
Abstract
Hepatocellular carcinoma (HCC), a major type of liver cancer, is a clinically and biologically heterogeneous malignancy. Recent advances in next-generation sequencing (NGS), a massively parallel nucleotide sequencing technology, allow researchers for cost-effective and simultaneous identification of genetic alterations, transcriptomic changes at much greater sensitivity, and accuracy. A large scale of mutational screening, gene expression examination, and their integration via NGS have been done in HCCs. These studies may enable the identification of new HCC genetic drivers and provide clues for HCC treatment using molecular-targeted therapies. The recent HCC NGS studies in this decade and their potential clinical utilization, with a focus on HCC heterogeneity, are discussed in this chapter.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Hepatocellular carcinoma
- Tumor heterogeneity
- Next-generation sequencing
- DNA deep sequencing
- RNA deep sequencing
- Small RNA deep sequencing
- Genetic alteration
4.1 Hepatocellular Carcinoma and Its Heterogeneity
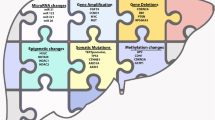
Hepatocellular carcinoma (HCC) accounts for 80% of all primary liver cancers worldwide and is a clinically and biologically heterogeneous disease. It ranks at the fifth most prevalent cancer and the third most deadly malignancy globally. Although most HCC patients are resided in eastern Asia and sub-Saharan Africa, it is along the way to be more prevalent in Western countries [1,2,3,4,5].
4.1.1 HCC Heterogeneity in Etiology
Various risk factors give rise to HCC, and the tumorigenesis of HCC exerts distinct regional differences [5]. Hepatitis B virus (HBV) infection and hepatitis C virus (HCV) infection are the two major risk factors of HCC, accounting for about 80% of HCC tumorigenesis globally. HBV infection is the dominant risk factor in eastern Asia and sub-Saharan Africa. While in Europe and North America, more than half of HCC are due to HCV infection [6,7,8]. Analysis of HBV genome sequencing data identifies eight distinct genotypes (A–H), which also exert obvious geographical and ethnic distributions [7]. HBV infection generally gives rise to the integration of viral DNA into the host genome, especially in hosts with attenuated immunity. While as a RNA virus, HCV is not able to integrate into host genome. HCV tends to escape from host’s immune responses, implicates in chronic infection, and gives rise to liver cirrhosis. HCV proteins seem to change many potentially oncogenic pathways and promote the malignant transformation of hepatocytes [1, 9]. In addition, hepatitis delta virus (HDV) is a defective RNA virus, which could also contribute to hepatic carcinogenesis [10, 11].
Many metabolic-related diseases also contribute to HCC development, which includes alcoholic fatty liver disease (AFLD), non-AFLD (NAFLD), diabetes, obesity, etc. Alcohol consumption and alcoholism is prevalent worldwide. Excessive alcohol consumption may cause AFLD, which ultimately gives rise to HCC [12,13,14]. NAFLD is one of the prevalent clinicopathological syndromes associated with insulin resistance and dyslipidemia. Synergistically with chronic HCV infection, alcoholic liver injury, or risk factors, NAFLD evolves into cirrhosis and HCC ultimately. As the prevalence of obesity in the industrialized countries, NAFLD has been a dominant element for chronic liver disease [15, 16]. Obesity itself is also proposed to relate with increased risk of HCC. In patients with chronic viral infection, obesity synergistically increases the risk of HCC by 100 folds [17, 18]. Meanwhile, diabetes is an independent risk factor for the development of HCC and ranks second of the most prevalent cause for HCC in the USA after viral infection [19,20,21].
In addition, HCC has been also associated with the increased exposure to aflatoxin B1, which mainly happened in Africa and Asia. Other HCC risk factors include neonatal hepatitis, autoimmune hepatitis, hereditary diseases (hemochromatosis, a-1AT deficiency, tyrosinemia, and Wilson’s disease), other immune disorders, etc. [2, 22, 23].
4.1.2 HCC Heterogeneity in Clinical Presentation
HCC develops generally in a previously diseased liver that is related to various HCC risk factors as above. Either HCC tumor or previously diseased liver can be at different disease-progressing stage at the time of diagnosis and has diverse therapeutic perspectives. HCC population is therefore very heterogeneous. According to tumor size, differentiation grate, and nodular type, HCC could be presumably divided into early HCC and progressed HCC [24, 25]. The global HCC BRIDGE study scientifically documents various therapeutic approaches across regions and/or countries [26]. Tumor classification can help us identify the difference between various subtypes of HCC with comparable criteria and possibly provide specific subgroup candidate patients with appropriate therapeutic interventions [27].
A number of HCC staging systems and related treatment algorithms have been developed, which facilitate prognosis and guide clinical practice, prolonging survival of HCC patients eventually. These systems include Barcelona-Clinic Liver Cancer (BCLC) staging system, the Cancer of the Liver Italian Program (CLIP) score system, Okuda system, the Hong Kong Liver Cancer (HKLC) classification system, etc. [28,29,30,31]. Among them, HCC BCLC staging and treatment algorithm has been widely applied. At early stage, HCC patients with carcinoma in situ can be reliably curable. Curative therapies include liver transplantation, tumor resection, and local ablation. However, the number of nodules is relevant to resectability, and post-resection survival rates are variant [32]. Most HCC patients are diagnosed at intermediate stage and even advanced stage. In this case, trans-arterial chemoembolization (TACE) is the first-line option to extend live-life expectancy, but the overall survival is variable and limited. For those failed by TACE, sorafenib (a multi-tyrosine kinase inhibitor) and radioembolization merit consideration [32]. Sorafenib currently is the only FDA-approved systemic therapy for advanced HCC. However, it only has a modest 2-month survival benefit for HCC patients without any preselection in several clinical trials.
