
Abstract
In certain settings, chemotherapy can trigger an immunogenic form of tumor cell death. More often, however, tumor cell death after chemotherapy is not immunogenic, and may be actively tolerizing. However, even in these settings the dying tumor cells may be much more immunogenic than previously recognized, if key suppressive immune checkpoints such as indoleamine 2,3-dioxygenase (IDO) can be blocked. This is an important question, because a robust immune response to dying tumor cells could potentially augment the efficacy of conventional chemotherapy, or enhance the strength and duration of response to other immunologic therapies. Recent findings using preclinical models of self-tolerance and autoimmunity suggest that IDO and related downstream pathways may play a fundamental role in the decision between tolerance versus immune activation in response to dying cells. Thus, in the period of tumor cell death following chemotherapy or immunotherapy, the presence of IDO may help dictate the choice between dominant immunosuppression versus inflammation, antigen cross-presentation, and epitope spreading. The IDO pathway thus differs from other checkpoint-blockade strategies, in that it affects early immune responses, at the level of inflammation, activation of antigen-presenting cells, and initial cross-presentation of tumor antigens. This “up-stream” position may make IDO a potent target for therapeutic inhibition.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Indoleamine 2,3-dioxygenase
- IDO
- Tolerance
- Tumor microenvironment
- Tumor
- Immunotherapy
- Checkpoint
- Chemotherapy
- Radiation
7.1 Introduction
When established tumors are treated with chemotherapy many tumor cells die, and multiple tumor-associated antigens are released. It is becoming increasingly clear that tumors contain many immunogenic antigens [1, 2]; so, ideally, tumor cell death after chemotherapy should be an opportunity for immune activation [3,4,5]. Unfortunately, under most circumstances, the default response to death of nucleated cells tends to be immunologic tolerance, rather than immune activation. In particular, apoptotic cell death often elicits potent immune suppression, by activating natural tolerogenic mechanisms that normally maintain tolerance to self. Thus, while certain types of chemotherapy, in certain settings, may be spontaneously immunogenic [6], in most cases the immune response following chemotherapy is weak and disappointing. In this chapter we will discuss the possibility that the indoleamine 2,3-dioxygenase (IDO) enzyme may be one tolerogenic pathway that limits the immune response to dying tumor cells.
IDO is one of the regulatory mechanisms that contributes to immune suppression and tolerance in the tumor microenvironment. Like many suppressive pathways that are co-opted by tumors, IDO is a natural mechanism of counter-regulation and tolerance in the immune system. In tumors, IDO can be aberrantly expressed by the tumor cells themselves [7]; or, importantly, IDO can also be naturally induced in host antigen-presenting cells (APCs) by a variety of pro-inflammatory signals. IDO can be induced in response to signals from the adaptive immune system such as IFNγ [8]; or to signals from the innate immune system such as type I interferons [9, 10]; and to pattern-recognition receptors such as TLR4 and TLR9 [11,12,13]. These IDO-inducing signals may be constitutively present in the inflammatory microenvironment of the tumor [8]; they may be actively up-regulated by the dying cells and release of tumor antigens that occurs after chemotherapy; or they may be actively induced by exogenous immunotherapy (checkpoint blockade, adoptive cellular therapy, vaccines or other modalities). In all of these cases, IDO and its related downstream pathways may help create an undesirable tolerogenic milieu, in which the immune system is prevented from responding to antigens released from dying tumor cells.
7.1.1 Natural Role of IDO
IDO is an immunoregulatory enzyme that exerts its biologic effects by degrading the essential amino acid tryptophan [14]. The IDO family includes two closely-related genes, IDO1 and IDO2 [15, 16], both of which catalyze the degradation of tryptophan along the kynurenine pathway. The biologic function of IDO2 is less well studied [17], and in this review we will use the general term “IDO” to include both genes, unless otherwise specified. IDO affects the immune system in two ways: first, by reducing the local concentration of tryptophan; and second, by producing biologically active tryptophan metabolites. Depletion of local tryptophan activates the GCN2 kinase pathway in neighboring cells [18]. GCN2 is a stress-response pathway that is sensitive to depletion of amino acids. Activation of GCN2 inhibits effector cell proliferation and differentiation, and it biases naive CD4+ T cells toward Treg differentiation [18, 19]. In addition, secreted tryptophan metabolites are produced by IDO, comprising kynurenine and its subsequent breakdown products. These metabolites bind to the aryl hydrocarbon receptor (AhR) [20]. Signaling via the AhR can promote Treg differentiation [20], and bias dendtritc cells (DCs) toward an immunosuppressive/tolerogenic phenotype [21, 22]. Thus, IDO acts by multiple pathways to inhibit immune responses.
7.1.1.1 IDO and Acquired Peripheral Tolerance
The IDO pathway is both anti-inflammatory (i.e., it suppresses inflammation from the innate immune system) and tolerogenic (i.e., can create antigen-specific unresponsiveness in T cells). IDO does not participate in central tolerance in the thymus; rather, it acts in the periphery to keep inflammation in check, and to create acquired tolerance to new antigens. Thus, for example, IDO is expressed in the placenta, and pregnant mice treated with an IDO-inhibitor drug spontaneously reject allogeneic fetuses, driven by paternal alloantigens [23,24,25]. In a variety of experimental models of acquired peripheral tolerance, blocking IDO prevents the induction of mucosal tolerance [26, 27], tolerance created by CTLA-4/B7 or CD40 blockade [28,29,30,31], and other forms of acquired peripheral tolerance [32, 33]. Tissue allografts engineered to overexpress the IDO gene are accepted across fully-mismatched MHC barriers without immunosuppression [31, 34, 35]. Conversely, blocking or ablating IDO makes autoimmunity and inflammation markedly worse. Ablating IDO in mouse models of graft-versus-host disease increases lethality [36, 37], and blocking IDO in models of autoimmunity [38,39,40,41] or chronic infection [42, 43] markedly increases inflammation and exacerbates disease severity. In all of these models, the role of IDO is narrow and selective. IDO-deficient mice do not have the broad, spontaneous autoimmunity that is seen with mice lacking CTLA-4 or Tregs. But in the settings where IDO is relevant, this pathway can create potent de novo tolerance.
