shih et al. 2014).
Group 3 medulloblastoma. Accounting for 25–30% of all medulloblastomas, Group 3 tumors exclusively affect infants and children, predominantly boys, and have the worst outcome as they commonly present with leptomeningeal metastatic dissemination at diagnosis. The cell of origin remains unclear and the mechanisms underlying tumorigenesis are less well defined than in the two previous subgroups. Recurrent mutations are uncommon and transcriptional profiling analysis failed to identify alterations in a common pathway, although an overall enrichment for genes involved in protein translation, nucleotide metabolism, photoreceptor, and GABAergic functions were noted. Morphologically, classic histology or LCA are common. Crucial cytogenetic features are the presence of isochromosome 17q and MYC amplifications, which are associated with a poor outcome in this subgroup. Other cytogenetic features include 1q amplification and 17q deletion.
Group 4 medulloblastoma. Group 4 tumors account for 40–50% of all medulloblastomas and, as such, represent the most common subgroup. Group 4 tumors have a 2:1 male predominance, are commonly metastatic at diagnosis and have an intermediate outcome. Histology is often classic but LCA may be seen. The cell of origin is unclear and transcriptional profiling revealed enrichment for genes involved in epigenetic regulation. Isochromosome 17q is found in the majority of these tumors but, unlike group 3 tumors, it is not predictive of outcome. Other cytogenetic alterations include amplification of MYCN and CDK6, loss of chromosome 11, and most females lose one copy of chromosome X.
Diagnostic Work-Up, Staging, and Risk Classification
Diagnostic work-up includes an MRI of the whole brain and spine and a lumbar puncture. Metastatic disease is classified according to the modified Chang criteria (Table 1). The baseline contrast-enhanced MRI for evaluation of metastasis is crucial and should ideally be done preoperatively. If not done prior to surgery, MRI should be obtained either within 48 h of resection (blood-brain barrier remains relatively intact in this time period, but nonspecific contrast enhancement is often seen thereafter) or at least 2 weeks post-surgery. Lumbar puncture is generally deferred to 2 weeks post-surgery, due to the risks related to increased intracranial pressure at diagnosis and to the presence of debris in the initial weeks post-surgery. Mild pleocytosis may be present, but nonspecific, cytopathology analysis of the CSF evaluates presence of cluster of medulloblastoma cells.
Because extraneural metastases are exceedingly rare, systemic staging with bone marrow examinations, bone scans, cross-sectional imaging of the neck/chest/abdomen/pelvis, or PET scans is not routinely recommended, unless specific clinical findings suggest involvement of other organs.
A postoperative surveillance MRI to evaluate the extent of resection (Table 1) is recommended within 48 h after surgery. After 48–72 h, enhancement due to inflammation or tissue granulation hinders the demarcation of residual and metastatic tumor tissue.
Given their prognostic and treatment implications, it is currently recommended that all patients with SHH medulloblastomas should be screened for TP53, PTCH1, and SUFU mutations (both tumors and germline) and offered genetic counseling.
Clinical Prognostic Factors and Risk Stratification Based on Clinical Features
Over previous decades medulloblastoma patients were stratified for treatment according solely to clinical criteria (Table 1): age at diagnosis, extent of surgical resection, presence of metastatic disease and, on some occasions, histological features such as anaplasia. Children under 36 months are treated, regardless to extent of disease, in separate protocols due to their young age and contraindication for craniospinal radiotherapy. Children with metastatic disease at diagnosis are at higher risk of relapse, as well as children with residual disease after surgery.
At present, patients older than 36 months are classified as high-risk patients if they have incomplete surgical resection (more than 1.5 cm2 in postsurgical imaging) and/or the presence of metastatic disease. Those without these features are stratified as average-risk (Table 1).
Risk Stratification of Non-infant Medulloblastoma: Integrating Clinical and Molecular Criteria
Recently a group of experts reviewed the level of evidence for clinical and molecular biomarkers and reached consensus regarding a new approach to risk stratification of childhood medulloblastoma (ages 4–17 years) (Ramaswamy et al. 2016b). The panel defined patient risk groups based on survival outcomes on current treatment protocols and outlined recommendations for the design of the next generation of clinical trials.
Low-risk patients with predicted overall survival above 90% – children with completely resected nonmetastatic WNT medulloblastoma and nonmetastatic Group 4 tumors harboring loss of chromosome 11 – should be prioritized for de-escalation of therapy with the goal to minimize long-term sequelae while maintaining survival outcomes. On the other hand, the panel defined a very high-risk group of patients (<50% survival) comprising children with TP53-mutated SHH medulloblastomas and metastatic Group 3 tumors, for which new therapies are urgently needed.
Treatment
Numerous studies have shown that multimodal treatment approaches with surgery, followed by radiation (for patients over the age of 3 years) and chemotherapy result in improved disease-free and overall survival. Targeted therapies are expected to be incorporated in the next generation of clinical trials and will be briefly discussed here.
Surgery
Surgery plays a key role in the treatment of medulloblastoma. The goals of surgical treatment are safe total or near total tumor resection, hydrocephalus management, and tissue collection for diagnosis, histopathology, and molecular studies.
Management of Hydrocephalus
High-pressure hydrocephalus due to obstruction of CSF flow at the level of the fourth ventricle is often associated with medulloblastoma at time of diagnosis, and it may persist in up to 30% of patients after tumor resection (Riva-Cambrin et al. 2009). Current therapeutic options for management of hydrocephalus include: endoscopic third ventriculostomy (ETV), external ventricular drainage (EVD), the implantation of a ventriculoperitoneal shunt (VP-shunt), and early surgery under treatment with steroids.
As the majority of patients with posterior fossa tumor will have resolution of hydrocephalus after resection, stratification of patients into low- and high-risk groups is crucial to optimize the treatment of these children. In 2009, Riva-Cambrin et al. introduced the prediction tool Canadian Preoperative Prediction Rule for Hydrocephalus (CPPRH), which is based on seven different criteria as outlined in Table 2. In the modified predictive model (modified Canadian Preoperative Prediction Rule for Hydrocephalus, mCPPRH), the criteria, presence of papilledema, was replaced by presence of transependymal edema, which can be evaluated by imaging (Foreman et al. 2013). This tool provides guidance for pre-resectional CSF diversion and postoperative monitoring for hydrocephalus, helping to improve patient counseling and surgical planning (Riva-Cambrin et al. 2009). Children with scores ≥5 are considered high-risk patients. Whereas low-risk patients may be monitored conservatively, with or without placement of an EVD, high-risk patients warrant an intraoperative EVD as well as intensive postoperative surveillance. In this patient group, a preoperative ETV should also be considered (Lin and Riva-Cambrin 2015). Comparing the rate of CSF diversion surgery among medulloblastoma subgroups, patients with SHH, Group 3, and Group 4 tumors are more likely to require CSF diversion compared to patients with WNT medulloblastoma. This is possibly related to the older age of the patients and the lack of metastases in the WNT subgroup (Schneider et al. 2015).
