
Abstract
The need to record directly the sensing target of interest in the vicinity of where a physiological and clinically relevant event takes place, rather than indirectly or through surrogate measures, has led to the need for implantable monitoring devices. In addition to ensuring the sensitivity and specificity of sensor responses, issues related to sensor fouling, drift , biocompatibility , and hermeticity of the packaging are important considerations. This chapter examines the current state of the art of sensing techniques, focusing on electrochemical methods (potentiometry , amperometry , and voltammetry ), due to their simplicity in design and fabrication [1], as well as low-power operation .
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
The need to record directly the sensing target of interest in the vicinity of where a physiological and clinically relevant event takes place, rather than indirectly or through surrogate measures, has led to the need for implantable monitoring devices. In addition to ensuring the sensitivity and specificity of sensor responses, issues related to sensor fouling, drift , biocompatibility , and hermeticity of the packaging are important considerations. This chapter examines the current state of the art of sensing techniques, focusing on electrochemical methods (potentiometry , amperometry , and voltammetry ), due to their simplicity in design and fabrication [1], as well as low-power operation . The basic principles of sensor operation and fabrication, with due consideration of these sensors when embodied as an implantable system, will be discussed.
2.2 Potentiometric Ionic Sensing
Potentiometric sensors represent an important family of sensors typically used for the detection of ions. Monitoring of various ion concentrations can provide valuable insights into the state of tissue and the underlying biochemical processes. Here we will discuss different potential ionic targets and the basic theory of potentiometric sensing , as well as clinical examples.
2.2.1 Ionic Targets
The most common application of ion sensors is their use in the clinical analysis for example in blood. Electrolytes such as Na+, K+, Cl−, Ca2+, Mg2+ and pH are routinely measured using ion sensors in clinical analyzers (HTA) and point-of-care (POC) discrete instruments [2]. Magnesium (Mg2+) is the eleventh most common element, the fourth most common cation in the human body, and the second most common intracellular cation [3]. It is essential to all cells, as more than 300 enzymes need magnesium ions for their catalytic action (including all enzymes synthesizing or utilizing ATP, or those that synthesize DNA or RNA). Intracellular Mg2+ correlates with intracellular potassium (K+), which is also essential for the function of all living cells. Figure 2.1 illustrates the intracellular and extracellular ions, their concentrations and ionic shifts taking place.

Reprinted from [4], © 2001 with permission from Elsevier
Intracellular and extracellular ions and shifts.
Another common element in the human body is K+. It is the main intracellular cation and is essential in neuronal function and in the osmotic balance between cells and the extracellular fluid. K+ is the predominant ion inside a cell (typically 140 mM [4]), being one to several orders of magnitude higher than Na+, Ca2+, and Cl−. Thus, intracellular K+ essentially determines cytoplasm volume. The extracellular space, however, contains low levels of K+ [5, 6]. Cellular loss of K+, leading to its extracellular accumulation, is one of the most prominent ionic shifts in early ischemia [7]. In general, cellular loss of K+ takes place in three distinct phases: (1) an initial rapid increase of extracellular K+ is followed by (2) a plateau and (3) a secondary increase. The latter is only partially reversible and hence marks the beginning of irreversible ischemic damage [7]. Thus, extracellular K+ increases due to the loss of the intracellular K+. According to [8], dysfunction of the sodium–potassium pumps due to ATP reduction and activation of potassium channels where the intracellular compartments are filled with water are key factors for the increase of K+ in extracellular space during ischemia . According to [9], in cellular cultures, potassium leaks out of dying cells, leading to elevated extracellular potassium ranging between 1 and 150 mM K+.
On the other hand, Na+ is the main extracellular cation. The concentration difference between K+ and Na+ leads to a potential difference, known as the membrane potential . Na+ regulates blood volume, pressure, osmotic equilibrium, and pH . “ Cellular and whole body Na:K ratios are crucial to the maintenance of normal blood pressure” [3]. The Na:K ratio may become too high via high Na+ or low K+, or indirectly by Mg2+ deficiency, leading to hypertension.
Extracellular Ca2+ concentration increases at sites of infection from its typical baseline, which ranges between 0.9 and 1.2 mM [10]. In addition, calcifications at sites of chronic inflammation or ischemic necrosis have led to the conclusion that extracellular Ca2+ concentrations also increase in sterile inflammation in addition to infectious inflammation [10]. According to [10], increased extracellular Ca2+ concentration can lead to the amplification of the inflammatory response .
Monitoring of tissue pH can be used as a means of assessing the ischemic state of tissues, especially since its value follows cell metabolism, and it can thus be considered as a direct method for monitoring ischemia [11,12,13,14]. A lack of oxygen delivery to the tissue , due to ischemia , can lead to a decrease of intracellular pH . This is due to glycolytic lactate acid generation and ATP depletion processes [8]. The tissue acid–base balance is maintained by the release of energy by ATP (which releases protons) and the re-synthesis of ATP (which consumes protons). When oxygen delivery to the tissue is inadequate and fails to synthesize the amount of ATP to address the metabolic needs of the tissue , the rate of proton release exceeds the rate of proton consumption. As a result, the pH decreases proportionally. A significant blood loss induced by trauma, for example, can lead to a reduced perfusion of tissue . This reduced supply of blood to tissue has as a consequence the increase of CO2 and lactic acid , leading inevitably to a pH decrease. Measurement of tissue pH provides localized assessment of metabolic and blood flow abnormalities. If the body pH is significantly reduced below 7.4, acidosis takes place. Many chemical reactions taking place in the human body, especially those related to proteins, are pH dependent. If the pH decreases below 6.8 or increases above 7.9, cell death occurs. As discussed in [15], once the tissue becomes acidotic, pH cannot offer any more information regarding the progression of ischemic injury. According to [8], it is the end of the active metabolic processes (glycolytic lactate acid generation and the ATP depletion ) that gives rise to the decrease in pH that leads to a plateau in the measured pH value. Measurements of pH are regularly used to monitor tissue hypoxia [8], while low pH values have also been found in tumor tissue . Nevertheless, it is affected by other parameters, such as temperature, and its application is further restricted as it provides information only for the area in contact with the pH sensor. From the above, it is evident that human health is closely related to pH homeostasis, and its fluctuations, in addition to indicating ischemia , are also related to atherosclerotic plaque development, inflammation , and tumor growth [16]. Ionic sensors are therefore an important class of sensors for providing valuable information regarding the state of tissues and patients.
2.2.2 Basic Theory of Ion Sensing
The first significant commercial contribution in the field of potentiometric sensing was in 1932, when Arnold Beckman developed the pH glass-based liquid electrode [17]. pH provides a measure of the amount of hydrogen ions (H+) and hydroxide ions (OH−) in an aqueous solution. A solution is defined as being neutral when the hydrogen ion concentration is equal to the hydroxide ion concentration. When this is the case, the pH is equal to 7. This is the case for water at 25 °C, where the concentrations of hydrogen and hydroxide ions are both 1 × 10−7 mol l−1. If the H+ ion concentration increases, pH decreases and when the concentration of OH− ions increases pH increases. A low pH value (between 0 and 7) describes an acidic solution, while a high pH describes an alkaline solution (between 7 and 14). pH is given by the decimal logarithm of the reciprocal of the hydrogen ion activity, aH+, in a solution.
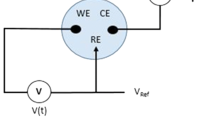
A standard pH potentiometric system consists of a reference electrode (RE) and a working electrode (WE). The former generates a constant potential irrespective of the ionic composition of the solution, while the latter generates a voltage which is a function of the hydrogen activity of the solution. Thus, a pH sensor can be considered as a voltage source, i.e. a battery. The series resistance of the source depends on the electrodes composition and configuration. The lower the pH , the larger the potential difference measured between the two electrodes and vice versa.
When equilibrium has been reached, the expressions for the ion-selective membrane phase (usually organic) and electrochemical potential in the aqueous phase are equal, so the phase-boundary potential (EPB) can be expressed as:
and when \( \mu_{\text{org}}^{0} = \mu_{\text{aq}}^{0} \)
where µ is the chemical potential (µ0 under standard conditions), kI desctribes the phase transfer energy, zI is the charge , and αI is the activity of ion I, ϕ is the electrical potential , T is the temperature, F = 96,485 C mol−1 is the Faraday constant, R = 8.314 J (K mol)−1 is the gas constant and kI is the phase transfer energy. The Nernst equation is expressed as:
E0 includes all the constant contributions, where the phase boundary potential is proportional to the logarithmic value of the activity ideally with a slope of 59.154 mV per decade for monovalent ions and for divalent ions 29.6 mV per decade at 25 °C. The modified Nernst equation is usually called the Nicolskii–Eisenman equation , and it is given by
where, αI,(IJ) and αJ,(IJ) are the activities of the primary (I) and interfering ions (J), \( K_{\text{IJ}}^{\text{pot}} \) represents the selectivity coefficient . The activity of αI,(I) is related to the mixed ion activity αI,(IJ) according to
The smaller the coefficient, the better the selectivity of the sensor is. When \( K_{\text{IJ}}^{\text{pot}} \) is very small, the part on the right-hand side of the equation approaches the primary ion activity αI,(I) without interfering ions, and therefore interference is negligible. If there is more than one interfering ion in the solution, the sum of the selectivity coefficients \( \left( {\mathop \sum \nolimits_{\text{J}} K_{\text{IJ}}^{\text{pot}} \alpha_{\text{J}} } \right) \) must be used. In mixed solutions, the potential response of an electrode obtained according to the Nicolskii–Eisenman equation will be different depending on the charges of primary and interfering ions [18].
If we consider pH sensors according to the Nernst equation , a pH sensor generates an ideal 59.154 mV pH −1 slope at 25 °C
where E is the voltage of the hydrogen electrode and pH is the hydrogen ion concentration being measured. E0 is the standard electrode potential . In metal –metal -oxide (MMO) electrodes, E0 is a constant that lumps together the standard potentials, the metal oxide solubility product, and the water ionization product [19]. Generally speaking, it is a constant potential difference that is independent of the sample composition, but depends on the temperature and the type of the reference electrode [20]. According to [21], in the case of a standard hydrogen electrode (SHE) E0 has a value of 926 mV and in case of using a Ag/AgCl RE has a value of 577 mV [22]. The intercept of the mV versus pH curve of the sensors response with the y-axis for pH of zero indicates the value of E0. Variations in E0 can in the case of IrO x electrodes, for example, be “due to variations in the stoichiometry of oxide compounds and the difference in oxidation states of iridium oxides” [22]. Additional factors related to the mechanical and chemical properties of the films as well as the hydrodynamics related to the redox processes taking place will influence the response of the sensors and lead to deviations between identical sensors [22]. The slope of the measured response is calculated by
where V1 is the potential difference measured with a solution with pH 1 and V2 is the potential difference measured with a solution of pH 2. The intercept for a temperature (T) in kelvin is found by:
where V″ is the potential difference measured in the solution being characterized.
According to the Nernst equation , at T = 25 °C, where the sensitivity of the sensor is equal to ±59.14 mV pH −1, for pH between 0 and 14, the voltage measurement can range between ±414 mV, and at T = 80 °C, where the sensitivity of the probe is ±70 mV pH −1, between ±490 mV. A typical pH measurement system produces at pH = 7 a zero voltage, has an accuracy of 0.05 pH between T = 20–25 °C, a resolution of 0.01 pH (=>0.1 mV) and a reaction time less than 1 s for 95% of the final value [23]. The pH sensitivity of −59 mV pH −1 at room temperature is well known as a Nernstian slope . However, depending on the surface and the formed microstructures and oxidation states of the ion -sensitive films and membranes, and thus the fabrication process, this slope may vary. Therefore, in [21] a sensitivity of −49.7 mV pH −1 was demonstrated, while, as will be discussed in following sections, certain films such as IrO x produce super-Nernstian responses.
All interfaces in the galvanic cell must be dominated by fast, reversible , and well-established faradaic charge transfer [2]. A practical potentiometric measurement of the potential difference across a galvanic cell requires an open circuit potential (OCP) measurement. Potentiometric measurements are performed with a zero faradaic current. Thus, high input impedance is mandatory for the measurement. This is in contrast with the amperometric , conductimetric, and impedimetric type of sensors.
The definition of the response time of a pH sensor is given as the time needed for the potential change induced by a pH unit change to reach 90% of its final equilibrium value. Issues regarding stability and repeatability in the measurements are related to potential fluctuation (∆V), potential deviation (δV) and potential drift (V′) and hysteresis (dV) as discussed in [22]. These are illustrated in Fig. 2.2. ∆V is a small non-random voltage fluctuation caused by electronic noise or interference and motion artifacts such as liquid motion. This can be in the range of ±0.3 mV and ±1 mV. δV is defined as the difference in measured potential responses between different tests when using the same electrode in the same solution. Reported values are less than 5 mV [22]. For recorded potentials varying between −0.07 and 0.46 V for pH between 1.5 and 12.1, the proposed ideal resolution is 0.02 pH mV−1. Nevertheless, due to a δV = 5 mV, the minimum pH sensing resolution is 0.1 unit of pH . The difference between the peak recorded potential and the 90% value of the saturated recorded potential is defined as the V′. This can range between 3 and 10 mV, with the potential stabilizing within 5 s. Hysteresis , is defined as the dV obtained when comparing measurements obtained from solutions with the same value of pH within the same experiment. Reported values range from 0.3 mV and can be as large as 23.7 mV [22]. In iridium-based pH sensors, dV can be minimized by creating high-quality IrO x films in terms of thickness, amorphousness and porosity [22]. The theoretical temperature dependence of potentiometric pH sensors is 0.3, 0.8, 1.3, and 2 mV °C−1 for pH of 2, 4, 7, and 10, respectively. Drift is a change in baseline potential over time, while the loss of sensitivity has a decreasing slope [20].