7.1.1.2 Acquired Tolerance to Apoptotic Cells
One striking example of the tolerogenic role of IDO occurs when mice are exposed to apoptotic cells. When apoptotic cells are injected intravenously they are cleared by specialized macrophages and dendritic cells in the spleen. This process normally produces robust antigen-specific tolerance [44, 45]. In this model, apoptotic cells were found to be potent inducers of IDO expression by CD169+ macrophages in the spleen [46]. Blocking or genetic ablation of IDO prevented the immune system from creating the normal tolerance to antigens associated with apoptotic cells, leading to progressive development of a lethal lupus-like autoimmunity after repeated challenge [46]. Importantly, in this model the apoptotic cells were normal, syngeneic thymocytes, and thus contained no mutational neoantigens. Nevertheless, just the normal array of self antigens associated with apoptotic cells was sufficient to drive rapid breakdown of self-tolerance if the immunosuppressive IDO signal was removed. This natural tolerogenic function of IDO during apoptosis suggests that the IDO pathway might become especially important in tumors during the wave of cell death and antigen release following chemotherapy.
7.1.2 Downstream Mechanisms: IDO-Induced Activation of Tregs
The signals generated by IDO are inherently local and short-range, based on local tryptophan depletion and secretion of bioactive metabolites. Therefore, beyond the immediate vicinity of the IDO-expressing cell these effects would rapidly abate. In tumors and tumor-draining lymph nodes, the number of IDO-expressing host cells is quite small, comprising at most a few percent of total immune cells [47]. Even if the tumor cells themselves express IDO, the distribution is patchy and local. These same observations are also true of IDO expression during infection, autoimmunity, or tolerance to apoptotic cells: in each case, the actual number of IDO-expressing APCs is small. Yet despite this inherently restricted and localized distribution, IDO is able to create robust effects throughout entire lymph nodes, spleen, tumors, and at the systemic (whole-animal) level [9, 12, 13, 18, 31, 46,47,48]. These widespread and systemic effects appear to rely not upon IDO itself, upon the ability of IDO to activate the potent and mobile regulatory T cell (Treg) population.
IDO can drive naive CD4+ T cells to differentiate into Foxp3+ “inducible” Tregs in vitro [19]. In vivo, IDO expressed by CD103+ DCs in the gut was found to be required for de novo generation of Tregs from naive CD4+ T cells during mucosal tolerance [26]. In human cells, plasmacytoid DCs from peripheral blood up-regulate IDO in vitro in response to CpG oligonucleotides [49] or HIV infection [50], and this can induce differentiation of CD4+ cells into Foxp3+ Treg-like cells. Similar findings have been reported using human monocyte-derived DCs [51, 52]. Thus, IDO can bias CD4+ T cells to differentiate towards a regulatory phenotype.
Tumors are dominated by large numbers of Tregs, with a highly activated phenotype [53, 54]. The role of “inducible” (peripherally-generated) Tregs against unique tumor-specific neo-antigens remains somewhat controversial [55, 56]. However, it is not necessary that tumors create their associated Tregs de novo. Even if most of the Tregs in tumors are thymically-derived, and recognize the same set of self antigens found in normal tissues [57], these Tregs may still be recruited to the tumor in abnormally large numbers. More importantly, tumor-associated Tregs may become potently activated by the conditions of the tumor microenvironment. Consistent with this possibility, highly activated Tregs appear rapidly in growing tumors [58], and Tregs in human tumors have high levels of CTLA-4, PD-1 and other markers of activation [54]. Functionally, Tregs isolated from mouse tumor-draining LNs are constitutively pre-activated for in vitro suppression, without requiring any additional signals [12], and similar constitutive Treg activation seems to occur in human tumors [59].
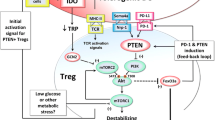
We have shown that mouse plasmacytoid DCs isolated from tumor-draining LNs express IDO, and potently activate resting Tregs in vitro, in an IDO-dependent fashion [12]. This activation was rapid (occurring within hours) and affected pre-existing, fully mature Tregs. In vivo, Tregs from tumor-draining LNs displayed similar potent, IDO-induced suppressor activity. Tregs activated by IDO acquired a characteristic form of suppressor activity characterized by strict dependence on the PD-1/PD-ligand pathway [12]. While IDO is only one of multiple upstream signals by which Tregs may become activated [60,61,62], it is a mechanism that is frequently found in the tumor microenvironment.
Finally, IDO appears to stabilize the suppressive phenotype in Tregs so that they do not become destabilized (lose their suppressor activity) during inflammation. It has been somewhat controversial whether mature, thymic-derived Tregs can ever actually lose their suppressive phenotype [63, 64], but a number of studies now suggest that this may indeed occur in certain biologically-relevant settings of inflammation [65,66,67]. It is certainly true that artificial genetic ablation of key pathways that maintain Treg stability will cause Tregs to convert into pro-inflammatory effector cells, leading to progressive autoimmunity [67,68,69]. We have shown that IDO stabilizes the Treg phenotype in the face of inflammation, by maintaining high levels of the Foxp3 co-repressor Eos (Ikzf4) and preventing IL-6-driven conversion into “helper-like” pro-inflammatory cells [70,71,72,73]. Under normal circumstances, this stabilizing effect of IDO on Tregs is beneficial for maintaining self-tolerance, but in the context of tumors it may instead help maintain the suppressive intra-tumoral milieu, and prevent desirable immune activation during immunotherapy.