4.1.3 HCC Heterogeneity at the Biological Level
Multiple array technologies including expression examination methods (cDNA/oligo/noncoding RNA arrays) and genetic assays (CGH/methylation arrays) have provided powerful tools to amend our understanding on the tumorigenesis, progression, and metastasis of HCC and hold potential of improving HCC therapeutic efficacy [33,34,35,36,37,38,39,40,41,42,43,44]. The accumulated studies have revealed that diverse changes in HCCs were associated with different viral backgrounds as well as different HCC subtypes.
Chronic HBV and HCV hepatic lesions present differential gene expression profiles. Genes being affected by HBV were related to inflammation, while HCV-associated genes were involved into the anti-inflammatory process [45]. Studies have also revealed increases of DNA copy number at 10p and decreases at 10q in HCV-HCCs [46], while gains on 1q, 6p, 8q, 9p, and losses at 1p, 16q, and 19p in many HBV-HCCs [47].
Several array studies have compared numerous HCC tumors to distinguish HCC subtypes. Via the combination of genomic and transcriptomic analysis, researchers have identified six robust HCC subclasses with distinct activation of biological pathways and therapeutic implications [48, 49]. Primary HCC tissues with a propensity for metastasis had a significantly different cDNA and microRNA expression profiles compared to profiles of relapse-free HCC tissues [40, 41, 43]. A 20-miRNA metastasis signature was significantly associated with recurrence in HCC at early stage [34].
In our group, we have found that a subgroup of HCCs with EpCAM and alpha-fetoprotein (AFP) high expression displayed a high rate of metastasis and poor outcome and were biologically different from other HCCs based on their stem cell-related gene expression profiles and singling pathway analysis [37, 42, 50]. Stem cell-related signaling pathways were highly active in these HCC cases, and isolated EpCAM + AFP + HCC cells were enriched hepatic cancer stem cells. In addition, our studies also revealed that HCC patients with low level of microRNA-26 had a significant prolonged survival after adjuvant interferon alpha (IFNα) therapy compared to the control treatment group. However, HCC patients with high level of microRNA-26 did not have survival benefit from adjuvant IFNα therapy [51, 52].
In this vein, HCC is a heterogeneous disease. It is thus important to identify the HCC tumor subtypes, in-depth study of each subtype using an unbiased high-throughput method to explore the potential early diagnostic, prognostic, as well as therapeutic biomarkers. The array technologies have been well established and widely used. However, they mainly focus on gene expression, only to detect known genes, and are at one genomic level per technology at a time. Recent advances in massively parallel nucleotide sequencing technologies allow for simultaneous identification of genetic substitutions, insertion/deletions, expression changes, and structural alterations with high accuracy and sensitivity. It is so-called next-generation sequencing (NGS) technology.
4.2 Next-Generation Sequencing System
NGS is now known as high-throughput parallel nucleotide sequencing. In 2005, a new massively parallel sequencing technique emerged and sequenced over 20 megabase data in a single run, which eventually launched the “next-generation” of genomic science [53]. Since that, NGS has largely revolutionized omics study in this decade. Now, the wide application of NGS has transformed the way scientists think about genetic information. Currently there are a number of different modern sequencing technologies including three dominate commercial platforms, i.e., Roche Genome Sequencer, Illumina Genome Analyzer, and Life Technologies Sequencing by Oligo Ligation Detection (SOLiD) System (Table 4.1).
4.2.1 Roche 454 Sequencing
In 2005, 454 Life Sciences (now Roche) developed the first commercially available NGS platform. Now the 454 family of platforms has been utilized for many applications due to its long reads. The overall approach for 454 is pyrosequencing based. It depends on the detection of pyrophosphate release on nucleotide incorporation [54]. The library DNAs with adapters are prepared using PCR primers or by ligation. These DNAs are then fixed to amplification beads followed by emulsion PCR. An emulsion PCR step produces a set of beads, and each set contains many cloned copies from the same DNA. The beads are loaded into PicoTiterPlate with one bead per well, and the sequencing by synthesis begins in a system with a group of enzymes and the substrates. The four DNA nucleotides are added sequentially in a fixed order. A nucleotide complementary to the template strand triggers pyrophosphate release, which generates a signal being recorded to infer the sequence of the DNA fragments as each base type is added. During the nucleotide flow, millions of copies of DNA are sequenced in parallel.
Currently Roche sequencing platforms are mainly GS-FLX and GS Junior Systems, which may be a good choice for certain applications where long read lengths are needed [55]. GS-FLX Titanium could generate about one million reads with the length of 700 bp per run within 24 h [53], while GS Junior Systems simplifies the library preparation and data processing.
The advantages of Roche sequencing are long read length (which generally offering higher accuracy) and sequencing speed (about 10 h). One shortcoming for this approach is the misidentification of homopolymers length [56]. In addition, 454 is relatively cost-ineffective compared to other sequencing platforms such as Illumina and SOLiD [57]. For downstream analysis, the GS Data analysis software packages are also available.