Reprinted from [22], © 2011, with permission from Elsevier
a Potential fluctuation (δV), deviation (ΔV) and drift (V′) are characteristics defining stability and repeatability, while b hysteresis (dV) a defines the reversibility of the sensor.
2.2.3 Ion Selective Electrodes
There are many different ways to develop an ion sensor. The classical pH electrode is the glass-based electrode sensor, which, however, has a number of drawbacks when considering its use for biomedical applications. These include slow response times, instability in fluoride, hydrofluoric acid, and silane solutions, the need for recalibration, and their susceptibility to membrane fouling, leading to loss of precision and accuracy. In physiological measurements, rapid pH measurements are of interest, as is the steady state value. Because of the very high impedance of these electrodes (multiple GΩs), these sensors require a high impedance meter [24], complicating the recording electronics. Due to their mechanical fragility, large size, and lack of deformability [22], they are unsuitable for in vivo and implantable applications.
Ion Selective Electrodes (ISEs) were first described by Cremer [25]. The discovery of antibiotics and selective binding cations gave a push to the field of ISEs. In the last two decades, solid state pH sensors have been developed in an effort to tackle the issues associated with glass-based electrodes [26]. The ionic markers discussed earlier can be monitored using solid contact ISE sensors instead of glass-based ones. Similarly to standard glass-based sensors, these are transducers , which produce a change in their equilibrium d.c. electrical potential , which is related to the activity of a specific ion in the solution. The potential is measured as the difference between an RE of constant potential and a working electrode , the surface of which is modified by an ion -specific membrane , since this process has to be selective. ISEs are very simple, robust sensors, which can be easily miniaturized. In addition, they are very sensitive, reaching sensing limits as low as parts-per-billion (ppb) [20]. They are based on the ion transfer processes taking place between the solution and the ion -sensitive membrane . The interaction of the analyte with the membrane leads to a non-uniform charge distribution, which gives rise to a change in electrode potential . As discussed in [27], the response of potentiometric sensors, i.e. the recorded signal , does not depend on the size of the active area of the sensor. Thus, due to their size independence, the response of these sensors will not change as their dimensions are reduced to micro and nano scales. It is important to note that when multiple ISEs are used there is a need for only one reference electrode . Nevertheless, each ISE will have its own baseline and slope [20].
Noble metals with stable oxides and reasonable conductance are one possible implementation of the ion-selective membrane , forming what are known as metal /metal -oxide and metal -oxide /metal -oxide electrodes [28]. They have an intrinsic mechanical stability and they can be easily miniaturized using semiconductor fabrication technology, making them CMOS compatible. Metal oxides absorb hydrogen atoms at the surface sites, changing the valency of the oxygen atom. This leads to the formation of an OCP difference between the two electrodes, the magnitude of which is proportional to the solutions pH [24]. Iridium oxide is a well-documented material [28,29,30,31,32,33], which can be formed hydrated or unhydrated [28, 30], with the hydrated form exhibiting super-Nernstian responses.
An alternative to MMO electrodes is solid-contact conductive polymer electrodes. These have been extensively reviewed in [34, 35]. Functional polymers (such as conductive polymers like polyaniline and polypyrrole) can be designed to selectively swell and shrink so that, depending on the analyte’s concentration. They can change their mass and elasticity. Polymer pH sensors react to these changes due to the protonation and deprotonation of nitrogen atoms in the polymer film. In conductive polymers , protons added to the material make them more conductive. Linear polyethylenimine (L-PEI) and linear polypropylenimine (L-PPI) polymers are particularly suitable for biosensing applications due to their strong bonding to electrode surfaces [24].
For the detection of K+, Na+ and ammonium (NH4+), valinomycin, monensin and nonactin are respectively used [17, 36,37,38]. In comparison to glass electrodes, in ionophore-based ISEs the chemical structure of the ionophore can be tailored such that a very high selectivity is achieved [39, 40]. Today there are ISEs for more than 60 different ions. ISEs can be used for the measurement of polyions, such as heparin and protamine, inorganic ions [41, 42], and also neutral species such as CO2, O2, SO2 [43,44,45], water, ammonia, organic amines, alcohol and non-ionic surfactants [41]. Advances in the host–guest chemistry for ISEs and the understanding of their theoretical response mechanisms led to the development of the first clinical analyser and its commercialization in 1972. Nowadays ISEs are almost irreplaceable in clinical analysis for detection of ions of clinical relevance, such as K+, Na+, Ca2+, Mg2+, and Cl−. ISEs determine the concentration of free ions in comparison with other methods, such as atomic absorption spectrophotometry (AAS) or inductively coupled plasma mass spectrometry (ICP-MS) techniques used to determine total ion concentration.
Lipophilicity of the ISE’s membrane is defined as p = log k, with k being the distribution coefficient of the different species between water and normal octanol [46]. Ionophores and ion -exchangers of low lipophilicity (p < 6), e.g. unsubstituted tetrakis(phenyl)borates, can leach from the ISE membranes relatively fast, so that the bulk resistance of the membranes increases 2–3 times within a few days, even at room temperature. Most of the used ionophores (charged or neutral), as well as ion exchangers, are highly lipophilic, with p = 8.
No matter what technique is used for the functionalization of the electrodes, deposited ion -sensitive membranes should have good adhesion to the sensor substrate. An issue with ISEs is the membrane lifetime. Membrane failure is mainly caused by partial detachment of the sensing membrane from the conductive surface or by the loss of the ionophore, plasticizer or carrier from the polymeric membrane film used due to leaching of some of the components into the surround solution. Leaching of the ionophore from its polymer matrix can influence the lifetime of a selective membrane . Insufficient lipophilicity of the membrane components is a dominant reason for the limited lifetime of ISEs; the more lipophilic the ionophore, the longer the lifetime of an ISE. Several approaches to how to improve it are discussed in [47]. For example, in the application of ISEs having a minimum lifetime of one month in clinical analysis, it requires a lipophilicity (log Pc) in regular 200 µm thick membranes of 11.3 and greater. It is possible to increase the lipophilicity with attachment of longer alkyl chains to the ionophore’s body. The attachment of an ionophore to a plasticizer -free polymer matrix has also been reported with minimal or no loss of the ionophore’s functionality [48,49,50]. The lower detection limit is dictated by the presence of the complexed primary ions in the membrane , where the upper limit is determined from the fact that the counterions must not enter from the sample side. When simultaneous extraction of a primary ion and its counterion (coextraction) occurs, the response slope is changed [51].
Ion exchangers allow for a proper electrode response and also have another important role. Membranes containing only ionic sites in a polymer matrix will still exhibit some selectivity based on the lipophilicity of ions due to their ion exchange properties. The more lipophilic ions have a lower hydration energy and partition more easily into the organic membrane phase from the aqueous solution [52, 53]. The physical and chemical properties, such as the polarity, and lipophilicity can have a significant effect on the ion -selective electrode selectivity and response time [54, 55]. Apolar and polar plasticizers can be distinguished based on the dielectric constant. Apolar plasticizers, such as bis(2-ethylhexyl)sebacate (DOS), promote the formation of ion pairs within the membrane [56].
The formation of ion pairs can significantly influence the slope of the response . If a divalent ion M2+ forms ion pairs with a monovalent ion A−, the phase transfer equilibria are dictated by MA+ as the predominant species, and the slope , characteristic for monovalent ion , can be obtained [57]. A polar membrane containing 2-nitrophenyloctyl ether (o-NPOE) shows an increase in selectivity for alkali ions due to decreasing coordinating ability [58], which is the reason for improvement of the detection limit [59]. There are several problems associated with using plasticized PVC. Some disadvantages are related to the possible leaching of the plasticizer from the membrane into the sample. While it shortens the lifetime of an electrode , it can significantly affect in vivo measurements due to inflammation and toxicity to the surrounding tissue [60]. In the case of microelectrodes, the specific resistance of the membrane can be increased, which can affect the electrode response [61]. The adhesion towards the matrix is important because the formation of an aqueous layer between the ion -selective solid-based membrane and metallic conductor can significantly impair the response [62]. The development of polymers that are self-plasticizing, such as polyurethanes [63], polysiloxanes [64], and polyacrylates [65], is a significant and active area of research. Other plasticizer -free polymers, prepared by one-step polymerization, are methacrylate and methacrilate–acrilate copolymers [66].
Nowadays ISEs are miniaturized, robust, and solid state and use integrated solid state REs. The main sensors developed for use in clinical environments are H+, Na+, K+, Ca2+, and Cl−; also used are Li+, Mg2+, NH4+, trace metal ion detection, and organic ion detection. Improved selectivity and sensitivity are achieved with new affinity ligands [67].
Another important family of potentiometric sensors are ion -sensitive field effect transistors (ISFETs). As discussed in [68], the line separating ISFETs (and thus also ChemFETs) from ISEs with integrated electronics is very thin. In an ISE, there is a conductor between the selective sensing membrane and the transistor gate (similarly to an extended gate ISFET), and an operational amplifier-based voltage follower is used instead of a single transistor . Active ISEs demonstrate improved thermal stability and reduced photo-induced junction currents. ISE membrane deposition is performed following the CMOS fabrication process. Active ISEs demonstrate an improved response time to chemical changes in contrast to passive ones. CMOS ISEs have been reported with a lifetime in excess of 120 days, with response time within 5% of the ideal and a 10 µM detection limit .
2.2.3.1 Iridium Oxide MMO pH Sensors
In vivo pH sensing is an important target for implantable systems. In general, pH sensing can be achieved by using iridium oxide metal –metal -oxide (MMO) sensors or polymer-based pH sensors. The ideal characteristics of these MMOs are discussed in [19]. According to [16, 19, 69], the advantages of IrO x films over other pH -sensitive oxides include a wide response range , fast response time, high pH sensitivity (a super Nernstian response , i.e. greater than 59 mV pH −1), minimal potential drift , low sensitivity to redox pair interference (thus high chemical selectivity), and finally high durability. IrO x has also demonstrated an outstanding stability over wide pH ranges and in different solutions. They are also characterized by a low impedance and low temperature dependence [33]. Thus far, there are many methods for the fabrication of IrO x functionalized electrodes for pH sensing . Sputtered IrO x (SIROF) is costly and the fabrication protocol is hard to optimize. Thermal oxidation requires very high temperatures (500–800 °C) and the film may crack if it is incompatible with a CMOS process. Anodic deposition based on electrolysis is an economic solution; however, process parameters can easily affect the deposition efficiency. Electroplating allows the formation of hydrous (electrochemical growth in aqueous solutions) IrO x on target planar microelectrodes selectively, using relatively inexpensive compounds of iridium. Hydrous IrO x leads to higher sensitivities [16]. According to [69], the above make IrO x “one of the best choices as sensitive material for pH electrodes”. Its use has also been discussed within the context of muscle and nerve electrical stimulation. Another advantage of (electrochemically generated, AIROF, not thermally) iridium-based pH sensing is the compatibility of the material with CMOS processes.
The authors in [70] discuss in detail how different applied potentials for the oxidation and formation of the IrO x film change the oxidation state of the film. At a constant pH the change is reflected in the OCP. A higher OCP corresponds to an oxide with higher valence and leads to higher sensitivity . Thus, the OCP at pH 0 increased from 490 to 825 mV and the slope from 62 mV pH −1 to 73.6 mV pH −1. In this way, the OCP recorded can be fine-tuned, e.g., for a specific power supply, so that the sensor response can be compatible with the output swing of the pH recording amplifier. In addition, the thicker the oxide the slower the response of the sensor to a pH change (350 vs. 40 ms), while in terms of drift the sensors are characterized as electrochemically stable, with a drift less than 300 µV h−1 [70].
The characteristics of these devices and the variety of different fabrication methods of IrO x pH sensors are discussed and summarized in [71]. As discussed in this paper, the potential drift , which causes errors in the measurement of pH , depends on the oxidation state and hydration of the oxide film and the preparation method used for the film. The fabricated sensor exhibited OCP changes between 700 and −100 mV for pH 0 to pH 14 and thus a slope of 59 mV pH −1, with no hysteresis and no changes in sensitivity , but a small 5 mV drift within 100 days. During a two-day continuous experiment at a pH of 6.6 the sensor demonstrated a relatively constant potential of 321.2 mV with a deviation of less than ±0.2 mV. Part of this could be due to temperature variations. The response time of the sensor to a pH change was considered to be less than 0.2 s. In [72] a drift of ±0.03 pH h−1 was reported for AEIROF and responses of −71 mV pH −1 and −61 mV pH −1 varying from 700 to 120 mV between pH of 3 and 10.
Sol-gel is a simpler and economical approach. Sol-gel based IrO x has low temperature dependence, low interference and low voltage drifts. Potential differences from 500 to −100 mV between pH 0 and 12 and a 58.5 mV pH −1 sensitivity is demonstrated. However, both AIROF and SIROF films are more sensitive due to their higher porosity. In [22, 73], response times from 2 to 60 s were reported for flexible iridium oxide fabricated with a sol-gel process. The response time is mainly affected by the porosity of the sensing membrane . The sol-gel process results in a lower porosity (reduced response time). Lower porosity, however, means lower sensitivity . The measured potential differences ranged from about 500 to −200 mV and sensitivities from 57 mV pH −1 to 63.4 mV pH −1.
In [74], the authors again used an IrO x pH electrode and a Ag/AgCl reference with sensitivities of 69–71 mV pH −1 and OCP varying between 500 mV and 0 V. In order to protect the electrode from corrosion by physiological electrolytes a biocompatible ionophoric coating was used (Nafion). In [75], implanted pH sensors were used to assess in vivo tissue trauma in the brain . Following characterization, the sensors were dipped in Nafion. AIROF electrodes were also coated with Nafion in [19] in order to stabilize the response of the sensors and to protect them from chloride and protein adsorption. Nafion coatings were also used in [76] and once again improved the stability and selectivity of the sensor. IrO x was once again used with a 80–90 mV pH −1 sensitivity and an E0 from 800 to 400 mV. Nafion was also used in [77] in AIROF electrodes. According to [78], most metal oxides, such as IrO x , are electronic conductors and thus they will respond to solution redox species. This will lead to large measurement errors. Many of these interferences can be eliminated or attenuated by coating the electrode with Nafion. There is, however, a tradeoff. The thicker the Nafion coating is, the greater the attenuation and thus elimination of the effect of interference, but the slower the response time of the electrode will be to a change in pH . This equilibration time was found to be a function of pH and it can reach a maximum value of 2 min [78].
The cytotoxicity of IrO x has been examined and has been found not to be toxic, with cells adhering and surviving on Ir surfaces. Table 2.1 compares and summarizes the published literature in terms of MMO pH sensor characteristics and fabrication methodology. Different methods are used, such as electrochemical growth (AIROF), electrodeposition, sputtered coating (SIROF), thermal, and printing methods.
2.2.3.2 Polymer-Based pH Sensing
Polyvinylchloride (PVC) is the most used polymer for solid state ISEs. Immobilization of the PVS membrane is via adsorption through Van der Waals interactions; thus adhesion to the surface is poor. In this case, stability and reproducibility could be a problem [91]. Leaching of components from the membrane will lead to a short lifetime and a loss of functionality. Leaching from covalently linked benzo-18-crown-6 with multi-walled carbon nanotubes (MWCNT) was studied in [92]. It is known that PVC is not haemocompatible [93]. Possible rejection and passivation of the sensor can be observed during blood fouling [94]. Thick PVC layers can hinder ion diffusion through the membrane . The thermal stability of these is low; thus one must consider the application they are intended to be used for [90].
Modification of the ISE surface is discussed in [95]. Poly(ethylene glycol) (PEG) and phosphorylcholine [96, 97] are used as hydrophilic materials so as to improve the sensor biocompatibility . Phosphorylcholine mimics the zwitterion nature of phospholipid moieties which is observed on the cell membrane surface [98]. PEG binds water molecules and creates a hydrophilic barrier which prevents protein adsorption.
It is evident from the above discussions that the field of polymeric pH sensors is an active research field. In [83], a Nernstian slope of 59.9 mV pH −1 was achieved between pH 2 and 9 with a measured potential difference between 200 and −150 mV. In [20], a conductive polymer pH sensor had a response time of t90% = 45 s and slopes between 59.8 and 62 mV pH −1. In [84], a pH sensor was developed using a metal Pt electrode electropolymerized by a pyrrole conductive polymer and a drop-cast PVC neutral carrier membrane . This sensor exhibited a Nernstian response , a response time less than 1 s, an impedance of 10 MΩ (the measurements are performed without sophisticated shielding and integrated amplifiers ), and a potential drift of less than 250 µV d−1, corresponding to a pH drift of 5/1000 pH units day−1.
2.2.3.3 In Vivo pH Sensing
There is significant interest in the use of pH sensing for tissue ischemia , as pH can be used as a marker for ischemia . In [13], tissue pH due to ischemia changes from about 7 to 6.5 within an hour of induced ischemia . In [99], within 45 min of induced ischemia , the measured pH changed by 0.8 (from 7.4 to 6.6) and K+ by 30 mM. Measurements were performed using commercially available ion -selective electrodes. In [8], pH changed from 7.4 to 6.4 within 40 min of induced ischemia . In a number of papers (e.g. [8, 15, 99]), the measured tissue pH initially decreased rapidly on the onset of ischemia and it subsequently stabilized to a constant value. This is presumably due to the tissue becoming acidic, marking the end of all metabolic processes giving rise to the pH decrease. This was argued to be a disadvantage of pH measurement when compared to tissue bio-impedance . Intracellular pH falls by 0.5–1 pH units during cerebral ischemia and the extracellular space becomes more acidic within 20 s of ischemia . According to [100,101,102], one of the main problems with tissue pH measurements is that they are not sufficiently stable during long-term measurements. Drifts, which have been described as unacceptable, greater than 0.48 pH units per 100 h were reported in [101]. According to [101, 103], the deposition of proteins on the surface of glass pH sensors during tissue pH measurements leads to measurement errors, producing a rise in measured pH . This protein deposition potentially creates an additional junction potential , which in turn leads to this pH increase. Other sources of drift are temperature variations, patient movement, and RE junction potential variation. A second pH sensor was used to simultaneously measure the pH of healthy muscle tissue (pH between 7.38 and 7.44) in order to compare this with ischemic muscle tissue pH . The pH measured from the healthy site remained somewhat constant throughout the experiment, between pH 7 and 7.2. The pH of the compromised tissue was initially at a lower pH value of 6.9. At the moment of occlusion, the pH started to decrease. Within one hour of occlusion, the pH dropped to 6.5. Within minutes from the release of occlusion, the pH increased to 6.9. In [101], 120 h of pH measurements from a patient with good post-operative recovery remained essentially constant and averaged a pH of 7.4, which is within healthy levels. Results obtained from a patient with an ischemic episode indicated a pH decrease 5 h following surgery. During this period, pH decreased by 0.4 pH units. Paper [104] reports pH changes of pH 0.73 between pH 7.08 to pH 6.35 within three hours (i.e. −0.0041 pH min−1) of tourniquet on canine hind limbs, a drop of pH 0.66 within an hour (i.e. −0.011 pH min−1) via arterial occlusion, a drop of pH 0.27 within an hour (i.e. −0.0045 pH min−1) via venous occlusion and a drop of pH 0.55 within an hour (i.e. −0.0092 pH min−1) via total occlusion of rat anterior thigh flaps. Other reported values are a drop of pH 0.80 in tissue pH within 240 min (i.e. 0.0033 pH min−1) in an amputated rat hind limb and a decrease of pH 0.35 from pH 7.4 to pH 7.05 (i.e. −0.0058 pH min−1) following occlusion of the femoral artery of a rabbit hind limb. An average decline of −0.00677 pH min−1 was also reported using a similar setup. An average per minute change of muscle tissue pH ranging between pH −0.33 to pH −0.11 is possible. In [105], the pH probe used exhibited a drift of less than 0.01 pH units over a six-hour period.
Of interest is the intestinal luminal pH of the normal healthy gastrointestinal (GI) tract. The colonic mucosa is covered by a surface of mucus with a thickness of 100–800 µm [106]. As discussed in [107], the colonic epithelium maintains a constant pH up to 840 µm above the cell lining. Failure of the anastomosis in the GI tract following colorectal resection, for example, leads to anastomotic leakage , which is considered one of the most challenging complications in GI surgery. Intramucosal pH at the anastomosis was found to be an independent factor for leakage development [108]. In patients with an anastomotic intramucosal pH which was less than 7.28 units within the first 24 h after surgery a 22 times higher risk of leakage is reported [108]. Patients with anastomotic leak had a mean pH of 7.05, while patients without a leak had a pH of 7.32 in the first 24 h [108]. In [105], the threshold value of intramural pH for ischemia and thus failure in the anastomosis was found to be less than pH 7. Compared to laser Doppler velocimetry, which correctly predicted the outcome of the anastomosis by 70%, tissue pH measurements succeeded in 93% of the cases [105]. It is, however, important to note that normal intestinal mucosal pH levels are not constant throughout the length of the intestines. In [107] the authors assessed the pH in different areas of the colon leading to what they termed as “the closest approximation of pH values of superficial colonic epithelium”. The measurements follow luminal colonic measurements (such as those obtained with pH sensing capsules), which nevertheless are more acidic in all colonic segments. It was found that the rectum (pH 7.15 ± 0.44) and the right colon (pH 7.05 ± 0.32) were relatively acidic, while the transverse (pH 7.42 ± 0.51), left (pH 7.46 ± 0.60), and sigmoid (pH 7.38 ± 0.59) colon were more alkaline. According to [107], the colonic mucosa tends to be more alkaline relative to the luminal colonic pH . In [15], pH in the intestine significantly dropped after ischemia , from 7.6 to 6.8 within 180 min of induced ischemia . Arterial pH followed a similar trend (7.44–6.95). According to [108], a pH less than 7.28 in the first 24 h following surgery is associated with a 22 times higher risk of anastomotic leakage .
In addition, continuous pH measurements provide an important parameter in detection of gastroesophageal reflux disease (GERD) [109] and also drug activity, which depend on saliva pH [110]. In [85], a system for GERD employing both impedance and pH measurements was demonstrated. The pH sensor was based on [22], which provided a slope of 51 mV pH −1, a voltage ranged between 0.5, and −0.1 V for pH values between 1 and 12. A second example of an ingestible capsule incorporating both impedance and pH for GERD is presented in [111]. The pH sensor had a sensitivity of 33.3 mV pH −1 and recorded voltages of 350 mV for pH 7 and 250 mV for pH 4. The combination of multichannel impedance with pH monitoring helps to detect nonacid reflux.
2.2.4 K+ Sensors
As discussed earlier, K+ is an important ion in biomedical applications. The cellular K+ level is a clinically important parameter for assessing cellular function. In ischemic myocardium for example, K+ loss is observed [112]. A connection between the cellular K+ loss and the balance in the charge during lactate transport has been hypothesized [113]. Passive movement of K+ in response to cellular loading of Na+ may enhance the loss of cellular potassium ions [114]. Cellular K+ loss in the ischemic heart is also related to the pH level [115].
An enhanced selectivity to K+ was achieved using a solid state-based internal electrolyte, such as polypyrrole stabilized with cobalt organic counterion and using calyx[4]arene ester [116]. In [117], micromolar detection was achieved where the silicon atom is part of the ether ring. In [91], the use of valinomycin for highly selective measurements of K+ is discussed, where different changes in the internal layer can give an extended sensitivity limit. Another internal solid contact electrode is based on the use of poly(3-octylthiophene) [118]. Using a lipophilic ion conductor avoids the drift related to the development of a water layer between the ionophore layer and the substrate. Graphene has also been proposed as the solid contact material [119]. An example of early multiparametric chemical tissue sensing is shown in Fig. 2.3.

Reprinted from [82], © 2002, with permission from Elsevier
Isolated rabbit papillary muscle, which was arterially perfused, with lactate , pH , K+ sensors and with both the reference and counter electrodes positioned at the septum.
In [120], a K+ solid contact microelectrode is described. The sensor is based on a neutral carrier and demonstrated operation within a pH range of 3–11, detection limit of 1.8 × 10−6 M, and working range between 6 × 10−6 to 1 × 10−1 M with a Nernstian slope of 52 mV decade−1. Valinomycin-based electrodes for online detection of intravascular and myocardial K+ ions was described in [91]. In [82], K+ sensitive electrodes were presented with a slope of 51 mV per decade (potential differences between 0 and 250 mV) for concentrations between 6 µM and 100 mM, a life time exceeding 40 days and a response time of t95% = 14.2 s.
2.2.5 Na+ Sensors
Na+ selective electrodes were traditionally made using variant glass electrodes. However, nowadays one uses PVC-based membranes loaded with ionophores like monensin, an antibiotic, or its derivatives. Urine can be tested for Na+ in the case that a nonpolar pre-extraction is done, because the lipophilic PVC membrane can extract nonpolar species that interfere with the response towards the primary ion , which leads to a drift and loss of selectivity [91].
2.2.6 In Vivo Ionic Sensing Using ISEs
In the last few decades, extensive progress has been made through the development of portable blood-gas and electrolyte devices. However, these remain unsuitable for continuous in vivo measurements and thus unable to provide real-time diagnostic information for rapid therapeutic decisions. Current ambulatory pH recordings require the passing of the sensor transanally or transnasally using a catheter. These are uncomfortable and conspicuous, thus affecting the daily life of patients. The Medtronic Bravo pH monitoring system is a solution which is attached to the mucosal wall to measure its pH and transmits data wirelessly to an external receiving device [121, 122]. Its dimensions are 6 mm × 5.5 mm × 25 mm and has an antimony pH sensor and a RE. The entire system is contained within epoxy.
A number of flexible implantable ion -sensitive electrode arrays mainly based on polymeric membranes have also been fabricated for myocardial measurements. In [79], flexible arrays of eight ion -sensitive 250 µm diameter electrodes were presented. The electrodes were functionalized such that they are sensitive to pH (55 mV pH −1), K+ (58.1 mV/K+) and Ca2+ (28.9 mV/Ca2+). The sensors demonstrated a reduced lifetime when used in serum and whole blood measurements. According to the authors, interferences in living tissue are more complex due to the inflammatory response . Protein adsorption and other effects in tissues can reduce the sensitivity of the sensors. In vivo experiments were performed in rabbits.
In [80], in vivo experiments were performed in porcine models. pH sensors with 500 µm diameter electrodes demonstrated linear responses between pH 4 and 12, with a slope of 58.5 mV pH −1. The reported lifetime of the electrodes was about 3 months while the reported drift was equal to 0.12 mV h−1. The authors reported a myocardial pH decrease during induced ischemia from pH 7.4 to 7.25 and from 7.4 to 7.05 (at a second site closer to the ischemic region). In [81], the same authors reported results from a similar electrode array for pH and K+ sensing (Fig. 2.4b, c). A slope of 58.9 mV pH −1 with a drift of 0.25 mV h−1 and 58.3 mV/K+ with a drift of 0.08 mV h−1 were reported and the electrodes exhibited equivalent resistances of less than 10 MΩ. The platform was used to detect acute ischemia in several sites of the heart. During an 18 min experiment in porcine hearts, pH changed from about 7.3 to 6.25 and K+ from 3 to 12 mM. Once again, electrodes closer to the ischemic region demonstrated a greater change in tissue pH . The same group reported on the use of metal –metal -oxide (MMO) anodic electrodeposited iridium oxide film (AIROF) pH electrodes which exhibited a super-Nernstian response of −63.5 mV pH −1, linear between pH of 2–10, a lifetime of one month, a fast response and an accuracy of 0.02 pH units [77] (Fig. 2.4a). The measured potentials varied between about 300 mV for pH 2 to −100 mV for pH 10. During ischemia , a significant increase of CO2 takes place. This can diffuse through the polymeric membrane to the inner hydrogel layer of polymeric pH sensors leading to pH measurement errors. This is potentially eliminated by MMO pH sensors. Generally, during ischemia , pH may decline from 7.2 to 6.3 and even as low as 5.7 [77, 82]. Simultaneously, extracellular K+ may vary between 3.5 and 9 mM [77, 82]. In [8, 99, 125,126,127,128] the authors built upon previous work by incorporating in their system impedance measurements in addition to pH and K+. Nevertheless, the ionic and impedance sensors were on two different substrates. The ionic sensors were realized with ISFET-based structures (Fig. 2.4e).

a A flexible IrO x pH sensor for measurements at the myocardium. Reprinted with permission from [77], © American Chemical Society 1998. b Top: illustration of a flexible platform for pH and K+ measurements with a common reference electrode for in vivo measurements. Bottom: Sensor cross-section. Reprinted with permission from [123], © American Chemical Society 2001 and c its realization, with connecting wires. Reprinted with permission from [81], © American Chemical Society 1995. The sensors of (a–c) are some examples of the first generation of flexible sensors for in vivo measurements developed by R. Buck’s group in the late 1990s for heart ischemia monitoring. d Ionic sensor array-enabled endoscopic probe developed for measurements in the upper GI tract. Reprinted from [124], © 2014, with permission from Elsevier. e A pH and K+ ISFET and temperature sensor array probe for ischemia monitoring from the early 2000s. Reprinted from [125], © 2001, with permission from Elsevier. f A flexible and stretchable array of IrO x pH sensors developed by J. Rogers’ group for heart ischemia monitoring. Reprinted with permission from [16], © Wiley 2013
Rogers’ group [16, 129] also uses IrO x , for pH measurements. They reported super-Nernstian sensitivities of 68.9 mV pH −1 in [129] (Fig. 2.5) and 69.9 mV pH −1 in [16] (Fig. 2.4f) and a temperature dependence of −1.6 mV °C−1, corresponding to a change of pH of 0.02 for a 1 °C change in temperature. Thus temperature changes within typical physiological temperature variations will not affect pH measurements significantly. Measurements on a rabbit heart gave a baseline pH of 7.4 and 7.32 in [129] and [16] respectively. During induced ischemia , this dropped to 6.22 in [129] and to 5.6 in [16] within 30 min. A significant advance in the field is presented in Fig. 2.5 [129], where an array employing a variety of sensing methodologies from electrical, chemical, and optical was developed for cardiac monitoring.

Multisensor array for monitoring of the epicardium for ischemia and other applications from Rogers’ group. Reprinted with permission from [129]. © Wiley 2015
An endoscopic sensor array (Fig. 2.5d) for use with a laparoscopic teleoperated robot gastroendoscope was presented in [90, 124, 130] for the detection of ischemia in the gastrointestinal system. The electrodes used were commercially available 3 mm long, 600 µm diameter beryllium copper alloy pins. For the RE the pin was covered by a carbon ink and subsequently by Ag/AgCl and finally by Nafion. The authors studied the interference of K+ and Na+ and the adhesion of the sensing films to the substrate at low pH . In in vivo applications, membrane stability and leaching of membrane constituents are important issues which must be addressed. A response time of about 18 s and a maximum sensitivity of 60 mV pH −1 were reported in [130], while sensitivities of 13.07 mV pH −1 and 17.8 mV/K+ were reported in [124] and 43.23 mV pH −1, 52.62 mV pH −1 and 53.88 mV pH −1 in [90]. Polymeric sensing films were used in all of these ISEs using PVC, PEDOT, and PEDOT-PEG [90] membranes and the authors report that the eletropolymerized PEDOT membranes exhibit a significantly smaller impedance (about 100 times less, 1.3 × 106 Ω vs. 1.4 × 104 Ω). PEDOT is strongly attached on Au surfaces used as a substrate through the dative binding of the sulfur groups in the PEDOT molecule [90], making the membrane very stable. In another approach the authors used as a crosslinker poly (ethylene glycol) diglycidilether (PEG) mixed with PEDOT.
2.2.7 ISFETs
An extension to potentiometric sensors, such as those discussed in previous sections, is the ISFET. This was first proposed in the 1970s by Piet Bergveld as a MOSFET without a gate metallization [131], as shown in Fig. 2.6. The exposed oxide is then placed in direct contact with the solution/sample under test. Charge accumulates at the exposed oxide insulator of the ISFET via the specific adsorption of hydrogen ions available in the solution [132]. This build-up of charge is a function of ionic concentration (pH or others depending on the sensing membrane ) and leads to the formation of an electrical double layer capacitance at the oxide /solution interface due to charge separation and polarization . Because of this, a potential difference is established across the interface and thus at the transistor’s gate oxide . The ISFET equivalent circuit will hence include a number of different capacitances and voltage sources.

a Simplified schematic of a MOSFET. b The ISFET structure. Reprinted from [131], © 2003, with permission from Elsevier
CMOS transistors can be operated in two modes. In strong inversion the device is operated above its threshold voltage, and its operation is characterized by the drift of electrons across an electric field established in the transistor channel. Its current-voltage characteristic when the device is operated in saturation is defined by the classic square law equation:
If it is operated in the strong inversion triode region, then it is characterized by
In weak inversion, the operation of the device is characterized by the diffusion of electrons across an electric field established in the transistor channel due to the very low gate voltage (e.g. VGS < VT). The drain current in this regime is typically in the nano-ampere scale or lower and the transconductance to current ratio is maximized and a constant for a given current, leading to a maximum intrinsic voltage gain for the device . Due to the low currents, this regime is unsuitable for high-frequency application, but for potentiometric sensing , which involves the recording of d.c. voltages, it is ideal, leading to ultra-low-power instrumentation. The low terminal voltage required for operation in weak inversion and the gate capacitance being minimized in this regime leads to a minimum input noise density for a given drain current and a minimum input offset voltage due to low voltage mismatch [133]. However, the output noise current is maximized and so is the current mismatch, which is mainly due to VT mismatch between identical devices. The current-voltage characteristic follows an exponential relationship similarly to bipolar transistors, thus allowing translinear and log domain processing circuit techniques to be employed. If the transistor source is tied to the bulk, the current-voltage characteristic of a MOS operated in weak inversion is approximated by [134]
and if VDS > 4Ut, the transistor is in saturation and the last term is ignored since e−4 = 0.018, leading to
where I0 is also known as the specific current (Is or Ispec), defined as the current when VGS = VT. This is equal to
Parameter \( I_{\text{spec}}^{\prime} \) is the specific current of a square device and is technology dependent, and n is known as the slope or substrate factor or gate coupling coefficient, and is typically considered to be constant. It represents the loss of “coupling efficiency between gate and channel caused by the substrate or body, which act as a back gate ” [134]. The slope factor in weak inversion corresponds to the capacitive division between the gate voltage and the silicon surface potential resulting from the gate oxide (Cox), the depletion below the channel (Cd) and the interface state (Cint) capacitances, with the effect of the latter being negligible; thus, this is expressed as [134]
Its value can vary between 1 and 1.5 depending on the technology used. Parameter Cox is the oxide capacitance per unit area, which is transistor (as a process may have transistors with different gate oxide thicknesses) and process-dependent; thus the gate capacitance is defined by
where \( \varepsilon_{{{\text{SiO}}_{2} }} \) is the SiO2 permittivity , ε0 is the permittivity of free space and tox is the thickness of the gate oxide SiO2 layer ; µn,p is the carrier mobility (electrons for NMOS and holes for PMOS, µn > µp), W and L are the width and length of the transistor , respectively, VGS is the transistor gate-to-source voltage, VDS the drain to source voltage and VTn,p is the transistor’s threshold voltage. The subscript n, p indicates that these parameters will be different for NMOS and PMOS devices. Cox and µn,p are typically provided by the CMOS foundry for a specific process as is VTn,p for a regular MOSFET. The product \( \mu_{{{\text{n}},{\text{p}}}} C_{\text{ox}} \) is known as the transistor gain factor and is typically given as Kn,p or as βn,p. All other variables are design parameters. If VDS is kept constant by circuit design techniques, the only device input variable is VGS. It has thus been debated whether the sensitivity of the device to ionic changes should be defined as an additional variable changing VGS or VT [131]. The measurement is performed against a RE, such that a two electrode potentiometric cell is formed. Typically, a non-polarizable RE is used, such as Ag/AgCl, in order to form a stable potential difference at its interface with the solution in contact. If the RE biasing the otherwise floating solution, is defined as a remote transistor gate connected to the gate oxide via the intermediate ionic solution, then the ionic sensitivity of the device can be described as a change in VT, changing as a function of the concentration of a target ion [131]. The perception that the device threshold modulation leads to detectable changes in the I-V characteristic of the device has been the dominant method to describe the device operation. The RE voltage is capacitively coupled thought the electrolyte to the ion sensitive passivation surface. The ion -dependent charge separation of this interface modulates the threshold voltage of the device . A MOSFET’s VT (VT_MOS) is defined by [131]
The first term defines the difference between the gate metal (ΦM) and silicon (ΦSi) work functions, with q being the electron charge . The second term is due to the total semiconductor charge density (QTot), which is equal to the sum of the accumulated charge in the oxide insulator (Qox), the trapped charge in the oxide -silicon interface (QSS) and the depletion charge in the silicon bulk (QB), i.e. QTot = Qox + QSS + QB. The final term determines the onset of inversion depending on the silicon doping level [131, 135]. In the ISFET structure, however, the VT expression is defined by [133]
where Vref is the constant potential formed at the RE/solution interface , Ψ is the chemical potential across the electrolyte/passivation interface , which is a function of pH and χSol is the surface dipole potential of the electrolyte in contact with the sensing passivation layer , a constant. Thus, Ψ is the only pH dependent term modeled by the combination of the site binding theory and the Gouy–Chapman–Stern double-layer model. According to the former, the insulator surface becomes charged once an equilibrium is reached between the insulator surface binding sites and the bulk solution hydrogen ions. The charge on both sides of the interface is matched and on the solution side it spreads across the double layer . According to the double layer model, there is a tightly packed compact layer close to the interface . This is known as the Helmholtz layer , the potential across which rises linearly. This is followed by a diffuse outer layer of charge which follows a Boltzmann distribution. This is known as the Gouy–Chapman layer , the potential across which rises exponentially. This eventually is replaced by an even distribution of charge in the bulk of the electrolyte. The double layer can thus be modeled by a linear constant capacitance CHelm and a nonlinear capacitance CGouy-Chap, as shown in the ISFET macro-model of Fig. 2.7. Following the above, it can be shown [131, 136] that Ψ is given by