In the following sections, we will consider the potential role of IDO in the tumor microenvironment following chemotherapy, during the time that the immune system faces the fundamental decision whether or not to respond to dying tumor cells.
7.2 Tolerance Is a Choice: The Response to Dying Cells Is Dictated by the Local Milieu
In a normal organism, cells are constantly dying and being replaced. Under homeostatic conditions, most of these cells will die by apoptosis, which is classically considered immunologically “silent”. But this silence is not because apoptotic cells are invisible or inherently non-immunogenic; rather, it is because apoptotic cells generate specific signals that actively suppress the immune response and create tolerance [74]. IDO is one of these active tolerogenic signals elicited by apoptotic cells [46, 75]. The IDO pathway in turn is closely linked to production of TGFβ, activation of Tregs, and other known immunosuppressive responses to dying cells [12, 46, 75]. This concept of active immunosuppression by apoptotic cells has an important corollary, which is that tolerance to apoptotic cells is not inherent and inevitable—rather, it is a choice. If the suppressive mechanisms that enforce tolerance are blocked, then the same dying cells may now become spontaneously immunogenic. In the following discussion, we will consider primarily the case of chemotherapy, because this modality is widely used. However, similar molecular mechanisms may apply to the dying tumor cells released by immunologic therapy as well; so the discussion may be equally relevant to epitope-spreading after immunotherapy.
7.2.1 Tolerance to Tumor Cells After Chemotherapy Is Not Inevitable
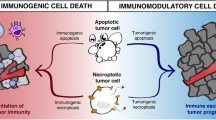
Originally, chemotherapy was assumed to kill tumor cells solely by apoptosis [76]. This implied that cell death after chemotherapy would not be immunogenic. And indeed, in clinical practice this often appears to be the case: e.g., even large chemotherapy-sensitive tumors may melt away without evidence of inflammation or antigen-specific immune response. More recently, however, Drs. Zitvogel, Kroemer and colleagues have shown that, in at least in certain situations, chemotherapy can cause tumor cells to die by much more immunogenic forms of cell death, characterized by exposure of calreticulin and release of HMGB1 or ATP [77,78,79]. This discovery led to the speculation that the immune system might therefore be a fundamental contributor to the overall efficacy of chemotherapy [80]. While this would be an exciting possibility, the contribution of immunogenic cell death to chemotherapy has not been a universal finding in all tumor models, or with all chemotherapy drugs [81]. Immunogenic cell death has been more evident with anthracyclines or oxaliplatin than with other agents; and it is primarily observed in certain transplantable tumors. In the more refractory autochthonous tumors, which have “co-evolved” throughout their existence with the host immune system to create profound immunosuppression and tolerance, the immune system does not appear to contribute to the effects of chemotherapy [82]. Thus, in many settings, the immune system does not seem to play the hoped-for role in the response to chemotherapy.
However, from a therapeutic perspective, the key question is not whether the immune system spontaneously contributes to the effect of standard chemotherapy. Indeed, we know that such spontaneous immune activation is probably often suppressed by endogenous counter-regulatory mechanisms. Rather, the relevant question for therapy is whether dying tumor cells would potentially immunogenic, if these endogenous tolerogenic pathways could be blocked. If the relevant endogenous suppressive pathways can be identified and understood, then these pathways present a rich therapeutic opportunity to capitalize upon the wave of antigens released after chemotherapy. By extension, this same opportunity may arise when tumor cells are killed by adoptive transfer of CAR-T cells, or by active immunization or other immunotherapy (although this setting has not been as well studied).
7.2.1.1 After Chemotherapy, both Tolerogenic and Immunogenic Cell Death Can Occur
The classical form of cell death induced by chemotherapy is apoptosis [83]. This should lead to exposure of phosphatidylserine on the outer leaflet of the cell membrane, which triggers production of immunosuppressive TGFβ by the macrophages that phagocytose the debris. The result—at least in theory—is immune suppression and tolerance. However, not all tumor cells die in such a well-behaved fashion. Depending on the type of cytotoxic insult and the nature of the tumor, dying cells may release pro-inflammatory factors such as HMGB1, ATP or free DNA. These can be sensed by cognate receptors (e.g., TLRs, purinergic receptors or STING) leading to inflammation and immune activation. With certain chemotherapy drugs, in certain tumor models, this immunogenic cell death may be quite robust [77, 78]. However, in most tumors the picture is probably mixed, with much immunosuppressive apoptosis occurring side-by-side with more immunogenic forms of cell death. The question therefore becomes which set of signals exerts the dominant effect on the local immune system.
Unfortunately, tumor-cell death takes place in an environment that is already heavily biased toward immune-suppression. Even prior to chemotherapy, the tumor milieu is usually rich in TGFβ and IL-10, and suppressive Tregs dominate over effector T cells. Similarly, the local macrophage population is biased toward an immunosuppressive “M2”-like phenotype, and many of the local myeloid cells are inhibitory myeloid-derived suppressor cells (MDSCs) rather than pro-inflammatory DCs and monocytes. Further, the tumor cells or host APCs may constitutively over-express IDO, and tumor-draining LNs may be dominated by IDO-expressing APCs. Given this extensive pre-existing bias toward suppression, it is not surprising that the degree of immune response following chemotherapy often appears sub-optimal.
