
Abstract
An important aspect of Chlamydomonas reinhardtii’s metabolism is its ability to produce molecular hydrogen (H2) from protons and electrons. Hydrogen production is catalysed by two [FeFe]-hydrogenases, HYDA1 and HYDA2, although HYDA1 is the main isoform, accounting for ∼75% of the H2 produced. Hydrogen production can be light dependent, with the hydrogenase receiving electrons from the photosynthetic electron transport chain via the ferredoxin PETF, or light independent, where H2 is produced via fermentation in the dark. Hydrogen production was first reported in microalgae in the early 1940s; however, due to HYDA gene expression being induced by anaerobiosis and the extreme oxygen sensitivity of the enzyme, this process only occurred transiently at low levels when the algae were subjected to anaerobic or hypoxic conditions. It was thus considered nothing more than a biological curiosity until the early 2000s, when a method temporally separating oxygenic photosynthesis and H2 production was developed, which allowed sustained H2 production in the light over the course of a few days. Light-driven H2 production has the highest theoretical photon conversion efficiency and is thus of considerable biotechnological interest. However, the calculated theoretical efficiencies are still not achievable in practice, despite the implementation of a wide range of engineering strategies. For an improved H2 production, a better understanding of the underlying biology is needed. C. reinhardtii is the ideal organism in which to study H2 production, due to the many molecular tools available and the simplicity and long history of study of its hydrogenase.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
Rising global population growth and concomitant increases in demand for food, water and energy, as well as renewed efforts by governments to reduce carbon emissions, have all heightened interest in hydrogen (H2) as a renewable fuel. The main attraction of H2 is that the only by-product of its combustion is water, meaning that it can be carbon neutral or even carbon negative when produced renewably. Hydrogen also has an ∼3× higher energy density compared to hydrocarbon fuels (Gupta et al. 2013). Currently, H2 is produced via expensive energy-intensive fossil fuel-based methods such as steam reformation of natural gas, industrial oil and naphtha reforming, coal gasification and fossil fuel-driven water electrolysis (Gupta et al. 2013). However, it can also be produced under less energy-intensive conditions in cyanobacterial or microalgal systems, which use sunlight as their energy source (Oey et al. 2016).
To date, high bio-H2 production efficiencies have been reported for eukaryotic microalgae. This is partly due to the high efficiency of the algal [FeFe]-hydrogenase, which is 100-fold higher than that of cyanobacterial hydrogenases, having a turnover rate of up to 104 H2 molecules s−1 (Lubitz et al. 2014; Volgusheva et al. 2013). Chlamydomonas reinhardtii has emerged as the model organism for the study of photobiological H2 production in green microalgae. This is likely due to the fact that the C. reinhardtii [FeFe]-hydrogenase HYDA1 was the first eukaryotic hydrogenase to be isolated (Roessler and Lien 1984; Happe and Naber 1993) but is also due to the large knowledge base and molecular toolset available for this organism. Since the initial purification of HYDA1, much progress has been made characterising the enzyme and elucidating its maturation, as well as in understanding the underlying regulatory pathways and improving H2 production efficiencies. The structure, catalysis and oxygen (O2) sensitivity of the enzyme have all been analysed, and many hydrogen mutants and higher H2-producing algal strains have been created (see reviews Kruse et al. 2005; Ghirardi et al. 2007; Dubini and Ghirardi 2015; Antal et al. 2015; Torzillo et al. 2015; Oey et al. 2016). In this chapter, we review H2 production in C. reinhardtii with a focus on HYDA1. We detail the induction, structure, maturation and catalytic mechanism of this enzyme and summarise strategies that have been used to improve microalgal H2 production efficiencies.
2 Microalgal Hydrogen Metabolism
Microalgal H2 production was first reported for Scenedesmus sp. in 1942 (Gaffron and Rubin) and later for C. reinhardtii and other species (Stuart and Gaffron 1972). Hydrogen evolution was observed at low levels under dark anaerobiosis and at higher levels in the light (Gaffron and Rubin 1942). Photosystem I (PSI) was identified as being essential for light-dependent H2 production, whereas photosystem II (PSII), although a contributor of electrons, was found to be dispensable (Stuart and Gaffron 1972).