where pHpzc is the pH value with which the oxide surface is electrically neutral, Ut is the thermal voltage (Ut = kT/q ≈ 25.85 mV at room temperature), while α is related to the intrinsic surface proton buffer capacity and the double layer capacity and can take a value between 0 and 1 [136]. Parameter α is a dimensionless sensitivity parameter describing the reduction of sensitivity by relating the ISFET sensitivity (SISFET) to the ideal Nernstian response (SNernst = 2.303Ut ≈ 59.2 mV pH −1 at 25 °C).
For α = 1, the device has the ideal Nernstian sensitivity , while for α = 0, the device is insensitive to pH changes. Using the above, VChem can be simplified by introducing the term γ, which groups all the pH -independent terms [133, 136], leading to
According to which, if α is considered a constant, VT and pH are linearly related. The temperature dependence of this should not be overlooked.
As mentioned previously, the sensitivity of the device to ionic changes can also be defined as an additional variable changing VGS instead of a change in VT [131, 135]. This can be performed by considering the ISFET as a floating -gate MOS (FG-MOS) device with a single input capacitively coupled through the passivation and defined by the passivation interface chemistry to the floating gate and considering VT as a process-dependent constant; thus, in this scenario VT = \( V_{{{\text{T}}_{\text{MOS}} }} \). An ISFET macro-model is shown in Fig. 2.7. There is a potential applied to the RE (VR). The RE/solution interface is characterized by a double layer model. Depending on the electrode material used (polarizable , e.g. Pt, or non-polarizable , e.g. Ag/AgCl), different elements of the electrode’s equivalent circuit model may dominate. A polarizable electrode is characterized by charge storage, demonstrating a high resistance to charge transfer through the interface ; thus the model is described by the double layer capacitance. In a non-polarizable electrode , charge transfer dominates, in which the interface is characterized by a small resistance . In either case, a potential drop across the interface will be developed, and in the case of a non-polarizable electrode , an (ideally) constant potential difference with the solution (Vref) is formed across the interface , unaffected by changes in solution composition, temperature or other factors. At the interface between the solution and the ISFET sensing membrane , there is a second double layer capacitive interface , elements of which, however, are pH dependent, leading to a pH dependent potential formed at this interface (VSense, which we termed earlier as Ψ), i.e. across the interfacial capacitances CHelm and CGouy-Chap. The series combination of these two capacitances (CChem) is estimated to be equal to 0.14 pF µm−2 [133] and 1 pF µm−2 [137]. Subsequently, there is the passivation capacitance (CPass) coupling the latter potential to the floating gate . There are trapped charges both in the passivation (VPass-TC) and in the floating gate structure (VFG-TC) due to the fabrication process. Additional parasitic capacitances exist between the transistor’s electrical gate and its other nodes (body, CFGB, substrate, CFGSub, drain, CFGD, and source, CFGS) due to the overlap of conductive areas between the gate and these nodes and the operating region of the device [138], as well as between the chemical gate passivation and these same nodes (CPassB, CPassSub, CPassD, CPassS), which are all process and device geometry dependent. Finally, there is also Cox and the bias dependent depletion capacitance (Cd) of the device (which is a constant in the deep weak inversion region of operation) [133, 137]. Taking into account the above, it leads to the equivalent circuit of Fig. 2.7. All these capacitances collectively influence the sensor performance nonlinearly [137]. The passivation capacitance couples the chemical dependent interface potential to the floating gate and influences the chemical response of the sensor. This leads to a capacitive division of the chemical dependent voltage developed at the interface and explains the reduced transconductance of ISFETs when compared to a MOSFET with the same dimensions [133, 137]. This capacitance depends on the top metal layer geometry which defines the chemical sensing area of the device , i.e., the chemical gate . It can be modeled, to a first level approximation, as a series of capacitances due to the two dielectrics (e.g. SiO2 and Si3N4), using (2.22), however, the fringing field at the edges will essentially modify the geometric factor of the equation and can be more accurately modeled using finite element analysis (FEM) [137]. This fringing field effect is modeled by the δ power factor (2.22) and it was experimentally found to be in agreement with FEM simulations which indicated that it is equal to 0.7. Approximate values for CPass given in [138] are 100 aF for 0.35 and 0.18 µm and 200 aF for 0.13 and 0.09 µm CMOS processes. The value of CPass can be in the region of 22.65 µF m−2 for a standard 0.35 µm process according to [139, 140], giving for a WS = LS = 2 µm a capacitance of 90.6 aF. Thus, since capacitance is defined by \( C_{x} = \varepsilon_{0} \varepsilon_{x} W_{\text{s}} L_{\text{s}} /t_{x} \), where ε x the permittivity of the x dielectric layer , t is the thickness of the x dielectric layer , with x being either SiO2 or Si3N4, and WS and LS are the width and length of the top metal layer defining the chemical gate [133, 137, 138]. CPass is given by
Cd is given by [139]
From the above discussion and the equivalent model, we can calculate the voltage at the interface as [139]
As mentioned previously, there are trapped charges in the passivation and in the floating gate [137]. If these are modeled as a single voltage source (\( V_{{{\text{TC}} - {\text{total}}}} \)) prior to the passivation capacitance, then from [133] we have
From the simple equivalent model, the potential formed at the floating gate (VFG) can be deduced from the following, if the total capacitance (Ctotal) seen between the nodes is defined as Ctotal–1:
For more discussions regarding the term ζ, refer to [139, 140]. The model can become increasingly complex if the intrinsic parasitics of the MOSFET are considered. In this case Ctotal is equal to Ctotal–2 below
In which case VFG becomes [135]
Typically, the last two terms in the numerator are ignored, as they may be grounded, while CFGSub may be one and the same with CFGB if the device is not on a separate well. An additional capacitive term, CP, was introduced in [137] to account for the increase in floating gate capacitance due to the larger top metal layer defining the chemical gate of the device . The ratio of CPass to Ctotal (the total capacitance seen by the floating gate ) scales down the effect of VChem, and thus the device’s pH sensitivity [135]. Maximizing this ratio by increasing the chemical gate area formed by the top metal layer (WSLS) and making it much larger than the transistor’s electrical gate area (WL) improves the pH sensitivity . In this case, the ISFET characteristics will approach those of the underlying MOSFET, since VFG will become equal to VPass [133]. This is one of the reasons for the sub-Nernstian pH sensitivity of unmodified CMOS process-compatible ISFETs [135]. This capacitive division due to passivation thus leads to a reduction of the device transconductance when referred to the remote gate (i.e. the RE). Increasing CPass will increase it and will also decrease VT [140]. In addition, the subthreshold slope will also be affected, shown to be inversely proportional to the dimensions of the chemical gate and thus CPass [133, 137]. In [137], this was found to be larger in ISFETs than in MOSFETs with the same electrical dimensions. Thus, a smaller WSLS will lead to a larger linear voltage operating range , requiring larger voltages to generate the same drain current [133]. The factor n is modified due to the additional capacitances present and thus according to [133] it becomes
The first two terms of Ceff are several orders of magnitude smaller than the other two associated with the chemically sensitive interface , and thus they dominate.
The noise of an ISFET is considered to be the same as that of a MOSFET in terms of the intrinsic device properties. Chemical noise, which is attributed to electrode degradation over time and surface chemical noise, will add up to this, as reported in [137, 138]. This additional noise contribution was defined as K/f, where f is the frequency and K was found experimentally to be equal to 1 nV2 Hz. The ISFET noise was found thus to be an order of magnitude larger than that of a MOSFET. Increasing the chemical gate dimensions was suggested as a means of reducing the random offset associated with trapped charge in [137]. The interested reader may refer to [137, 138] for further analysis, discussion, and equations, while the effect of the capacitances between passivation and all transistor nodes to VFG are also taken into account in [137]. When silicon nitride is expose in an aqueous solution, a thin hydrated surface layer is formed as hydrogen ions diffuse into the material. This changes the effective layer of the passivation and thus also the potential drop across it. Changes of this over time lead to drift in the recorded signal . In [137], it was found that drift had a negligible dimensional relationship to physical dimensions, ranging between 1 and 1.4 µV s−1, three orders of magnitude larger than that of fabricated MOSFETs of the same dimensions, while it exhibited a relaxed exponential characteristic.
2.2.7.1 CMOS-Compatible ISFETs
In [141], ISFET structures where developed using an unmodified standard commercial CMOS process. The polysilicon gate was not removed and it formed a floating gate electrode together with the two metal layers of the process on top of the gate as in Fig. 2.8. Above the top metal layer , the insulating pH sensitive oxynitride layer was located [131, 141]. In this way, the top layer nitride passivation was connected to the gate oxide through the stack of the process’s metal layers. The top metal layer essentially defines the chemical sensing area of the device . Thus two gates may be defined; one is the electrical gate , defined by the transistor polysilicon gate and the other is the chemical sensing gate defined by the top metal layer [135]. This separation allows the two to be dimensionally different [135]. The top passivation layer , which is typically silicon nitride on top of silicon oxynitride in commercially available CMOS processes, is used to protect the silicon chip from the environment [132]. It is deposited using low-temperature (300–400 °C) chemical vapor deposition (PECVD) [142]. Non-CMOS-compatible ISFETs typically use low-pressure chemical vapor deposition (LPCVD) to deposit the silicon nitride pH sensing film. This process allows a higher deposition rate and leads to dense films with superior purity and uniformity and few pinholes. However, it is incompatible with film deposition on metals as a result of the high temperatures required (700–800 °C). Si3N4 films deposited with PECVD, on the other hand, lead to films with lower oxynitride content, i.e. low density and consequently fewer hydrogen ion binding sites, and thus a sub-Nernstian pH sensitivity [133], while they also contain pinholes. Silicon nitride provides a linear response with pH with a sensitivity typically within 45–56 mV pH −1, with the sensitivity as a function of the oxide’s stoichiometry [142]. These led to a linear sub-Nernstian surface potential /pH slope (less than 59 mV pH −1) [143]. This structure is essentially an ISE with FET detection via an electrically floating transistor polysilicon gate [141]. Another advantage of keeping the polysilicon gate is reduced light sensitivity [141]. A structure where the chemically sensitive layer is deposited on a metal , which is located at a distance from the MOS structure and is connected to the transistor gate through a metal line, is known as an extended gate ISFET [141]. However, if the length of this wire is reduced and is connected vertically to the transistor gate the compact device of Fig. 2.8 is obtained. The use of the passivation opening mask typically used in standard CMOS processes to define bond-pad openings in the top passivation layer was proposed in [144]. This was used as a means of minimizing the use of a full stack of metals and inter-metal insulators in a technology. In this way, the ISFET consisted of the gate oxide , the polysilicon gate , the first aluminum metal layer and the first inter-metal silicon dioxide dielectric, and thus the second and third metal and their intermediate inter-metal dielectric as well as the top layer insulation were removed without additional fabrication steps. This led to ISFETs with similar chemical sensitivity , improved effective transconductance (approximately three times larger), and reduced threshold voltage (approximately a third smaller). Classic CMOS compatible ISFETs exhibit a low transconductance due to the capacitive voltage divider formed by the stack of capacitances between the gate and the sensing membrane . In [144], this was reduced by reducing the sensing membrane by 5–11%. Nevertheless, the device noise was increased by two orders of magnitude, but still within acceptable levels for chemical sensing application according to [144], while the drift was dramatically increased, making the device inappropriate for absolute concentration measurements and limiting its usefulness for detecting the occurrence of an event (such as ssDNA binding).

Another issue with CMOS-compatible ISFETs is related to the low-temperature PECVD process used in CMOS for the deposition of the passivation sensing layers. While the quality of the deposited passivation may be adequate for typical CMOS device and circuit performance, it may potentially fall short in chemical sensing applications. This is due to the low density and the porosity of the resulting sensing film. Because of these characteristics, the passivation layer may over time “absorb and become saturated with analytes or other substances in the solution, which may in turn cause an undesirable time-varying drift in the chemFETs” VT [145, 146]. Consequently, this may lead to a loss of accuracy in the measurement and monitoring of the target analyte .
2.2.7.2 ReFETs and Differential Measurements
An ISFET insensitive to any ions is known as a reference FET, i.e. a ReFET, and such a device offers important advantages. Identical ISFET and ReFET pairs can allow differential measurements to be performed, leading to common mode errors, such as drift and temperature effects, to be eliminated while also relaxing the RE specifications. Thus pseudo-reference electrodes, i.e. polarizable electrodes, made out of noble materials such as Au or Pt, can be used and the non-constant interface potential formed at their interface with the solution/sample under test will also be a common mode signal that can be removed by the instrumentation. One such circuit is shown in Fig. 2.10a. A ReFET must be as similar as possible to the ISFET it is being used with; it should thus have the same electrical and chemical gate dimensions and it should be laid out similarly, such that common mode interferences influence the devices similarly. The challenge is in making it insensitive to ions. Many methods have been proposed. One option is to apply a membrane on top of the chemical gate such that the device’s pH sensitivity is reduced ideally to zero. Hydrophobic membranes with very low buffer capacity, such that α = 0, have been proposed [131]. Examples of such membranes are parylene and Teflon. Another approach is to add a diffusion barrier, such that the ISFET illustrates a delayed response [131]. According to [132], a polymer membrane must be used which prevents hydrogen ions from reaching the pH -sensitive membrane . However, this should not block any other ions to “preserve the electrical characteristics of the underlying ISFET” [132], such that there is no potential drop across this additional membrane . In [132] a PVC-based membrane was used which has a very low sensitivity to H+, Na+, and K+. The ISFET and the device to be used as a ReFET were matched through UV exposure; however, a VT mismatch between the two was established once the PVC membrane was deposited. Another approach used to perform differential measurements is to have two ISFETs with different sensitivities. In [147] for example, the first ISFET had a Ta2O5/SiO2 gate with a sensitivity of 58–59 mV pH −1 and the second one used a SiO x N y /Si3N4/SiO2 gate with a sensitivity of 18–20 mV pH −1. Each of the two devices was used as one of the input transistors in the differential pair of an operational amplifier configured as a source follower and their outputs were subsequently subtracted by a differential amplifier resulting in an overall sensitivity of 40–43 mV pH −1. Another approach is to use the same ISFETs, with the same sensitivity and sensing films , but to use circuit techniques to decrease the sensitivity of one of the two devices. This was implemented in [148], where the authors used different gains for two ISFET amplifiers by controlling the width of the ISFET pMOS load. The two topologies resulted in sensitivities of 50 and 30 mV pH −1. Similarly to [147], a differential amplifier subtracted the signals from the two ISFET amplifiers and amplified the resulting signal leading to an overall sensitivity of 40–45 mV pH −1. These techniques are not ideal; they may relax the specifications of the RE, but since the devices are not identical and their sensitivities different [147] or because their response is scaled [148], the effect of common mode interferences will not be completely eliminated.
2.2.7.3 Sensing Oxides
The use of different insulators and other layers allows sensitivities to different target analytes, similarly to standard potentiometric sensors. The first ISFET gate material used was silicon dioxide [131]. This was followed by double layer insulator structures which resulted in better pH sensitivities and stability. The sandwich of silicon dioxide with silicon nitride (Si3N4) as the top insulator is an example [131]. Other top layer insulators used include tantalum oxide (Ta2O5) and aluminum oxide (Al2O3) [132]; these are examples of oxide insulators used for pH sensitive ISFETs. This standard ISFET, however, is not CMOS process compatible. Early examples of ISFET systems with recoding electronics on the same substrate required additional fabrication steps; e.g. in [147] four extra masks were required for the ISFET fabrication. In CMOS processes a polysilicon electrode is required in the gate region to determine the self-aligned source and drain regions of the transistors [141]. Since this is not in agreement with the original ISFET device structure, e.g. in [147], the polysilicon gate and the underlying gate oxide were removed by wet etching and a thinner gate oxide was regrown in the gate region of the device followed by the deposition of different gate dielectrics to form ISFETs with different pH sensitivities.
2.2.7.4 Trapped Charge
One of the main advantages of the ISFET is high miniaturization not only of the sensor (allowing measurements of nano and picoliter volumes [139]), but also of the complete system, since sensor and recording (typically analog) front-end electronics and back-end digital circuits can coexist on the same silicon chip . Exploiting CMOS technology economies of scale with ISFETs in unmodified CMOS processes can lead to mass-produced and low-cost miniature systems for point-of-care and implantable systems. ISFETs are, however, associated with a number of disadvantages. One parameter which is typically ignored according to [131] is the potential electrostatic damage of the gate oxide due to high external electric fields [131]. Electrostatic discharge (ESD) may cause charge to be trapped in the floating gate . This will lead to large shifts in the ISFET’s VT (residual charges may remain on different structures comprising the ISFET, e.g. oxides) and may even permanently damage (physically and chemically) the device . Steps in the CMOS fabrication process such as plasma metal etching , wafer cleaning, dicing, packaging/wire bonding, and handling may negatively affect the fabricated devices [146]. The etching process, for example, used to define the metals of the floating gate may induce the accumulation of charge trapped in the metal layers that constitute the floating gate . This is known as the antenna effect [145, 146]. If there are no paths for these to be removed the charge may be injected to the oxide . Thus, charge can be trapped in the passivation and/or on the floating gate . Charge in the gate , being floating , has no path to ground and remains there similarly to EEPROM [132]. Trapped charge at the typically dual passivation layer stack has mechanisms similar to the one used in metal -nitride-oxide -silicon (MNOS) transistors used for non-volatile memory [133]. Fixed charge in the passivation is due to the intrinsic dangling bonds within the silicon nitride sensing films and buried sites or surface defects due to extrinsic dangling bonds [137]. The trapped charge , and thus the VT shift, is indeterminable, leading to random offsets and a large VT, which may lead to an inability of the instrumentation to record the sensor output signal within its recording range . For example the p-type ISFET of [132] was found to have a VT of −5 V, when the proposed system was designed for 3 V supply operation, while in [142] the VT of an array of ISFETs was found to range from −4 to −1 V. In addition, geometrically identical ISFETs comprising arrays on the same die will demonstrate different VT. Thus, ISFETs comprising large sensor arrays will also behave differently among them. An ESD event may occur also due to the solution under test floating , and thus it is recommended to ground the solution via the RE prior to ISFET contact, or to insert the RE and ISFET within the solution simultaneously [131].
-
Fabrication-based solutions to trapped charge
A thinner gate oxide can allow an adequate amount of charge accumulated on the floating gate to move to the substrate [145]. Such a device can be used either as an ISFET or as an additional device connected to the gate of an ISFET. In order to pattern metal structures using plasma etching , a photoresist is deposited on the metal . An additional dielectric, such as an oxide between metal and photoresist, can be used in order to reduce the charge trapped during the plasma etching process. This dielectric may be left behind and form part of the sensing membrane of the final device . Alternatively, the etching rate may be modified by using e.g. lower power densities and thus potentially lead to reduction in the trapped charge . Instead of plasma etching , wet chemistry or ion -beam milling may be used [145, 146], leading to no charge being trapped during the metal patterning step since these techniques do not rely on an electric field. However, these require alterations to the CMOS process.
Modifications in the wafer post-fabrication handling and packaging steps may also be employed in order to reduce trapped charge [145]. Examples include using anti-static dicing tape to hold wafers in place and replacing conventional deionized water for cooling the dicing saws, which is of high resistivity , with lower resistivity ones [146].
-
Post-processing solutions to trapped charge
Post-processing techniques include gas annealing using a heated hydrogen and nitrogen gas mixture following wafer dicing into individual chips. This neutralizes trapped charges. Another approach is using UV radiation. For this to be effective, a hole or window, i.e. an aperture in the top metal layer within the vicinity of the floating gate is required, such that the radiation reaches the gate oxide and other layers [142]. An EPROM eraser was used, and following three hours of UV exposure the VT was increased from −5 to −1 V in [132]. In [142] the UV wavelength used was 253.7 nm with an exposure level of 4 mW cm−2. Appropriate biasing of the device during UV illumination facilitates the reduction of the trapped charge . Applying a positive voltage to the bulk with respect to the substrate (VBS = 5 V), and thus with the floating gate , during UV exposure, allowed further increase of VT in [132] to about 3 V. This voltage changes the conduction and valence band level in the bulk of the device . UV light excites the charges trapped at the gate such that they overcome the oxide energy barrier and discharge the gate to reach equilibrium with those in the bulk, permanently changing the ISFET’s VT [132]. The effect of the device biasing during UV illumination to VT modification demonstrated that it is in fact the removal of trapped charge in the floating gate rather than the fixed charge in the passivation layer that mainly affects the VT. UV exposure of 17 h led to a minimum 15 mV VT mismatch between identical ISFETs in [132]. In [142] during UV exposure a constant 20 µA current was supplied to the source of every ISFET and the drain and bulk voltage was set to ground. After 10 h the VT of all the ISFETs converge to about −1 V with an offset less than 10 mV.
-
Design-based solutions to trapped charge
A number of solutions have been proposed, which focus on the use of on-chip ESD protection techniques. Such solutions may involve the use of protection diodes (Zener or avalanche diode structures) connected to electrode (metal ) structures partially or entirely encompassing (rings or other geometries) the floating gate . The diodes can allow conduction above a certain predetermined breakdown voltage [131]. The metal structure should be in close proximity to the floating gate ; this may be connected to ground, to a power supply directly, or through a passive device (resistor or capacitor) or diodes and a rail clamp circuit, which may be internal or external to the chip . Thus, a path of least resistance to a large ESD current is formed which will be preferred against the path towards the floating gate [149]. Alternatively a diode may be connected directly between the floating gate and the substrate [146]. Another solution to ESD was presented in the early paper of [150]. An ISFET structure was presented, which is similar to those proposed in unmodified CMOS processes, as there is a polysilicon gate electrode , which is in turn in contact with a silicon nitride pH -sensitive top passivation layer . The polysilicon gate is connected to a MOSFET switch which allows the connection of the ISFET gate to a potential , in which case the device can be operated as a regular MOSFET. However, the off resistance of the switch is finite and there will be a leakage current. This leakage current needs to be minimized. If the switch is located in the immediate vicinity of the gate and connected to the diffusion of the switch’s source, then a pn-junction diode exists between gate and substrate. This diode provides additional ESD protection as its breakdown voltage (35–45 V) can be substantially smaller than the gate oxide breakdown voltage (100 V) [150]. According to [141], if an ISFET gate is connected to a diode, switch [150, 151] or other structures, then it is not completely floating . The solution proposed in [149] allows the gate to remain completely floating .
-
Floating gate circuit design solutions to trapped charge
Nevertheless, the ISFET VT can be programmed without the need for device post-processing techniques. The use of well-known techniques developed for the programmability of FG-MOS devices and systems can be used. FG-MOS transistors can have multiple capacitively coupled input nodes resulting in a floating gate potential determined by their weighted sum. The effective weight of each input is proportional to its coupling capacitance. The operating point of the device can hence be controlled by applying a voltage to one such control gate . In [152] a second capacitively coupled input was used for the floating gate of the ISFET, the capacitance of which was made large such that it may be programmed by a voltage within the rails of the proposed system. Thus, in an array of ISFETs, different programming voltages may be used in order to match their VTS, while known drift signals may be cancelled out. The proposed device allowed a 2.16 V decrease in VT for an increase of 500 mV in the programming voltage [152]. This, however, can lead to a reduction in the strength of the chemical signal , thus reducing the chemical sensitivity of the device [153]. Hot-electron injection [154] and electron tunneling [153] are techniques that have been proposed as alternatives enabling VT programmability while the device is in a pH -buffered saline solution. Hot-electron injection can take place in a p-type ISFET when a large VSD across the device channel is applied, such that a large lateral electric field is established. If such a current injection is induced, charge storage takes place on the gate , counteracting the trapped charge . In [154], the solution RE was grounded and a VSD ranging from 500 mV to 8 V was applied for 1 min. In this way the VT was increased and shifted by up to 2.8 V. This method cannot decrease the VT by removing electrons; electron tunneling, however, allows this. This was demonstrated in [153, 155] where the use of additional diodes and voltages capacitively coupled to the gate allowed electrons to be removed from the floating ISFET gate . These capacitively coupled inputs are similar to the one used in FG-MOS structures and in [152]. During normal device operation they are grounded and all other device inputs are used as usual. During threshold programmability the RE, drain and source voltages are grounded and the programming input nodes are set to the desired voltage for 80 s in [155] and 5 min in [153]. Tunneling voltages between 6.9 and 8.5 V allowed a VT shift of −3.45 V in [155].
-
ISFET-based biomedical devices
A popular recent application of ISFETs is in large arrays for DNA detection. In [156], a system with 1.2 million sensing wells was demonstrated which allowed the sequencing of three bacterial and one human genomes. In [157] the authors presented a system that not only performed DNA detection but also real-time DNA amplification using PCR and isothermal amplification. DNA amplification was monitored and performed using on-chip temperature sensors and resistive heating. Differential measurements were performed by using a regular ISFET identical with the rest but on a separate well, such that the ISFET can record baseline signals and reference measurements can be performed to eliminate common mode interferences. The device was used to genotype and discriminate unique single-nucleotide polymorphism (SNP) from human saliva. A recent system for DNA detection which combined a CMOS image sensor with the ISFET array was presented in [158]. By deposition of ion -selective membranes on the insulator sensing membrane , structures sensitive to ions such as Na+, K+ and Ca2+ can be obtained. These devices are known as ChemFETs. More relevant to implantable applications is the work presented in [125, 159,160,161]. Flexible organic ISFETs are presented in [159] for the detection of pH and K+ changes induced by myocardial ischemia in the extracellular matrix of the myocardium. Once again, valinomycin was used for the K+ sensor. A wireless diagnostic capsule for ISFET-based pH monitoring in the GI tract was presented in [161] for Gastroesophageal Reflux Disease (GERD) and inflammatory bowel diseases such as Crohn’s and ulcerative colitis. The system had two ISFETs and a ReFET and could perform single-ended and differential measurements with the use of multiplexers. The system also incorporated an 8-bit ADC and a microcontroller. The ISFET demonstrated a 48 mV pH −1 sensitivity . Another pH capsule was presented in [160], where the ISFET demonstrated a 44.95 mV pH −1 sensitivity . Surrounding an ISFET by a thin film of an electrolyte solution and by enclosing this in a gas-permeable membrane allows the development of gas-sensitive structures known as GasFETs. The immobilization of enzymes or enzyme -containing layers directly on the inorganic gate insulator of an ISFET leads to a device known as the EnFET, originally proposed in 1976 [162]. EnFETs for the detection of penicillin, glucose , sucrose, urea, and creatinine, among others, have been developed over the years [162]. If the enzymatic reaction leads to the production of hydrogen or other ions, these can then be subsequently detected by the underlying ISFET of ChemFET. Major problems associated with EnFETs are related to enzyme immobilization (covalent attachment and polymer entrapment) and associated nonlinearities, with stability and reproducibility of the results being major issues [162]. Recently an EnFET for ATP detection was presented in [163]. Similarly to the EnFET, ISFETs can be used for label-free DNA hybridization detection and immunological sensing , e.g. through antibodies [162].
2.2.8 Reference Electrodes
It is evident from previous discussions that in electroanalytical measurements the RE plays a very important role. Such an electrode should be insensitive to changes in the ionic concentration, be reversible (thus a low charge transfer resistance is required), and demonstrate a stable constant reference potential and a reproducible response [27, 164, 165]. A well-defined RE is necessary in all potentiometric measurements. As discussed in [27], there is always a potential at any interface ; however, if this potential is not dominated by a reaction with high exchange current density it cannot be stable. The traditional glass-based Ag/AgCl RE addresses the above. However, careful control of its working conditions is required. This is because it requires a liquid reservoir with a positive hydrostatic pressure of the inner electrolyte. The inner solution is moving in one direction through a liquid junction so as to keep a high and constant concentration of the electrolyte at the solution interface .
Thus, one of the main limitations of potentiometric sensors lies in the fundamental need of this family of electrochemical sensors for such an RE. This is particularly problematic in miniaturized biocompatible applications. In such applications, the use of conventional glass-based REs is not possible due to their large size, fragility and use of the liquid filling solution. Such conventional REs cannot be miniaturized and are unpractical. The RE has to have similar dimensions to the ion sensors used in order to be practical.
Emerging technologies, such as wearable and embedded electrochemical sensors, point-of-care, and telehealth have driven the development of suitable REs. Several approaches are suggested: using solid polymers [83, 166], solid KCl [167], micro-fabricated liquid junctions, agarose gels with KCl and heterogeneous membranes separating a KCl reservoir. Solid-state technologies [168, 169] have played an important role in RE technology. Several solutions have been proposed, such as solid-state junctions based on carbon-fiber nanocomposite membranes incorporating poly(methyl methacrylate) and carbon graphene stacked nanofibers [170], membranes prepared by aromatic polyurethane [171], metal alloys and composite materials [172]. A solid-state RE made with vinyl ester resin doped with KCl has been reported in [173]. Combination of PVC membrane and ionic liquid is reported in [174, 175], while other groups [176] used a screen-printed insulation paste. In all these approaches there is always a range in which the performance of the sensor is interfered with by lipophilic species or even Ag ions in solution. Different types of polymers , such as polyacrilate microspheres and polypyrrole microcapsules, were used to improve the stability of the potential [177, 178]. Single-wall carbon nanotubes (CNTs) are used [179, 180] as potentiometric transducers so that the Ag/AgCl/Cl– system is encapsulated in a polyacrylate membrane [181, 182]. Recently, a new reference system was developed in [183], where polyvinyl acetate with KCl was used.
The development of an RE based on polyvinyl butyral (PVB) membranes is reported in [184]. The membrane was made by dispersing Ag, AgCl, and NaCl in a PVB solution with methanol. The observed stability of the signal is related to the formation of nanopores on the surface. PVB has high polarity, making it highly resistant to fouling, which can be caused by non-polar substances [185]. Polar substances and ionic salts can be incorporated in PVB, which can increase the ionic conductivity of the polymer [186]. In another example, the use of REs prepared using a poly(vinyl chloride) membrane on top of conductive poly(3, 4-ethylenedioxythiophene) was reported in [177]. In [177] the authors used poly(n-butyl acrylate). Polyacrilates are promising materials showing a lower detection limit and lower ionic activity compared with PVC, and they can be produced by photopolymerization.
In [187], four chemical designs are compared as REs. In one of the approaches the membranes were non-responsive to ions, but the system wasn’t evaluated. A different concept involves the doping of the membrane with an inert electrolyte, such as ions with medium lipophilicity , which can partioning in the sample and become the potential determining ion . In this case the membrane responds only to the membrane ions and does not respond to ions in the sample. Kakiuchi et al. [188] used Ag/AgCl electrodes and IL as a salt bridge. The authors reported that the saturation of the ionic liquid with AgCl is crucial for proper functioning of the RE. In [175] the authors reported a liquid-junction-free polymer membrane, where an intermediate layer is used together with a PVC-IL membrane on top as reference. Another type of reference electrode was proposed using ionic liquid [175, 188] where gelled and non-gelled ionic liquid is used so as to form a liquid junction reference electrode .
Solid-contact RE based on a PEDOT layer electrochemically deposited on a carbon layer and an ionogel membrane consisting of tris(penta–fluoroethyl)trifluorophosphate [HMIM]+[FAP]− in poly(butyl-co-decyl–methacrylate) is discussed in [189]. Other reported ionic liquids are tributyl (2-methoxyethyl) phosphonium bis(penthafluoroethanesulfonyl)amide [190] and bis(tritrifluoromethanesulfonyl) amides with 1-alkyl-3-methyl-imidazolium, phosphonium or ammonium cations [175]. Another type of RE was proposed using poly(n-butyl acrylate)-based membrane on top of glassy carbon electrode with polypyrrole as a transducing layer [177]. REs based on a polymer, an inorganic salt, and an Ag/AgCl element showed excellent stability, long lifetime, and insensitivity towards the ions in the sample. The basic principles of REs are discussed in [191] and shown in Fig. 2.9.