4.2.2 Illumina Sequencing
Illumina sequencing, Solexa platform, was first introduced in 2006. Now Illumina produces the most widely used platforms and have been used by numerous researchers due to its production of a large amount of data in a cost-effective manner. Similar to 454 technology, Illumina sequencing also uses a sequencing-by-synthesis method [58,59,60]. There are two major differences of Illumina technology from 454 sequencing. One is that Illumina uses a flow cell with coated oligoes instead of microwells with beads. As the DNA enters the flow cell, one of the adapters attaches to a complementary oligo. The other is that Illumina sequencing uses fluorescent reversible termination approach instead of pyrosequencing. A reversible terminator is on every nucleotide to prevent multiple additions in one round, one base per round and one unique emission for each of the four bases. After each round, the added base is recorded.
Now Illumina has many different sequencing platforms on the market including Genome Analyzer, HiSeq, MiSeq, and NextSeq [58, 59]. Each one has several different versions. Among them, MiSeq could produce the longest reads, about 300 bp and use the minimum sequencing time. It could sequence one sample in 10 h including sample and library preparation. MiSeqDx platform is the only one approved by the FDA for in vitro diagnostics. HiSeq could produce the greatest output amount. For example, HiSeq 2500 could generate four billion reads with 125 bp/read in a single run.
The superiorities of the Illumina system include small sample requirements, simple process, short run time, and high-quality data [58, 59]. It also has the proper data analysis flow and tools developed by Illumina so that the researchers can easily analyze and manage genome data. The major flaw is false positive when identifying sequence variations [60]. At this stage, more than 90% of sequencing data have been produced via Illumina technology.
4.2.3 SOLiD Sequencing
SOLiD system is appeared in 2006 and commercialized in 2009. This technology uses a sequencing-by-ligation method. After adaptor ligation and emulsion PCR, the library DNAs are sequenced in an entirely different method compared to 454 and Illumina. In SOLiD there are a set of four fluorescently labeled di-base probes and four di-base probes per dye. It uses DNA ligase for di-base incorporation, which makes a “sequencing by ligation” approach. Following each ligation cycle, the system removes the extension product and resets DNA template with a primer complementary to the n − 1 position for the next round ligation. There are several rounds of reset, by which each base is interrogated in two independent ligation reactions. Due to two-base probing and two-color encoding per base, the sequencing accuracy is very high (about 99.99%), and the systemic noise is very low [61, 62].
Now the updated version of SOLiD system can produce a mass of data, and the cost is substantially low. Since its read has relatively short length (up to 75 bp) but high accuracy, SOLiD technology has some advantages in detecting single-nucleotide polymorphism (SNP), small RNA sequencing, and ChIP-seq over other sequencers [55].
4.2.4 Ion Personal Genome Machine
Ion Torrent entered the sequencing market in 2010. The first semiconductor sequencing device was Ion Personal Genome Machine. This technology is mainly used for small genome sequencing and exome sequencing. It begins with a similar method with 454, which uses a chip of microwells containing beads being fixed with DNA fragments [63]. However, this chip is a semiconductor chip with micro-detectors sensitive to pH. As a base incorporation, a proton is released and alters the surrounding pH, and micro-detector could record the change. As each base type is added in turn and washed sequentially, the sequence is informed. Now, the accuracy rate of this instrument on a per read basis averages approximately 99%.
Ion Torrent Personal Genome Machine technology does not require fluorescence and camera recording, which leads to a higher speed, smaller instrument size, and lower cost. It now produces the highest output. Ion Torrent Sequencer can complete a DNA sequencing workflow in just 1 day. Since Ion AmpliSeq being launched in 2012, this technology has seen broad global adoption. However, similar to 454, Ion Torrent suffers homopolymer-related errors.
4.2.5 The Third-Generation Sequencers
Above NGS technologies start by fragmenting and amplifying DNA, which often sacrifices vital long-range connectivity. The third-generation sequencing is then developed to possibly overcome such defects. It has two major characteristics differing from the above sequencing technologies. One is that amplification of DNA fragments is not needed before sequencing. The other is that the base signal is obtained in real time during the enzyme reaction of adding nucleotide. Thus, the third-generation sequencing gains the advantages of high speed and long read length. Currently it mainly concentrates in the optical or electrical signal detection at single molecular level, such as single-molecule real-time (SMRT) and MinION system [64].
SMRT was developed by Pacific Biosciences, which uses nanotechnology (zero-mode waveguide, ZMW) [54]. The ZMW is a structure that is small enough to observe only a dye-labeled single nucleotide of DNA being incorporated by DNA polymerase. Four different fluorescent dyes are attached to A, T, C, and G. The sequencing is performed on a chip containing numerous ZMW detectors. As DNA strands are synthesized, the dye-labeled nucleotide incorporation is imaged in real time. SMRT completely depends on the role of DNA polymerase that enabled the length of sequencing.
The MinION system was released by Oxford Nanopore Technologies in 2014, which delivered long read real-time sequencing of individual molecules. It is the first commercial nanopore-based sequencer, small and portable. Nanopore is a tiny biopore with diameter in nanoscale and can facilitate ion exchange. Current MinION nanopore sequencing methods rely on the measurement of changes in ionic current at the time of a DNA molecule translocating through a protein nanopore. Biological nanopores aim at single nucleotide, so that this technique has good continuity and accuracy. Moreover, there is no need DNA polymerase ligase or dNTPs or complex optical detection system. It could potentially reach long read at length over 5 kb.
Fluorescence resonance energy transfer (FRET) is another third-generation sequencing technology, which is developed by VisiGen (now Life Tech). It uses a four-color set of FRET dideoxy nucleotide terminators. The fluorescence is cleaved off during the base incorporation and generates an optical signal to achieve the purpose of testing the sequence of DNA bases. The obvious advantages of FRET sequencing are simple and straightforward; the speed can reach one million bases per second.