7.2.1.2 In the Absence of Inducible Counter-Regulatory Mechanisms, Dying Cells Can be Highly Immunogenic
In the absence of elicited suppressive signals, however, dying cells themselves can be highly inflammatory. Cells that die by either necrosis or necroptosis release multiple pro-inflammatory mediators and danger signals [84]. Even cells that die by apoptosis can be immunogenic if they are phagocytosed by the right APC populations [85]. Indeed, spontaneous cross-presentation of antigens from necroptotic or apoptotic cells can be important in host defense against viral infections [86]. Thus, the underlying (intrinsic) nature of dying cells may actually be immunogenic, and would bias the immune response toward inflammation and immune responses, unless this process is actively suppressed by counter-regulation.
Consistent with this possibility, studies using in vivo challenge with apoptotic cells have revealed a potent regulatory role for IDO in controlling the choice between tolerance and immunity to dying cells [46, 75]. As described above in Sect. 1.1.2, when the IDO pathway was active then challenge with apoptotic cells led to tolerance induction, with high TGFβ and IL-10, and activation of Tregs. In contrast, when IDO was genetically ablated or blocked with indoximod (D-1MT) then apoptotic cells elicited high levels of IL-6, IL-12 and TNFα, and mice developed lupus autoimmunity. Likewise, genetic ablation of the key IDO-expressing cell type in this system—a population of CD169+ macrophages in the splenic marginal zone—resulted in failure to recruit suppressive Tregs, and inability to create acquired systemic tolerance to neo-antigens delivered on apoptotic cells [87]. Thus, IDO acted as a pivotal regulatory “switch” controlling the natural physiologic response to apoptotic cells. If they were allowed to induce IDO then apoptotic cells were tolerogenic, but if IDO was blocked then the same cells were immunogenic.
It is not yet known whether IDO plays a similar controlling role in the response to dying cells after chemotherapy. However, the importance of IDO in the normal physiologic response to apoptotic cells, and the fact that IDO is already either expressed or rapidly inducible in many tumors, suggest that this could be an important regulatory pathway in this setting.
7.2.1.3 Immunologic Contribution to the Effectiveness of Chemotherapy
Exactly how the immunosuppressive milieu in tumors affects responses to dying tumor cells has been difficult to study. Experimental systems using nominal antigens and TCR-transgenic T cells have yielded mixed results, which are sometimes contradictory. Some mouse models suggest that T cell responses to nominal tumor antigens are robust [88], but others suggest that they are poor and difficult to achieve [89]. One confounding factor in many mouse models is that they do not seem to recapitulate the profound degree of immune-suppression associated with actual human tumors. TCR-transgenic T cells often activate and proliferate robustly just by encountering the tumor, even without chemotherapy or other manipulation. It is unclear whether this occurs because the transplantable mouse tumor cells are not suppressive enough, or because the TCR-transgenic T cells are high-affinity and not readily tolerized. But whatever the cause, this does not at all resemble the situation in real human tumors [90]. Thus, results from experimental models that do not recapitulate this baseline level of immune suppression should probably be interpreted with caution.
This is not to say, however, that the immune system does not influence the response to chemotherapy in humans. Patients with large numbers of tumor-infiltrating T cells have a more favorable response to chemotherapy in breast and colon cancer [91, 92]. While this does not necessarily prove a mechanistic link, it is tempting to speculate that the immune system in these patients responds more robustly after chemotherapy, and this improves the outcome. Attempts are being made to exploit the immunogenicity of chemotherapy in the clinic [93]. Nonetheless, with or without a pre-existing immune infiltrate, the tumor milieu in human patients remains dominated by an array of immunosuppressive factors.
7.2.1.4 Breaking Tolerance to Tumor-Associated Antigens
Fortunately, therapeutic tools for reducing tumor-associated immunosuppression are now becoming available. Blocking antibodies against the CTLA-4 pathway and PD-1/PD-L pathway are approved or in development, and IDO-inhibitors are progressing through Phase I and II trials. Other agents are in the pipeline. Thus, the immunosuppressive nature of the tumor microenvironment is no longer an inevitable condition. However, the array of suppressive and counter-regulatory pathways in the tumor is still daunting, and much additional research is needed to understand how these pathways can best be overcome.
One important conceptual breakthrough has been the growing evidence that human tumors inherently possess immunogenic antigens. As genomic sequencing is increasingly used to predict immunogenic mutations, tumors are found to express multiple potential neo-antigens (reviewed in [1]). Importantly, in several studies the number of these putative neo-antigens appears to correlate with the likelihood of response to checkpoint blockade of CTLA-4 or PD-1 [94,95,96]. This last point is important, because it implies a paradigm shift in how we think about “immunogenic” tumors. In the clinical studies cited, the presence of mutational neo-antigens was not, in and of itself, associated with an obviously “good-risk” subgroup. All of the patients had progressive disease at study entry; and, left untreated, all would have presumably succumbed. Thus, the presence of neo-antigens was not, by itself, protective against the tumor. The benefit accrued only when the patients received a therapeutic checkpoint inhibitor to help overcome immune suppression. Thus, the potential immunogenicity of the mutations was transformed into actual benefit only when the tumor-induced immunosuppression was removed. Conceptually, tolerance to these neo-antigens was broken by the therapy.
To extend this paradigm-shift further, it is now clear that tolerance can also be broken even to authentic, unmodified self antigens. This was demonstrated experimentally in the studies described above in Sect. 1.1.2, in which injection of unmodified “self” cells (syngeneic thymocytes) could break tolerance against even ubiquitous self antigens such as histones and DNA, as long as two conditions were met: the cells had to be induced to die, and the IDO pathway had to be blocked at the time of antigen presentation [46]. Thus, while self antigens from dying cells may be tolerogenic under normal circumstances, this apparent tolerance may be only contingent and conditional. The same antigens may become highly immunogenic if the relevant regulatory pathways are blocked.