Principle of RE (Ag/Ag/Cl reference electrode ), 1: metal lead (Ag), 2: metal salt (AgCl), 3: aqueous solution of KCl, 4: hydrogel-containing KCl solution, 5: solid melt of KCl, 6: junction containing diaphragm, porous ceramic or opening, 7: solid state reference element, 8: insulating encapsulation. Reprinted from [191] with permission of Springer. © 2008
In [78], an Ag/AgCl RE was coated with a polymer containing chloride ions, which was encapsulated by a Nafion outer layer . Nafion blocks the chloride anion diffusion to the test solution maintaining a constant chloride concentration on the AgCl electrode surface. An Ag/AgCl RE with a Nafion coating to increase its stability was reported in [124, 130].
Despite all the aforementioned effort in the development of a miniature RE, many issues related to the long-term behavior of these have not been solved yet. The electrochemical kinetics need to be investigated further to clarify the processes related with the mobility and exchange between the solids and dissolved ions. Figure 2.9 summarizes and illustrates the various methodologies followed for RE development.
2.2.9 Read-Out Circuits and Instrumentation
In the preceding sections, the technology regarding the potentiometric sensing element was discussed. It is obvious that these are not standalone devices, but they rather require additional circuitry in order to record their output signal . The following is a very brief discussion of the challenges in recording from potentiometric sensors and some examples of recording electronics used in the literature using commercially available discreet electronic components.
A practical potentiometric measurement of the potential difference across a galvanic cell requires an OCP measurement. Typical glass electrode pH probes have a very high output impedance (R0 > 1 GΩ), which is in series with the pH voltage source. This is an important parameter, as the input impedance of the circuits interfacing with ion -selective sensors must have an input impedance which is at least three orders of magnitude higher than the electrode resistance [20], sinking minimal current (Iin) to prevent loading. This is because potentiometric cells are galvanostatic cells and no current should flow through the ISE or RE used in the cell . If not, an unwanted potential drop will be created leading to a measurement error: e.g. for a glass electrode with R0 = 1 GΩ and Iin = 200 fA, a voltage offset error of 0.2 mV is generated which is translated into an error of 0.0037 pH [23]. Thus, recording from such electrodes can be achieved with the use of high input impedance (>1014 Ω) and hence low input bias currents (<100 fA) amplifiers . Input currents greater than a pA will disturb the electrochemical potential established. An instrumentation amplifier typically satisfies this condition. Polymer membrane ISEs have a membrane resistance of the order of 1–10 MΩ. Thus, in such a case, the input impedance of the circuit used to interface with the electrode must be larger than 1010 Ω. The signal of interest is at DC. A low-pass filter (LPF) at the output of the recording amplifier reduces the harmonic content of the signal . This is especially important with human body measurements, as physiological signals in this case will act as interference and thus need to be cancelled out, as do motion artifacts.
In [100], the voltage of the pH electrode was first buffered by a high input impedance , low input bias current operation amplifier (op-amp), in order to transform the high output impedance signal to a low impedance signal . Subsequently, the signal was amplified differentially with respect to the RE potential by a gain of 14.7 with an instrumentation amplifier. The IA was used to reduce common-mode interference. An active Butterworth LPF with a cutoff frequency of 1 Hz was used before digitization.
The polymer-based pH sensor demonstrated in [20] was sensitive between pH 2 and pH 9.5. This leads to a change of measured potential of from 400 to −20 mV and from 770 to 320 mV for the two different types of electrodes used. Hence, the signal conditioning circuitry needs a dynamic range of 450 mV. The circuit must be able to adjust the baseline and thus offset the signal such that the lowest potential difference is about 0 V and also to amplify the signal by 10 V/V for a 5 V supply. Because negative potential differences could be generated, a split ±5 V supply was used. In order to offset the signal so that it is within the supply of the subsequent stages (microcontroller), which had a 0–5 V supply, the offset pin of the IA was used to apply a d.c. voltage offset generated through a trimmer.
In [21], the pH electrode is connected to a relaxation oscillator to convert the electrochemical potential into a frequency varying signal . This leads to a varying modulating frequency from 22 to 15.9 kHz for pH levels of 2–12. This system was inductively powered. As discussed in [27], drift can be minimized by decreasing the distance between the sensor and the amplifier, as the parasitics will be reduced . Measurements using a porcine heart were collected in [77] using an amplification of 10 V/V and an LPF with a cutoff of 1 Hz. For a resolution of 0.1 pH units a voltage resolution of 5.9 mV is required for the typical Nernstian response of 59 mV pH −1.
In practice, the readout circuits commonly used for ISFETs are different. An ISFET is an impedance transformer, so the high impedance to low impedance node transformation is by default dealt with. As discussed in previous sections, one approach to examining the pH sensitivity of an ISFET is to consider the pH needed to alter the VT of the device . In order to properly assess this it is evident from the transistor equations that the device needs to be biased at a constant ID and VDS, such that as VT varies, the RE to source potential (VRS) changes by an equal amount to compensate, since ID is constant. VR is constant, thus VS will change and consequently also VD will change, tracking VS to maintain a constant VDS. This is achieved by the circuit of Fig. 2.10a [132]. The current source on the top forces a constant current through the ISFET and the bottom one forces the same current to flow through resistor RDS. The constant voltage across this resistor is forced across the ISFET via the two operation amplifier buffers. This circuit has been used with both with an ISFET and a ReFET and the difference between the two was obtained using an instrumentation amplifier (IA). In [132] the ISFET was operated in the saturation region, while in [192] it operated in the linear region. A variation of this uses the instrumentation amplifier concept as in [131]. Both these circuits can be implemented using discrete component or on-chip solutions. Another classic ISFET circuit is the source-drain follower of [141] (Fig. 2.10b). More advanced circuit techniques implemented in CMOS are out of the scope of this chapter and will not be discussed. The interested reader can refer to the referenced ISFET literature for recent circuit implementations.

2.3 Amperometric Sensors
The development of amperometric sensors focuses on different electrode materials, enzyme immobilization techniques, mediators and coenzymes, sensor stability and lifetime, miniaturization, and implementation. The operation of amperometric sensors is based on applying a potential between two electrodes and measuring the resulting current [193], typically via a third electrode . The working electrode (typically denoted as W or WE), is the electrode that is being characterized or that on which specific processes are characterized. The purpose of the second electrode is to balance the charge added or removed by redox processes taking place at the WE and also to maintain a constant interfacial potential . However, it is challenging to perform both these tasks; thus these are separated between two electrodes, with the task of maintaining a contact interfacial potential being carried out by the reference electrode (R or RE) and separated by charge transfer kinetics, which are dealt with by the third electrode , known as the counter electrode (C or CE) [194, 195]. The latter has typically a much larger area compared to the WE, such that the effect of its interfacial impedance is minimal. No current should pass through the RE if it is to act as a true reference electrode . The most popular amperometric sensors are sensors for the detection of glucose . Such sensors are also the most popular and successful electrochemical sensors in general. As such, the following discussion of amperometric sensors will begin with glucose before expanding to other popular target analytes such as oxygen and lactate .
2.3.1 Glucose
Often glucose concentration is determined using the immobilization of glucose oxidase in a membrane . The current between the electrodes is measured as a function of the oxygen reduction concentration as it diffuses through the bioactive membrane to the cathode as in (2.31) and (2.32).
One approach is to measure the production of H2O2 by applying a potential of +0.65 V versus a Pt electrode . A major drawback is the dependence of the biosensor from the dissolved oxygen concentration. A possible method to overcome this issue is to use mediators playing the role of transfering electrons directly to the electrode . These mediators are important to react quickly with the reduced form of the enzyme and to be sufficiently soluble in both the oxidized and reduced form, such that they diffuse rapidly between the electrode surface and the active site of the enzyme . One very important aspect of using mediators is associated with their solubility; it must be non-toxic if there is a possibility of losing the mediator in the biosensor microenvironment. Some of the requirements are related to the reduced form of the mediator, which should not readily react with oxygen ; also the generation of the oxidized mediator at the electrode surface should be low and independent of pH . Ferrocene is a commonly used mediator. A schematic figure of the different techniques in the development of amperometric biosensors is shown in Fig. 2.11 [196].

Reprinted with permission from [196]. © 2003 from JBCS
Schematic representation of different biosensor generations.
Currently, electrodes are developed so that the electrons are directly removed from the reduced enzymes without the need for mediators. The current (i) is related to the rate of reaction (vA) by:
where n represents the number of electrons transferred, A is the electrode area, and F is the Faraday constant. External membranes are used to achieve a diffusional controlled rate of reaction . The electrical current produced is proportional to the analyte concentration and independent of the electrochemical kinetics.
Roche Diagnostics [197] advanced the realization of redox enzyme -based systems for glucose and lactate . They developed stable, thick films with good adhesion over screen-printed working electrodes using the oxidase enzyme . A biocompatible membrane consisting of a PVC copolymer with vinyl acetate/vinyl maleine was used. Minimization of the electrochemical interferences through measurements at reduced polarized voltages was achieved using an MnO2-carbon ink composite electrode , where oxygen from the enzyme reaction aids in replenishing the substrate such that the linear working range is extended.
In [198], the authors used SiO2 nanoparticles (NPs ) to control the transmembrane potential by using a combination of an enzyme , an electron mediator, and a selectivity membrane barrier. Permeability is one of the important features for sensor performance. In a modeling study [199] it was found that a greater barrier is achieved with layer -by-layer deposition related to a bigger response . The reason is the reduced permeability to H2O2 and its accumulation in the sensing layer .
It was observed that using single-wall CNTs and glucose oxidase entrapped within a polypyrrole layer considerably extends the upper linear range , with detection limits down to the micromolar level [200]. In another case, lowering the polarization voltage was achieved using carboxylated graphene , enabling self-assembly (enzyme prosthetic group with direct electron exchange) of the glucose oxidase.
The pyrroloquinoline quinone (PQQ) cofactor is also used as an electron carrier to a mediator or directly to the polarized electrode , where O2 dependence is not observed. Ruthenium hexamine and osmium complexes are used to construct commercial glucose strips [201]. Direct electron transfer is also achieved by using nanoporous carbon electrodes. The reported PPQ-glucose dehydrogenase enzyme is not as selective as glucose oxidase, but it was modified genetically. FAD-glucose dehydrogenase is modified to give better glucose sensitivity [202].
A nonenzymatic glucose sensor with wide linear range , low detection limit and good selectivity, based on in situ synthesis of Au NPs on reduced graphene oxide (rGO) was reported in [203]. In [204], polymer-enzyme -metallic NP composite films are prepared for glucose and galactose. In [205] perovskite-type oxide La0.88Sr0.12MnO3 (LSMO) nanofibers made by electrospinning and calcination, demonstrated wide linear range , high sensitivity , and a rapid response time.
A xerogel-based amperometric sensor was developed in [206] (Fig. 2.12), which demonstrated a linear range of detection (>24–28 mM glucose ), fast response time, and discrimination against interferences, such as acetaminophen, ascorbic acid (AA), sodium nitrite, oxalic acid, and uric acid.

Schematic of a layered xerogel-based amperometric sensor consisting of enzyme doped, diffusion limiting xerogel layers, semipermeable eletropolymerized polyphenol and a polyurethane outer membrane . Reprinted with permission from [206]. © American Chemical Society 2015
A remotely powered telemetric implantable sensor chip (Fig. 2.13) for subcutaneous applications with integrated electronics was presented in [207]. The potentiostat developed generated an output current which in turn was converted into an output frequency. The signal was used together with a load modulator circuit to transmit the data. The sensor was comprised of a Au counter electrode and an Au working electrode with an enzyme -immobilized layer on top and an Ag RE. The sensors had a glucose detection range of 0–40 mM.

a The telemetrically powered and interrogated implantable continuous blood glucose monitoring system with a three-electrode amperometric glucose sensor and ASIC and the front-end potentiostat electronics implemented as in b using a novel current mirror-based topology. Reprinted with permission from [207]. © IEEE 2009
2.3.2 Commercially Available Implantable Glucose Sensors
Monitoring glucose levels of diabetic patients is an enormous market, which is constantly increasing. As a result, the implantable sensors research field as well as the market is driven by the development of glucose monitoring systems. It comes thus as no surprise that the first FDA-approved and CE-marked commercially available implantable sensing products are for glucose monitoring. Such an approach is particularly important for Type I diabetes patients, who depend on daily insulin injections. These patients require rigorous monitoring of their glucose levels (ideally about every 5 min), as large fluctuations of blood glucose levels take place due to the insulin therapy. The Medtronic Enlite [208], the Dexcom S5 [209], and the Abbott Freestyle Libre [210] (Fig. 2.14) are examples of such commercially available implantable glucose sensing systems. The sensors use glucose oxidase catalyzed oxidation of glucose and are implanted transcutaneously so they do not measure blood glucose directly. Instead they measure the interstitial fluid of the subcutaneous tissue . Nevertheless, prick finger stick tests are still necessary, as they are required to calibrate the implanted sensor. The Enlite recommends 3–4 prick tests a day and the Dexcom at least 2 prick tests a day, while the Libre claims that it does not need any. Nevertheless, according to the product’s website: prick tests are required “during times of rapidly changing glucose levels when interstitial fluid glucose levels may not accurately reflect blood glucose levels, or if hypoglycemia or impending hypoglycemia is reported or the symptoms do not match the system readings” [210]. The Enlite sensor can be implanted for up to 6 days, the Dexcomm for up to 7 days and the Libre for up to 14 days. These sensors can be linked to mobile phone and tablet applications and also have their own devices with which they can communicate if needed to provide the patient with warning and glucose level trends and recommendations. Such implantable systems can be linked with insulin pump systems to essentially create an artificial pancreas, which (depending on the implant sensor readings) can deliver the necessary amount of insulin immediately when needed. One example of such a system is the Medtronic MiniMed 530G pump, which works together with the Medtronic Enlite sensor. The system provides patients with a 30 min warning as to whether their glucose levels are going up or down. Information is updated every 5 min to provide a real-time and realistic image of the trends and the insulin delivery is controlled according to the sensor readings and preset settings.