Now sequencing technologies are widely utilized for mutation profiles, gene expression analysis, methylation analysis, metagenomics, disease-related gene identification, etc. [65]. It has also started to provide service of establishing personal genome information as well as noninvasive prenatal testing [66]. Since these, NGS has accelerated biological research by providing researchers a better understanding of the biology of diseases including carcinogenesis.
4.3 HCC Genomics Studies via NGS Technology
NGS has provided a sensitive, accurate, and cost-effective method to uncover the genetic basis of human disease including cancer at a single-nucleotide resolution [67, 68]. The tumorigenesis and progression of HCC are accompanied by the accumulation of somatic genetic variations. Previous microarray-based technologies have analyzed variations of HCC genome, transcriptome, epigenome, etc., which improved our understanding on HCC tumorigenesis, progression, and inter-/intra-heterogeneity as well as promoted HCC translational research. Here we summarized the original NGS studies in HCC as well as its potential clinical utilization, with an emphasis on understanding HCC heterogeneity (Table 4.2).
4.3.1 HCC Viral Risk Factors in Viral-Related HCCs
4.3.1.1 HBV Integration in HBV-Related HCCs
HBV is the most prevalent risk factor of HCC and the integration of HBV DNA into the host genome has been reported in 1980s [69]. Now, deep sequencing has been successfully applied to the study of HBV genome integration.
Whole-genome sequencing of 11 HBV-related HCC samples has revealed the HBV genome integration to the telomerase reverse transcriptase (TERT) locus [70], which is consistent with previous reports [71]. After that, a study of whole-genome sequencing using 88 HCC patients (81 HBV+ and 7 HBV−) further noted that HBV integration was more significantly frequent in tumors compared to control liver tissues and that about 40% of HBV breakpoints were around the HBV X and core genes [72]. Moreover, via a comparison of gene expression array data between tumors with and without HBV integration, the authors reported that five genes (TERT, MLL4, CCNE1, SENP5, and ROCK1) were recurrently affected by HBV integration. HCC patients with several HBV integration sites had shorter overall survival time [72].
One group recently reported that HBV was apt to integrate into promoters of genes, and recurrent integration into the promoter of TERT seemed to increase the expression level of TERT. HBx 3′-end was preferred to involve into integration, resulting into the expression of HBV-human chimeric proteins [73]. Li et al. developed high-throughput viral integration detection method to enrich and sequence HBV fragments. They identified 246 integration breakpoints in the gene TERT, MLL4, and CCNE1 [74]. Meanwhile, the whole-exome sequencing and oncovirome sequencing in 68 HBV positive HCC cases have also revealed a group of genes close to the recurrent HBV integrations, including TERT, MLL4, ALOX5, etc. [75].
4.3.1.2 HCV Quasispecies Diversity in Chronic Liver Disease and HCC
In human, HCV presents as a group of genetic variants, which is known as “quasispecies.” HCV quasispecies has been revealed to act as an important factor in HCC pathogenesis [76]. Park et al. performed pyrosequencing to compare the structural protein-coding genes of HCV genome between patients with chronic hepatitis C (n = 26) and HCC patients (n = 23) [77]. Data analysis revealed that quasispecies diversity in HCV E1 was significantly lower in HCC patients compared to patients chronic HCV, and 14 amino acid positions significantly differed between two groups. Miura et al. also conducted deep sequencing of serum samples from 79 HCV-infected patients (25 of chronic hepatitis, 29 of liver cirrhosis, 25 HCCs) to examine the association of HCV quasispecies with HCC [78]. They have found the HCV core amino acid that 70 residue sequencing data could reflect the status of liver disease. The ratio of mutant residue to wild-type one in HCV core was increased as liver disease advanced to liver cirrhosis and HCC.
4.3.2 Mutational Landscapes in HCC
NGS technologies have discovered many known and novel genetic alterations in numerous cancers including HCC. Recently, accumulated NGS studies in HCC have suggested that some genetic alterations being grouped in several important HCC oncogenic pathways are likely to be oncogenic driver mutations [70, 75, 79,80,81,82].
4.3.2.1 WNT/Beta-Catenin Pathway and P53 Pathway
It has been known that CTNNB1 (encoding protein beta-catenin) and TP53 are two frequently mutated genes in HCC [83, 84]. Many recent NGS studies in HCCs have confirmed CTNNB1 as the most frequently mutated oncogene while TP53 as the most frequently mutated tumor suppressor [74, 75, 80,81,82, 85]. These studies have also consistently revealed that CTNNB1 mutation was more frequent in HCV and non-viral-related HCC cases (20–40%) but less frequent in HBV-related HCC cases (~10%). In a recent study, Schulze et al. performed a large scale of whole-exome sequencing in 243 HCCs with different etiologic background [82]. In their study, CTNNB1 mutation was associated with alcohol-related HCCs, while Tp53 mutation was related with HBV-HCCs. Further, two studies reported that AXIN1, one of WNT pathway regulators, had a high mutation rate in HCCs [75, 81]. Its mutation was more frequent in HBV-HCCs compared to HCV-HCCs and non-viral-related HCCs [75]. These results indicate that different viral etiologies might activate WNT pathways in distinct ways. Strikingly, about 66% of HCCs presented WNT pathway-related genetic alterations [75]. In P53 pathway, besides TP53 mutation, CDKN2A/CDKN2B, MDM2, and IRF2 mutation have also been noticed in a rate of over 1% in HCC, respectively. Together, about 49% of HCCs presented P53 pathway-related genetic alterations [75, 82].