Tumor Resection
Maximum safe resection is a key aspect of the current treatment of medulloblastoma. The prone position is often preferred for pediatric patients due to a lower risk of air embolism, systemic hypotension, and postoperative pneumocephalus when compared to the sitting position. However, the sitting position lowers intracranial pressure during surgery and supports gravity drainage of cerebral spinal fluid and blood. Pin fixation is typically used to stabilize the head in a flexed position; moderate head flexion improves the surgical exposure and is beneficial for venous drainage. Alternatively, a padded horseshoe headrest can be used for young kids with a thin cranium.
A standard posterior fossa approach consists of a midline skin incision and median suboccipital craniotomy. The craniotomy should extend from just below the transverse sinuses to the opisthion and reach widely bilaterally. A navigation system can be helpful for orientation. The posterior arch of C1 is removed (C1 laminectomy) and the dura opened with a Y-shaped incision, followed by the exposure of the cisterna magna. A transvermian approach – incision in the inferior cerebellar vermis – or telovelar approach – split of the superior medullary velum in the cerebello-mesencephalic fissure – are commonly performed to expose a tumor in the fourth ventricle. A cavitron ultrasonic surgical aspirator (CUSA) is used for tumor removal. The floor of the fourth ventricle should be maintained in order to prevent direct surgical damage to the brainstem and, to avoid neurological complications, tumor tissue directly infiltrating the floor of the fourth ventricle should not be resected. Before closing, the resection cavity has to be checked for sufficient hemostasis and residual tumor tissue (Sutton et al. 1996). To reduce the risk of postoperative pseudomeningocele, a suboccipital craniotomy with replacement of the bone flap is preferred to a craniectomy.
Extent of Resection
According to a recent study, there is no statistically significant difference of overall survival between gross total (no residual tumor) and near-total resection (<1.5 cm2 tumor remaining) in pediatric medulloblastoma patients. Gross total resection compared to subtotal resection (≥1.5 cm2 tumor remaining) increases progression-free survival in patients with Group 4 medulloblastoma; however, there is no improvement of overall survival. Therefore, although maximum safe surgical resection remains standard of care, aggressive surgical resection should not be performed at the risk of postoperative neurological morbidity (Thompson et al. 2016).
Intraoperative Imaging
Intraoperative MRI (ioMRI) can be helpful in detecting the presence and extent of residual tumor, thus increasing the chances for a safe gross total resection, minimizing morbidity and reducing the need for early reoperation (second-look surgery). However, these advantages come along with increased anesthesia and operating room time (Choudhri et al. 2014).
Intraoperative ultrasonography is a safe and radiation-free imaging method, which can be used as an alternative to MRI to acquire real-time information about neuroanatomy and tumor location. Intraoperative ultrasonography supports the neurosurgeon in terms of orientation in the surgical area, planning of the approach for tumor removal, and verifying the completion of resection before closing (Ulrich et al. 2012).
Postoperative Care
Compared to the supratentorial space, the posterior fossa consists of a relatively small volume and even minor complications can cause severe neurological deficiencies. Close monitoring of vital signs and frequent neurological examinations in a specialized intensive care unit should be ensured for all patients. Delayed extubation is an option for patients with a severe preoperative health condition, extensive manipulation of cranial nerves during surgery, and/or a long operation time.
If a patient wakes up from anesthesia with unexpected deficits or presents postoperatively with neurological deterioration, a CT scan is necessary to rule out postoperative hemorrhage, acute hydrocephalus, and/or extensive cerebellar edema. An implantation of an EVD during surgery can be helpful for intracranial pressure (ICP) monitoring and allows drainage of cerebrospinal fluid in case of elevated ICP, which can improve recovery. In case of elevated ICP associated with acute hydrocephalus, an early intervention, ETV or a shunt implantation, may be indicated to control postresection hydrocephalus.
Steroids are usually administered in the immediate postoperative period with the goal of reducing the peritumoral edema, and doses are tapered within the first postoperative days, but may be given longer if there is evidence of extensive edema on postoperative imaging.
Postoperative Morbidity and Complications
Complications after posterior fossa surgery are common, warranting close monitoring of these patients. In the following section, we discuss specific neurosurgical complications after posterior fossa tumor resection in pediatric patients. We do not describe any general surgical complications or risks of anesthesia.
Posterior fossa syndrome (PFS) is a complication in children following posterior fossa surgery and is characterized by deficits in speech and language, behavioral changes with labile affect and reduced social interaction, ataxia, as well as deficits in volitional behavior. There is typically a latency of 1–7 days to onset of PFS after surgery and deficits can be transient but are more often long-lasting (e.g., speech difficulties, ataxia).
Aggressive neurosurgical treatment with the goal of gross total resection increases the incidence of this complication (Robertson et al. 2006; Korah et al. 2010); however, the pathophysiological mechanism of PFS is not fully understood. A high risk for development of PFS has been described following injury of the dentatothalamocortical pathways, which project to and from the dentate nucleus of the cerebellum. Though some studies report conflicting results, several predictive factors have been suggested, such as tumor invasion of the brainstem and involvement of the cerebellar peduncles, among others.
Patients with a posterior fossa tumor often present with ataxia, typically accompanied by dysarthria and nystagmus. Given the lack of studies addressing the extent of ataxia pre- and post-surgery, it is difficult to determine the incidence of the cerebellar syndrome as a surgical complication.
To mitigate the risk of aspiration and respiratory complications, it is important to diagnose dysphagia and other bulbar cranial nerve palsies immediately after surgery. Dysphagia increases the requirement for extended postoperative ventilation and tube feeding and is often present with other cranial nerve palsies (e.g., dysarthria and facial weakness). The use of electrophysiological monitoring and neuronavigation during surgery decreases the incidence of cranial nerve palsies.