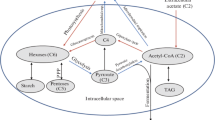
Three H2 production pathways have since been identified in green microalgae: two that produce H2 in the light and a third, dark fermentation pathway. The two light-dependent pathways (Fig. 1) include a direct pathway and an indirect pathway. In the direct pathway, HYDA1 receives electrons generated by the splitting of water at PSII via the photosynthetic electron transfer (PET) pathway. This involves the light-dependent excitation of electrons by PSI, the reduction of the ferredoxin PETF and the subsequent transfer of electrons from PETF to the hydrogenase (Melis and Happe 2001; Melis et al. 2000). In the indirect pathway, which is independent of PSII, electrons are also received via PETF but are derived from the catabolism of starch. In this pathway, the plastoquinone pool is reduced by NAD(P)H in a reaction mediated by the type II NADH dehydrogenase NDA2 (Desplats et al. 2009; Fouchard et al. 2005; Godde and Trebst 1980; Maione and Gibbs 1986; Chochois et al. 2009; Baltz et al. 2014; Jans et al. 2008; Mignolet et al. 2012; Mus et al. 2005). However, in this pathway, H2 production is ∼10× lower than in the direct pathway (Chochois et al. 2009). In the dark fermentation metabolism, electrons are also derived from the catabolism of starch but do not enter the PET pathway. Instead, it is thought that they are transferred to pyruvate, which can be oxidised by a pyruvate ferredoxin oxidoreductase and which can then reduce PETF in a process similar to H2-producing fermentation pathways found in other microorganisms (Grossman et al. 2011; Atteia et al. 2013; van Lis et al. 2013; Noth et al. 2013).

Overview of the direct and indirect light-dependent hydrogen (H2) production pathways. In the direct pathway, electrons are derived from the water-splitting reaction at photosystem II (PSII), while in the indirect pathway, electrons are derived from starch catabolism. Both pathways involve the capture of light, either by both light-harvesting complexes I and II (LHCI and LHCII, respectively) or by LHCI, and the transfer of electrons to the hydrogenase HYDA1 through the electron transport chain via the plastoquinone pool (PQ), cytochrome b6f (Cytb6f), plastocyanin (PC), PSI and the ferredoxin PETF. The indirect pathway additionally involves the NADPH-dehydrogenase NDA2
It would appear slightly paradoxical that oxygenic green algae contain fermentative pathways such as the H2 production pathway, which are more typical of strictly anaerobic microbes. However, green microalgae such as C. reinhardtii are in fact frequently exposed to anaerobic or hypoxic conditions and therefore need to be able to readily adapt their metabolisms (Mus et al. 2007; see chapter “Chlamydomonas: Anoxic Acclimation and Signaling” of Volume 1). Although the exact physiological role of H2 production in microalgae remains unclear, it likely functions as a protection mechanism against the over-reduction of the chloroplast (Hemschemeier et al. 2008a). Under aerobic conditions, photosynthesis produces the carbohydrates that are required for respiration and cell growth. However, under anaerobiosis in the light, oxidative phosphorylation in the mitochondria is largely inhibited by a lack of O2, leading to the over-reduction of the chloroplast and a reduced electron transfer, which ultimately results in photodamage and decreased levels of ATP. Under these conditions, the protons and electrons derived from water by the remaining PSII in the direct pathway or from starch in the indirect pathway can be fed to the hydrogenase via the PET chain, which recombines the protons and electrons to produce H2. Therefore the hydrogenase is thought to act as a proton/electron release valve, removing excess protons and electrons by recombining them to produce H2 gas, which can be easily released from the cells.