2.3.3 Lactate
Lactate is another important analyte . Lactic acid production can be increased in the case of haemorrhagic shock or pulmonary embolism [211] related to anaerobic metabolism. Increased levels of lactate can be observed in pathological conditions such as respiratory failure [211], liver disease [212], endotoxin [213] or cardiogenic [214] shocks, and tissue hypoxia [215]. Lactate levels can increase and lead to acidosis in the event of insufficient oxygen supply during childbirth. Hypoxic/acidotic distress in fetuses during labour [216] has been detected from fetal scalp blood measurements of lactate and pH . The possibility of developing metastasis in cancer and tumour cells is associated with high levels of lactate [217]. Acidification around the cancer cells and tumours is related to lactate levels because of changes in the ratio of lactate production to lactate consumption [218]. Lactate is related to brain metabolism and plays an important role in brain tissue , [219] and its measurement would provide important information regarding stroke and head trauma [220, 221]. It was demonstrated in [222] that the extracellular lactate level is related to sleep: it is increases while awake and decreases during sleep. Commonly used enzymes for lactate sensors are lactate oxidase and lactate dehydrogenase. It is very important to have a high transport rate of oxygen versus substrate so as to maximize the linear operational range . A study [223] showed that oxygen concentration in the enzyme layer is minimally changed when compared with the bulk solution. The operating voltage was reduced when using a cobalt phthalocyanine as an electron mediator [224] on modified screen-printed electrodes. Stabilization of the enzyme can be achieved by crosslinking . Enhanced enzyme stability when mucin is employed in the crosslinking has been reported [225]. Immobilized lactate oxidase on a 3D macroporous Au construct showed that electrode architecture and surface area are important [226].
A possible way to increase the sensitivity of enzymatic biosensors is to increase the surface area using nanomaterials such Au NPs [227], Pt NPs [228, 229], CNTs [230, 231], NPs of metal oxides such as molybdenum oxide [232] or niobium oxide [233] and NPs of semiconducting materials like zinc oxide [234]. An alternative is to use a bioenzyme system, where for example horseradish peroxidase is combined with lactate oxidase [235]. For long-term use, enzyme stability is critical. There are different ways to achieve this, such as crosslinking [236, 237], entrapment in conductive or nonconductive polymers [236, 238], entrapment in sol-gel [239, 240], and immobilization with another biological component, such as bovine serum albumin and covalent attachment of the enzyme to the electrode [241]. Leakage can be prevented by using a combination of a sol-gel matrix and covalent attachment of the enzyme [242], where the carboxylic group of lactate dehydrogenase is covalently linked to the amino group in the organoalkoxysilane precursor . Higher sensitivity and stability are reported based on molecular interactions with chitosan [243]. A number of different approaches have been investigated to reduce the interfering species, one of which involves the use of a mediator, such as phenazine methosulfate [244]. Alternatively, protection and reduction of interference can be achieved by the use of permselective membranes [245, 246] or multilayer polymers [247, 248]. The membranes will serve as a barrier and also as an antifouling component [249]. Such approaches also aid in increasing the linear response of the sensor. Nafion is one such commonly used polymer [223, 250,251,252].
Thanks to improvements in sensitivity , portability and low detection limits a number of commercial electrochemical lactate sensors are available [223]. Implantable sensors sampling interstitial fluids are less invasive than those sampling blood. Nevertheless, this is still an invasive process. It has been found that lactate levels in tear fluid correlate well with the blood lactate level, which, provided there is a safe interface between the eye and the sensor, has the potential of providing completely non-invasive measurements [223].
L-lactate oxidase catalyses the oxidation of L-lactate to pyruvate in the presence of dissolved oxygen . The hydrogen peroxide formed is electrochemically active and the produced current is proportional to the L-lactate concentration as in
The electrons are transferred in the enzyme structure through a FAD cofactor. Because of the oxygen participation in the reaction and the related difficulties of additional methods to detect the oxygen level, another approach was investigated. This involved the use of L-lactate dehydrogenase instead of L-lactate oxidase. This type of enzyme needs a coenzyme (NADH or NADPH):
The coenzyme acts as a mediator and transports the electrons between the electrode surface and the enzyme . However, the use of coenzymes in vivo is related to challenges associated with high oxidation potential and an additional need for optimization [233].
2.3.4 Oxygen
Physiological oxygenation is fundamental to tissue metabolism, providing important information regarding the oxygen supply and local tissue oxygen uptake. Oxygen partial pressure is the standard measure of the oxygenation state. The common pathway of oxygen supply is from the arterial circulation via cellular and extracellular capillaries through diffusive transport in the interstitial tissue space to individual cells. Hb saturation measurement using pulse oximetry has proved a valuable, non-invasive measurement of the oxygenation state, but it is focused on the arterial compartment.
The Clark oxygen electrode is well known, where reduction of oxygen is detected on Pt surface as in (2.37) and (2.38). The applied potential is −0.65 V versus Ag/AgCl RE and the detected current is proportional to the oxygen concentration. Both electrodes are separated from the solution by an oxygen -permeable plastic membrane such as polytetrafluoroethylene or Teflon.
The rate of the electrochemical reaction depends on the diffusion rate of oxygen from the bulk of the solution, which is related to the concentration gradient and bulk oxygen concentration. Oxygen electrodes measure the rate of the process, that is far from equilibrium (in contrast to ISEs which measure close to equilibrium conditions), because they are more sensitive to temperature, as discussed in [193].
The Clark oxygen electrode has addressed the major challenges of biofouling, selectivity and sample convection through the use of a hydrophobic gas permeable membrane with an internal electrolyte film for stable electrochemistry. However, miniaturization of this type of electrode is challenging. This is mainly due to the problem of bonding and integrating the hydrophobic gas membrane with the body of the device , where the internal electrolyte can be removed during the sterilization process. A microfabricated version containing three electrodes, a glass substrate with a fluorinated ethylene propylene oxygen -permeable membrane , and a PDMS (polydimethylsiloxane) tank used for storing solution has been described [253]. Miniaturization using a glass capillary platform has been achieved [254]; however, due to the fragility of the glass used these are not suitable for in vivo use. Other polarographic electrodes, e.g. measuring current that is generated when pair a cathode and anode pair are immersed in an electrolyte solution [255], have been described, where oxygen diffuses through a polyethylene wall into the electrolyte solution, but relative long response times are observed here. A commercial variant of the Clark electrode has been produced for brain monitoring [256]. The sensor is based on an O2 permeable polypropylene tube, rather than a planar membrane , to house the inner electrolyte solution.
Various experimental physiological studies have been conducted using Clark electrode variants. Muscle tissue has been monitored to assess treatment in septic shock, as well as human head and neck solid cancer tissue . The Eppendorf electrode consists of a central Au and Pt wire encased in glass [257] with the overall sensing electrode constructed within a protective metal needle of 300 µm o.d. Another Licox 0.8 mm tip diameter electrode has been used to monitor pO2 of central retinal vein occlusion during vitrectomy [258, 259].
A non-membrane alternative for protecting the electrochemical tip surface is to incorporate it into a recess. Recessed polarographic cathode Au microelectrodes have been reported in [260]. The construct used was a metal alloy within a glass micropipette; the device is quite complicated and in vivo use would be difficult because of likely damage during percutaneous insertion. Additional problems with the etched needle microelectrode design include hydration and weakening of the glass insulation over time and membrane disruption during insertion [261].
To limit the diffusion of oxygen and control the oxygen consumption, a p(HEMA)-based hydrogel was used. The constructed sensor was used in combination with other sensors for detection of analytes in tissue brain . In [262], a sensor for oxygen detection in the brain using carbon paste electrodes was presented. Such oxygen sensors have the advantage of being less prone to surface poisoning, thus requiring (in contrast to metal -based sensors) no additional protective membranes. The sensors were fabricated by using Teflon-covered silver wires and carbon paste prepared with graphite powder and silicone oil. Electrochemical reduction of O2 took place at a potential of −650 mV and the sensor demonstrated a sensitivity of −1.49 nA µM−1. In vivo experiments in rat brains for monitoring tissue O2 in the brain’s extracellular fluid, where hypoxia and hyperoxia were artificially induced, validated the sensor operation. The sensor performance demonstrated no sensitivity to pH , temperature, or flow and an in vivo stability over 12 weeks.
2.3.5 Oxygen Dependence
The majority of amperometric sensors depend on oxidase enzymes which catalyze analyte oxidation to form hydrogen peroxide, which is in turn amperometrically recorded at the working electrode . This transduction process in most oxidase enzymes depends on the local oxygen concentration [1]. In implantable applications this is an important sensor failure parameter, as the dissolved body oxygen is significantly lower than that of target analytes. Exogenous environmental conditions (e.g. anesthesia), endogenous physiological responses (e.g. due to physical exercise) and local inflammatory response during implantation, reduce oxygen levels further. The latter is a significant source of oxygen exhaustion [263]. This will be an issue when ischemic events are studied, as by definition tissue oxygen levels are low. These will affect the accuracy of the sensor, as oxygen inadequacy will saturate the sensor at high concentrations of the analyte [1]. The concentrations of glucose and lactate in the brain and in the interstitial fluid of the transcutaneous tissue are high and in the mM range . According to [264], tissue oxygen concentration is an order of magnitude lower than glucose .
Many techniques have been proposed which aim at decreasing the dependence of the transduction process on the local oxygen concentration and can be separated into three generations of sensors Fig. 2.11. Second-generation sensors decrease oxygen dependence by means of synthetic redox mediators to compete with oxygen concentration. In third-generation sensors, the redox cofactor of the enzyme is bound directly to the working electrode to re-reduce or re-oxidize the enzyme . According to [1], the second and third generations have never been tested in vivo due to toxicity concerns. In addition, the need for additional outer membranes to reduce the risk of biofouling and leaching makes first-generation sensors more appealing. This family of sensors uses such membranes in order to reduce the flux of the target analyte to the sensor enzyme layer by, for example, 100, while at the same time the flux of oxygen is effectively unaffected, leading to an enzyme layer rich in oxygen . An additional advantage of this is that the sensor’s response time is no longer dependent on the kinetics of the enzymatic reaction , which is temperature and concentration dependent, but rather by mass transfer , i.e. the influx of the target analyte into the sensor’s enzyme layer [1, 263]. While these analyte flux-limiting membranes address the issue with oxygen , they lead to decreased sensor sensitivity and an increased response time [1, 263].
2.3.6 Multi-parametric Amperometric Systems
Guiseppi-Elli et al. have presented a number of amperometric systems for implantable applications [265,266,267,268]. In [265] the authors present a 2 × 4 × 0.5 mm chip with two Pt working electrodes (Fig. 2.15a). Each working electrode is covered by a passivation layer , which has a circular opening to form Pt microdiscs. This minimizes the diffusional limitation associated with enzyme kinetics [265]. Each working electrode is complemented by a large Pt counter electrode while a single Ag/AgCl electrode works as a common RE to both electrode sensing sets. A 1 µm thick layer was deposited on the working electrode , comprised of “tetraethylene glycol (TEGDA) cross-linked poly(2-hydroxyethyl methacrylate) that also contained a derivatized polypyrrole component and a biomimetic methacrylate component with pendant phosphorylcholine groups” [265], providing interference screening and in vivo biocompatibility , and used to immobilize glucose oxidase and lactate oxidase to create glucose and lactate sensors. The glucose sensor had a linear range of 0.1–13 mM and the lactate sensor up to 90 mM. The lactate linear range sensing response was 30-fold greater than that obtained from a sensor without the microdisc passivation openings. The sensors retained 80% of their initial performance after 5 days of continuous measurements in vitro. A similar topology was presented in [266, 267], with the relevant potentiostat electronics (Fig. 2.15b) in [266] and its use in lactate and glucose sensing in [267]. The same electrode array was used in [268, 269] together with a commercially available wireless potentiostat from Pinnacle Technology, Inc. [270] designed for tethered implantable in vivo central nervous system (CNS) applications in rats (Fig. 2.15c). The authors focused in [269] on trauma-induced hemorrhage applications and the system was validated in vivo with a hemorrhage model, where tissue lactate rose more rapidly. Immobilized on each working electrode were glucose oxidase or lactate oxidase within a biorecognition layer with a thickness of 1.0–5.0 μm. This was a 3 mol% tetraethyleneglycol diacrylate cross-linked p(HEMA-co-PEGMA-co-HMMA-co-SPA)-p(Py-co-PyBA) electroconductive hydrogel. The device was subsequently coated with a bioactive hydrogel layer containing phosphoryl choline and polyethylene glycol pendant moieties [p(HEMA-co-PEGMA-co-HMMA-co-MPC)] to increase the biocompatibility of the implanted sensor platform. The sensors demonstrated linear ranges of 0.1–13 mM concentration range for glucose and 1–7 mM for lactate with response times of 50 and 35–30 s respectively. Extensive in vitro experimentation demonstrated the biocompatibility of the implanted sensors, while in vivo experiments of up to two weeks demonstrated only a thin band of encapsulation and reduced inflammation of poly(2-hydroxyethyl methacrylate) (p(HEMA)) hydrogels containing phosphorylchlorine in contrast to unmodified p(HEMA).

a The three-electrode amperometric sensing device with two sets of Pt CE and micropore WEs and a common Ag/AgCl RE. Reprinted with permission from [265]. © IEEE 2005. b Amperometric sensors. Reprinted with permission from [266]. © Springer 2008. Each amperometric sensing system has its own RE, which are, however, shorted together. Similarly to (a), these are microdisc WEs; in this case there are 37 microdisc openings in the passivation 50 µm in diameter and with a pitch of 100 µm. c Lactate and glucose measurements in the trapezius muscle of a rat via sensors similar to those of (b) and with a head-mounted wireless dual channel potentiostat. Reprinted with permission from [268]. © Springer 2010
A multisite microelectrode array for in vivo measurements was presented in [252] (Fig. 2.16a) where the authors demonstrated its use for monitoring rapid endogenous changes in brain tissue . The polyimide platform of Fig. 2.16b was used for detection of glucose , lactate , glutamate , and O2 in soft tissues, such as muscles or abdominal tissues and also brain tissue [271].

a Ceramic-based multisite microelectrode with 50 µm × 150 µm recording sites. The various sensor layers are shown on the right. Reprinted from [252], © 2005, with permission from Elsevier. b Cross-sectional view of the multi-layer structure, which is composed of polyimidesubstrate, deposited Pt electrodes, insulation from spin-coated epoxy and a laminated dry-film resist. All electrodes are modified with hydrogel membranes. Reprinted with permission from [271]. © IEEE 2014
Amperometric sensors are also applicable to experimental neuroscience applications, where for example researchers are interested in the CNS [262, 272, 273] and where microdialysis systems lack the temporal resolution necessary for the rapid changes in brain metabolism [272]. The brain requires up to 25% of total glucose consumption, being the main energy substrate for glial cells and neurons, with lactate and pyruvate following at a smaller extent in the adult brain [272]. Disturbances in the regulation of the brain energy metabolism are associated with learning and memory impairments and neuropathologies such as epilepsy, meningitis, and ischemia [272]. Brain glucose and lactate concentrations are typically between 1–2 mM and pyruvate in the region of 200 µM. An implantable system for amperometric detection of glucose , lactate , and pyruvate using needle Pt electrodes (Fig. 2.17) was presented in [272]. Electrodes were coated via cyclic voltammetry with a permselective membrane (PmPD). These were then coated manually with a hydrogel of either GOx, LOx, or POx, cross-linked with glutaraldehyde (GA) and bovine serum albumin (BSA). An electrode with just the PmPD layer was also included to record the background (BG). All sensors demonstrated limits of detection (LOD) lower than 5 µM. The lactate sensor used, was loaded by a 0.4 U µL−1 of LOx and had an additional external layer of polyacrylonitrile (PAN) and demonstrated a LOD of 0.86 µM, a linear range of 1.13 mM and a sensitivity of 241.14 nA mM−1. The system was tested in vivo with rats to monitor the levels of these markers in their brains during induced hypo- and hyper-glycaemia, with all sensors retaining high sensitivity and selectivity after implantation. The lactate sensors were the least affected by the in vivo environment due to the additional PAN layer . Similarly in [273], carbon and Pt-based sensors were used for the amperometric detection of AA, O2 and glucose in the striatum of untethered, freely moving rats using a biotelemetric device . One of the most important low molecular weight antioxidants in the brain is vitamin AA. Electrodes at a mild anodic potential (+120 mV) can detect AA amperometrically. The AA sensors were fabricated using a graphite-loaded epoxy which was patterned with a drill to form a needle with a tip diameter of 25 µm. The sensor demonstrated a sensitivity of 7.3 pA µM−1. The O2 sensor was fabricated similarly to the AA sensors, with an additional surface treatment of cellulose nitrate. The O2 sensors achieved a 213 pA µM−1 sensitivity . The glucose sensor was fabricated using a Teflon-covered Pt wire. GOx was deposited on the Pt and poly(ortho-phenylenediamine) (p-OPD) was electrosynthesized on the sensor against an Ag/AgCl reference and H2O2 electro-oxidation was subsequently carried out, finalizing the glucose sensor fabrication. The sensor demonstrated a linear region with a slope of 15.2 nA mM−1 at concentrations between 0 and 2 mM.

The multisensory amperometric platform developed. Reprinted with permission from [272], © Elsevier 2015
2.3.7 Voltammetry
Voltammetry is an analytical method that, similarly to amperometry , measures the current through an electrochemical cell under an applied potential . The difference is that while in amperometry the applied potential is constant, in voltammetry the applied potential changes linearly over time. This is known as linear sweep voltammetry (LSV). The linear rate of change of the applied potential can vary from a few µV s−1 to mV s−1 and it is known as the scan rate . The sensitivity of the measurement can be improved by increasing the scan rate , as more redox reactions will take place per second on the WE. For example, the peak current may be proportional to the square root of the scan rate . As in amperometry , a three-electrode system is typically used. The concentration of the material undergoing the redox reaction and the faradaic current are according to
where A is the area of the electrode involved in the reaction , D is the diffusion coefficient, and C(x,t) is a function of concentration of the analyte at a distance from the electrode at time t. During the measurements, redox reactions must occur at both the WE and CE. In most cases, the WE is a polarizable electrode (i.e. from a noble metal , such as Au or Pt, or carbon-based materials, such as glassy carbon and graphite), which must also be electrochemically inert, unless it is the electrode itself that is to be characterized. The voltage drop across the double layer provides the energy for the electron transfer . Considering a generic redox reaction , then:
where O is the oxidized form, R is the reduced form of the analyte , and n is the number of electrons (e−) exchanged in the reaction . When equilibrium is achieved, the potential difference, EM, between the solution and the electrode depends on electrode material and the concentration of the analyte . The potential is the Nernst potential and given by the Nernst equation :
where E0 is the standard potential of the reaction , R is the molar gas constant (8.3145 J Kmol−1), T is temperature, n is the number of electrons exchanged in the reaction , F is Faraday’s constant (9.6485 × 104 C mol−1), C0 is the concentration of the oxidized form, and CR is the concentration of the reduced form [274]. In more concentrated solutions the concentrations CR and C0 must be replaced by chemical activities . For solid phase reactants at T = 298 K, the Nernst equation becomes:
For a normal hydrogen electrode , E0 is defined to be 0 V. When the cell is away from equilibrium:
Different parameters control the rate of the reaction , such as the applied potential between the working and counter electrodes, the mass transfer to the surface of the electrode and of the product, adsorption of ions on the electrode surface, and the electron kinetics of the redox reaction . The Cottrell equation is used for quantiative analysis and to measure the electrode area, where the Ipa is related to the concentration of the measured redox molecule [275]
where n is the number of electrons interchanged on the redox reaction , F is the Faraday constant (C/mol), A is the area of the working electrode (cm2), C is the concentration (mol/cm3), D is the diffusion coefficient of the redox species (cm2/s), and t is time (s).
There are many variations of this electroanalytical technique that fall under the term “voltammetry ”. Different voltammetric techniques arise, depending on how the applied potential changes. Thus, in addition to LSV, techniques include staircase voltammetry , square wave voltammetry (SWV), differential pulse voltammetry , cyclic voltammetry (CV), and fast-scan cyclic voltammetry (FSCV). Staircase voltammetry is a variation of LSV, where, as the name suggests, the applied voltage is increased in steps. Measurements are recorded at the end of each step. This allows enough time for the signal to settle, in order to minimize contributions from capacitive charging (i.e. non-faradaic currents). A variation of these methods is SWV, which is a type of differential pulse voltammetry , where the pulse width is 50% of the cycle time of the square wave used. Basically, a square wave stimulus is superimposed on a linear staircase waveform. Similarly to staircase voltammetry , non-faradaic current contributions are minimized. Measurements are performed at the end of the forward and reverse half cycles of the stimulus. The two recorded currents are similar to those obtained from CV, as will be explained subsequently. However, in differential pulse voltammetry , the difference of these currents is plotted instead. Peaks and troughs are associated with redox reactions taking place at specific potentials and the magnitude of the current is proportional to the concentration of the redox active species.
CV is an extension of LSV where the voltage is immediately swept back at the same rate instead of sweeping the voltage in one direction, as in Fig. 2.18a. A typical cyclic voltammogram is shown in Fig. 2.18b. CV has several advantages; the reverse sweep gives additional information regarding the reactions taking place within the electrochemical cell . These include whether the reaction is reversible (in which case the reverse sweep converts the material back to its initial form), information about the transfer kinetics of the reaction , and the intermediate products of the reaction [274, 276, 277]. As shown in Fig. 2.18b, troughs and peaks in the recorded current response of a typical CV voltammogram correspond to the potential at which oxidation or reduction of species takes place, respectively. The peak currents increase with the increasing concentration of the target analyte and with increasing scan rate if the concentration is kept constant. FSCV is used as a term to described measurements undertaken with scan rates in the mV s−1 region. Such high scan rates are used to perform rapid measurements for high temporal resolution. As such, FSCV is useful for in vivo measurements for the detection of neurotransmitters, metabolites and hormones. This is because at these scan rates, FSCV can perform voltammetric measurements at ~100 ms which is on the same timescale as that of neurotransmission, thus enabling real-time measurements of neurochemical dynamics. Since a cyclic voltammogram is obtained, FSCV offers greater specificity , as the chemical signatures of different species at different potentials can be identified. In such applications, carbon fiber microelectrodes are typically used and since each species of interest will have unique redox potentials, a high specificity is achieved. Nevertheless, the applied potential must not exceed 1.23 V, where water electrolysis takes place. On the same time, the current flowing though the cell , which is a function of the applied potential and the electrode tissue interface impedances, among others, must also remain low to avoid cell lysis and depolarization.

a Black: LSV, blue: staircase voltammetry , green: SWV stimulus waveforms. b CV stimulus waveform
From the above, we can see that voltammetry provides us with important information regarding the kinetic and thermodynamic parameters of electron-transfer events. In more detail, CV is typically used for the characterization of an electrochemical system and provides information regarding electron stoichiometry, diffusion coefficients, reversibility, and the redox potential of the process being examined. Identifying the latter allows one to properly perform amperometric measurements. The two techniques, in terms of signals applied and measured, are similar in that the current is measured while a voltage is applied, but in amperometry the applied voltage is a constant d.c. potential , while in cyclic voltammetry it is a triangular waveform. Thus, the same front-end electronics can be used in both applications.
An important implantable application in which CV and amperometry are used is in neurotransmitter sensing , where they can be combined with traditional electrical action potential recordings for neuroprosthetics applications and basic neuroscience research.
2.3.8 Neurotransmitter Sensing
The majority of the communication in the nervous system is via electrical impulses. Many systems have been developed for the recording of these for various implantable and benchtop/basic research applications [278]. An example is shown in Fig. 2.19a, b. Communication between neurons is performed at sites known as synapses. These allow a neuron to transfer information to another neuron. At a synapse the plasma membranes of the two neurons are in close proximity, and various molecular machinery link the two membranes together for the purposes of the signaling process. The neuron transferring the signal is known as the presynaptic neuron (the transmitter in electronic engineering terms) and the target neuron is known as the postsynaptic cell (i.e. the receiver). This is illustrated in Fig. 2.19c. There are two types of synapses, electrical and chemical. In the former, cell membranes are connected via gap junctions. These are conductive channels capable of passing electrical current. In chemical synapses, however, there are no conductive pathways, and communication is established via the release or uptake of chemicals known as neurotransmitters. Neurotransmitters play the role of chemical messengers which send the signal between neurons and other cells. The activation of voltage-gated calcium channels allows the conversion of electrical activity in the presynaptic neuron into the release of excitatory or inhibitory neurotransmitters into the synapse. Thus, electrical and chemical neural activities are tightly coupled together [279]. The released neurotransmitter diffuses towards the postsynaptic neuron and binds to specific protein receptors in the plasma membrane of the postsynaptic cell . This causes excitatory or inhibitory postsynaptic potentials. These are integrated by the neuron and lead to physiological changes which will determine whether an action potential will be fired by the postsynaptic neuron.

Examples of multichannel CMOS ASICs for the measurement of action potentials from electrogenic cells of a brain slices and b neuronal cell cultures in vitro and directly on the ASIC via on-chip electrode arrays and recording electronics. Reprinted with permission from [278]. © IEEE 2011. c Neurochemical signaling between two neurons at a synapse. Reprinted with permission from [279]. © IEEE 2009
Neurotransmitters play a very important role in controlling behavioral and physiological conditions. They are involved in the processes of sleep, learning, appetite, and memory [280]. Detection and monitoring of neurotransmitters, such as nitric oxide , glutamate , gamma-aminobutyric acid(γ-aminobutyric acid) (GABA), acetylcholine, norepinephrine, and dopamine , is extremely important for research in the neurochemistry of the nervous system as it can reveal how information is transmitted and processed. Recording these at different areas of the brain would help us understand further the neurophysiology of sensory-motor systems and encoding in the auditory nerve and visual cortex [281]. This would be also important since various neuropathologies, such as Parkinson’s disease , epilepsy, schizophrenia, substance addiction, and stroke are associated with neurotransmitters, while it may lead to advances also in neural prostheses (closed-loop systems), artificial limbs, brain -computer interfaces (BCI), and tissue engineering [282].
Electroactive monoamines such as serotonin, norepinephrine, epinephrine, and dopamine are of interest. Because these neurotransmitters are redox active , they can be measured by electrochemical methods at their oxidation or reduction potential . In the brain other electroactive neurochemicals are present, which can interfere with the detection of neurotransmitters. Common inteferants include AA and uric acid. The extracellular concentration of neurotransmitters is low and their concentrations can change rapidly, which is why their in vivo measurement is challenging [283]. Optical and chromatographic techniques, positron emission tomography (PET), and single photon emission computerized tomography (SPECT) have been used for the detection and analysis of neurotransmitters. These are, however, large, high-power, complicated, and expensive techniques, most capable of detecting the reaction products of neurotransmitters, with low quantitative, temporal, and spatial resolution [272, 282].
Depending on the target analyte , highly sensitive front-end sensor recording electronics (potentiostats) with current measuring capabilities ranging from fA to mA may be required; thus the recording electronics need to have a wide dynamic range . At the same time, the sensors themselves must demonstrate fast response times and low hysteresis . As mentioned previously, in a typical amperometric /voltammetric cell there are three electrodes. No current should flow through the RE, to prevent an ohmic drop forming across the solution which would influence the potential between reference and working electrodes. However, in most neurotransmitter detection applications, the currents can be very low, leading to negligible ohmic drops across the solution. Hence, reference and counter electrode terminals can be short-circuited and two electrodes can thus be used instead of three [281, 282]. Thakor et al. have developed a number of CMOS-based integrated potentiostats for implantable neurochemical sensing and neuroprosthetics [279, 281, 282, 284]. A two-electrode system was similarly used in [285]. The telemetrically operated system was used to record extracellular levels of dopamine in the caudate-putamen of an anesthetized rat, elicited via electrical stimulation of the medial forebrain bundle. The system was capable of performing both chrono-amperometric measurements and fast-scan cyclic voltammetry (FSCV). CMOS on-chip recordings of transmitter release were presented in [286], via post-processing of the chips such that aluminum electrodes are converted into Ti/Au electrodes. A 96 channel potentiostat with on-chip Au electrodes and microfluidics (Fig. 2.20a–c) incorporated on the CMOS die was demonstrated in [287]. Flat 2D electrodes (Fig. 2.20a) were developed on chip for neural culture measurements, while 3D electrode spikes (Fig. 2.20b) were developed for brain slice measurements. An amperometric CMOS ASIC was presented in [288] together with a microfabricated multi-electrode platform (Fig. 2.20d–f) for neurotransmitter sensing . The Au working electrodes of the array (Fig. 2.20d) were functionalized with additional layers for the detection of dopamine and glutamate (Fig. 2.20e, f). Since dopamine is electroactive, a MWCNT-loaded Nafion layer is sufficient for its detection at the appropriate redox potential . AA and uric acid have similar redox potentials and this layer increases the specificity to dopamine . Glutamate is not electroactive; thus, an enzymatic approach was implemented with the use of glutamate dehydrogenase (GLDH) enzyme that produces electroactive nicotinamide adenine dinucleotide (NADH). A composite film was developed with MWCNT, chitosan (CHIT), meldola’s blue (MDB), and GLDH. An automatic spotting machine was used to dropcast these membranes onto the Au electrodes.