Vomiting, headaches, and neck pain are common in children after posterior fossa surgery, they are likely multifactorial. Vomiting can be due to anesthesia, acute postresection hydrocephalus, or related to adjuvant chemo- and radiotherapy. Postoperative neck pain is commonly explained by direct surgical trauma to the muscle. Headaches can occur due to intraventricular blood collection or pneumocephalus, as well as related to metastatic leptomeningeal involvement. Patients should be examined regularly for further postoperative complications, such as CSF leakage, wound infection, meningitis, and acute hydrocephalus .
Radiotherapy
Medulloblastoma is a radiosensitive tumor and the first long-term survivors of childhood medulloblastoma were reported only after introduction of radiation therapy (Paterson and Farr 1953). In order to cure medulloblastoma, irradiation of the entire neuraxis (whole brain and spine) with 23.4 or 36 Gy – depending on the extent of disease – and a local boost up to 54 Gy to the tumor bed are necessary. Attempts at restricting the craniospinal field sparing the supratentorial compartment resulted in increased rate of relapse in the nonirradiated areas and inferior outcomes (Bouffet et al. 1992). Although radiotherapy is a cornerstone of treatment, it is not used in children under 3 years due to the severe side effects to the developing brain. In order to minimize treatment-related toxicities, there are efforts under way to increase this age threshold and – given that sequelae are dose-dependent (Moxon-Emre et al. 2014) – reduce the total dose and volume of irradiated tissue.
Radiation therapy is usually started within 4–6 weeks post-surgery, with or without concomitant chemotherapy. Multiple studies have shown that delays in the initiation or progress of radiation correlate with worse outcome (Lannering et al. 2012), with improved outcome if initiated within 28 days post-surgery (Rieken et al. 2011) and completed within 50 days (Taylor et al. 2004).
For children older than 3 years of age at diagnosis with average-risk disease, the current approach for adjuvant radiotherapy includes 23.4 Gy of craniospinal irradiation (CSI) and a boost up to 54 Gy of the tumor bed. Though initial attempts at reducing the craniospinal dose for average-risk patients from the standard 36 Gy to 23.4 Gy without chemotherapy resulted in higher rate of leptomeningeal relapse (Thomas et al. 2000), a subsequent Children’s Oncology Group trial showed an encouraging 5-year EFS of 81% for average-risk patients treated with reduced-dose craniospinal irradiation in combination with an adjuvant chemotherapy regimen (Packer et al. 2006). Another approach reduced CSI dose using a cyclophosphamide-based high dose chemotherapy regimen with autologous stem cell rescue and resulted as well in EFS above 80% at 5 years (Gajjar et al. 2006).
Irradiation protocols for children older than 3 years of age with high-risk disease include similar fields but differ in the dose of craniospinal irradiation, using the standard dose of 36 Gy to the neuraxis and 54 Gy to the tumor bed. The Milan group reported promising outcomes in patients with metastatic medulloblastoma using hyperfractionated accelerated radiotherapy (HART) – consisting of smaller radiation fractions given twice per day with the goal of increasing the antitumor effect – combined with intensive chemotherapy, and myeloablative chemotherapy in selected cases, the 5-year event-free and overall survival rates were 70% and 73%, respectively (Gandola et al. 2009). The UK experience with this approach failed to replicate the good survival outcome, with an estimated 3-year overall survival of 56%; this is likely related to differences in patient subgroups and regional protocol administration (Vivekanandan et al. 2015), highlighting the importance of evaluating new therapies in multi-institutional clinical trials and, especially as we move forward in an era of molecular diagnosis, carefully correlating treatment results with tumor subgroups and known clinical biomarkers.
In the randomized, European multicenter HIT-SIOP PNET 4 trial, hyperfractionated radiotherapy was compared to the conventional approach with daily fractions with the same total dose, followed by maintenance chemotherapy for both groups. There was, however, no survival advantage of hyperfracionated radiotherapy compared to conventional fractionated radiotherapy, and as such, the latter remains the standard of care (Lannering et al. 2012).
While the minimal dose of radiation that is necessary for disease control is still unknown, a major breakthrough in radiation therapy has been a reduction of the posterior fossa volume boost. Initially, a broad field encompassing the whole posterior fossa was used, resulting in a significant volume of the brain receiving high dose radiation. In fact, for this reason the first attempts at reducing the dose of CSI in the context of whole posterior fossa boost failed to translate into considerable improvement in IQ. However, progressive reduction of the boost field to the tumor with lower margins, driven mostly by the St. Jude’s Children’s Research Hospital group, resulted in good disease control with no increase in local recurrence and improved neurocognitive outcomes.
In the past few years there has been an increased use of proton radiotherapy in favor of conventional photon radiotherapy. Compared to photons, protons deposit the maximum dose at the desired volume with less entrance dose and no exit dose, thereby reducing the irradiation of the healthy surrounding tissues. With many claiming it unethical to compare the modalities in a randomized clinical trial, good quality data about efficacy are lacking. A phase II single arm study demonstrated similar survival outcomes in medulloblastoma patients treated with proton-beam radiotherapy compared to the historical controls treated with conventional photon-beam radiation therapy, with acceptable toxicity but treatment-related hearing, endocrine and neurocognitive sequelae (Yock et al. 2016). Comparison of IQ scores between patients with CNS tumors that received proton radiotherapy or conventional photon therapy revealed no difference in the IQ slopes for the patients that received CSI; it therefore remains unclear whether protons result in improved neurocognitive outcomes, particularly for those patients that require irradiation of the whole neuraxis, as is the case for children with medulloblastoma (Kahalley et al. 2016) .
Chemotherapy
Adjuvant chemotherapy is an integral part of contemporary treatment protocols for children with medulloblastoma, across all ages and risk groups. When the first prospective trials introducing adjuvant chemotherapy were designed in the 1970s in Europe and North America, the futility of surgery alone had been established and postoperative craniospinal irradiation was the standard of care with 5-year survival rates around 50%. The initial trials used vincristine- and lomustine-based chemotherapy during and after radiation therapy, resulting in an advantage for adjuvant chemotherapy when compared to radiation alone, particularly for those children with more advanced disease (Evans et al. 1990; Tait et al. 1990). Later, addition of cisplatin led to further improvement in outcome. Other drugs have shown efficacy against medulloblastoma, including cyclophosphamide, etoposide, and topotecan.