3 The Chlamydomonas reinhardtii Hydrogenase HYDA1
3.1 Hydrogenases
Three phylogenetically distinct classes of hydrogenases have been identified to date in H2-consuming and H2-evolving prokaryotes: [NiFe]-hydrogenases, which contain a nickel (Ni) and an iron (Fe) atom in their active site; [FeFe]-hydrogenases, which contain two Fe atoms; and [Fe]-hydrogenases, which have a mononuclear Fe active site but which are only found in a few methanogenic archaea (Vignais et al. 2001; Meyer 2007). Eukaryotic green microalgae have only been found to contain [FeFe]-hydrogenases, while cyanobacteria only contain [NiFe]-hydrogenases (Ludwig et al. 2006). However, [FeFe]-hydrogenases have also been found in eubacteria such as Clostridium sp. and Desulfovibrio sp., as well as in anaerobic protozoa such as Trichomonas sp. and Nyctotherus sp. (Vignais et al. 2001).
3.2 HYDA1 Gene
Two [FeFe]-hydrogenase genes have been identified in C. reinhardtii: HYDA1 (Happe and Kaminski 2002) and HYDA2 (Forestier et al. 2003). HYDA1 appears to be the primary isoform as it produces ∼75% of the H2 in the light (Meuser et al. 2012). Both genes are nuclear encoded; however HYDA1 contains a transit peptide that facilitates its translocation to the chloroplast (Happe and Kaminski 2002). HYDA2 is also predicted to contain a chloroplast transit peptide and is thus also likely to function in the chloroplast (Forestier et al. 2003). Expression of HYDA1 and HYDA2, as well as the hydrogenase specific maturase genes HYDEF and HYDG, is induced by anaerobiosis (Happe and Kaminski 2002; Happe and Naber 1993; Posewitz et al. 2004; Mus et al. 2007; Hemschemeier et al. 2013). HYDA1 transcript abundance also increases after copper deprivation (Castruita et al. 2011), and the HYDA1 promoter has been found to contain two GTAC motifs that are recognised by the copper response regulator 1 (CRR1) transcription factor (Pape et al. 2012), which regulates several genes under both copper deprivation and hypoxia/anaerobiosis (Kropat et al. 2005). In fact, anaerobiosis and copper deprivation appear to be linked, with many genes being expressed under both conditions (Castruita et al. 2011; see chapter “Chlamydomonas: Anoxic Acclimation and Signaling” of Volume 1).
3.3 HYDA1 Structure
The C. reinhardtii hydrogenase is a small (∼48 kDa) monomeric soluble [FeFe]-hydrogenase located in the chloroplast (Happe and Naber 1993; Happe et al. 1994). Its structure consists of two lobes, each containing a beta-sheet surrounded by several alpha-helices, with the active site located between the lobes and interacting with amino acids from seven different protein stretches (Mulder et al. 2010) (Fig. 2b). The structure of HYDA1 and nearly all known [FeFe]-hydrogenases from microalgae features only this one domain, known as the H-domain, which is conserved in all [FeFe]-hydrogenases (Peters et al. 2015). Since additional N-terminal domains with further electron transporting FeS-clusters are present in all non-eukaryotic [FeFe]-hydrogenases, the relative simplicity of HYDA1 contributes to it being one of the model enzymes for hydrogenase research (Lubitz et al. 2014).