In the setting of human cancer, it has long been observed that patients with immunogenic tumors such as melanoma often have circulating T cells against self antigens associated with the tumor [97]. The relevance (and potential danger) of such self antigens as therapeutic targets is supported by the occurrence of cross-reactive autoimmunity such as vitiligo and uveitis during immunotherapy for melanoma [98]. But the risk of autoimmunity, while real, does not mean that self antigens are not potentially useful targets in cancers. Tumors are very different from normal tissues: they are often much more chronically inflamed [99]; they may re-express antigens not normally found in the adult host (oncofetal antigens); they may process and present even normal self antigens in aberrant and immunogenic ways [100, 101]; and they have a constant level of cellular stress, autophagy and ongoing apoptosis that may render them more immunogenic than normal tissues [85, 102, 103]. These unique attributes of the tumor may allow certain self antigens to become important tumor-associated targets, with a manageable degree of selectivity for tumor over normal tissue. The relative contribution of mutational neo-antigens versus self antigens in anti-tumor therapy is currently unknown. But the key point for this discussion is that both sets of antigens may potentially be immunogenic, if the suppressive pathways in tumors can be blocked. And, unlike the case with a defined vaccine antigen, the optimally immunogenic antigens do not need to be known in advance. If the tumor milieu can be rendered immunogenic rather than immunosuppressive, then the patient’s own immune system will identify the immunogenic antigens.
7.3 IDO as a Clinically Relevant Target
The preceding discussion introduces the concept of dying tumor cells as a rich source of antigens that are potentially immunogenic, but which cannot become actually immunogenic unless the relevant inhibitory pathways in the tumor are blocked. Therefore, it becomes important to identify which are the relevant pathways that control immunity versus tolerance to dying tumor cells. At present, this is incompletely understood.
The tumor microenvironment is filled with multiple immunosuppressive pathways. However, only certain of these mechanisms will be relevant to the uptake and cross-presentation of antigens from dying tumor cells. The CTLA-4 and PD-1/PD-L pathways, which are very important for the control of T cells, are not major direct regulators of antigen-presenting cells, or the innate inflammatory milieu. In contrast, IDO has a major effect on the biology of APCs, and in controlling innate inflammation (see Sect. 1.1.1). Thus, IDO and its associated downstream pathways may represent important therapeutic targets for modulating the key initial immune response to tumor-associated antigens.
7.3.1 The Inflammatory Signals Produced by Dying Cells May Elicit IDO
One of the defining attributes of the IDO gene is that it is highly inducible in response to inflammation. Depending on the context, both IFNγ and type I IFNs can be physiologic inducers of IDO, as can signals via the TLR/MyD88 pathway [14]. The degree to which dying tumor cells drive up-regulation of IDO in the tumor and tumor-draining LNs has not been well studied. However, it is known that tumors can be rich in type I IFNs (IFNα and IFNβ), driven in part by “danger” signals released by dying tumor cells [104]. Likewise, following chemotherapy, dying tumor cells may release HMGB1, a ligand for TLR4 [77], or extracellular DNA, which can be sensed via the pro-inflammatory STING pathway [105]. Like IFNs and TLR ligands, in other settings, STING has been shown to be a potent inducer of IDO [106,107,108], with consequent suppression of T cell responses. IDO can also be induced by prostaglandins such as PGE2 [109], which can be produced by stressed cells. Thus, dying tumor cells potentially have multiple pathways by which they might induce IDO.
Any chemotherapy or immunologic therapy will, if successful, kill some fraction of the tumor cells, and thus release an array of tumor antigens. It would be highly desirable if the immune system could generate a productive response against this wave of endogenous tumor antigens. One of the important unanswered questions for the field is the extent to which counter-regulatory IDO may suppress immune responses to these endogenous antigens following conventional chemotherapy or immunotherapy; and how this may be targeted for therapy.
7.3.2 IDO and Counter-Regulation
At present, the extent to which IDO is induced and up-regulated in tumors following chemotherapy or immunotherapy remains unknown. In practice, this has been a difficult question to answer in humans, because it requires on-treatment biopsies of the tumor (or tumor-draining LNs) following therapy. To date, however, all studies of IDO have been in untreated tumors, prior to therapy. This is useful for identifying which tumors constitutively express or elicit IDO as part of their underlying biology, but it gives no information about how much reactive (counter-regulatory) IDO may have been elicited in response to cell death and inflammation. This “reactive” IDO may be a critical and highly relevant target for therapy, but it can only be detected by obtaining on-treatment biopsies. The fact that a patient’s tumor cells were initially IDO-negative at diagnosis does not mean that the immunosuppressive host APCs will not subsequently up-regulate IDO in response to therapy.
The role of this reactive or counter-regulatory IDO becomes particularly germane in the case of clinical immunotherapy, such as T cell adoptive-transfer or checkpoint blockade. Indeed, preclinical models suggest that even the spontaneous, low-level endogenous T cell response against the tumor may generate enough inflammation to drive counter-regulatory IDO expression [8]. This level of inducible IDO might be greatly increased by interventions such as T cell adoptive transfer or checkpoint blockade. Not only do such treatments cause tumor cell death, but—as a consequence of their own success—they also create intense inflammation within the tumor. Both the cell death and this local inflammation may induce counter-regulatory IDO, and thus blunt the desired effect of therapy. Counter-regulatory IDO would not abrogate the effect entirely (the treatment would still show some efficacy), but there might be substantially more efficacy potentially available if the counter-regulatory IDO were blocked. Emerging evidence from mouse preclinical models suggests that this hypothetical concern may indeed be the case [110, 111]. In these studies, the efficacy of both CTLA-4 blockade and PD-1 blockade were enhanced by adding an IDO-inhibitor drug (indoximod or INCB23843). How much of this effect was due specifically to reactive (counter-regulatory) IDO was not determined, but the effect was recapitulated by genetic deficiency of IDO1 in the host [110], suggesting that the target was host IDO rather than tumor. Recently, using a mouse xenograft model, it was shown that human CD19 CAR-T cells were strongly inhibited in vivo by IDO expression in the target B cell malignancies; and inhibition was reversed by administering oral indoximod [112]. Here again, the contribution of reactive versus constitutive IDO was not ascertained, but the study shows that human CAR-T cells are susceptible to the effects of IDO.