Chemotherapy has not only greatly improved overall outcomes but also facilitated a reduction of the dose of craniospinal radiation used to treat average-risk patients. For these children, the benefit of postradiation chemotherapy has been demonstrated in several studies and has become the standard of care. Protocols with cisplatin, lomustine, and vincristine after reduced-dose CSI (23.4 Gy instead of standard 36 Gy) and a boost to the primary site resulted in a 5-year overall-survival above 85% (Packer et al. 2006; Lannering et al. 2012). The St. Jude’s trial SJMB96 adopted a shorter, dose-intense regimen with four cycles of high dose cyclophosphamide-based chemotherapy followed by stem cell reinfusion with similarly favorable outcomes (Gajjar et al. 2006).
For high-risk patients, the craniospinal radiation dose remains critical at achieving cure. The SJMB96 post-radiation chemotherapy approach – which was similar for average and high-risk patients – resulted in overall survival of 70% in the high-risk group (Gajjar et al. 2006). There are data suggesting that carboplatin may be effective as a radiosensitizer if given during radiation for patients with metastases (Jakacki et al. 2012); this approach was prospectively studied in the COG trial ACNS0332 and the results are pending.
Despite being a chemosensitive tumor, strategies using chemotherapy prior to surgery have not been explored, and preradiation chemotherapy schedules have generally not shown benefit. In the German Hirntumoren study HIT’91, giving chemotherapy earlier with the intent of postponing radiation therapy resulted in treatment delays (mostly due to prolonged myelosuppression) and poorer outcome overall (Kortmann et al. 2000).
Treatment of Young Children with Medulloblastoma
Due to the devastating side effects of radiotherapy in the developing brain, different radiation-sparing strategies were introduced in the 1980s and 1990s for post-surgical treatment of young children with medulloblastoma. The first studies used alternating cycles of cisplatin/etoposide and cyclophosphamide/vincristine (BabyPOG) or intensified multi-agent induction chemotherapy (vincristine, etoposide, cisplatin, carboplatin, cyclophosphamide, ifosphamide) followed by maintenance chemotherapy of carboplatin, etoposide, vincristine, and cyclophosphamide, with the last approach leading to a 5-year radiation free survival of 32% (Geyer et al. 2005). The French BabySFOP study also suggested that a significant proportion of young children with nonmetastatic, completely resected medulloblastoma could be treated with chemotherapy alone (Grill et al. 2005).
High dose, myeloablative chemotherapy for young patients has been evaluated by several cooperative group studies. The Headstart studies were designed to avoid radiotherapy using induction chemotherapy (cisplatin, etoposide, cyclophosphamide and vincristine, with or without methotrexate) followed by one cycle of myeloablative chemotherapy (carboplatin, etoposide, and thiotepa) and autologous stem cell rescue. This led to 3-year event-free survivals approaching 50% for all patients (Chi et al. 2004); however, some patients were irradiated based on physician discretion. The Children’s Cancer Group then initiated the CCG99703 study of three induction cycles of cisplatin, cyclophosphamide, etoposide, and vincristine followed by three consolidation cycles of carboplatin and thiotepa with autologous stem cell support. The three tandem transplants were shown to be safe and tolerated, and results of this study suggested overall survival outcomes for nonmetastatic patients of 67.5% and metastatic (M1+) patients having outcomes of 30% (Cohen et al. 2015).
The HIT-SKK’92 study from Germany evaluated systemic chemotherapy (including high-dose methotrexate) with intraventricular therapy (methotrexate delivered via Rickman or Ommaya reservoir), omitting radiotherapy for those in complete remission after chemotherapy. The 5-year progression-free survival rate was 82% for children who had complete resection, 50% for children with residual tumor, and 33% for children with macroscopic metastasis.
Multiple studies have shown that infants with tumors with desmoplastic histology – which account for approximately one third of the tumors in this age group – had superior outcomes when compared to classic histology, both with conventional and high-dose chemotherapy approaches (Rutkowski et al. 2005; Cohen et al. 2015). A Canadian study confirmed the excellent outcome of patients with desmoplasia (5-year PFS of 92.3%), which belong exclusively to the SHH subgroup, and found that patients with SHH-medulloblastoma and classic histology had a similarly good outcome (5-year PFS of 87.5%) (Lafay-Cousin et al. 2016). However, the prognostic value of molecular subgroups and, importantly, the genetic alterations common in young children with SHH-medulloblastoma – such SUFU mutations – remain poorly characterized and should be evaluated in prospective clinical trials.
Targeted Therapies and Future Directions
Incorporation of molecular profiling will be mandatory in all future trials to properly stratify patients, allocate treatment, and prospectively evaluate the impact of molecular subgroups in outcome. WNT and SHH medulloblastomas have been well characterized at a molecular level, whereas there is a paucity of targetable molecular drivers known for Group 3 and Group 4 tumors.
WNT tumors in children have an excellent outcome and although several drugs targeting the WNT pathway have been tested in preclinical models, the next trials will de-escalate therapy rather than introduce molecular therapies.
SHH tumors were the first to benefit from targeted therapies. The drug most extensively studied so far has been the SHH/smoothened inhibitor vismodegib (GDC-0449), after a patient with refractory medulloblastoma showed an impressive, though transient, response (Rudin et al. 2009). In phase I/II clinical trials, vismodegib was well tolerated and exhibited activity against recurrent SHH-medulloblastoma but not against recurrent non-SHH-medulloblastoma, emphasizing the need to molecularly stratify patients for targeted therapies (Robinson et al. 2015). Smoothened inhibitors are unlikely to be effective in SHH-tumors harboring downstream alterations, such as GLI2 amplifications. Moreover, several resistance mechanisms have been described, such as the development of mutations that block the activity of smoothened inhibitors or oncogenic bypass by upregulation of other survival pathways, such as the PI3K/mTOR pathway.
Separate protocols should be designed for TP53-mutated SHH-medulloblastomas, a very high-risk group of patients with a bleak prognosis. Possible agents with therapeutic efficacy for these tumors include lithium (a GSK3β inhibitor that in preclinical models acts as a radiosensitizer), arsenic trioxide (GLI2 inhibitor), and bromodomain inhibitors.