Structure and maturation of the H-cluster. (a) HYDA1 expressed in a background devoid of specific maturases yields protein with the 4FeH-subcluster only, which is supplied by the ISC, SUF or CIA systems. The maturases HYDG and HYDE synthesise parts of the 2FeH-subcluster, which is probably assembled on HYDF. Transfer of the preassembled 2FeH-subcluster from HYDF to HYDA1 completes the H-cluster. (b) Structure of HYDA1ΔEFG in cartoon representation with the beta-sheets in dark grey and the 4FeH-subcluster as spheres in yellow (S) and orange (Fe), respectively (PDB ID 3lx4) (Mulder et al. 2011)
Protons can reach the active site of the hydrogenase from the surface by a phylogenetically conserved proton transfer pathway through the H-domain consisting of an arginine, two glutamates, a serine, a water molecule and a cysteine (Peters et al. 1998). Variations of these amino acids were shown to drastically alter both the catalytic competence and the pH optimum of bacterial [FeFe]-hydrogenases (Cornish et al. 2011; Morra et al. 2012). Channels for the diffusion of H2 and O2 through the protein towards the active site were discovered in the central domain of the bacterial [FeFe]-hydrogenase CPI from Clostridium pasteurianum by in silico approaches (Hong and Pachter 2012; Cohen et al. 2005). A complex [6Fe6S]-cluster, known as the H-cluster, forms the active site of [FeFe]-hydrogenases (Fig. 2a). It comprises a standard [4Fe4S]-cluster (the 4FeH-subcluster) coordinated by four cysteine residues, which is linked to a unique [2Fe]-cluster (the 2FeH-subcluster) through one of the cysteines (Peters et al. 1998, 2015; Nicolet et al. 1999). The iron atoms of this 2FeH-subcluster are bridged by both thiolates of a singular aza-dithiolate (adt) ligand with further ligands being one CN− and one CO molecule per iron, as well as another CO in a bridged coordination between the two Fe atoms (Fig. 2a). While a number of amino acids are well positioned to form stabilising hydrogen bonds with the CN− ligands in an otherwise hydrophobic active site pocket, the thiolate bond to the cysteine linking the two subsites is the only covalent connection between the 2FeH-subcluster and the protein (Peters et al. 1998) (Fig. 4). In the case of the iron atom located proximal to the 4FeH-subcluster (Fep), the linking cysteine is the sixth ligand in an octahedral coordination geometry, while the more distal iron (Fed), although restrained by the surrounding amino acids to the same octahedral geometry, is missing one of the axial ligands (Peters et al. 2015). This open coordination site and the amine group of the adt ligand pending above it have been identified as the key reasons for the striking difference in activity between the H-cluster within the protein and chemical compounds of similar composition (Berggren et al. 2013; Simmons et al. 2014; Esselborn et al. 2016).
3.4 Enzyme Maturation
The assembly of the H-cluster of [FeFe]-hydrogenases takes place in two stages. The 4FeH-subcluster is assembled first by the standard FeS-cluster assembly machinery (Broderick et al. 2014), and then the 2FeH-subcluster is added by the specific maturation enzymes HYDEF and HYDG (Posewitz et al. 2004) (Fig. 2). Recently, it became evident that the source of both CO and CN− is tyrosine, which is split by radical SAM chemistry in HYDG on a [4Fe4S]-cluster (Peters et al. 2015). The products are then assembled on the other end of a beta-barrel in HYDG to an Fe(CO)2(CN)-cysteine-cluster at a second [4Fe4S]-cluster, turning it transiently into a [5Fe5S]-cluster (Suess et al. 2016; Pagnier et al. 2016). HYDE (in most organisms HYDE and HYDF are separate genes) also relies on radical SAM chemistry and is believed to contribute the aza-dithiolate ligand, as its substrate was shown to be a small sulphur compound (Betz et al. 2015). The product of HYDE appears to come together with two of the mono-iron clusters from HYDG onto HYDF, which serves as a scaffold for the complete [2Fe]-cluster. The interaction between the three maturases is not yet fully understood, but most likely involves GTP hydrolysis, as HYDF has been reported to have GTPase functionality (Peters et al. 2015). Once the [2Fe]-cluster is assembled on HYDF linked to a [4Fe4S]-cluster, the transfer of the [2Fe]-subcluster into the hydrogenase and assembly of the complete H-cluster takes place without additional energy input. However, it requires the 4FeH-subcluster to be present within the hydrogenase (Shepard et al. 2010).
Importantly for hydrogenase research, the specific maturation machinery can be bypassed, as synthetic [2Fe]-clusters can be incorporated spontaneously into [FeFe]-hydrogenases in vitro once the 4FeH-subcluster is formed either by the standard machinery in vivo or by reconstitution in vitro (Berggren et al. 2013; Esselborn et al. 2013). This yields semi-synthetic enzymes, which are indiscernible from their completely in vivo maturated counterparts.