a On-chip 2D and b 3D Au electrodes for neural cell and brain slices, respectively, c neurotransmitter sensing and microfluidic chamber on the CMOS die. Reprinted with permission from [287]. © IEEE 2013. d The sensors with five Au WEs, common Pt RE and Au CE. e The Au electrodes are covered with CNT-based films . Electrodes 1, 3, and 5 with MWCNT-loaded Nafion and electrode 2 and 4 with MDB-CHIT-MWCNT-GLDH. f Schematic sideviews of the glutamate and dopamine sensors. Reprinted with permission from [288]. © IEEE 2016
Some of the most used electrochemical sensors for neurotransmitter analysis are carbon-based, because it has been shown that the surface oxides facilitate the electrostatic interactions [289] and since carbon biocompatibility nanomaterials with enhanced electrochemical properties [290]. The nanostructured surface is related with larger specific surface area, increased adsorption capabilities, and enhanced electro catalytic activity. For neurotransmitter detection the use of the CNT-based electrodes is related to increased sensitivity and selectivity rapid electron transfer and reduced electrode fouling [291]. New carbon-hybrid materials like CNTs or carbon nanofibers directly on tetrahedral amorphous carbon have also been proposed. These demonstrated high sensitivity and selectivity in the presence of AA [292]. The authors discussed the possibility of non-enzymatic detection of glutamate at physiological pH .
Similarly to glutamate , lactate is also an electrochemically inactive molecule. Specific enzymes to generate redox active molecules can be used to determine it. Glutamate measurements with a high sensitivity of 1.95 nA mm−2 µM−1 were demonstrated in [271], with a linear range of up to 100 µM. It was discussed that using glutaraldehyde for cross-linking results in a thinner membrane and that a higher concentration of enzyme could be immobilized. This increased the sensitivity , but reduced the linear working range and long-term stability. The sensor was used in measurements of extracellular concentration in the brain over 3 days. Acetylcholine (ACh) is another important neurotransmitter . Choline (Ch) is a neurotransmitter at nicotinic receptor sites [293]. Detection of Ch is done by using detectable peroxidase through the use of choline oxidase [294, 295]. Determination of ACh is connected with conversion of Ch, which leads to peroxide in two steps.
Determination of ACh needs to be corrected for the endogenous Ch, which means that two sensors are needed, one for total ACh + Ch and one for Ch. Some sensors are reported to measure neurotransmitters released in individual tissues, such as the hippocampus [296, 297]. Multielectrode arrays of planar CNTs on indium tin oxide microelectrodes have also been proposed for long-term recording at nM concentrations of dopamine and changes in the extracellular chemical microenvironment in mouse striatal brain slices [297]. Some issues are related to non-specific adsorption and reduced tissue reaction and also interference from electroactive molecules in the physiological environment. There is thus a need for the design and development of novel approaches and materials for selective membranes.
Implantable neural signal recording for neuroprosthetic applications and diagnostics is a large research area that has drawn considerable attention in recent years. Such devices are used in both the CNS and the peripheral nervous system (PNS). At the core of these technologies lie the electrodes necessary for recording action potentials and for electrical stimulation. Pt and Au, conducting polymers (e.g. PEDOT), CNTs, and graphene are some of the materials currently being used for the electrodes. A discussion on the technology of these is out of the scope of this chapter; the reader is referred to [298] on recent advances on flexible neural interfaces using organic and inorganic materials.
2.3.9 Read-Out Circuits for Amperometric and Voltammetric Sensors
As mentioned earlier, three-electrode systems are typically used in amperometric applications. For steady state amperometry a stable voltage source is required that can respond rapidly to current loading of many orders of magnitude. The circuit needs to ensure that the cell potential is kept constant with respect to the RE, and feedback is used to achieve this irrespective of any changes in the cell impedance . In other words, the cell potential , i.e. the potential difference between WE and RE, needs to remain constant [195], and this is done by controlling the cell current (Icell) [299]. The basic setup of the driving circuit of a potentiostat is shown in Fig. 2.21a. The equivalent circuit of the three electrode electrochemical cell is shown in (b) [299]. RCE, RWE, and RRE are the interfacial charge transfer resistances at the CE/solution, WE/solution, and RE/solution interfaces, respectively. CCE and CWE are the double layer capacitances at those interfaces. RS1 is the solution resistance between CE and RE and RS2 is the solution resistance between RE and WE. As can be seen, a voltage signal is applied to the CE and the circuit also needs to supply the required current (Icell) for the electrochemical reaction being studied. This is so that the CE can provide sufficient current to compensate for the redox reaction currents at the WE. The circuits of Fig. 2.21 thus compensate for the potential drops across the impedances along the CE-RE path [195]. Placing the RE close to the WE minimizes potential errors due to the voltage drop across RS2, which is the only uncompensated impedance [195]. If the CE is large enough compared to the WE, then the effects of its interfacial impedance on the circuit operation and redox processes taking place there are minimized and can be ignored. As mentioned earlier, no current should flow through the RE, to avoid RE polarization , which would influence its potential , which should be constant. To achieve this, the input impedance of the amplifier in Fig. 2.21a should be very large and its input current should be minimal. Depending on the application, a number of other design parameters may become important. These include output voltage swing, input referred offset and noise, gain, bandwidth, and slew rate. The circuit of Fig. 2.21c is a common potentiostat configuration. A2, A3, and A4 are unity gain buffers used to isolate the electrochemical cell from the rest of the circuit, and R1 and R2 are summing resistors, with a low resistance in the range of 15 kΩ to minimize thermal noise [195].