4.3.2.2 Chromatin Regulators
Using exome sequencing of 24 HCC samples, researchers demonstrated that chromatin regulation pathway was commonly altered by genetic alterations including somatic mutations and gene deletions [80]. They noticed the frequent mutation of ARID1A, a chromatin-remodeling gene, in alcohol-related HCC. This study was further confirmed by Huang and his colleagues [47]. ARID1A mutations were found in 13% of HBV-related HCCs, and mutated ARID1A played an important role in HCC invasion and migration [47]. Interestingly, ARID2 mutation was also identified in HCCs, and its mutation was significantly enriched in HCV-HCC cases compared to HBV-HCCs (14% vs. 2%) [86]. Furthermore, using whole-genome sequencing of HCC samples, Fujimoto et al. found recurrent somatic mutations in a group of chromatin regulation-related genes [70]. These genes were ARID1A, ARID1B, ARID2, MLL, MLL3, etc. In more than 50% of HCC tissues, they noticed mutations in at least one of these chromatin regulator genes. Therefore, dysregulated chromatin remodeling might play a key role in HCC.
Several studies have also revealed that genetic alterations of transcription modulators. Cleary et al. performed whole-exome sequencing for 87 pairs of HCC tumors and adjacent normal tissues and identified several significantly mutated transcription modulators, including genes in NFE2L2-KEAP1 pathways [85]. Totoki’s study has also revealed the frequent alterations in NFE2L2 [75].
4.3.2.3 PI3K/Akt/mTOR-Pathway and MAPK Pathway
Two groups have consistently reported that about 50% of HCC cases have genetic alterations in mTOR/PI3K pathway [75, 82]. They have noticed recurrent inactivating mutations in tuberous sclerosis 1 (TSC1) (3%) and TSC2 (5%), activating mutations and copy again in PIK3CA (2%), and mutations in other modulators including RPS6KA3 (7%), PTEN (3%), DAPK1 (3%), MTOR (2%), etc. In MAPK pathway, a group of growth factors and their receptors have shown mutations in HCCs, including FGF3 (4%), FGF4 (5%), FGF19 (19%), HGF (3%), PDGFRs (3%), IGF1R (2%), etc. Meanwhile, Lin et al. have also detected three cancer-related alternative splicing events including FGFR2, ADAM15, and abundance of FGFR2-IIIc (one of FGFR2 isoform) that were associated with tumor recurrence [87]. Mutations of IGFALS and JAK1 have also been found as key genetic determinants in HCC [88, 89].
4.3.2.4 Others
Fernandez-Banet et al. provided a comprehensive set of somatic genomic rearrangement and gene fusion predictions in HCCs by performing whole-genome sequencing with 88 pairs of primary HCC tumor and non-tumor tissues [90]. They predicted 4314 genomic rearrangements and 260 gene fusions that frequently result in aberrant overexpression of the 3′ genes in tumors. Further, 18 gene fusions, including recurrent fusion (2/88) of ABCB11-LRP2, were validated in HCCs. Xu et al. analyzed copy number variations using DNA sequencing in plasma samples from 31 HCC patients and 8 patients with chronic hepatitis or cirrhosis [91]. They found that copy number variations were recognizable in the majority of HCC plasma samples with large tumor size, and in few HCCs with small tumor size, but not in samples from chronic hepatitis/cirrhosis-related patients. Chen et al. sequenced the exomes of three pairs of HBV-HCC tumor and normal tissues and identified 59 original genes mutated in HBV-associated HCCs [92]. In combination with whole-genome sequencing data from the European Genome-phenome Archive database, 33 of these 59 genes were confirmed, and variants of two mutated genes, ZNF717 and PARP4, were detected in more than 10% of samples from this database. In addition, high-proportion mutations of LAMA2 (encoding an extracellular matrix protein), BAP1, and IDH1 in HCCs have also been reported [81]. The sequencing in three HCCs and adjacent tissue pairs revealed five non-synonymously mutational genes (IRS1, HMGCS1, ATP8B1, PRMT6, and CLU), which were associated with metabolic diseases diabetes and obesity [93].
4.3.3 Expression Profiles of HCC
Besides the genetic changes, whole transcriptome sequencing also reveals gene expression levels from mapped RNA-seq reads. Compared to microarray, RNA sequencing could identify low copy and novel transcripts and is affected at a minimum level by probe efficacy and hybridization condition. Murakami et al. have performed both small RNA sequencing and microarray in 11 HCCs and found that microRNA profiling from sequencing is comparable and reproducible to that from microarray. Moreover, RNA sequencing discovered novel microRNAs (such as miR-9985 and miR-1843) that were otherwise undetectable by array [94]. Wojcicka et al. performed microRNA transcriptome sequencing on total RNAs from 24 paired HCC tumors and adjacent non-tumor tissues [95]. Among all 374 detected microRNAs, miR-122-5p was the most abundant, 64 miRNAs were differentially expressed, and almost every microRNA generated isomiRNAs. Among the most deregulated miRNAs, miR-199a-3p/miR-199b-3p was significantly downregulated in HCCs compared to adjacent non-tumor tissues, expressed in nine isoforms with three different seeds, dramatically activated TGF-β signaling pathway [95].