Although much progress has been made using recombinant proteins in in vitro assays (Peters et al. 2015), neither the exact cellular mechanism, nor the cellular compartment in which the incorporation of either subcluster occurs, is known. In eukaryotic microalgae, proteins involved in the assembly of proteins with standard FeS-clusters are located in all cellular compartments, i.e. in the cytosol, plastid stroma and mitochondrial matrix (Balk and Schaedler 2014). Each plant cell compartment is able to synthesise FeS-clusters, but the machineries employed differ: the mitochondria and cytosol contain the FeS-cluster (ISC) and cytosolic FeS-protein assembly (CIA) systems, respectively, while the chloroplast utilises the sulphur mobilisation (SUF)-like machinery (Balk and Schaedler 2014). However, it is likely that the chloroplast-localised HYDA1 enzyme is assembled in the plastid compartment, as current knowledge indicates that only unfolded proteins are transported across the chloroplast outer membranes (Paila et al. 2015). Also, both HYDEF and HYDG contain putative chloroplast transit peptides, and HYDG was detected in the chloroplast in a proteomics study (Terashima et al. 2010), suggesting that at least the addition of the [2Fe]-cluster occurs in the chloroplast.
3.5 HYDA1 Catalysis
A comparative electrochemical analysis of representative [FeFe]- and [NiFe]-hydrogenases concluded that the catalytic competence of the H-cluster is inherently superior to the one from the [NiFe]-cofactor (Hexter et al. 2012). Mössbauer spectroscopy and pulsed ENDOR/HYSCORE experiments with 57Fe-enriched [FeFe]-hydrogenase showed that both subsites of the H-cluster are electronically coupled and show strong exchange interactions. Substrate binding (H+/H2) occurs at the open coordination site at the distal Fe centre (Fed) of 2FeH while being in the most oxidised “active ready” state denoted as Hox (Fig. 3,1). The catalytic cycle, which either oxidatively degrades H2 to H+ or reductively converts two protons into H2, presumably comprises three further catalytic main states (Fig. 3,1–4) while successively taking up or releasing two protons and electrons. The exact sequence of electron and proton transfer steps occurring during catalysis is unknown and has only been speculated to follow an ECEC mechanism in which electron- and chemical proton-transfer steps alternate. However, electron paramagnetic resonance (EPR) spectroscopy and Fourier transform infrared (FTIR) spectroscopy have enabled the detailed characterisation of the redox and spin state properties for states 1–3 (Hox, Hred and Hsred). Monitoring H2 evolution, the first reduction step occurs at the 2FeH-site, thus transforming the paramagnetic Hox state (4FeH(II)-Fe(I)-Fe(II)) into the EPR silent Hred state (4FeH(II)-Fe(I)-Fe(I)). The second reduction step is restricted to the 4FeH-site, yielding the super-reduced 4FeH(I)-Fe(I)-Fe(I) state (Hsred), which accumulates under redox potentials below −500 mV. While Hsred has not been found as an active state for the bacterial [FeFe]-hydrogenase DDH and is rather assumed to be an instable artificial product of over-reduction (Roseboom et al. 2006), this state has been demonstrated to be stable and catalytically relevant for the small M1-type enzymes of green algae (Adamska-Venkatesh et al. 2014; Adamska et al. 2012). The final state carrying the hydride intermediate of the heterosynthetic H2-evolution mechanism (Fig. 3,4) has been presupposed but not yet experimentally verified for any wild-type [FeFe]-hydrogenase. However, for the virtually inactive HYDA1 variant C169S, which displays a severely compromised proton transfer efficiency, two slightly deviating unknown states have been reported which might correspond to the protonated and unprotonated derivative of the hydride state (HH−) (Mulder et al. 2014).