7.3.3 IDO-Inhibitor Drugs in the Clinic
A number of drugs targeting the IDO pathway are now in early-phase clinical trials, or in preclinical development. Drugs in trials include indoximod (1-methyl-D-tryptophan) and NLG919 (both from NewLink Genetics, Inc.) and INCB024360 (from Incyte Corp.). Published data currently are limited to interim abstracts from on-going trials, so efficacy data are not yet available. However, toxicity profiles have been generally favorable, which has facilitated combinations with additional agents.
Preclinical mouse models show that IDO-inhibitor drugs are synergistic with a variety of chemotherapeutic agents in a number of different tumor models (transplantable and autochthonous) [24, 113, 114]. Based on this, several of the ongoing trials of indoximod are structured to combine this agent with conventional chemotherapy (docetaxel in breast cancer; temozolomide in brain tumors; or gemcitabine/abraxane in pancreatic cancer). Trials are also open combining either INCB024360 or indoximod with CTLA-4 blocking antibody. Combinations with inhibitors of the PD-1/L pathway are also in progress, and are entering Phase 3 trials.
Open questions in the field of IDO drug-development currently include the relative contribution of IDO1 and IDO2 genes to tumor-induced immunosuppression, and the potential contribution of the unrelated enzyme TDO (tryptophan dioxygenase). IDO2 has been much less extensively studied than IDO1, and its biologic role remains unclear. One study found that tumors grown in IDO1-deficient mice had increased levels of IDO2 [115], suggesting that IDO2 may compensate for lack of IDO1. Therefore, inhibitor drugs with dual specificity for both IDO1 and IDO2 may be of benefit. TDO is an unrelated enzyme that catalyzes the same conversion of tryptophan to N-formyl-kynurenine. TDO is constitutively expressed in liver and brain, and it can also be an autocrine growth pathway for brain tumors [116]. Although there is no physiologic role known for TDO in the immune system (in contrast to IDO), there is concern that some tumors may be able up-regulate TDO as an immunosuppressive pathway (or as an escape pathway when IDO is blocked). Hence, there is interest in TDO-inhibitors, and in dual-specificity inhibitors that could inhibit both IDO and TDO.
7.4 Conclusions
Inducible counter-regulation by IDO may be an important inhibitory pathway during chemotherapy and immunotherapy. IDO can be elicited as a natural tolerogenic pathway in response to signals from dying tumor cells. As such, IDO may bias the immune response toward tolerance rather than immune activation following chemotherapy. IDO can also be elicited as a counter-regulatory response to attempted inflammation and immune activation. This is of concern in settings of active immunotherapy, where desirable immune responses may be inadvertently suppressed because the elicit counter-regulatory IDO. However, these effects of IDO also represent a therapeutic opportunity. IDO is emerging as a mechanism that influences the fundamental choice of whether dying cells will be perceived by the immune system as tolerogenic or immunogenic. Thus, if the tolerogenic IDO pathway can be blocked, then conventional chemotherapy may be more spontaneously immunogenic than previously appreciated. Likewise, active immunotherapy may become able to elicit a more robust immune response, with epitope-spreading to additional endogenous tumor antigens. These areas represent topics for future basic research, and therapeutic opportunities for synergistic combinatorial regimens in the clinic.
References
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135–46.
Medler TR, Cotechini T, Coussens LM. Immune response to cancer therapy: mounting an effective antitumor response and mechanisms of resistance. Trends Cancer. 2015;1:66–75.
Belvin M, Mellman I. Is all cancer therapy immunotherapy? Sci Transl Med. 2015;7:315fs48.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28:690–714.
Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187.
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van Den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–74.
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5:200ra116.
Baban B, Hansen A, Chandler P, Manlapat A, Bingaman A, Kahler D, Munn D, Mellor A. A minor population of splenic dendritic cells expressing CD19 mediates IDO-dependent T cell suppression via type 1 interferon-signaling following B7 ligation. Int Immunol. 2005;17:909–19.
Manlapat AK, Kahler DJ, Chandler PR, Munn DH, Mellor AL. Cell-autonomous control of interferon type I expression by indoleamine 2,3-dioxygenase in regulatory CD19(+) dendritic cells. Eur J Immunol. 2007;37:1064–71.
Hwu P, MX D, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. 2000;164:3596–9.
Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–82.
Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN type 1 signaling. J Immunol. 2005;175:5601–5.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–43.
Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7.
Ball HJ, Yuasa HJ, Austin CJ, Weiser S, Hunt NH. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol. 2009;41:467–71.
Fatokun AA, Hunt NH, Ball HJ. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: characteristics and potential roles in health and disease. Amino Acids. 2013;45:1319–29.
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42.
Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–61.
Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8.
Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–73.
Jaronen M, Quintana FJ. Immunological relevance of the coevolution of IDO1 and AHR. Front Immunol. 2014;5:521.
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3.
Muller AJ, Duhadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–9.
Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, Munn DH. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–8.