We have also performed small RNA deep sequencing using isolated EpCAM+ cancer cells with stem cell features and EpCAM− cancer cells with mature hepatocyte features, as well as EpCAM+ normal hepatic stem cells and EpCAM− hepatocytes from health liver donors [39]. Through the comprehensive comparison, we have discovered a group of microRNAs with a specific altered level in purified EpCAM+ hepatic cancer stem cells but not in other cells. The expression of miR-155 in EpCAM+ hepatic cancer stem cells was further validated, and the putative miR-155 targets were correlated with overall survival or time to recurrence [39]. Other groups have revealed the upregulation of a new PIWI-interacting RNA (piR-Hep1) via small RNA deep sequencing using RNAs from an immortalized hepatocyte and two HCC cell lines [96]. The functional study has also discovered that piR-Hep1 was involved in the regulation of HCC cell viability, proliferation, and invasiveness. Selitsky et al. performed small RNA sequencing on liver samples from advanced hepatitis B or C and HCC patients [97]. Compared to microRNAs, small RNAs derived from tRNAs, specifically 5′ tRNA-halves (5′ tRHs), were more abundant in nonmalignant liver. However, 5′ tRH abundance was reduced in matched cancer tissue.
As the development and progression of HCC is a multistage process, Thorgeirsson’s group has performed sequential transcriptome analysis with liver samples in various HCC stages [89]. These samples include tumor-free surrounding liver (n = 7), low (n = 4)- and high (n = 9)-grade dysplastic lesions, early HCCs (n = 5), and progressed HCC (n = 3) from a total of eight HBV-HCC patients. They further integrated genetic and transcriptomic changes during hepatic carcinogenesis to characterize the genomic alteration. In their study, transcriptomes changes of early lesions (from low-grade dysplastic lesion to early HCC) were modest and homogenous. Extensive genetic and transcriptomic alterations occurred at late stage during hepatic carcinogenesis. The deregulated pathways were centered on TGF-beta, WNT, NOTCH, MYC, and EMT-related genes highlighting HCC molecular diversity. Meanwhile, other researchers reported that Aurora B signaling, Wnt pathways, and FOXM1 transcription factor network were altered in HCC via transcriptome sequencing [93]. In addition, two groups have performed transcriptome sequencing on RNAs obtained from rats with or without Aflatoxin B1 (a potent HCC carcinogen) treatment. A group of known and novel transcripts were identified to be differentially expressed under Aflatoxin B1 stress [98, 99].
4.3.4 Epigenetic Alterations in HCC
It has been shown that HCC has large panels of genes with aberrant DNA methylation. Whole-genome bisulfite sequencing could provide a comprehensive view of methylation patterns at single-base resolution across the genome. Chan et al. first explored the detection of genome-wide methylation in plasma from HCC patients using shotgun bisulfite sequencing [100]. Plasma DNAs from 26 HCC patients and 32 non-tumor control subjects were submitted to bisulfite conversion and then massively parallel sequencing. Meanwhile, available tumor DNAs and buffy coat DNAs from 15 HCC cases were also subjected to massively parallel bisulfite sequencing. Analysis of sequencing data revealed hypomethylation was pervasive across the genome. The hypomethylation pattern has high sensitivity and specificity for HCC diagnosis. They further applied the same analysis using copy number variation. However, the diagnostic role of tumor-associated copy number variation is much more dependent on the depth of sequencing. Meanwhile, Shen et al. performed targeted bisulfite sequencing with 24 pairs of HCC tumor and adjacent non-tumor tissues, to investigate associations of DNA methylation and mRNA expression in HCC [101]. In this study, they reported that downregulation of GRASP and TSPYL5 in HCC were regulated by DNA hypermethylation.
4.3.5 Potential Clinical Utilization in HCC
Inter- and intra-tumor heterogeneity has been observed in HCC tumors from both array-based technology and NGS-based technology. Thus HCC patient stratification is important for the introduction of precision medicine for clinical cancer care. Large-scale NGS mutational screening approaches have revealed some key driving signaling pathways in HCC based on the most frequent mutation profiles. Since these, HCC might be subjected to different subgroups based on their genetic alterations in TP53 pathway, WNT pathway, chromatin-remodeling regulation, PI3K/mTOR signaling, MAPK pathway, etc. [75, 82, 89]. For patients who have distinct genetic profiles, different treatment might be required for the best care. It is expected that patient survival will be largely improved with molecular-targeted therapies directed against these pathways. Encouraging data have been shown in a small-scale study. Two HCC patients at advanced stage had genetic alteration in PI3K/AKT/mTOR pathway being identified via targeted NGS and benefited from the treatment of an mTOR inhibitor, everolimus [102]. Schulze et al. reported that 28% of HCC patients harbored at least one damaging genetic alteration potentially targetable by one FDA-approved drug [82]. Meanwhile, some genetic alterations might be potentially related to the drug sensitivity in HCC cells such as NQO1 mutation increasing the sensitivity of HCC cell growth inhibition with HSP90 inhibitor treatment.
In addition, Kelley et al. performed Personal Genome Machine sequencing using DNAs from circulating HCC cells and showed that analysis of genomic interrogation of circulating tumor cells could provide precise information for stratifying patients with metastatic HCC [103]. Ouyang et al. performed whole-genome sequencing with primary HCC and paired lung metastases samples and identified very similar genomic variations including genetic mutations and copy number alterations between primary and metastatic pairs [104]. These indicate the possibility of using the genomic variations to identify the primary tumor site for patients having cancer in multiorgans.