a The basic grounded WE topology, b The equivalent circuit of a three-electrode electrochemical cell [300], c The same topology with buffers for isolating the reference electrode and a TIA for recording of the current through the cell . d Recording the current through a current-sense resistor with an instrumentation amplifier (IA)
In the circuits of Fig. 2.21, there is a current flow (Icell) from the CE to the WE, which needs to be recorded. This is typically done by converting this into a voltage, which is linearly proportional to Icell, by a transimpedance amplifier (TIA), implemented in Fig. 2.21c by A5, R3, and C. This comes with the benefit of holding the working electrode at virtual ground, measuring with reference to ground and measuring small currents by switching between different values of feedback resistors. The output voltage of the TIA (Vout-TIA) is given by
In order for A5 to provide a good virtual ground, the input-referred offset voltage and noise current need to be minimized. The latter is particularly important as the electrode dimensions are reduced . This is because electrode impedance increases and the current being recorded may be very low (fA levels). In these cases active shielding is necessary and current follower circuits are advantageous, as leakage currents to ground are minimized. In addition, since Icell must all flow through R3, the input bias current of the amplifier should be minimized and the input impedance maximized. The input impedance of a TIA is low at low frequencies, thus behaving inductively. At higher frequencies it increases and is in series with the impedance of the electrochemical cell , which, as shown in Fig. 2.21b, is mainly capacitive. This may lead to instabilities in the feedback circuit [299]. The feedback capacitor, C, is used to stabilize the amplifier by reducing the bandwidth. The input impedance issue on both A4 and A5 is particularly important when measuring very low currents, which will require large amplifications via a large R3. As discussed in [299], in single-supply systems the non-inverting terminal of TIAs must be biased at an appropriate level to allow all voltages to lie within the power supply range.
The current can also be recorded by adding a resistor between the driving circuit output and the CE or between WE and ground and measuring the potential drop across the resistor. The former is preferable, as the direct connection of the WE to ground ensures that its potential is constant. Also, because this is a true ground connection, as opposed to that in the TIA circuits , this topology is insensitive to noise and EMI. An instrumentation amplifier (IA) is used as in Fig. 2.21d to measure the potential difference between the two terminals of the resistor. In addition to input impedance , input bias current, and offsets, which are once again important, resistor tolerance and resistor thermal noise must also be considered. Because there are no active components in the feedback path, this topology is more stable, while both voltage and current are measured with respect to ground [299]. A disadvantage of this topology, is that R is part of the feedback loop, which limits the voltage compliance across the cell [195, 299]. When connecting the resistor between WE and ground, the recorded voltage needs to be fed back to the drive circuit to ensure that the WE potential is constant as discussed in [299].
It should also be noted that apart from the circuits described above, which consider only the scenario of having a grounded WE, other topologies also exist, where the CE can be grounded as discussed briefly in [299] and in more detail in [301]. According to these papers, when shielding and screening of the WE to reduce the effect of electromagnetic interference (EMI) is challenging, the grounded CE approach can achieve a better performance.
2.4 A Note on Affinity-Based Biosensing
Affinity -based measurement techniques, where a binding molecule [e.g. antibodies or other proteins, aptamers, ss-DNA (single stranded deoxyribonucleic acid), or RNA (ribonucleic acid)] is used as part of the transduction process to enhance selectivity and specificity , are mainly single use disposable types of sensors. A discussion on these is thus excluded from this chapter, as are sensors requiring agents and labels for detection of target analytes (thus only reagentless and label-free techniques are discussed). This is due to the inherent limitation of these devices in their implementation as implantable sensors. These hurdles are associated with the in situ regeneration of antigen binding sites once a detection event has taken place; i.e. the antigen–bio-recognition element binding is very specific with strong bond formation and is essentially irreversible [263]. Breaking that bond to free up the detection site and reuse the biosensor (without damaging the binding sites) is extremely challenging. Applying high temperatures, chemicals, and electromagnetic fields are some options examined in the literature. Nevertheless, it is obvious that these are not suitable for implantable applications. In addition, using (redox ) agents and labels for detection is also not possible in an implantable in vivo environment. For the sake of completeness we mention a variety of methods that can be used in affinity -based sensing . Affinity based-sensing can be performed via quartz crystal microbalance (QCM), surface acoustic wave (SAW), surface plasmon resonance (SPR), impedance spectroscopy (EIS), voltammetry or amperometry , micro-electro-mechanical systems (MEMS) based cantilever sensors read out electronically (resistor bridge or capacitive sensing ), or optically and atomic force microscopy (AFM). Developing complex microfluidics for affinity -based sensors could potentially allow the use of these in implantable applications. Affinity -based sensing can be used for detecting a plethora of clinically relevant targets, such as bacteria (e.g. for infection diagnostics) and cancer biomarkers (e.g. certain proteins [302]); thus their potential use in implantable applications should not be overseen.
2.5 Conclusion
Electrochemical sensors are an important family of sensing techniques invaluable for the detection of a wide range of physiologically critical markers. Their advantages include their ability to be highly miniaturized, high dynamic range and sensitivity , high specificity through biorecognition elements and membranes, and their low-power operation . These make them vital for biomedical applications and especially for implantable systems . Potentiometric sensors are two-electrode galvanostatic systems used for the detection of vital ions such as pH , Na+, Ca2+, Mg2+, NH3 and K+. Polymeric membranes are typically used on metal electrodes to create miniaturizable electrodes known as ISEs, which circumvent the disadvantages of traditional glass-based sensors such as fragility, slow response , and large size. MMO pH sensors are an alternative with IrO x being a common MMO used, demonstrating super-Nernstian responses (sensitivities above the Nernstian 59 mV pH −1). The galvanostatic operational mode of potentiometric sensors requires high input impedance electronics to minimize the current through the electrochemical cell . ISFETs are an important family of potentiometric chemical sensors that readily provide this functionality, as an ISFET is by default an impedance converter. ISFET technology allows high-density miniaturized chemical arrays to be developed using common VLSI technologies for mass-produced, low-cost chemical analysis systems. Charge trapped during fabrication hinders the performance of ISFETs and their matching; however, various techniques can be used during and following fabrication together with circuit design techniques to minimize the effects of trapped charge . Voltammetric techniques are essential in the characterization and development of sensors and materials, especially since it allows the identification of the redox potential of target species and a voltammetric sensor’s response at that potential . These are typically three-electrode systems, and in contrast to potentiometric sensors, there is a current flow through the electrochemical cell that is being recorded. CV is the most common voltammetric technique . FSCV is a high-speed variant which, when used in combination with carbon-based electrodes, is a powerful technique for the detection of electroactive species and in particular various neurotransmitters. Such sensors can be used to monitor the nervous system chemically and can also be used in combination with action potential electrical recordings. Knowledge of the redox potential allows simple chrono-amperometric measurements at a constant potential . A wide range of amperometric sensors have been developed for the detection of important analytes, such as lactate , O2, and glucose , the latter type being the most successful, with a wide range of commercially available systems.
Electrochemical sensors are constantly evolving. In recent years, the introduction of various nanomaterials, such as graphene , CNTs, and NPs has led to new developments. These together with flexible, stretchable, and transient/resorbable materials are paving the way for further developments in the field, with a particular focus on implantable medical devices.
Abbreviations
- AIROF:
-
Anodic iridium oxide film
- Al2O3:
-
Aluminium oxide
- ATP:
-
Adenosine triphosphate
- CMOS:
-
Complementary metal-oxide-semiconductor
- CNS:
-
Central nervous system
- DOS:
-
Bis(2-ethylhexyl)sebacate
- ESD:
-
Electrostatic discharge
- FEM:
-
Finite element analysis
- FET:
-
Field-effect transistor
- FG-MOS:
-
Floating-gate MOS
- GA:
-
Glutaraldehyde
- GERD:
-
Gastroesophageal reflux disease
- GI tract:
-
Gastro intestinal tract
- IrOx:
-
Iridium oxide
- ISE:
-
Ion-selective electrode
- ISFET:
-
Ion-sensitive field-effect transistor
- LPCVD:
-
Low pressure chemical vapour deposition
- L-PEI:
-
Linear polyethylenimine polymer
- LPF:
-
Low-pass filter
- L-PPI:
-
Linear polypropyleneimine polymer
- MMO:
-
Metal-metal-oxide
- MNOS:
-
Metal-nitride-oxide-silicon
- MOSFET:
-
Metal oxide semiconductor field effect transistor
- MWCNT:
-
Multi-walled carbon nanotubes
- NMOS:
-
N-type metal-oxide-semiconductor
- OCP:
-
Open circuit potential
- o-NPOE:
-
2-nitrophenyloctyl ether
- p(HEMA):
-
Poly(2-hydroxyethyl methacrylate)
- PECVD:
-
Low temperature chemical vapour deposition
- PEDOT:
-
Poly(3,4-ethylenedioxythiophene) polymer
- PEG:
-
Poly(ethylene glycol)
- PMOS:
-
P-type metal-oxide-semiconductor
- PVC:
-
Polyvinylchloride
- RE:
-
Reference electrode
- ReFET:
-
Reference FET
- Si3N4:
-
Silicon nitride
- SIROF:
-
Sputtered Ir(Ox) films
- SNP:
-
Single-nucleotide polymorphism
- SsDNA:
-
Single-Stranded Deoxyribonucleic acid
- Ta2O5:
-
Tantalum oxide
- UV exposure:
-
Ultraviolet exposure
- WE:
-
Working electrode
References
S. Vaddiraju, I. Tomazos, D.J. Burgess, F.C. Jain, F. Papadimitrakopoulos, Emerging synergy between nanotechnology and implantable biosensors: a review. Biosens. Bioelectron. 25(7), 1553–1565 (2010)
A. Lewenstam, Routines and challenges in clinical application of electrochemical ion-sensors. Electroanalysis 26(6), 1171–1181 (2014)
K. Ueshima, Magnesium and ischemic heart disease: a review of epidemiological, experimental, and clinical evidences. Magnes. Res. 18(4), 275–284 (2005)
S.P. Yu, L.M.T. Canzoniero, D.W. Choi, Ion homeostasis and apoptosis. Curr. Opin. Cell Biol. 13(4), 405–411 (2001)
S.P. Yu, Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 70(4), 363–386 (2003)
J. Flores, D.R. DiBona, N. Frega, D.A. Leaf, Cell volume regulation and ischemic tissue damage. J. Membr. Biol. 10(1), 331–343 (1972)
W.E. Cascio, G.X. Yan, A.G. Kléber, Early changes in extracellular potassium in ischemic rabbit myocardium. The role of extracellular carbon dioxide accumulation and diffusion. Circ. Res. 70(2), 409–422 (1992)
A. Sola et al., Multiparametric monitoring of ischemia-reperfusion in rat kidney: effect of ischemic preconditioning. Transplantation 75(6), 744–749 (2003)
O.T. Guenat, S. Generelli, N.F. de Rooij, M. Koudelka-Hep, F. Berthiaume, M.L. Yarmush, Development of an array of ion-selective microelectrodes aimed for the monitoring of extracellular ionic activities. Anal. Chem. 78(21), 7453–7460 (2006)
M. Rossol et al., Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 3, 1329 (2012)
S. Kun, B. Ristic, R.A. Peura, R.M. Dunn, Algorithm for tissue ischemia estimation based on electrical impedance spectroscopy. IEEE Trans. Biomed. Eng. 50(12), 1352–1359 (2003)
J. Wtorek, L. Jozefiak, A. Polinski, J. Siebert, An averaging two-electrode probe for monitoring changes in myocardial conductivity evoked by ischemia. IEEE Trans. Biomed. Eng. 49(3), 240–246 (2002)
B. Ristic, S. Kun, R.A. Peura, Muscle tissue ischemia monitoring using impedance spectroscopy: quantitative results of animal studies, in Proceedings of the 19th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, vol. 5 (1997), pp. 2108–2111, 5
S. Kun, R.A. Peura, Tissue ischemia detection using impedance spectroscopy, in Proceedings of the 16th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 1994. Engineering Advances: New Opportunities for Biomedical Engineers, vol. 2 (1994), pp. 868–869
C.A. González, C. Villanueva, S. Othman, R. Narváez, E. Sacristán, Impedance spectroscopy for monitoring ischemic injury in the intestinal mucosa. Physiol. Meas. 24(2), 277 (2003)
H.-J. Chung et al., Stretchable, multiplexed pH sensors with demonstrations on rabbit and human hearts undergoing Ischemia. Adv. Healthc. Mater. 3(1), 59–68 (2014)
M.S. Frant, Historical perspective. History of the early commercialization of ion-selective electrodes. Analyst 119(11), 2293–2301 (1994)
E. Bakker, R. Meruva, E. Pretsch, M. Meyerhoff, Selectivity of polymer membrane-based ion-selective electrodes: self-consistent model describing the potentiometric response in mixed ion solutions of different charge. Anal. Chem. 66(19), 3021–3030 (1994)
D. O’Hare, K.H. Parker, C.P. Winlove, Metal–metal oxide pH sensors for physiological application. Med. Eng. Phys. 28(10), 982–988 (2006)
C. Fay et al., Wireless ion-selective electrode autonomous sensing system. IEEE Sens. J. 11(10), 2374–2382 (2011)
W.-D. Huang, S. Deb, Y.-S. Seo, S. Rao, M. Chiao, J.C. Chiao, A passive radio-frequency pH-sensing tag for wireless food-quality monitoring. IEEE Sens. J. 12(3), 487–495 (2012)
W.-D. Huang, H. Cao, S. Deb, M. Chiao, J.C. Chiao, A flexible pH sensor based on the iridium oxide sensing film. Sens. Actuators Phys. 169(1), 1–11 (2011)
CN–0326: isolated low power pH monitor with temperature compensation. Analog Devices, Inc., 2013
O. Korostynska, K. Arshak, E. Gill, A. Arshak, Review paper: materials and techniques for in vivo pH monitoring. IEEE Sens. J. 8(1), 20–28 (2008)
M. Cremer, Origin of electromotor properties of tissues, and instructional contribution for polyphasic electrolyte chains. Z. Für Biol. 47, 562–608 (1906)
J. Ruzicka, The seventies—golden age for ion selective electrodes. J. Chem. Educ. 74(2), 167 (1997)
J. Janata, Potentiometric microsensors. Chem. Rev. 90(5), 691–703 (1990)
P. Steegstra, E. Ahlberg, Influence of oxidation state on the pH dependence of hydrous iridium oxide films. Electrochim. Acta 76, 26–33 (2012)
A. Fog, R.P. Buck, Electronic semiconducting oxides as pH sensors. Sens. Actuators 5(2), 137–146 (1984)
L.D. Burke, J.K. Mulcahy, D.P. Whelan, Preparation of an oxidized iridium electrode and the variation of its potential with pH. J. Electroanal. Chem. Interfacial Electrochem. 163(1), 117–128 (1984)
K. Kinoshita, M.J. Madou, Electrochemical measurements on Pt, Ir, and Ti oxides as pH probes. J. Electrochem. Soc. 131(5), 1089–1094 (1984)
E. Kinoshita, F. Ingman, G. Edwall, S. Thulin, S. Gła̧b, Polycrystalline and monocrystalline antimony, iridium and palladium as electrode material for pH-sensing electrodes. Talanta 33(2), 125–134 (1986)
M.L. Hitchman, S. Ramanathan, Evaluation of iridium oxide electrodes formed by potential cycling as pH probes. Analyst 113(1), 35–39 (1988)
J. Bobacka, Conducting polymer-based solid-state ion-selective electrodes. Electroanalysis 18(1), 7–18 (2006)
J. Bobacka, A. Ivaska, A. Lewenstam, Potentiometric ion sensors. Chem. Rev. 108(2), 329–351 (2008)
Z. Štefanac, W. Simon, Ion specific electrochemical behavior of macrotetrolides in membranes. Microchem. J. 12(1), 125–132 (1967)
Z. Stefanac, W. Simon, Highly selective cation electrode systems based on in-vitro behavior of macrotetrolides in membranes. Chimica 20 (1966)
L.A.R. Pioda, H.A. Wachter, R.E. Dohner, W. Simon, Complexes of nonactin and monactin with sodium, potassium and ammonium ions. Helv. Chim. Acta 50, 1373–1375 (1967)
A.C. Ion, E. Bakker, E. Pretsch, Potentiometric Cd2+-selective electrode with a detection limit in the low ppt range. Anal. Chim. Acta 440(2), 71–79 (2001)
A. Ceresa, E. Bakker, B. Hattendorf, D. Günther, E. Pretsch, Potentiometric polymeric membrane electrodes for measurement of environmental samples at trace levels: new requirements for selectivities and measuring protocols, and comparison with ICPMS. Anal. Chem. 73(2), 343–351 (2001)
E. Bakker, M.E. Meyerhoff, Ionophore-based membrane electrodes: new analytical concepts and non-classical response mechanisms. Anal. Chim. Acta 416(2), 121–137 (2000)
A. Shvarev, E. Bakker, Reversible electrochemical detection of nonelectroactive polyions. J. Am. Chem. Soc. 125(37), 11192–11193 (2003)
M.E. Collison, G.V. Aebli, J. Petty, M.E. Meyerhoff, Potentiometric combination ion-carbon dioxide sensors for in vitro and in vivo blood measurements. Anal. Chem. 61(21), 2365–2372 (1989)
D.L. Simpson, R.K. Kobos, Potentiometric microbiological assay of gentamicin, streptomycin, and neomycin with a carbon dioxide gas-sensing electrode. Anal. Chem. 55(12), 1974–1977 (1983)
A.F. Bradley, Determination of blood-gases utilizing specially designed electrodes for PCO2, PO2, PO2 and pH. Biomed. Sci. Instrum. 3, 181–188 (1966)
R. Zahradník, P. Hobza, Z. Slanina, Calculations of Henry constants and partition coefficients using quantum chemical approach, in Quantitative Structure-Activity Relationships, ed. by M. Tichý (Birkhäuser Basel, 1976), p. 217–230
I.A. Pechenkina, K.N. Mikhelson, Materials for the ionophore-based membranes for ion-selective electrodes: problems and achievements (review paper). Russ. J. Electrochem. 51(2), 93–102 (2015)
A. Radu, Y. Qin, S. Peper, A. Ceresa, E. Bakker, Improving the low detection limit of polymer-based ion selective electrodes with a plasticizer-free polymer containing a covalently immobilized Ca2+-selective ionophore. Abstr. Pap. Am. Chem. Soc. 226, U105–U105 (2003)
L.Y. Heng, E.A.H. Hall, One-step synthesis of K+-selective methacrylic-acrylic copolymers containing grafted ionophore and requiring no plasticizer. Electroanalysis 12(3), 178–186 (2000)
E. Malinowska, L. Gawart, P. Parzuchowski, G. Rokicki, Z. Brzózka, Novel approach of immobilization of calix[4]arene type ionophore in ‘self-plasticized’ polymeric membrane. Anal. Chim. Acta 421(1), 93–101 (2000)
E. Bakker, P. Bühlmann, E. Pretsch, Carrier-based ion-selective electrodes and bulk optodes. 1. General characteristics. Chem. Rev. 97(8), 3083–3132 (1997)
W.E. Morf, The Principles of Ion-Selective Electrodes and of Membrane Transport (Elsevier, New York, 2012)
E. Bakker, M. Willer, M. Lerchi, K. Seiler, E. Pretsch, Determination of complex formation constants of neutral cation-selective ionophores in solvent polymeric membranes. Anal. Chem. 66(4), 516–521 (1994)
P. Anker, E. Wieland, D. Ammann, R.E. Dohner, R. Asper, W. Simon, Neutral carrier based ion-selective electrode for the determination of total calcium in blood serum. Anal. Chem. 53(13), 1970–1974 (1981)
D. Ammann et al., Preparation of neutral ionophores for Alkali and Alkaline earth metal cations and their application in ion selective membrane electrodes. Helv. Chim. Acta 58(6), 1535–1548 (1975)
Y. Qin, E. Bakker, Evaluation of the separate equilibrium processes that dictate the upper detection limit of neutral ionophore-based potentiometric sensors. Anal. Chem. 74(13), 3134–3141 (2002)
E. Lindner, K. Toth, E. Pungor, Lead-selective neutral carrier based liquid membrane electrode. Anal. Chem. 56(7), 1127–1131 (1984)
E. Bakker, A. Xu, E. Pretsch, Optimum composition of neutral carrier based pH electrodes. Anal. Chim. Acta 295(3), 253–262 (1994)
M. Telting-Diaz, E. Bakker, Effect of lipophilic ion-exchanger leaching on the detection limit of carrier-based ion-selective electrodes. Anal. Chem. 73(22), 5582–5589 (2001)
E. Lindner et al., Ion-selective membranes with low plasticizer content: electroanalytical characterization and biocompatibility studies. J. Biomed. Mater. Res. 28(5), 591–601 (1994)
R. Lenigk, H. Zhu, T.-C. Lo, R. Renneberg, Recessed microelectrode array for a micro flow-through system allowing on-line multianalyte determination in vivo. Fresenius J. Anal. Chem. 364(1–2), 66–71 (1999)
R.E. Gyurcsányi, N. Rangisetty, S. Clifton, B.D. Pendley, E. Lindner, Microfabricated ISEs: critical comparison of inherently conducting polymer and hydrogel based inner contacts. Talanta 63(1), 89–99 (2004)
S.Y. Yun et al., Potentiometric properties of ion-selective electrode membranes based on segmented polyether urethane matrices. Anal. Chem. 69(5), 868–873 (1997)
D.N. Reinhoudt et al., Development of durable K+-selective chemically modified field effect transistors with functionalized polysiloxane membranes. Anal. Chem. 66(21), 3618–3623 (1994)
G.J. Moody, B. Saad, J.D.R. Thomas, Glass transition temperatures of poly(vinyl chloride) and polyacrylate materials and calcium ion-selective electrode properties. Analyst 112(8), 1143–1147 (1987)
L.Y. Heng, E.A.H. Hall, Methacrylic–acrylic polymers in ion-selective membranes: achieving the right polymer recipe. Anal. Chim. Acta 403(1–2), 77–89 (2000)
E. Bakker, P. Bühlmann, E. Pretsch, Polymer membrane ion-selective electrodes-what are the limits? Electroanalysis 11(13), 915–933 (1999)
S. Joo, R.B. Brown, Chemical sensors with integrated electronics. Chem. Rev. 108(2), 638–651 (2008)
I.A. Ges, B.L. Ivanov, D.K. Schaffer, E.A. Lima, A.A. Werdich, F.J. Baudenbacher, Thin-film IrOx pH microelectrode for microfluidic-based microsystems. Biosens. Bioelectron. 21(2), 248–256 (2005)
W. Olthuis, M.A.M. Robben, P. Bergveld, M. Bos, W.E. van der Linden, pH sensor properties of electrochemically grown iridium oxide. Sens. Actuators B Chem. 2(4), 247–256 (1990)
S. Yao, M. Wang, M. Madou, A pH electrode based on melt-oxidized iridium oxide. J. Electrochem. Soc. 148(4), H29–H36 (2001)
J. Kieninger, A. Marx, F. Spies, A. Weltin, G.A. Urban, G. Jobst, pH micro sensor with micro-fluidic liquid-junction reference electrode on-chip for cell culture applications. IEEE Sens. 2009, 2009–2012
C.M. Nguyen et al., Sol-Gel iridium oxide-based pH sensor array on flexible polyimide substrate. IEEE Sens. J. 13(10), 3857–3864 (2013)
M. Kubon et al., A microsensor system to probe physiological environments and tissue response. IEEE Sens. 2010, 2607–2611 (2010)
M.D. Johnson, O.E. Kao, D.R. Kipke, Spatiotemporal pH dynamics following insertion of neural microelectrode arrays. J. Neurosci. Methods 160(2), 276–287 (2007)
X. Yue et al., A real-time multi-channel monitoring system for stem cell culture process. IEEE Trans. Biomed. Circuits Syst. 2(2), 66–77 (2008)
S.A.M. Marzouk, S. Ufer, R.P. Buck, T.A. Johnson, L.A. Dunlap, W.E. Cascio, Electrodeposited iridium oxide pH electrode for measurement of extracellular myocardial acidosis during acute ischemia. Anal. Chem. 70(23), 5054–5061 (1998)
P.J. Kinlen, J.E. Heider, D.E. Hubbard, A solid-state pH sensor based on a Nafion-coated iridium oxide indicator electrode and a polymer-based silver chloride reference electrode. Sens. Actuators B Chem. 22(1), 13–25 (1994)
E. Lindner et al., In vivo and in vitro testing of microelectronically fabricated planar sensors designed for applications in cardiology. Fresenius J. Anal. Chem. 346(6–9), 584–588 (1993)
V.V. Cosofret, E. Lindner, T.A. Johnson, M.R. Neuman, Planar micro sensors for in vivo myocardial pH measurements. Talanta 41(6), 931–938 (1994)
V.V. Cosofret, M. Erdosy, T.A. Johnson, R.P. Buck, R.B. Ash, M.R. Neuman, Microfabricated sensor arrays sensitive to pH and K+ for ionic distribution measurements in the beating heart. Anal. Chem. 67(10), 1647–1653 (1995)
S.A.M. Marzouk, R.P. Buck, L.A. Dunlap, T.A. Johnson, W.E. Cascio, Measurement of extracellular pH, K+, and lactate in ischemic heart. Anal. Biochem. 308(1), 52–60 (2002)
S. Anastasova-Ivanova et al., Development of miniature all-solid-state potentiometric sensing system. Sens. Actuators B Chem. 146(1), 199–205 (2010)
G. Urban et al., Miniaturized multi-enzyme biosensors integrated with pH sensors on flexible polymer carriers for in vivo applications. Biosens. Bioelectron. 7(10), 733–739 (1992)
H. Cao et al., An implantable, batteryless, and wireless capsule with integrated impedance and pH sensors for gastroesophageal reflux monitoring. IEEE Trans. Biomed. Eng. 59(11), 3131–3139 (2012)
E. Bitziou, D. O’Hare, B.A. Patel, Spatial changes in acid secretion from isolated stomach tissue using a pH-histamine sensing microarray. Analyst 135(3), 482–487 (2010)
E. Bitziou, D. O’Hare, B.A. Patel, Simultaneous detection of pH changes and histamine release from oxyntic glands in isolated stomach. Anal. Chem. 80(22), 8733–8740 (2008)
I.A. Ges, B.L. Ivanov, A.A. Werdich, F.J. Baudenbacher, Differential pH measurements of metabolic cellular activity in nl culture volumes using microfabricated iridium oxide electrodes. Biosens. Bioelectron. 22(7), 1303–1310 (2007)
I.A. Ges, I.A. Dzhura, F.J. Baudenbacher, On-chip acidification rate measurements from single cardiac cells confined in sub-nanoliter volumes. Biomed. Microdevices 10(3), 347–354 (2008)
M. Mir, R. Lugo, I.B. Tahirbegi, J. Samitier, Miniaturizable ion-selective arrays based on highly stable polymer membranes for biomedical applications. Sensors 14(7), 11844–11854 (2014)
W. Qin, T. Zwickl, E. Pretsch, Improved detection limits and unbiased selectivity coefficients obtained by using ion-exchange resins in the inner reference solution of ion-selective polymeric membrane electrodes. Anal. Chem. 72(14), 3236–3240 (2000)
E.J. Parra, P. Blondeau, G.A. Crespo, F.X. Rius, An effective nanostructured assembly for ion-selective electrodes. An ionophore covalently linked to carbon nanotubes for Pb2+ determination. Chem. Commun. 47(8), 2438–2440 (2011)
M.A. Simon, R.P. Kusy, Plasticizer-level study of poly(vinyl chloride) ion-selective membranes. J. Biomed. Mater. Res. 30(3), 313–320 (1996)
V.G. Gavalas, M.J. Berrocal, L.G. Bachas, Enhancing the blood compatibility of ion-selective electrodes. Anal. Bioanal. Chem. 384(1), 65–72 (2005)
M. Pawlak, E. Bakker, Chemical modification of polymer ion-selective membrane electrode surfaces. Electroanalysis 26(6), 1121–1131 (2014)
S. Yajima, Y. Sonoyama, K. Suzuki, K. Kimura, Ion-sensor property and blood compatibility of neutral-carrier-type poly(vinyl chloride) membranes coated by phosphorylcholine polymers. Anal. Chim. Acta 463(1), 31–37 (2002)
M.J. Berrocal, R.D. Johnson, I.H.A. Badr, M. Liu, D. Gao, L.G. Bachas, Improving the blood compatibility of ion-selective electrodes by employing poly(MPC-co-BMA), a copolymer containing phosphorylcholine, as a membrane coating. Anal. Chem. 74(15), 3644–3648 (2002)
J.A. Hayward, D. Chapman, Biomembrane surfaces as models for polymer design: the potential for haemocompatibility. Biomaterials 5(3), 135–142 (1984)
A. Ivorra et al., Minimally invasive silicon probe for electrical impedance measurements in small animals. Biosens. Bioelectron. 19(4), 391–399 (2003)
G.P. Gumbrell, R.A. Peura, S. Kun, R.M. Dunn, Development of a pH based tissue ischemia monitor: hardware realization, in Proceedings of the 1996 IEEE Twenty-Second Annual Northeast Bioengineering Conference, 54–55 (1996)
G.P. Gumbrell, R.A. Peura, S. Kun, R.M. Dunn, Development of a minimally invasive microvascular ischemia monitor: drift reduction results, in Proceedings of the 19th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, vol. 1, 25–27 (1997)
G.P. Gumbrell, R.A. Peura, S. Kun, R.M. Dunn, Development of a pH based tissue ischemia monitor: preliminary clinical results, in Proceedings of the 18th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 1996. Bridging Disciplines for Biomedicine, vol. 1, 40–41 (1996)
A.H. Auerbach, B.R. Soller, R.A. Peura, Sources of error in measuring tissue pH with microsensors, in Proceedings of the 20th Annual Northeast Bioengineering Conference, 108–109 (1994)
J. Songer, Tissue ischemia monitoring using impedance spectroscopy. MSc. Thesis, Worcester Polytechnic Institute, 2001
A. Senagore, D.J.W. Milsom, R.K. Walshaw, R. Dunstan, W.P. Mazier, I.H. Chaudry, Intramural pH: a quantitative measurement for predicting colorectal anastomotic healing. Dis. Colon Rectum 33(3), 175–179 (1990)
S.G. Nugent, D. Kumar, D.S. Rampton, D.F. Evans, Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 48(4), 571–577 (2001)
D.C.J. McDougall, R. Wong, P. Scudera, M. Lesser, J.J. DeCosse, Colonic mucosal pH in humans. Dig. Dis. Sci. 38(3), 542–545 (1993)
M. Millan, E. García-Granero, B. Flor, S. García-Botello, S. Lledo, Early prediction of anastomotic leak in colorectal cancer surgery by intramucosal pH. Dis. Colon Rectum 49(5), 595–601 (2006)
C.A. Eckley, H.O. Costa, Comparative study of salivary pH and volume in adults with chronic laryngopharyngitis by gastroesophageal reflux disease before and after treatment. Rev. Bras. Otorrinolaringol. 72(1), 55–60 (2006)
S. Ghimenti et al., Measurement of warfarin in the oral fluid of patients undergoing anticoagulant oral therapy. PLoS ONE 6(12), e28182 (2011)
J.L. Gonzalez-Guillaumin, D.C. Sadowski, K.V.I.S. Kaler, M.P. Mintchev, Ingestible capsule for impedance and pH monitoring in the esophagus. IEEE Trans. Biomed. Eng. 54(12), 2231–2236 (2007)
G.X. Yan, K.A. Yamada, A.G. Kléber, J. McHowat, P.B. Corr, Dissociation between cellular K+ loss, reduction in repolarization time, and tissue ATP levels during myocardial hypoxia and ischemia. Circ. Res. 72(3), 560–570 (1993)
J.N. Weiss, S.T. Lamp, K.I. Shine, Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. Am. J. Physiol. Heart Circ. Physiol. 256(4), H1165–H1175 (1989)
B.F. Palmer, Regulation of potassium homeostasis. Clin. J. Am. Soc. Nephrol. 10(6), 1050–1060 (2015)
A.G. Kléber, C.B. Riegger, M.J. Janse, Extracellular K+ and H+ shifts in early ischemia: mechanisms and relation to changes in impulse propagation. J. Mol. Cell. Cardiol. 19(Suppl 5), 35–44 (1987)
I.A. Marques de Oliveira et al., Sodium ion sensitive microelectrode based on a p-tert-butylcalix[4]arene ethyl ester. Sens. Actuators B Chem. 130(1), 295–299 (2008)
S. Chandra, H. Lang, A new sodium ion selective electrode based on a novel silacrown ether. Sens. Actuators B Chem. 114(2), 849–854 (2006)
K.Y. Chumbimuni-Torres, N. Rubinova, A. Radu, L.T. Kubota, E. Bakker, Solid contact potentiometric sensors for trace level measurements. Anal. Chem. 78(4), 1318–1322 (2006)
F. Li et al., All-solid-state potassium-selective electrode using graphene as the solid contact. Analyst 137(3), 618–623 (2012)
N. Zine et al., Potassium-ion selective solid contact microelectrode based on a novel 1,3-(di-4-oxabutanol)-calix[4]arene-crown–5 neutral carrier. Electrochim. Acta 51(24), 5075–5079 (2006)