Molecular profiling studies failed to identify mutations and alterations responsible for tumor initiation and drug resistance of Group 3 and Group 4 tumors. Multiple drugs have been tested in preclinical models; pemetrexed and gemcitabine have emerged as potential therapeutic agents (Morfouace et al. 2014) and will be incorporated in the upcoming St. Jude’s trial for a subset of Group 3 and Group 4 tumors. Other approaches under development include BET-bromodomain inhibition in MYC-amplified medulloblastoma and induction of neuronal differentiation using retinoic acid or histone deacetylase inhibitors.
Recurrent Medulloblastoma
Recurrent medulloblastoma is largely incurable and remains a major challenge in pediatric neuro-oncology. Relapse occurs usually within the first 2 years after completion of therapy and may be localized to the primary site or metastatic (brain, spine, CSF or, rarely, extraneural), isolated or in combination. Medulloblastoma subgroup affiliation remains unchanged at relapse but the patterns of disease spread at recurrence differ among them; SHH tumors relapse predominantly in the primary tumor bed in the posterior fossa, whereas Group 3 and Group 4 tumors present as disseminated leptomeningeal disease (Ramaswamy et al. 2013).
Though different approaches have been used, there is no ideal regimen or data to support a standard of care. At time of relapse, different treatment options should be carefully considered on a case-by-case basis and include repeated surgery, radiation, chemotherapy, high dose chemotherapy with stem cell rescue, and molecular targeted therapies, whenever possible enrolled in phase I/II clinical trials. However, the prognosis overall is bleak and most interventions have a palliative intent, transiently controlling tumor growth and ameliorating symptoms. The majority of children will progress within 18 months.
Some young children not irradiated upfront may be salvaged if they are eligible to receive radiation at time of relapse. There is some evidence of long-term tumor control in patients with Group 4, particularly those with supratentorial metastatic relapses that are resected.
Recent data show that medulloblastoma tumors progress and significantly diverge from the primary (pretreatment) tumor after therapy, thus targeted strategies tailored to the primary tumor will likely be unsuccessful (Morrissy et al. 2016). Though historically surgery has not been indicated at relapse, these data support a role for biopsy at relapse when targeted therapies are an option.
Long-Term Effects and Quality of Life
Improved outcomes led to an increasing recognition and concern about the debilitating, long-lasting tumor- and treatment-related sequelae that affect survivors of childhood medulloblastoma.
Long-term neurocognitive decline, particularly after craniospinal irradiation, is age and dose dependent, has no plateau and represents one of the most significant side effects, leading to learning difficulties in the vast majority of affected children. The degree of impairment is multifactorial, and related to tumor and patient factors; recent data suggest that SHH-tumors are associated with better functional outcomes (Moxon-Emre et al. 2016).
Other sequelae include neurological deficits related to the initial tumor extension or surgery (cerebellar dysfunction, posterior fossa syndrome), emotional and behavioral issues, endocrine dysfunction after craniospinal irradiation (panhypopituitarism, delayed puberty), infertility after high dose alkylating chemotherapy, secondary malignancies related to radiation (meningeomas and high-grade gliomas) and chemotherapy (hematological malignancies), and sensorineural hearing loss related to radiation and ototoxic chemotherapy with cisplatin.
Over the last decade there has been an increased focus on quality of life versus survival as a measure of outcome. Future studies should include measures of quality of life, in order to determine risk and resilience factors, long-term patterns of decline, and family and patient outcomes .
Conclusion
After decades of clinical and biological advances, medulloblastoma remains a remarkable challenge. As we continue into a molecular era, rethinking how we define, approach, and treat the disease is imperative and as such, the upcoming biologically informed clinical trials are greatly needed. Although there is still much to be learned, a compelling body of evidence supports the de-escalation of therapy for children with localized WNT-medulloblastoma and a subset of Group 4 tumors. Infants with nondesmoplastic and metastatic tumors, as well as children with TP53-mutated SHH and metastatic Group 3 tumors have a poor prognosis and should be prioritized for development of novel therapies. Neurocognitive and quality of life outcomes after treatment for medulloblastoma are often dismal. While striving for improved survival, future studies need to measure quality of life and long-term intellectual function, in order to improve patient care and provide appropriate support to the survivors of childhood medulloblastoma.
References
Amlashi SF, Riffaud L, Brassier G, Morandi X (2003) Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer 98(3):618–624
Bailey P, Cushing H (1926) A classification of the tumors of the glioma group on a histogenetic basis with a correlated study of prognosis. Lippincott, Philadelphia
Bouffet E, Bernard JL, Frappaz D, Gentet JC, Roche H, Tron P, Carrie C, Raybaud C, Joannard A, Lapras C et al (1992) M4 protocol for cerebellar medulloblastoma: supratentorial radiotherapy may not be avoided. Int J Radiat Oncol Biol Phys 24(1):79–85