4.4 Conclusion and Prospective
NGS analysis for identifying genetic profiles in human malignancies has become a research priority. It has enabled the identification of new cancer drivers in several solid tumors including lung and melanoma. Molecular-targeted cancer therapies against genetic alterations in oncogenic genes have prolonged patient survival, such as vemurafenib treatment in BRAF-mutated melanoma [105, 106], crizotinib in lung cancer with ALK rearrangements [107]. Unfortunately, the molecular-based HCC treatment stratification has not been fulfilled reached for HCC. Now, sorafenib is the only FDA-approved molecular drug in HCC and could only moderately improve survival of patients with advanced HCC [108]. It might be valuable to test whether HCC patients with genetic alterations in PI3K and MAPK signaling could gain significant survival benefit from sorafenib instead of nonselected HCC populations.
A group of NGS data have shown HCC heterogeneity and identified several possible drivers that might be useful for sub-classifying HCC populations. However, such subgrouping should be further confirmed through basic and clinical investigation. Meanwhile, therapies targeting the most prevalent genetic alterations including TERT, CTNNB1, and TP53 in HCC have not been clinically applied. It is also necessary to discover new therapeutic targets that come from genomic studies assessing chromosomal amplifications or deletions. Overall, HCC NGS studies have improved our understanding on HCC biological features and would eventually contribute to the treatment selection for heterogeneous HCC populations. Further efforts are needed to investigate the application the genomic information in patient decision-making and the utilization of molecular-targeted therapies against genetic alternations in some key signaling pathways.
References
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87.
Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46.
Llovet JM, et al. Hepatocellular carcinoma. Lancet. 2003;362:1907–17.
Lozano R, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–128.
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–73.e1.
Arzumanyan A, et al. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–35.
Pujol FH, et al. Worldwide genetic diversity of HBV genotypes and risk of hepatocellular carcinoma. Cancer Lett. 2009;286:80–8.
Forner A, et al. Hepatocellular carcinoma. Lancet. 2012;379:1245–55.
Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834–47.
Fattovich G, et al. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–50.
Romeo R, et al. High serum levels of HDV RNA are predictors of cirrhosis and liver cancer in patients with chronic hepatitis delta. PLoS One. 2014;9:e92062.
Boffetta P, Hashibe M. Alcohol and cancer. Lancet Oncol. 2006;7:149–56.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27.
Hartling L, et al. Association between alcohol consumption and cancers in the Chinese population—a systematic review and meta-analysis. PLoS One. 2011;6:e18776.
Baffy G, et al. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–91.
Nobili V, et al. A 360-degree overview of paediatric NAFLD: recent insights. J Hepatol. 2013;58:1218–29.
Calle EE, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med. 2003;348:1625–38.
Larsson S, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005–8.
Arase Y, et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology. 2013;57:964–73.
Hsiang JC, et al. Type 2 diabetes: a risk factor for liver mortality and complications in hepatitis B cirrhosis patients. J Gastroenterol Hepatol. 2015;30:591–9.
Lai MS, et al. Type 2 diabetes and hepatocellular carcinoma: a cohort study in high prevalence area of hepatitis virus infection. Hepatology. 2006;43:1295–302.
Feitelson MA, et al. Genetic mechanisms of hepatocarcinogenesis. Oncogene. 2002;21:2593–604.
Nault JC, Zucman-Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol. 2014;60:224–6.
Roncalli M, et al. Histopathological classification of hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S228–34.
Severi T, et al. Tumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targets. Acta Pharmacol Sin. 2010;31:1409–20.
Park JW, et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE Study. Liver Int. 2015;35:2155–66.
Villanueva A, et al. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med. 2010;61:317–28.
Bruix J, et al. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–9.
Yau T, et al. Development of Hong Kong Liver Cancer staging system with treatment stratification for patients with hepatocellular carcinoma. Gastroenterology. 2014;146:1691–700.e3.
Kudo M, et al. Prognostic staging system for hepatocellular carcinoma (CLIP score): its value and limitations, and a proposal for a new staging system, the Japan Integrated Staging Score (JIS score). J Gastroenterol. 2003;38:207–15.
Okuda K, et al. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer. 1985;56:918–28.
Forner A, et al. Treatment of intermediate-stage hepatocellular carcinoma. Nat Rev Clin Oncol. 2014;11:525–35.
Quetglas IM, et al. Integration of genomic information in the clinical management of HCC. Best Pract Res Clin Gastroenterol. 2014;28:831–42.
Budhu A, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907.
Budhu A, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–75.e1.
Ji J, Wang XW. New kids on the block: diagnostic and prognostic microRNAs in hepatocellular carcinoma. Cancer Biol Ther. 2009;8:1686–93.
Ji J, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472–80.
Ji J, et al. Let-7g targets collagen type I alpha2 and inhibits cell migration in hepatocellular carcinoma. J Hepatol. 2010;52:690–7.
Ji J, et al. Identification of microRNAs specific for epithelial cell adhesion molecule-positive tumor cells in hepatocellular carcinoma. Hepatology. 2015;62:829–40.
Kim JW, et al. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology. 2004;39:518–27.
Lee JS, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–76.
Yamashita T, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–24.
Ye QH, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23.
Parpart S, et al. Modulation of miR-29 expression by alpha-fetoprotein is linked to the hepatocellular carcinoma epigenome. Hepatology. 2014;60:872–83.
Honda M, et al. Differential gene expression between chronic hepatitis B and C hepatic lesion. Gastroenterology. 2001;120:955–66.
Hashimoto K, et al. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod Pathol. 2004;17:617–22.
Huang J, et al. Correlation between genomic DNA copy number alterations and transcriptional expression in hepatitis B virus-associated hepatocellular carcinoma. FEBS Lett. 2006;580:3571–81.
Boyault S, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52.