Catalytic cycle of hydrogen (H2) evolution and inhibitors of [FeFe]-hydrogenases. 1–4 Schematic working model of the reversible catalytic cycle of H2 evolution at the H-cluster. Local redox states are indicated in black and red numbers (transiently lowered redox state). Proton transfer steps are presented according to the favoured ECEC mechanism. The dotted line indicates a transition of μCO from the bridging coordination to a terminally bound state. 5–6 Reversible inhibition of the two H-cluster states Hox and Hred by CO binding to the open coordination site. Reductive reactivation is achieved via HredCO resulting in Hsred after another reduction step. 7 The irreversible process of oxygen (O2)-induced H-cluster degradation starts with the binding of O2 to the substrate coordination site, yielding a transient O2 adduct which presumably requires a protonation step to induce the degradative process. Although the sequence of steps during H-cluster degradation is still under debate, recent experimental data support an initial loss of the 2FeH-site before the oxidative degradation of the remaining 4FeH-cluster occurs.
The two types of diatomic ligands at the 2FeH-site (CO and CN−) participate in adjusting its redox chemistry and spin distribution and further provide individual qualities to the H-cluster which are essential for catalytic competence (Winkler et al. 2013).
The CN− ligands, which are coordinated in a trans orientation to each other, are embedded in strong H-bond networks on either side of the cofactor (Fig. 4, dotted lines), thereby stabilising the orientation and hexacoordinated configuration of the two Fe-sites in the 2FeH-cluster (Winkler et al. 2013).

First and second ligand sphere of the H-cluster in the active site of [FeFe]-hydrogenases. The model was generated using the CPI [FeFe]-hydrogenase holoprotein crystal structure (PDB ID 3C8Y) and depicts the H-cluster, as well as the most important polypeptide positions which provide the necessary environment for catalytic function (stick structures). The labelling corresponds to the homologous positions in HYDA1. Substrate/product pathways for electrons, protons and hydrogen (H2), leading to or coming from the site of catalytic turnover at Fed, are indicated by an orange, green or purple glow, respectively. Amino acid positions contributing to the H-bond networks that stabilise the CN− ligands are depicted in blue; those involved in proton transfer are presented in green, while the two methionine residues which allow catalytically relevant ligand movements are shown in red.
The latter aspect ensures a terminal coordination of the catalytically generated hydrido species (Fig. 3,4), which would otherwise swap to a kinetically stabilised and thus disadvantageous bridging coordination between the two Fe centres (Winkler et al. 2013). As they do not support H-bond interactions to the environment, CO ligands exhibit a higher degree of configurational flexibility, which is of significance for the role of the third CO ligand (μCO). In the oxidised state, μCO is located in a bridging configuration between both Fe sites (Fig. 3,1), while in the more reduced states (Hred; Hsred), it stabilises the higher electron density at Fed by switching to a terminally bound coordination (Adamska et al. 2012) (Fig. 3,2–3).
The close protein environment of the 2FeH-site provides a second ligand sphere which further participates in the fine-tuning of catalytic features. Apart from the above-mentioned role of the H-bond networks in coordinating the two CN− ligands, the second ligand sphere provides an efficient coupling to the proton transfer pathway (Fig. 4, green) and with precisely positioned methionine residues (Fig. 4, red), provides a suitable environment for structurally guided and reversible movements of individual ligands such as the proton-shuttling aza-dithiolate ligand or μCO (Knörzer et al. 2012).
Like the active sites of many enzymes, the ligand binding site on Fed can also be occupied by small inhibitor molecules such as formaldehyde (FA) or carbon monoxide (CO). While formaldehyde preferably binds to Hsred (Bachmeier et al. 2015), CO only interacts with Hox or Hred, yielding the catalytically inactive states HoxCO or HredCO (Fig. 3,5–6), which can be reductively reactivated to yield Hsred (Fig. 3,3) (Adamska-Venkatesh et al. 2014).