Bourdeaut F, Miquel C, Richer W, Grill J, Zerah M, Grison C, Pierron G, Amiel J, Krucker C, Radvanyi F, Brugieres L, Delattre O (2014) Rubinstein-Taybi syndrome predisposing to non-WNT, non-SHH, group 3 medulloblastoma. Pediatr Blood Cancer 61(2):383–386
Chi SN, Gardner SL, Levy AS, Knopp EA, Miller DC, Wisoff JH, Weiner HL, Finlay JL (2004) Feasibility and response to induction chemotherapy intensified with high-dose methotrexate for young children with newly diagnosed high-risk disseminated medulloblastoma. J Clin Oncol 22(24):4881–4887
Choudhri AF, Klimo P Jr, Auschwitz TS, Whitehead MT, Boop FA (2014) 3T intraoperative MRI for management of pediatric CNS neoplasms. AJNR Am J Neuroradiol 35(12):2382–2387
Cohen BH, Geyer JR, Miller DC, Curran JG, Zhou T, Holmes E, Ingles SA, Dunkel IJ, Hilden J, Packer RJ, Pollack IF, Gajjar A, Finlay JL, Children’s Oncology Group (2015) Pilot study of intensive chemotherapy with peripheral hematopoietic cell support for children less than 3 years of age with malignant brain tumors, the CCG-99703 phase I/II study. A report from the Children’s Oncology Group. Pediatr Neurol 53(1):31–46
Eran A, Ozturk A, Aygun N, Izbudak I (2010) Medulloblastoma: atypical CT and MRI findings in children. Pediatr Radiol 40(7):1254–1262
Evans AE, Jenkin RD, Sposto R, Ortega JA, Wilson CB, Wara W, Ertel IJ, Kramer S, Chang CH, Leikin SL et al (1990) The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg 72(4):572–582
Foreman P, McClugage S 3rd, Naftel R, Griessenauer CJ, Ditty BJ, Agee BS, Riva-Cambrin J, Wellons J 3rd (2013) Validation and modification of a predictive model of postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr 12(3): 220–226
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM, Wallace D, Gilbertson RJ (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7(10):813–820
Gandola L, Massimino M, Cefalo G, Solero C, Spreafico F, Pecori E, Riva D, Collini P, Pignoli E, Giangaspero F, Luksch R, Berretta S, Poggi G, Biassoni V, Ferrari A, Pollo B, Favre C, Sardi I, Terenziani M, Fossati-Bellani F (2009) Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol 27(4):566–571
Gerber NU, von Hoff K, von Bueren AO, Treulieb W, Deinlein F, Benesch M, Zwiener I, Soerensen N, Warmuth-Metz M, Pietsch T, Mittler U, Kuehl J, Kortmann RD, Grotzer MA, Rutkowski S (2012) A long duration of the prediagnostic symptomatic interval is not associated with an unfavourable prognosis in childhood medulloblastoma. Eur J Cancer 48(13):2028–2036
Geyer JR, Sposto R, Jennings M, Boyett JM, Axtell RA, Breiger D, Broxson E, Donahue B, Finlay JL, Goldwein JW, Heier LA, Johnson D, Mazewski C, Miller DC, Packer R, Puccetti D, Radcliffe J, Tao ML, Shiminski-Maher T, Children’s Oncology Group (2005) Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol 23(30):7621–7631
Giangaspero F, Perilongo G, Fondelli MP, Brisigotti M, Carollo C, Burnelli R, Burger PC, Garre ML (1999) Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91(6):971–977
Giangaspero F, Wellek S, Masuoka J, Gessi M, Kleihues P, Ohgaki H (2006) Stratification of medulloblastoma on the basis of histopathological grading. Acta Neuropathol 112(1):5–12
Grill J, Sainte-Rose C, Jouvet A, Gentet JC, Lejars O, Frappaz D, Doz F, Rialland X, Pichon F, Bertozzi AI, Chastagner P, Couanet D, Habrand JL, Raquin MA, Le Deley MC, Kalifa C, French Society of Paediatric (2005) Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6(8):573–580
Halperin EC, Watson DM, George SL (2001) Duration of symptoms prior to diagnosis is related inversely to presenting disease stage in children with medulloblastoma. Cancer 91(8): 1444–1450
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B et al (1995) The molecular basis of Turcot’s syndrome. N Engl J Med 332(13):839–847
Jakacki RI, Burger PC, Zhou T, Holmes EJ, Kocak M, Onar A, Goldwein J, Mehta M, Packer RJ, Tarbell N, Fitz C, Vezina G, Hilden J, Pollack IF (2012) Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children’s Oncology Group Phase I/II study. J Clin Oncol 30(21):2648–2653
Kahalley LS, Ris MD, Grosshans DR, Okcu MF, Paulino AC, Chintagumpala M, Moore BD, Guffey D, Minard CG, Stancel HH, Mahajan A (2016) Comparing intelligence quotient change after treatment with proton versus photon radiation therapy for pediatric brain tumors. J Clin Oncol 34(10):1043–1049
Korah MP, Esiashvili N, Mazewski CM, Hudgins RJ, Tighiouart M, Janss AJ, Schwaibold FP, Crocker IR, Curran WJ Jr, Marcus RB Jr (2010) Incidence, risks, and sequelae of posterior fossa syndrome in pediatric medulloblastoma. Int J Radiat Oncol Biol Phys 77(1):106–112
Kortmann RD, Kuhl J, Timmermann B, Mittler U, Urban C, Budach V, Richter E, Willich N, Flentje M, Berthold F, Slavc I, Wolff J, Meisner C, Wiestler O, Sorensen N, Warmuth-Metz M, Bamberg M (2000) Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys 46(2):269–279
Lafay-Cousin L, Smith A, Chi SN, Wells E, Madden J, Margol A, Ramaswamy V, Finlay J, Taylor MD, Dhall G, Strother D, Kieran MW, Foreman NK, Packer RJ, Bouffet E (2016) Clinical, pathological, and molecular characterization of infant medulloblastomas treated with sequential high-dose chemotherapy. Pediatr Blood Cancer 63(9):1527–1534
Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, Massimino M, Reddingius R, Benesch M, Carrie C, Taylor R, Gandola L, Bjork-Eriksson T, Giralt J, Oldenburger F, Pietsch T, Figarella-Branger D, Robson K, Forni M, Clifford SC, Warmuth-Metz M, von Hoff K, Faldum A, Mosseri V, Kortmann R (2012) Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30(26):3187–3193
Lin CT, Riva-Cambrin JK (2015) Management of posterior fossa tumors and hydrocephalus in children: a review. Childs Nerv Syst 31(10):1781–1789
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6): 803–820