Zucman-Rossi J, et al. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149:1226–39.e4.
Yamashita T, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–61.
Ji J, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–47.
Ji J, et al. Development of a miR-26 companion diagnostic test for adjuvant interferon-alpha therapy in hepatocellular carcinoma. Int J Biol Sci. 2013;9:303–12.
Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80.
Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8.
Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46.
Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nat Biotechnol. 2008;26:1117–24.
Ronaghi M, et al. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 1996;242:84–9.
Feng YJ, et al. Parallel tagged amplicon sequencing of relatively long PCR products using the Illumina HiSeq platform and transcriptome assembly. Mol Ecol Resour. 2016;16:91–102.
Jeon YS, et al. Improved pipeline for reducing erroneous identification by 16S rRNA sequences using the Illumina MiSeq platform. J Microbiol. 2015;53:60–9.
Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–64.
Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–9.
Valouev A, et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–63.
Quail MA, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341.
Schadt EE, et al. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–40.
Buermans HP, den Dunnen JT. Next generation sequencing technology: advances and applications. Biochim Biophys Acta. 2014;1842:1932–41.
Wheeler DA, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6.
Meyerson M, et al. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–96.
Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45.
Brechot C, et al. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–5.
Fujimoto A, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4.
Paterlini-Brechot P, et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–6.
Sung WK, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–9.
Toh ST, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34:787–98.
Li W, et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102:338–44.
Totoki Y, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–73.
Pawlotsky JM. Hepatitis C virus population dynamics during infection. Curr Top Microbiol Immunol. 2006;299:261–84.
Park CW, et al. Comparison of quasispecies diversity of HCV between chronic hepatitis c and hepatocellular carcinoma by Ultradeep pyrosequencing. Biomed Res Int. 2014;2014:853076.
Miura M, et al. Deep-sequencing analysis of the association between the quasispecies nature of the hepatitis C virus core region and disease progression. J Virol. 2013;87:12541–51.
Jiang JH, et al. Clinical significance of the ubiquitin ligase UBE3C in hepatocellular carcinoma revealed by exome sequencing. Hepatology. 2014;59:2216–27.
Guichard C, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–8.
Jhunjhunwala S, et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014;15:436.
Schulze K, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–11.
Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50.
Forbes SA, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–7.
Cleary SP, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–702.
Li M, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–9.
Lin KT, et al. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene. 2014;33:4786–94.
Kan Z, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–33.
Marquardt JU, et al. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J Hepatol. 2014;60:346–53.
Fernandez-Banet J, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103:189–203.
Xu H, et al. Non-invasive analysis of genomic copy number variation in patients with hepatocellular carcinoma by next generation DNA sequencing. J Cancer. 2015;6:247–53.
Chen Y, et al. Exome capture sequencing reveals new insights into hepatitis B virus-induced hepatocellular carcinoma at the early stage of tumorigenesis. Oncol Rep. 2013;30:1906–12.
Meerzaman DM, et al. Genome-wide transcriptional sequencing identifies novel mutations in metabolic genes in human hepatocellular carcinoma. Cancer Genomics Proteomics. 2014;11:1–12.
Murakami Y, et al. Comparison of hepatocellular carcinoma miRNA expression profiling as evaluated by next generation sequencing and microarray. PLoS One. 2014;9:e106314.
Wojcicka A, et al. Next generation sequencing reveals microRNA isoforms in liver cirrhosis and hepatocellular carcinoma. Int J Biochem Cell Biol. 2014;53:208–17.
Law PT, et al. Deep sequencing of small RNA transcriptome reveals novel non-coding RNAs in hepatocellular carcinoma. J Hepatol. 2013;58:1165–73.
Selitsky SR, et al. Small tRNA-derived RNAs are increased and more abundant than microRNAs in chronic hepatitis B and C. Sci Rep. 2015;5:7675.
Merrick BA, et al. RNA-Seq profiling reveals novel hepatic gene expression pattern in aflatoxin B1 treated rats. PLoS One. 2013;8:e61768.
Yang W, et al. Genome-wide miRNA-profiling of aflatoxin B1-induced hepatic injury using deep sequencing. Toxicol Lett. 2014;226:140–9.
Chan KC, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A. 2013;110:18761–8.
Shen J, et al. Integrative epigenomic and genomic filtering for methylation markers in hepatocellular carcinomas. BMC Med Genet. 2015;8:28.
Janku F, et al. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 2014;5:3012–22.
Kelley RK, et al. Circulating tumor cells in hepatocellular carcinoma: a pilot study of detection, enumeration, and next-generation sequencing in cases and controls. BMC Cancer. 2015;15:206.
Ouyang L, et al. Whole-genome sequencing of matched primary and metastatic hepatocellular carcinomas. BMC Med Genet. 2014;7:2.
Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16.
Chapman PB, et al. Frontline approach to metastatic BRAF-mutant melanoma diagnosis, molecular evaluation, and treatment choice. Am Soc Clin Oncol Educ Book. 2014;34:e412–21.
Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–703.
Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
Totoki Y, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–9.
Ding D, et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet. 2012;8:e1003065.
Huang J, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–21.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Wei, X., Liu, N., Wang, X.W., Ji, J. (2018). Genomics Studies in Hepatocellular Carcinoma via Next-Generation Sequencing. In: Liu, C. (eds) Precision Molecular Pathology of Liver Cancer. Molecular Pathology Library. Springer, Cham. https://doi.org/10.1007/978-3-319-68082-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-68082-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68080-4
Online ISBN: 978-3-319-68082-8
eBook Packages: MedicineMedicine (R0)