Matteoli G, Mazzini E, Iliev ID, Mileti E, Fallarino F, Puccetti P, Chieppa M, Rescigno M. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut. 2010;59:595–604.
van der Marel AP, Samsom JN, Greuter M, van Berkel LA, O'Toole T, Kraal G, Mebius RE. Blockade of IDO inhibits nasal tolerance induction. J Immunol. 2007;179:894–900.
Sucher R, Fischler K, Oberhuber R, Kronberger I, Margreiter C, Ollinger R, Schneeberger S, Fuchs D, Werner ER, Watschinger K, Zelger B, Tellides G, Pilat N, Pratschke J, Margreiter R, Wekerle T, Brandacher G. IDO and regulatory T cell support are critical for cytotoxic T lymphocyte-associated Ag-4 Ig-mediated long-term solid organ allograft survival. J Immunol. 2012;188:37–46.
Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, Puccetti P. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–101.
Mellor AL, Baban B, Chandler P, Marshall B, Jhaver K, Hansen A, Koni PA, Iwashima M, Munn DH. Cutting edge: induced indoleamine 2,3 dioxygenase expression in dendritic cell subsets suppresses T cell clonal expansion. J Immunol. 2003;171:1652–5.
Guillonneau C, Hill M, Hubert FX, Chiffoleau E, Herve C, Li XL, Heslan M, Usal C, Tesson L, Menoret S, Saoudi A, Le Mauff B, Josien R, Cuturi MC, Anegon I. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-gamma, and indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:1096–106.
Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity. 2010;32:568–80.
Lan Z, Ge W, Arp J, Jiang J, Liu W, Gordon D, Healey D, DeBenedette M, Nicolette C, Garcia B, Wang H. Induction of kidney allograft tolerance by soluble CD83 associated with prevalence of tolerogenic dendritic cells and indoleamine 2,3-dioxygenase. Transplantation. 2010;90:1286–93.
Swanson KA, Zheng Y, Heidler KM, Mizobuchi T, Wilkes DS. CDllc+ cells modulate pulmonary immune responses by production of indoleamine 2,3-dioxygenase. Am J Respir Cell Mol Biol. 2004;30:311–8.
Liu H, Liu L, Fletcher BS, Visner GA. Novel action of indoleamine 2,3-dioxygenase attenuating acute lung allograft injury. Am J Respir Crit Care Med. 2006;173:566–72.
Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, Blazar BR. Indoleamine 2,3-dioxygenase is a critical regulator of acute GVHD lethality. Blood. 2008;111:3257–65.
Lu Y, Giver CR, Sharma A, Li JM, Darlak KA, Owens LM, Roback JD, Galipeau J, Waller EK. IFN-gamma and indoleamine 2,3-dioxygenase signaling between donor dendritic cells and T cells regulates graft versus host and graft versus leukemia activity. Blood. 2012;119:1075–85.
Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125:1762–73.
Szanto S, Koreny T, Mikecz K, Glant TT, Szekanecz Z, Varga J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res Ther. 2007;9:R50.
Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, Cullimore ML, Rostami A, Xu H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol. 2010;185:5953–61.
Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, Puccetti P, Romani L, Grohmann U. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol. 2009;183:6303–12.
Romani L, Fallarino F, De Luca A, Montagnoli C, D'Angelo C, Zelante T, Vacca C, Bistoni F, Fioretti MC, Grohmann U, Segal BH, Puccetti P. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature. 2008;451:211–5.
Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, Orabona C, Belladonna ML, Ayroldi E, Nocentini G, Boon L, Bistoni F, Fioretti MC, Romani L, Riccardi C, Puccetti P. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. 2007;13:579–86.
Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest. 2007;117:2268–78.
McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood. 2011;117:5403–12.
Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MC, McGaha TL. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2012;109:3909–14.
Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor A. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–90.
Mellor AL, Chandler P, Baban B, Hansen AM, Marshall B, Pihkala J, Waldmann H, Cobbold S, Adams E, Munn DH. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int Immunol. 2004;16:1391–401.
Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol. 2008;181:5396–404.
Manches O, Munn D, Fallahi A, Lifson J, Chaperot L, Plumas J, Bhardwaj N. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J Clin Invest. 2008;118:3431–9.
Chung DJ, Rossi M, Romano E, Ghith J, Yuan J, Munn DH, Young JW. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood. 2009;114:555–63.
Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood. 2009;114:3235–43.
Savage PA, Malchow S, Leventhal DS. Basic principles of tumor-associated regulatory T cell biology. Trends Immunol. 2013;34:33–40.
Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7.
Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D, Tighe R, Lan Y, Lo KM, Sabzevari H, Hofmeister R, Wilson NS. Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans. J Immunol. 2015;194:878–82.
Huehn J, Floess S, Ohkura N, Sakaguchi S. Comment on “cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans”. J Immunol. 2015;194:3533.
Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, Amit AS, Kang C, Geddes JE, Allison JP, Socci ND, Savage PA. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. 2013;339:1219–24.
Darrasse-Jeze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, Cohen JL, Bellier B, Podsypanina K, Klatzmann D. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest. 2009;119(9):2648–62.
Menetrier-Caux C, Gobert M, Caux C. Differences in tumor regulatory T-cell localization and activation status impact patient outcome. Cancer Res. 2009;69:7895–8.
Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DA. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–6.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9.
Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest. 2013;123:939–44.
Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–7.
Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–71.
Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, Bluestone JA. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. 2013;39:949–62.
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20:62–8.
Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, Bluestone JA, Turka LA. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–96.
Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain Th1 and Tfh cell responses. Nat Immunol. 2015;16:178–87.
Lee Jee H, Elly C, Park Y, Liu Y-C. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity. 2015;42:1062–74.
Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, Mellor AL, He Y, Munn DH. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood. 2009;113:6102–11.
Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol. 2009;183:2475–83.
Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, Blazar BR, Mellor AL, Munn DH. Reprogrammed Foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity. 2010;33:942–54.
Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, Shi H, Munn DH. An inherently bifunctional subset of Foxp3 T helper cells is controlled by the transcription factor Eos. Immunity. 2013;38:998–1012.
Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–80.
Sharma MD, Shinde R, McGaha T, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe A, Francisco LM, Powell DJ Jr, Yagita H, Mellor AL, Blazar BR, Munn DH. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1:e1500845.
Hannun YA. Apoptosis and the dilemma of cancer chemotherapy. Blood. 1997;89:1845–53.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050.
Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, Martins I, Schlemmer F, Michaud M, Kepp O, Sukkurwala AQ, Menger L, Vacchelli E, Droin N, Galluzzi L, Krzysiek R, Gordon S, Taylor PR, Van Endert P, Solary E, Smyth MJ, Zitvogel L, Kroemer G. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–41.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–7.
Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118:1991–2001.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med. 2012;18:344–6. author reply 6
Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res. 1989;49:5870–8.
Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23.
Spel L, Boelens JJ, Nierkens S, Boes M. Antitumor immune responses mediated by dendritic cells: how signals derived from dying cancer cells drive antigen cross-presentation. Oncoimmunology. 2013;2:e26403.
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–69.
Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, Chandler P, Tanaka M, Munn DH, Mellor AL, McGaha TL. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci U S A. 2014;111:4215–20.
Anyaegbu CC, Lake RA, Heel K, Robinson BW, Fisher SA. Chemotherapy enhances cross-presentation of nuclear tumor antigens. PLoS One. 2014;9:e107894.
Zeelenberg IS, van Maren WW, Boissonnas A, Van Hout-Kuijer MA, Den Brok MH, Wagenaars JA, van der Schaaf A, Jansen EJ, Amigorena S, Thery C, Figdor CG, Adema GJ. Antigen localization controls T cell-mediated tumor immunity. J Immunol. 2011;187:1281–8.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80.
Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B, Feyen N, Martens UM, Beckhove P, Gnjatic S, Schirmacher P, Herpel E, Weitz J, Grabe N, Jaeger D. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011;71:5670–7.
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28:105–13.
Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautès-Fridman C, Fucikova J, Galon J, Spisek R, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. OncoImmunology. 2015;4:e1008866.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–50.
Germeau C, Ma W, Schiavetti F, Lurquin C, Henry E, Vigneron N, Brasseur F, Lethe B, De Plaen E, Velu T, Boon T, Coulie PG. High frequency of antitumor T cells in the blood of melanoma patients before and after vaccination with tumor antigens. J Exp Med. 2005;201:241–8.
Palmer DC, Chan CC, Gattinoni L, Wrzesinski C, Paulos CM, Hinrichs CS, Powell DJ Jr, Klebanoff CA, Finkelstein SE, Fariss RN, Yu Z, Nussenblatt RB, Rosenberg SA, Restifo NP. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci U S A. 2008;105:8061–6.
Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–91.
Savage PA, Vosseller K, Kang C, Larimore K, Riedel E, Wojnoonski K, Jungbluth AA, Allison JP. Recognition of a ubiquitous self antigen by prostate cancer-infiltrating CD8+ T lymphocytes. Science. 2008;319:215–20.
Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, van der Bruggen P, Boon T, Van den Eynde BJ. An antigenic peptide produced by peptide splicing in the proteasome. Science. 2004;304:587–90.
Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, Shen S, Marino G, Criollo A, Boileve A, Job B, Ladoire S, Ghiringhelli F, Sistigu A, Yamazaki T, Rello-Varona S, Locher C, Poirier-Colame V, Talbot M, Valent A, Berardinelli F, Antoccia A, Ciccosanti F, Fimia GM, Piacentini M, Fueyo A, Messina NL, Li M, Chan CJ, Sigl V, Pourcher G, Ruckenstuhl C, Carmona-Gutierrez D, Lazar V, Penninger JM, Madeo F, Lopez-Otin C, Smyth MJ, Zitvogel L, Castedo M, Kroemer G. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337:1678–84.
Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. 2009;31:184–96.
Fuertes MB, Woo SR, Burnett B, YX F, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34:67–73.
Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015;36:250–6.
Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, Pacholczyk G, Barber GN, Hayakawa Y, Munn DH, Mellor AL. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol. 2014;192:5571–8.
Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, Barber GN, Hayakawa Y, McGaha TL, Ravishankar B, Munn DH, Mellor AL. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol. 2013;191:3509–13.
Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, Mellor AL. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76(8):2076–81.
von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, Fiore F, Roth U, Beyer M, Debey S, Wickenhauser C, Hanisch FG, Schultze JL. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood. 2006;108:228–37.
Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–402.
Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3.
Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, Heslop HE, Brenner MK, Rooney CM, Ramos CA. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–16.
Hou DY, Muller AJ, Sharma MD, Duhadaway JB, Banerjee T, Johnson M, Mellor AL, Prendergast GC, Munn DH. Inhibition of IDO in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with anti-tumor responses. Cancer Res. 2007;67:792–801.
Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, Hoang K, Ashley C, McCall D, Rojiani AM, Maria BL, Rixe O, MacDonald TJ, Heeger PS, Mellor AL, Munn DH, Johnson TS. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer. 2014;2:21.
Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y, Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20:5290–301.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Johnson, T.S., Mcgaha, T., Munn, D.H. (2017). Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment. In: Kalinski, P. (eds) Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. Advances in Experimental Medicine and Biology, vol 1036. Springer, Cham. https://doi.org/10.1007/978-3-319-67577-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-67577-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67575-6
Online ISBN: 978-3-319-67577-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)