J.E. Pandolfino, J.E. Richter, T. Ours, J.M. Guardino, J. Chapman, P.J. Kahrilas, Ambulatory esophageal pH monitoring using a wireless system. Am. J. Gastroenterol. 98(4), 740–749 (2003)
G. Karamanolis et al., Bravo 48-hour wireless pH monitoring in patients with non-cardiac chest pain. Objective gastroesophageal reflux disease parameters predict the responses to proton pump inhibitors. J. Neurogastroenterol. Motil. 18(2), 169–173 (2012)
E. Lindner, R. Buck, Microfabricated potentiometric electrodes and their in vivo applications. Anal. Chem. 72(9), 336 A–345 A (2000)
I.B. Tahirbegi, M. Mir, S. Schostek, M. Schurr, J. Samitier, In vivo ischemia monitoring array for endoscopic surgery. Biosens. Bioelectron. 61, 124–130 (2014)
A. Errachid, A. Ivorra, J. Aguiló, R. Villa, N. Zine, J. Bausells, New technology for multi-sensor silicon needles for biomedical applications. Sens. Actuators B Chem. 78(1–3), 279–284 (2001)
R. Gómez et al., A SiC microdevice for the minimally invasive monitoring of ischemia in living tissues. Biomed. Microdevices 8(1), 43–49 (2006)
M. Genescà et al., Electrical bioimpedance measurement during hypothermic rat kidney preservation for assessing ischemic injury. Biosens. Bioelectron. 20(9), 1866–1871 (2005)
M. Tijero et al., SU–8 microprobe with microelectrodes for monitoring electrical impedance in living tissues. Biosens. Bioelectron. 24(8), 2410–2416 (2009)
L. Xu et al., 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat. Commun. 5, 3329 (2014)
I.B. Tahirbegi, M. Mir, J. Samitier, Real-time monitoring of ischemia inside stomach. Biosens. Bioelectron. 40(1), 323–328 (2013)
P. Bergveld, Thirty years of ISFETOLOGY: what happened in the past 30 years and what may happen in the next 30 years. Sens. Actuators B Chem. 88(1), 1–20 (2003)
P.A. Hammond, D. Ali, D.R.S. Cumming, Design of a single-chip pH sensor using a conventional 0.6 μm CMOS process. IEEE Sens. J. 4(6), 706–712 (2004)
P. Georgiou, C. Toumazou, ISFET characteristics in CMOS and their application to weak inversion operation. Sens. Actuators B Chem. 143(1), 211–217 (2009)
D. Binkley, Tradeoffs and optimization in analog CMOS design (Wiley, Chichester, West Sussex, UK, 2008)
A. Al-Ahdal, C. Toumazou, ISFET-based chemical switch. IEEE Sens. J. 12(5), 1140–1146 (2012)
L.M. Shepherd, C. Toumazou, A biochemical translinear principle with weak inversion ISFETs. IEEE Trans. Circuits Syst. Regul. Pap. 52(12), 2614–2619 (2005)
Y. Liu, P. Georgiou, T. Prodromakis, T.G. Constandinou, C. Toumazou, An extended CMOS ISFET model incorporating the physical design geometry and the effects on performance and offset variation. IEEE Trans. Electron Devices 58(12), 4414–4422 (2011)
N. Miscourides, P. Georgiou, Impact of technology scaling on ISFET performance for genetic sequencing. IEEE Sens. J. 15(4), 2219–2226 (2015)
M. Sohbati, Y. Liu, P. Georgiou, C. Toumazou, An ISFET design methodology incorporating CMOS passivation, IEEE Biomedical Circuits and Systems Conference (BioCAS), 65–68 (2012)
M. Sohbati, C. Toumazou, Dimension and shape effects on the ISFET performance. IEEE Sens. J. 15(3), 1670–1679 (2015)
J. Bausells, J. Carrabina, A. Errachid, A. Merlos, Ion-sensitive field-effect transistors fabricated in a commercial CMOS technology. Sens. Actuators B Chem. 57(1–3), 56–62 (1999)
M.J. Milgrew, D.R.S. Cumming, Matching the transconductance characteristics of CMOS ISFET arrays by removing trapped charge. IEEE Trans. Electron Devices 55(4), 1074–1079 (2008)
M.J. Milgrew, M.O. Riehle, D.R.S. Cumming, A large transistor-based sensor array chip for direct extracellular imaging. Sens. Actuators B Chem. 111–112, 347–353 (2005)
T. Prodromakis, Y. Liu, T. Constandinou, P. Georgiou, C. Toumazou, Exploiting CMOS technology to enhance the performance of ISFET sensors. IEEE Electron Device Lett. 31(9), 1053–1055 (2010)
J.M. Rothberg, W. Hinz, K.L. Johnson, J. Bustillo, Methods and apparatus for measuring analytes using large scale fet arrays. CA2672315 A1, 26 Jun 2008
M. Milgrew, J. Bustillo, T. Rearick, Chemically-sensitive field effect transistor based pixel array with protection diodes. US20130001653 A1, 03 Jan 2013
H.-S. Wong, M.H. White, A CMOS-integrated `ISFET-operational amplifier’ chemical sensor employing differential sensing. IEEE Trans. Electron Devices 36(3), 479–487 (1989)
V.P. Chodavarapu, A.H. Titus, A.N. Cartwright, Differential read-out architecture for CMOS ISFET microsystems. Electron. Lett. 41(12), 698–699 (2005)
D. Garner, H. Bai, Electrostatic discharge protection. US20130188288 A1, 25 July 2013
R. Smith, R.J. Huber, J. Janata, Electrostatically protected ion sensitive field effect transistors. Sens. Actuators 5(2), 127–136 (1984)
Y. Hu, P. Georgiou, A robust ISFET pH-measuring front-end for chemical reaction monitoring. IEEE Trans. Biomed. Circuits Syst. 8(2), 177–185 (2014)
P. Georgiou, C. Toumazou, CMOS-based programmable gate ISFET. Electron. Lett. 44(22), 1289–1290 (2008)
A. Al-Ahdal, P. Georgiou, C. Toumazou, ISFET’s threshold voltage control using bidirectional electron tunnelling, in 2012 IEEE Biomedical Circuits and Systems Conference (BioCAS), 172–175 (2012)
P. Georgiou, C. Toumazou, ISFET threshold voltage programming in CMOS using hot-electron injection. Electron. Lett. 45(22), 1112–1113 (2009)
A.G. Al-Ahdal, C. Toumazou, ISFET threshold voltage programming in CMOS using electron tunnelling. Electron. Lett. 47(25), 1398–1399 (2011)
J.M. Rothberg et al., An integrated semiconductor device enabling non-optical genome sequencing. Nature 475(7356), 348–352 (2011)
C. Toumazou et al., Simultaneous DNA amplification and detection using a pH-sensing semiconductor system. Nat. Methods 10(7), 641–646 (2013)
X. Huang, H. Yu, X. Liu, Y. Jiang, M. Yan, D. Wu, A dual-mode large-arrayed CMOS ISFET sensor for accurate and high-throughput pH sensing in biomedical diagnosis. IEEE Trans. Biomed. Eng. 62(9), 2224–2233 (2015)
P. Rai, S. Jung, T. Ji, V.K. Varadan, Drain current centric modality: instrumentation and evaluation of ISFET for monitoring myocardial ischemia like variations in pH and potassium ion concentration. IEEE Sens. J. 9(12), 1987–1995 (2009)
F. Xu, G. Yan, Z. Wang, P. Jiang, Continuous accurate pH measurements of human GI tract using a digital pH-ISFET sensor inside a wireless capsule. Measurement 64, 49–56 (2015)
P.A. Hammond, D. Ali, D.R.S. Cumming, A system-on-chip digital pH meter for use in a wireless diagnostic capsule. IEEE Trans. Biomed. Eng. 52(4), 687–694 (2005)
C.-S. Lee, S.K. Kim, M. Kim, Ion-sensitive field-effect transistor for biological sensing. Sensors 9(9), 7111–7131 (2009)
S. Migita, K. Ozasa, T. Tanaka, T. Haruyama, Enzyme-based field-effect transistor for adenosine triphosphate (ATP) sensing. Anal. Sci. 23(1), 45–48 (2007)
A. Bratov, N. Abramova, A. Ipatov, Recent trends in potentiometric sensor arrays—a review. Anal. Chim. Acta 678(2), 149–159 (2010)
J. Janata, Principles of Chemical Sensors (Springer Science & Business Media, 2010)
T.-W. Huang, J.-C. Chou, T.-P. Sun, S.-K. Hsiung, Fabrication of a screen-printing reference electrode for potentiometric measurement. Sens. Lett. 6(6), 860–863 (2008)
W. Vonau, W. Oelßner, U. Guth, J. Henze, An all-solid-state reference electrode. Sens. Actuators B Chem. 144(2), 368–373 (2010)
J. Sutter, E. Lindner, R.E. Gyurcsányi, E. Pretsch, A polypyrrole-based solid-contact Pb2+-selective PVC-membrane electrode with a nanomolar detection limit. Anal. Bioanal. Chem. 380(1), 7–14 (2004)
A. Cadogan, Z. Gao, A. Lewenstam, A. Ivaska, D. Diamond, All-solid-state sodium-selective electrode based on a calixarene ionophore in a poly(vinyl chloride) membrane with a polypyrrole solid contact. Anal. Chem. 64(21), 2496–2501 (1992)
G.D. O’Neil, R. Buiculescu, S.P. Kounaves, N.A. Chaniotakis, Carbon-nanofiber-based nanocomposite membrane as a highly stable solid-state junction for reference electrodes. Anal. Chem. 83(14), 5749–5753 (2011)
H.J. Yoon et al., Solid-state ion sensors with a liquid junction-free polymer membrane-based reference electrode for blood analysis. Sens. Actuators B Chem. 64(1–3), 8–14 (2000)
G. Valdés-Ramírez, G.A. Álvarez-Romero, C.A. Galán-Vidal, P.R. Hernández-Rodríguez, M.T. Ramírez-Silva, Composites: a novel alternative to construct solid state Ag/AgCl reference electrodes. Sens. Actuators B Chem. 110(2), 264–270 (2005)
D. Rehm, E. McEnroe, D. Diamond, An all solid-state reference electrode based on a potassium chloride doped vinyl ester resin. Anal. Proc. Anal. Commun. 32(8), 319–322 (1995)
R. Mamińska, A. Dybko, W. Wróblewski, All-solid-state miniaturised planar reference electrodes based on ionic liquids. Sens. Actuators B Chem. 115(1), 552–557 (2006)
D. Cicmil et al., Ionic liquid-based, liquid-junction-free reference electrode. Electroanalysis 23(8), 1881–1890 (2011)
Ł. Tymecki, E. Zwierkowska, R. Koncki, Screen-printed reference electrodes for potentiometric measurements. Anal. Chim. Acta 526(1), 3–11 (2004)
A. Kisiel, A. Michalska, K. Maksymiuk, E.A.H. Hall, All-solid-state reference electrodes with poly(n-butyl acrylate) based membranes. Electroanalysis 20(3), 318–323 (2008)
A. Kisiel, M. Donten, J. Mieczkowski, F.X. Rius-Ruiz, K. Maksymiuk, A. Michalska, Polyacrylate microspheres composite for all-solid-state reference electrodes. Analyst 135(9), 2420–2425 (2010)
G.A. Crespo, S. Macho, F.X. Rius, Ion-selective electrodes using carbon nanotubes as ion-to-electron transducers. Anal. Chem. 80(4), 1316–1322 (2008)
G.A. Crespo, S. Macho, J. Bobacka, F.X. Rius, Transduction mechanism of carbon nanotubes in solid-contact ion-selective electrodes. Anal. Chem. 81(2), 676–681 (2009)
F.X. Rius-Ruiz, D. Bejarano-Nosas, P. Blondeau, J. Riu, F.X. Rius, Disposable planar reference electrode based on carbon nanotubes and polyacrylate membrane. Anal. Chem. 83(14), 5783–5788 (2011)
F.X. Rius-Ruiz, A. Kisiel, A. Michalska, K. Maksymiuk, J. Riu, F.X. Rius, Solid-state reference electrodes based on carbon nanotubes and polyacrylate membranes. Anal. Bioanal. Chem. 399(10), 3613–3622 (2011)
Z. Mousavi, K. Granholm, T. Sokalski, A. Lewenstam, An analytical quality solid-state composite reference electrode. The Analyst 138(18), 5216–5220 (2013)
T. Guinovart, G.A. Crespo, F.X. Rius, F.J. Andrade, A reference electrode based on polyvinyl butyral (PVB) polymer for decentralized chemical measurements. Anal. Chim. Acta 821, 72–80 (2014)
A.K. Ghosh, V. Ramachandhran, M.S. Hanra, B.M. Misra, Studies on fouling and gel polarisation aspects of polyvinyl butyral blended cellulose acetate ultrafiltration membrane by resistance model approach. Indian J. Chem. Technol. 7(2), 55–60 (2000)
Y. Saito, M. Okano, K. Kubota, T. Sakai, J. Fujioka, T. Kawakami, Evaluation of interactive effects on the ionic conduction properties of polymer gel electrolytes. J. Phys. Chem. B 116(33), 10089–10097 (2012)
E. Bakker, Hydrophobic membranes as liquid junction-free reference electrodes. Electroanalysis 11(10–11), 788–792 (1999)
T. Kakiuchi, T. Yoshimatsu, N. Nishi, New class of Ag/AgCl electrodes based on hydrophobic ionic liquid saturated with AgCl. Anal. Chem. 79(18), 7187–7191 (2007)
C. Zuliani, G. Matzeu, D. Diamond, A liquid-junction-free reference electrode based on a PEDOT solid-contact and ionogel capping membrane. Talanta 125, 58–64 (2014)
M. Shibata, H. Sakaida, T. Kakiuchi, Determination of the activity of hydrogen ions in dilute sulfuric acids by use of an ionic liquid salt bridge sandwiched by two hydrogen electrodes. Anal. Chem. 83(1), 164–168 (2011)
U. Guth, F. Gerlach, M. Decker, W. Oelßner, W. Vonau, Solid-state reference electrodes for potentiometric sensors. J. Solid State Electrochem. 13(1), 27–39 (2008)
B. Palán, F.V. Santos, J.M. Karam, B. Courtois, M. Husák, New ISFET sensor interface circuit for biomedical applications. Sens. Actuators B Chem. 57(1–3), 63–68 (1999)
M.F. Chaplin, C. Bucke, Enzyme Technology (CUP Archive, 1990)
G.N. Meloni, Building a microcontroller based potentiostat: a inexpensive and versatile platform for teaching electrochemistry and instrumentation. J. Chem. Educ. 93(7), 1320–1322 (2016)
M.D.M. Dryden, A.R. Wheeler, DStat: a versatile, open-source potentiostat for electroanalysis and integration. PLoS ONE 10(10), e0140349 (2015)
R.S. Freire, C.A. Pessoa, L.D. Mello, L.T. Kubota, Direct electron transfer: an approach for electrochemical biosensors with higher selectivity and sensitivity. J. Braz. Chem. Soc. 14(2), 230–243 (2003)
B.P. Schaffar, Thick film biosensors for metabolites in undiluted whole blood and plasma samples. Anal. Bioanal. Chem. 372(2), 254–260 (2002)
Z. Chen, C. Fang, H. Wang, J. He, Disposable glucose test strip for whole blood with integrated sensing/diffusion-limiting layer. Electrochim. Acta 55(2), 544–550 (2009)
R.A. Croce, S. Vaddiraju, F. Papadimitrakopoulos, F.C. Jain, Theoretical analysis of the performance of glucose sensors with layer-by-layer assembled outer membranes. Sensors 12(10), 13402–13416 (2012)
F. Valentini, L. Galache Fernàndez, E. Tamburri, G. Palleschi, Single Walled Carbon Nanotubes/polypyrrole-GOx composite films to modify gold microelectrodes for glucose biosensors: Study of the extended linearity. Biosens. Bioelectron. 43, 75–78 (2013)
A. Heller, B. Feldman, Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 108(7), 2482–2505 (2008)
Z. Zhou, L. Xu, S. Wu, B. Su, A novel biosensor array with a wheel-like pattern for glucose, lactate and choline based on electrochemiluminescence imaging. Analyst 139(19), 4934–4939 (2014)
Y. Liu, Y. Dong, C.X. Guo, Z. Cui, L. Zheng, C.M. Li, Protein-directed in situ synthesis of gold nanoparticles on reduced graphene oxide modified electrode for nonenzymatic glucose sensing. Electroanalysis 24(12), 2348–2353 (2012)
Y. Fu et al., One-pot preparation of polymer–enzyme–metallic nanoparticle composite films for high-performance biosensing of glucose and galactose. Adv. Funct. Mater. 19(11), 1784–1791 (2009)
D. Xu, L. Luo, Y. Ding, P. Xu, Sensitive electrochemical detection of glucose based on electrospun La0.88Sr0.12MnO3 nanofibers modified electrode. Anal. Biochem. 489, 38–43 (2015)
N.G. Poulos, J.R. Hall, M.C. Leopold, functional layer-by-layer design of xerogel-based first-generation amperometric glucose biosensors. Langmuir 31(4), 1547–1555 (2015)
M.M. Ahmadi, G.A. Jullien, A wireless-implantable microsystem for continuous blood glucose monitoring. IEEE Trans. Biomed. Circuits Syst. 3(3), 169–180 (2009)
Medtronic MiniMed 530G. [Online]. Available: http://www.medtronicdiabetes.com/products/minimed–530g-diabetes-system-with-enlite. Accessed: 28 Jan 2016
Dexcom G5TM Mobile Continuous Glucose Monitoring (CGM) System. [Online]. Available: http://www.dexcom.com/g5-mobile-cgm. Accessed: 28 Jan 2016
FreeStyle Libre| FreeStyle Blood Glucose Meters. [Online]. Available: https://abbottdiabetescare.co.uk/our-products/freestyle-libre. Accessed: 26 Jan 2016
D. De Backer, J. Creteur, H. Zhang, M. Norrenberg, J.-L. Vincent, Lactate production by the lungs in acute lung injury. Am. J. Respir. Crit. Care Med. 156(4), 1099–1104 (1997)
J.A. Kruse, S.A.J. Zaidi, R.W. Carlson, Significance of blood lactate levels in critically III patients with liver disease. Am. J. Med. 83(1), 77–82 (1987)
S. Mm, M. Pn, Adenine nucleotide and lactate metabolism in the lung in endotoxin shock. Circ. Shock 8(6), 657–666 (1980)
J. Karlsson, J.T. Willerson, S.J. Leshin, C.B. Mullins, J.H. Mitchell, Skeletal muscle metabolites in patients with cardiogenic shock or severe congestive heart failure. Scand. J. Clin. Lab. Invest. 35(1), 73–79 (1975)
R. Rimachi, F. Bruzzi de Carvahlo, C. Orellano-Jimenez, F. Cotton, J.L. Vincent, D. De Backer, Lactate/pyruvate ratio as a marker of tissue hypoxia in circulatory and septic shock. Anaesth. Intensive Care 40(3), 427–432 (2012)
B. Carbonne, K. Pons, E. Maisonneuve, Foetal scalp blood sampling during labour for pH and lactate measurements. Best Pract. Res. Clin. Obstet. Gynaecol. 30, 62–67 (2016)
R.K.D. Suveera Dhup, Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 18(10), 1319–1330 (2012)
K.M. Kennedy, M.W. Dewhirst, Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 6(1), 127–148 (2009)
R.M.A. Bhatia et al., Application of rapid-sampling, online microdialysis to the monitoring of brain metabolism during aneurysm surgery. Neurosurgery 58(4) (2006)
J.C. Goodman, A.B. Valadka, S.P. Gopinath, M. Uzura, C.S.M. Robertson, Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. [Miscellaneous Article]. Crit. Care Med. 27(9), 1965–1973 (1999)
E.L. Cureton, R.O. Kwan, K.C. Dozier, J. Sadjadi, J.D. Pal, G.P. Victorino, A different view of lactate in trauma patients: protecting the injured brain. J. Surg. Res. 159(1), 468–473 (2010)
E. Naylor et al., Lactate as a biomarker for sleep. Sleep 35(9), 1209–1222 (2012)
L. Rassaei, W. Olthuis, S. Tsujimura, E.J.R. Sudhölter, A. van den Berg, Lactate biosensors: current status and outlook. Anal. Bioanal. Chem. 406(1), 123–137 (2013)
G.F. Manbeck, E. Fujita, A review of iron and cobalt porphyrins, phthalocyanines and related complexes for electrochemical and photochemical reduction of carbon dioxide. J. Porphyr. Phthalocyanines 19(01–03), 45–64 (2015)
M.R. Romero, F. Ahumada, F. Garay, A.M. Baruzzi, Amperometric biosensor for direct blood lactate detection. Anal. Chem. 82(13), 5568–5572 (2010)
S.A. Bhakta, E. Evans, T.E. Benavidez, C.D. Garcia, Protein adsorption onto nanomaterials for the development of biosensors and analytical devices: a review. Anal. Chim. Acta 872, 7–25 (2015)
M. Gamero, F. Pariente, E. Lorenzo, C. Alonso, Nanostructured rough gold electrodes for the development of lactate oxidase-based biosensors. Biosens. Bioelectron. 25(9), 2038–2044 (2010)
X.-R. He et al., Amperometric L-lactate biosensor based on sol-gel film and multi-walled carbon nanotubes/platinum nanoparticles enhancement. Chin. J. Anal. Chem. Chin. Version 38(1), 57–61 (2010)
Y. Yu, Y. Yang, H. Gu, T. Zhou, G. Shi, Size-tunable Pt nanoparticles assembled on functionalized ordered mesoporous carbon for the simultaneous and on-line detection of glucose and L-lactate in brain microdialysate. Biosens. Bioelectron. 41, 511–518 (2013)
J.M. Goran, J.L. Lyon, K.J. Stevenson, Amperometric detection of l-Lactate using nitrogen-doped carbon nanotubes modified with lactate oxidase. Anal. Chem. 83(21), 8123–8129 (2011)
L. Agüí, M. Eguílaz, C. Peña-Farfal, P. Yáñez-Sedeño, J.M. Pingarrón, Lactate dehydrogenase biosensor based on an hybrid carbon nanotube-conducting polymer modified electrode. Electroanalysis 21(3–5), 386–391 (2009)
I. Shakir, M. Shahid, H.W. Yang, S. Cherevko, C.-H. Chung, D.J. Kang, α-MoO3 nanowire-based amperometric biosensor for l-lactate detection. J. Solid State Electrochem. 16(6), 2197–2201 (2012)
A.C. Pereira, A. Kisner, C.R.T. Tarley, L.T. Kubota, Development of a carbon paste electrode for lactate detection based on Meldola’s blue adsorbed on silica gel modified with niobium oxide and lactate oxidase. Electroanalysis 23(6), 1470–1477 (2011)
Y.T. Wang et al., A novel l-lactate sensor based on enzyme electrode modified with ZnO nanoparticles and multiwall carbon nanotubes. J. Electroanal. Chem. 661(1), 8–12 (2011)
U. Spohn, D. Narasaiah, L. Gorton, The influence of the carbon paste composition on the performance of an amperometric bienzyme sensor for L-lactate. Electroanalysis 8(6), 507–514 (1996)
M.R. Romero, F. Garay, A.M. Baruzzi, Design and optimization of a lactate amperometric biosensor based on lactate oxidase cross-linked with polymeric matrixes. Sens. Actuators B Chem. 131(2), 590–595 (2008)
M.M. Rahman, M.J.A. Shiddiky, M.A. Rahman, Y.-B. Shim, A lactate biosensor based on lactate dehydrogenase/nictotinamide adenine dinucleotide (oxidized form) immobilized on a conducting polymer/multiwall carbon nanotube composite film. Anal. Biochem. 384(1), 159–165 (2009)
E. Al-Jawadi, S. Pöller, R. Haddad, W. Schuhmann, NADH oxidation using modified electrodes based on lactate and glucose dehydrogenase entrapped between an electrocatalyst film and redox catalyst-modified polymers. Microchim. Acta 177(3–4), 405–410 (2012)
E.I. Yashina et al., Sol-gel immobilization of lactate oxidase from organic solvent: toward the advanced lactate biosensor. Anal. Chem. 82(5), 1601–1604 (2010)
M. Tsuchiya, H. Matsuhisa, Y. Hasebe, Selective amperometric response to hydrogen peroxide at a protein-incorporated sol-gel hybrid film-modified platinum electrode. Bunseki Kagaku 61(5), 425–428 (2012)
F. Palmisano, G.E.D. Benedetto, C.G. Zambonin, Lactate amperometric biosensor based on an electrosynthesizedbilayer film with covalently immobilized enzyme. Analyst 122(4), 365–369 (1997)
C.-L. Lin, C.-L. Shih, L.-K. Chau, Amperometric l-Lactate sensor based on sol-gel processing of an enzyme-linked silicon alkoxide. Anal. Chem. 79(10), 3757–3763 (2007)
E. Szymańska, K. Winnicka, Stability of chitosan—a challenge for pharmaceutical and biomedical applications. Mar. Drugs 13(4), 1819–1846 (2015)
R. Garjonyte, V. Melvydas, A. Malinauskas, Mediated amperometric biosensors for lactic acid based on carbon paste electrodes modified with baker’s yeast Saccharomyces cerevisiae. Bioelectrochemistry 68(2), 191–196 (2006)
M. Piano, S. Serban, R. Pittson, G.A. Drago, J.P. Hart, Amperometric lactate biosensor for flow injection analysis based on a screen-printed carbon electrode containing Meldola’s Blue-Reinecke salt, coated with lactate dehydrogenase and NAD+. Talanta 82(1), 34–37 (2010)
N. Hamdi, J. Wang, H.G. Monbouquette, Polymer films as permselective coatings for H2O2-sensing electrodes. J. Electroanal. Chem. 581(2), 258–264 (2005)
K. Bridge, F. Davis, S. Collyer, S.P.J. Higson, Flexible ultrathin PolyDVB/EVB composite membranes for the optimization of a whole blood glucose sensor. Electroanalysis 19(4), 487–495 (2007)
K. Bridge, F. Davis, S.D. Collyer, S.P.J. Higson, Flexible ultrathin PolyDVB/EVB composite membranes for the optimization of a lactate sensor. Electroanalysis 19(5), 567–574 (2007)
S. Cosnier, Biosensors based on electropolymerized films: new trends. Anal. Bioanal. Chem. 377(3), 507–520 (2003)
C. Qin et al., Amperometric enzyme electrodes of glucose and lactate based on poly(diallyldimethylammonium)-alginate-metal ion-enzyme biocomposites. Anal. Chim. Acta 720, 49–56 (2012)
A. Radoi, D. Moscone, G. Palleschi, Sensing the lactic acid in probiotic yogurts using an L-Lactate biosensor coupled with a microdialysis fiber inserted in a flow analysis system. Anal. Lett. 43(7–8), 1301–1309 (2010)
J.J. Burmeister, M. Palmer, G.A. Gerhardt, l-lactate measures in brain tissue with ceramic-based multisite microelectrodes. Biosens. Bioelectron. 20(9), 1772–1779 (2005)
J. Park, J. Chang, M. Choi, J.J. Pak, D.-Y. Lee, Y.K. Pak, Microfabirated clark-type sensor for measuring dissolved oxygen. IEEE Sens. 1412–1415 (2007)
K.K. Tremper, T.W. Rutter, J.A. Wahr, Monitoring oxygenation. Curr. Anaesth. Crit. Care 4(4), 213–222 (1993)
C. Cody, D.J. Buggy, B. Marsh, D.C. Moriarity, Subcutaneous tissue oxygen tension after coronary revascularisation with and without cardiopulmonary bypass. Anaesthesia 59(3), 237–242 (2004)
I. Bromley, Transcutaneous monitoring—understanding the principles. Infant 4(3), 95–98 (2008)
L.S. Mortensen et al., Identifying hypoxia in human tumors: a correlation study between 18F-FMISO PET and the Eppendorf oxygen-sensitive electrode. Acta Oncol. 49(7), 934–940 (2010)
T.H. Williamson, J. Grewal, B. Gupta, B. Mokete, M. Lim, C.H. Fry, Measurement of PO2 during vitrectomy for central retinal vein occlusion, a pilot study. Graefes Arch. Clin. Exp. Ophthalmol. 247(8), 1019–1023 (2009)
Y.-H. Park, Y.-B. Shui, D.C. Beebe, Comparison of two probe designs for determining intraocular oxygen distribution. Br. J. Ophthalmol. p. bjo.2010.186064 (2010)
L. Toma-Daşu, A. Waites, A. Daşu, J. Denekamp, Theoretical simulation of oxygen tension measurement in tissues using a microelectrode: I. The response function of the electrode. Physiol. Meas. 22(4), 713–725 (2001)
R.A. Linsenmeier, C.M. Yancey, Improved fabrication of double-barreled recessed cathode O2 microelectrodes. J. Appl. Physiol. Bethesda Md 1985, 63(6), 2554–2557 (1987)
F.B. Bolger et al., Characterisation of carbon paste electrodes for real-time amperometric monitoring of brain tissue oxygen. J. Neurosci. Methods 195(2), 135–142 (2011)
G.S. Wilson, R. Gifford, Biosensors for real-time in vivo measurements. Biosens. Bioelectron. 20(12), 2388–2403 (2005)
G.S. Wilson, M. Ammam, In vivo biosensors. FEBS J. 274(21), 5452–5461 (2007)
A. Guiseppi-Elie, S. Brahim, G. Slaughter, K.R. Ward, Design of a subcutaneous implantable biochip for monitoring of glucose and lactate. IEEE Sens. J. 5(3), 345–355 (2005)
A.R.A. Rahman, G. Justin, A. Guiseppi-Elie, Towards an implantable biochip for glucose and lactate monitoring using microdisc electrode arrays (MDEAs). Biomed. Microdevices 11(1), 75–85 (2008)
A.R.A. Rahman, G. Justin, A. Guiseppi-Wilson, A. Guiseppi-Elie, Fabrication and packaging of a dual sensing electrochemical biotransducer for glucose and lactate useful in intramuscular physiologic status monitoring. IEEE Sens. J. 9(12), 1856–1863 (2009)
A. Guiseppi-Elie, An implantable biochip to influence patient outcomes following trauma-induced hemorrhage. Anal. Bioanal. Chem. 399(1), 403–419 (2010)
C.N. Kotanen, A. Guiseppi-Elie, Characterization of a wireless potentiostat for integration with a novel implantable biotransducer. IEEE Sens. J. 14(3), 768–776 (2014)
Pinnacle Technology, Inc. [Online]. Available: http://www.pinnaclet.com/. Accessed: 28 Jan 2016
A. Weltin, B. Enderle, J. Kieninger, G.A. Urban, Multiparametric, flexible microsensor platform for metabolic monitoring. IEEE Sens. J. 14(10), 3345–3351 (2014)
C.A. Cordeiro, M.G. de Vries, W. Ngabi, P.E. Oomen, T.I.F.H. Cremers, B.H.C. Westerink, In vivo continuous and simultaneous monitoring of brain energy substrates with a multiplex amperometric enzyme-based biosensor device. Biosens. Bioelectron. 67, 677–686 (2015)
G. Calia et al., Biotelemetric monitoring of brain neurochemistry in conscious rats using microsensors and biosensors. Sensors 9(4), 2511–2523 (2009)
A.J. Bard, L.R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn. (Wiley, USA)
P.T. Kissinger, W.R. Heineman, Cyclic voltammetry. J. Chem. Educ. 60(9), 702 (1983)
P. Kissinger, W.R. Heineman, Laboratory Techniques in Electroanalytical Chemistry, Second Edition, Revised and Expanded (CRC Press, New York, 1996)
A.J. Bard, M. Stratmann, F. Scholz, C.J. Pickett, Encyclopedia of Electrochemistry, 7A, Inorganic Chemistry
A. Hierlemann, U. Frey, S. Hafizovic, F. Heer, Growing cells atop microelectronic chips: interfacing electrogenic cells in vitro with CMOS-based microelectrode arrays. Proc. IEEE 99(2), 252–284 (2011)
M. Mollazadeh, K. Murari, G. Cauwenberghs, N. Thakor, Wireless micropower instrumentation for multimodal acquisition of electrical and chemical neural activity. IEEE Trans. Biomed. Circuits Syst. 3(6), 388–397 (2009)
M. Sprenger, Learning and Memory: The Brain in Action. ASCD (1999)
R. Genov, M. Stanacevic, M. Naware, G. Cauwenberghs, N.V. Thakor, 16-channel integrated potentiostat for distributed neurochemical sensing. IEEE Trans. Circuits Syst. Regul. Pap. 53(11), 2371–2376 (2006)
K. Murari, M. Stanacevic, G. Cauwenberghs, N.V. Thakor, Integrated potentiostat for neurotransmitter sensing. IEEE Eng. Med. Biol. Mag. 24(6), 23–29 (2005)
J. Lerma, A.S. Herranz, O. Herreras, V. Abraira, R.M. del Rio, In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 384(1), 145–155 (1986)
M. Stanacevic, K. Murari, A. Rege, G. Cauwenberghs, N.V. Thakor, VLSI potentiostat array with oversampling gain modulation for wide-range neurotransmitter sensing. IEEE Trans. Biomed. Circuits Syst. 1(1), 63–72 (2007)
M. Roham et al., A wireless IC for wide-range neurochemical monitoring using amperometry and fast-scan cyclic voltammetry. IEEE Trans. Biomed. Circuits Syst. 2(1), 3–9 (2008)
S. Ayers, K. Berberian, K.D. Gillis, M. Lindau, B.A. Minch, Post-CMOS fabrication of working electrodes for on-chip recordings of transmitter release. IEEE Trans. Biomed. Circuits Syst. 4(2), 86–92 (2010)
M.H. Nazari, H. Mazhab-Jafari, L. Leng, A. Guenther, R. Genov, CMOS neurotransmitter microarray: 96-channel integrated potentiostat with on-die microsensors. IEEE Trans. Biomed. Circuits Syst. 7(3), 338–348 (2013)
G. Massicotte, S. Carrara, G. Di Micheli, M. Sawan, A CMOS amperometric system for multi-neurotransmitter detection. IEEE Trans. Biomed. Circuits Syst. 99, 1–1 (2016)
C. Yang, M.E. Denno, P. Pyakurel, B.J. Venton, Recent trends in carbon nanomaterial-based electrochemical sensors for biomolecules: a review. Anal. Chim. Acta 887, 17–37 (2015)
S. Sainio et al., Integrated carbon nanostructures for detection of neurotransmitters. Mol. Neurobiol. 52(2), 859–866 (2015)
N. Xiao, B.J. Venton, Rapid, sensitive detection of neurotransmitters at microelectrodes modified with self-assembled SWCNT forests. Anal. Chem. 84(18), 7816–7822 (2012)
S.B. Hočevar, J. Wang, R.P. Deo, M. Musameh, B. Ogorevc, Carbon nanotube modified microelectrode for enhanced voltammetric detection of dopamine in the presence of ascorbate. Electroanalysis 17(5–6), 417–422 (2005)
K.M. Mitchell, Acetylcholine and choline amperometric enzyme sensors characterized in vitro and in vivo. Anal. Chem. 76(4), 1098–1106 (2004)
M.G. Garguilo, A.C. Michael, Enzyme-modified electrodes for peroxide, choline, and acetylcholine. TrAC Trends Anal. Chem. 14(4), 164–169 (1995)
J. Cui, N.V. Kulagina, A.C. Michael, Pharmacological evidence for the selectivity of in vivo signals obtained with enzyme-based electrochemical sensors. J. Neurosci. Methods 104(2), 183–189 (2001)
I. Suzuki, M. Fukuda, K. Shirakawa, H. Jiko, M. Gotoh, Carbon nanotube multi-electrode array chips for noninvasive real-time measurement of dopamine, action potentials, and postsynaptic potentials. Biosens. Bioelectron. 49, 270–275 (2013)
M. Ganesana, J.S. Erlichman, S. Andreescu, Real-time monitoring of superoxide accumulation and antioxidant activity in a brain slice model using an electrochemical cytochrome c biosensor. Free Radic. Biol. Med. 53(12), 2240–2249 (2012)
P. Fattahi, G. Yang, G. Kim, M.R. Abidian, A review of organic and inorganic biomaterials for neural interfaces. Adv. Mater. 26(12), 1846–1885 (2014)
M.M. Ahmadi, G.A. Jullien, Current-mirror-based potentiostats for three-electrode amperometric electrochemical sensors. IEEE Trans. Circuits Syst. Regul. Pap. 56(7), 1339–1348 (2009)
M.M. Ahmadi, G.A. Jullien, A very low power CMOS potentiostat for bioimplantable applications, in Fifth International Workshop on System-on-Chip for Real-Time Applications (IWSOC’05) (2005), pp. 184–189
L. Busoni, M. Carlà, L. Lanzi, A comparison between potentiostatic circuits with grounded work or auxiliary electrode. Rev. Sci. Instrum. 73(4), 1921–1923 (2002)
P. Kassanos, R.K. Iles, R.H. Bayford, A. Demosthenous, Towards the development of an electrochemical biosensor for hCGβ detection. Physiol. Meas. 29(6), S241 (2008)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Anastasova, S., Kassanos, P., Yang, GZ. (2018). Electrochemical Sensor Designs for Biomedical Implants. In: Yang, GZ. (eds) Implantable Sensors and Systems. Springer, Cham. https://doi.org/10.1007/978-3-319-69748-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-69748-2_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-69747-5
Online ISBN: 978-3-319-69748-2
eBook Packages: Computer ScienceComputer Science (R0)