Morfouace M, Shelat A, Jacus M, Freeman BB, Turner D 3rd, Robinson S, Zindy F, Wang YD, Finkelstein D, Ayrault O, Bihannic L, Puget S, Li XN, Olson JM, Robinson GW, Guy RK, Stewart CF, Gajjar A, Roussel MF (2014) Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 25(4):516–529
Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, Remke M, Cavalli FM, Ramaswamy V, Lindsay PE, Jelveh S, Donovan LK, Wang X, Luu B, Zayne K, Li Y, Mayoh C, Thiessen N, Mercier E, Mungall KL, Ma Y, Tse K, Zeng T, Shumansky K, Roth AJ, Shah S, Farooq H, Kijima N, Holgado BL, Lee JJ, Matan-Lithwick S, Liu J, Mack SC, Manno A, Michealraj KA, Nor C, Peacock J, Qin L, Reimand J, Rolider A, Thompson YY, Wu X, Pugh T, Ally A, Bilenky M, Butterfield YS, Carlsen R, Cheng Y, Chuah E, Corbett RD, Dhalla N, He A, Lee D, Li HI, Long W, Mayo M, Plettner P, Qian JQ, Schein JE, Tam A, Wong T, Birol I, Zhao Y, Faria CC, Pimentel J, Nunes S, Shalaby T, Grotzer M, Pollack IF, Hamilton RL, Li XN, Bendel AE, Fults DW, Walter AW, Kumabe T, Tominaga T, Collins VP, Cho YJ, Hoffman C, Lyden D, Wisoff JH, Garvin JH Jr, Stearns DS, Massimi L, Schuller U, Sterba J, Zitterbart K, Puget S, Ayrault O, Dunn SE, Tirapelli DP, Carlotti CG, Wheeler H, Hallahan AR, Ingram W, MacDonald TJ, Olson JJ, Van Meir EG, Lee JY, Wang KC, Kim SK, Cho BK, Pietsch T, Fleischhack G, Tippelt S, Ra YS, Bailey S, Lindsey JC, Clifford SC, Eberhart CG, Cooper MK, Packer RJ, Massimino M, Garre ML, Bartels U, Tabori U, Hawkins CE, Dirks P, Bouffet E, Rutka JT, Wechsler-Reya RJ, Weiss WA, Collier LS, Dupuy AJ, Korshunov A, Jones DT, Kool M, Northcott PA, Pfister SM, Largaespada DA, Mungall AJ, Moore RA, Jabado N, Bader GD, Jones SJ, Malkin D, Marra MA, Taylor MD (2016) Divergent clonal selection dominates medulloblastoma at recurrence. Nature 529(7586):351–357
Moxon-Emre I, Bouffet E, Taylor MD, Laperriere N, Scantlebury N, Law N, Spiegler BJ, Malkin D, Janzen L, Mabbott D (2014) Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol 32(17): 1760–1768
Moxon-Emre I, Taylor MD, Bouffet E, Hardy K, Campen CJ, Malkin D, Hawkins C, Laperriere N, Ramaswamy V, Bartels U, Scantlebury N, Janzen L, Law N, Walsh KS, Mabbott DJ (2016) Intellectual outcome in molecular subgroups of medulloblastoma. J Clin Oncol 34:4161–4170
Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, Muraszko K, Langston J, Sposto R (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24(25):4202–4208
Paterson E, Farr RF (1953) Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol 39(4):323–336
Perreault S, Ramaswamy V, Achrol AS, Chao K, Liu TT, Shih D, Remke M, Schubert S, Bouffet E, Fisher PG, Partap S, Vogel H, Taylor MD, Cho YJ, Yeom KW (2014) MRI surrogates for molecular subgroups of medulloblastoma. AJNR Am J Neuroradiol 35(7):1263–1269
Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, Schuller U, Gururangan S, McLendon R, Bigner D, Fouladi M, Ligon KL, Pomeroy SL, Dunn S, Triscott J, Jabado N, Fontebasso A, Jones DT, Kool M, Karajannis MA, Gardner SL, Zagzag D, Nunes S, Pimentel J, Mora J, Lipp E, Walter AW, Ryzhova M, Zheludkova O, Kumirova E, Alshami J, Croul SE, Rutka JT, Hawkins C, Tabori U, Codispoti KE, Packer RJ, Pfister SM, Korshunov A, Taylor MD (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14(12):1200–1207
Ramaswamy V, Remke M, Shih D, Wang X, Northcott PA, Faria CC, Raybaud C, Tabori U, Hawkins C, Rutka J, Taylor MD, Bouffet E (2014) Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr Blood Cancer 61(7):1190–1194
Ramaswamy V, Remke M, Adamski J, Bartels U, Tabori U, Wang X, Huang A, Hawkins C, Mabbott D, Laperriere N, Taylor MD, Bouffet E (2016a) Medulloblastoma subgroup-specific outcomes in irradiated children: who are the true high-risk patients? Neuro-Oncology 18(2): 291–297
Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, Kool M, Dufour C, Vassal G, Milde T, Witt O, von Hoff K, Pietsch T, Northcott PA, Gajjar A, Robinson GW, Padovani L, Andre N, Massimino M, Pizer B, Packer R, Rutkowski S, Pfister SM, Taylor MD, Pomeroy SL (2016b) Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 131(6):821–831
Raybaud C, Ramaswamy V, Taylor MD, Laughlin S (2015) Posterior fossa tumors in children: developmental anatomy and diagnostic imaging. Childs Nerv Syst 31(10):1661–1676
Rieken S, Mohr A, Habermehl D, Welzel T, Lindel K, Witt O, Kulozik AE, Wick W, Debus J, Combs SE (2011) Outcome and prognostic factors of radiation therapy for medulloblastoma. Int J Radiat Oncol Biol Phys 81(3):e7-e13
Riva-Cambrin J, Detsky AS, Lamberti-Pasculli M, Sargent MA, Armstrong D, Moineddin R, Cochrane DD, Drake JM (2009) Predicting postresection hydrocephalus in pediatric patients with posterior fossa tumors. J Neurosurg Pediatr 3(5):378–385
Robertson PL, Muraszko KM, Holmes EJ, Sposto R, Packer RJ, Gajjar A, Dias MS, Allen JC, Children’s Oncology Group (2006) Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the Children’s Oncology Group. J Neurosurg 105(Suppl 6):444–451
Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, Packer RJ, Goldman S, Prados MD, Desjardins A, Chintagumpala M, Takebe N, Kaste SC, Rusch M, Allen SJ, Onar-Thomas A, Stewart CF, Fouladi M, Boyett JM, Gilbertson RJ, Curran T, Ellison DW, Gajjar A (2015) Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol 33(24):2646–2654
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA (2009) Treatment of medulloblastoma with hedgehog pathWeway inhibitor GDC-0449. N Engl J Med 361(12): 1173–1178
Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N, Graf N, Emser A, Pietsch T, Wolff JE, Kortmann RD, Kuehl J (2005) Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352(10):978–986
Schneider C, Ramaswamy V, Kulkarni AV, Rutka JT, Remke M, Tabori U, Hawkins C, Bouffet E, Taylor MD (2015) Clinical implications of medulloblastoma subgroups: incidence of CSF diversion surgery. J Neurosurg Pediatr 15(3):236–242
Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M, Luu B, Yao Y, Wang X, Dubuc AM, Garzia L, Peacock J, Mack SC, Wu X, Rolider A, Morrissy AS, Cavalli FM, Jones DT, Zitterbart K, Faria CC, Schuller U, Kren L, Kumabe T, Tominaga T, Shin Ra Y, Garami M, Hauser P, Chan JA, Robinson S, Bognar L, Klekner A, Saad AG, Liau LM, Albrecht S, Fontebasso A, Cinalli G, De Antonellis P, Zollo M, Cooper MK, Thompson RC, Bailey S, Lindsey JC, Di Rocco C, Massimi L, Michiels EM, Scherer SW, Phillips JJ, Gupta N, Fan X, Muraszko KM, Vibhakar R, Eberhart CG, Fouladi M, Lach B, Jung S, Wechsler-Reya RJ, Fevre-Montange M, Jouvet A, Jabado N, Pollack IF, Weiss WA, Lee JY, Cho BK, Kim SK, Wang KC, Leonard JR, Rubin JB, de Torres C, Lavarino C, Mora J, Cho YJ, Tabori U, Olson JM, Gajjar A, Packer RJ, Rutkowski S, Pomeroy SL, French PJ, Kloosterhof NK, Kros JM, Van Meir EG, Clifford SC, Bourdeaut F, Delattre O, Doz FF, Hawkins CE, Malkin D, Grajkowska WA, Perek-Polnik M, Bouffet E, Rutka JT, Pfister SM, Taylor MD (2014) Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32(9): 886–896
Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, Cheesman E, Kelsey A, McCabe MG, Newman WG, Evans DG (2014) Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32(36):4155–4161
Smoll NR, Drummond KJ (2012) The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci 19(11):1541–1544
Sutton LN, Phillips PC, Molloy PT (1996) Surgical management of medulloblastoma. J Neurooncol 29(1):9–21
Tait DM, Thornton-Jones H, Bloom HJ, Lemerle J, Morris-Jones P (1990) Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer 26(4):464–469
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31(3):306–310
Taylor RE, Bailey CC, Robinson KJ, Weston CL, Ellison D, Ironside J, Lucraft H, Gilbertson R, Tait DM, Saran F, Walker DA, Pizer BL, Lashford LS, United Kingdom Children’s Cancer Study Group Brain Tumour, International Society of Paediatric (2004) Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys 58(4):1184–1193
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123(4):465–472
Terterov S, Krieger MD, Bowen I, McComb JG (2010) Evaluation of intracranial cerebrospinal fluid cytology in staging pediatric medulloblastomas, supratentorial primitive neuroectodermal tumors, and ependymomas. J Neurosurg Pediatr 6(2):131–136
Thomas PR, Deutsch M, Kepner JL, Boyett JM, Krischer J, Aronin P, Albright L, Allen JC, Packer RJ, Linggood R, Mulhern R, Stehbens JA, Langston J, Stanley P, Duffner P, Rorke L, Cherlow J, Friedman HS, Finlay JL, Vietti TJ, Kun LE (2000) Low-stage medulloblastoma: final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol 18(16): 3004–3011
Thompson EM, Hielscher T, Bouffet E, Remke M, Luu B, Gururangan S, McLendon RE, Bigner DD, Lipp ES, Perreault S, Cho YJ, Grant G, Kim SK, Lee JY, Rao AA, Giannini C, Li KK, Ng HK, Yao Y, Kumabe T, Tominaga T, Grajkowska WA, Perek-Polnik M, Low DC, Seow WT, Chang KT, Mora J, Pollack IF, Hamilton RL, Leary S, Moore AS, Ingram WJ, Hallahan AR, Jouvet A, Fevre-Montange M, Vasiljevic A, Faure-Conter C, Shofuda T, Kagawa N, Hashimoto N, Jabado N, Weil AG, Gayden T, Wataya T, Shalaby T, Grotzer M, Zitterbart K, Sterba J, Kren L, Hortobagyi T, Klekner A, Laszlo B, Pocza T, Hauser P, Schuller U, Jung S, Jang WY, French PJ, Kros JM, van Veelen ML, Massimi L, Leonard JR, Rubin JB, Vibhakar R, Chambless LB, Cooper MK, Thompson RC, Faria CC, Carvalho A, Nunes S, Pimentel J, Fan X, Muraszko KM, Lopez-Aguilar E, Lyden D, Garzia L, Shih DJ, Kijima N, Schneider C, Adamski J, Northcott PA, Kool M, Jones DT, Chan JA, Nikolic A, Garre ML, Van Meir EG, Osuka S, Olson JJ, Jahangiri A, Castro BA, Gupta N, Weiss WA, Moxon-Emre I, Mabbott DJ, Lassaletta A, Hawkins CE, Tabori U, Drake J, Kulkarni A, Dirks P, Rutka JT, Korshunov A, Pfister SM, Packer RJ, Ramaswamy V, Taylor MD (2016) Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. Lancet Oncol 17(4):484–495
Ulrich NH, Burkhardt JK, Serra C, Bernays RL, Bozinov O (2012) Resection of pediatric intracerebral tumors with the aid of intraoperative real-time 3-D ultrasound. Childs Nerv Syst 28(1):101–109
Vivekanandan S, Breene R, Ramanujachar R, Traunecker H, Pizer B, Gaze MN, Saran F, Thorp N, English M, Wheeler KA, Michalski A, Walker DA, Saunders D, Cowie F, Cameron A, Picton SV, Parashar D, Horan G, Williams MV (2015) The UK experience of a treatment strategy for pediatric metastatic medulloblastoma comprising intensive induction chemotherapy, hyperfractionated accelerated radiotherapy and response directed high dose myeloablative chemotherapy or maintenance chemotherapy (Milan strategy). Pediatr Blood Cancer 62(12): 2132–2139
Yock TI, Yeap BY, Ebb DH, Weyman E, Eaton BR, Sherry NA, Jones RM, MacDonald SM, Pulsifer MB, Lavally B, Abrams AN, Huang MS, Marcus KJ, Tarbell NJ (2016) Long-term toxic effects of proton radiotherapy for paediatric medulloblastoma: a phase 2 single-arm study. Lancet Oncol 17(3):287–298
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Section Editor information
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this entry

Cite this entry
Guerreiro Stucklin, A.S., Kuzan-Fischer, C.M., Taylor, M.D. (2020). Medulloblastomas. In: Di Rocco, C., Pang, D., Rutka, J. (eds) Textbook of Pediatric Neurosurgery. Springer, Cham. https://doi.org/10.1007/978-3-319-72168-2_91
Download citation
DOI: https://doi.org/10.1007/978-3-319-72168-2_91
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-72167-5
Online ISBN: 978-3-319-72168-2
eBook Packages: MedicineReference Module Medicine