3.6 Oxygen Sensitivity
The reversible inhibitory effect of CO and the irreversibly destructive influence of O2 on the activity of algal hydrogenases were first described for whole cell extracts of C. reinhardtii in the late 1970s. It was further demonstrated that the destructive influence of O2 is antagonised by pre-exposing the cell extract to CO (Erbes et al. 1979). The protective effect of CO gassing prior to O2 exposure was later systematically examined by protein film voltammetry, where C. reinhardtii HYDA1 was compared to different bacterial-type [FeFe]-hydrogenases (Goldet et al. 2009; Stripp et al. 2009). In contrast to [NiFe]-hydrogenases, all known [FeFe]-hydrogenases exhibit extraordinary high levels of O2 sensitivity; however different enzyme subtypes show different nuances of sensitivity resulting from differing accessibilities of the gas channel system for O2 and slight differences in the binding affinities of Fed for O2. The authors further concluded that CO competitively occupies the initial binding site of O2, thus preventing the following destructive process from being initiated.
Freeze drying renders [FeFe]-hydrogenases surprisingly insensitive towards O2 (Noth et al. 2015). X-ray absorption spectroscopy demonstrates that exposure of lyophilized [FeFe]-hydrogenase to O2 results in a stable O2 adduct which might correspond to a trapped version of the otherwise highly transient initial reaction product between Fed and O2. As proton transfer activity of hydrogenases has been demonstrated to be severely hampered in the freeze dried state, it can be speculated that protonation of the first O2 adduct is essential for initiating the destructive downstream processes that lead to the irreversible loss of both H-cluster components (Fig. 3,7) (Noth et al. 2015).
While the formation of an initial O2 adduct is uniformly accepted, the following sequence of events leading to the irreversible degradation of both H-cluster components is still under debate. XAS spectroscopy suggests the production of a reactive oxygen species which detaches from Fed and attacks the 4FeH-site before a loss of the 2FeH-cluster can be demonstrated (Stripp et al. 2009; Lambertz et al. 2011). A more recent examination instead favours a loss of the 2FeH-cluster prior to the oxidative degradation of the 4FeH-cluster (Fig. 3,7). The latter model is strongly supported by the fact that a large fraction of the O2-inactivated enzyme can be reactivated by supplying synthetic [2Fe]-cofactor as a substitute for the lost H-cluster component (Swanson et al. 2015).
4 Photobiological H2 Production in Practice
As O2 gradually accumulates during photosynthesis, photobiological H2 production requires the balancing of oxygenic photosynthesis and cell growth with anaerobic H2 production. The standard approach for this involves first growing the algae aerobically, to allow for carbon fixation, cell growth and the build-up of carbohydrates, before inducing anaerobiosis and therefore H2 production via nutrient deprivation. The most common induction method is sulphur deprivation (Melis et al. 2000). Sulphur deprivation results in anaerobiosis due to a reduction in the rate of repair of the PSII reaction centre protein, D1, as a result of a limitation in the sulphur-containing amino acids methionine and cysteine, which are required for D1 protein synthesis. This reduces the level of O2 production below respiration, resulting in anaerobiosis and HYDA1 gene expression. Sulphur deprivation also results in the build-up of starch (Zhang et al. 2002), which is subsequently degraded during H2 production (Hemschemeier et al. 2008a).
Anaerobiosis can also be achieved through the deprivation of other nutrients. This is possible as the limitation of any macronutrient results in a number of general nutrient deficiency responses in C. reinhardtii, many of which are conducive for H2 production, such as the accumulation of starch and a reduction in photosynthetic O2 evolution (Ball et al. 1990; Gonzalez-Ballester et al. 2015; Grossman et al. 2010; Zhang et al. 2002; Wykoff et al. 1998). The deprivation of nitrogen (Philipps et al. 2012), phosphorous (Batyrova et al. 2012) and magnesium (Volgusheva et al. 2015) have all been shown to induce H2 production. Nitrogen and phosphorous deprivation resulted in lower levels of H2 production compared to sulphur deprivation. In contrast, magnesium deprivation resulted in higher levels of H2 production (Volgusheva et al. 2015). This was thought to be due to a prolonged H2 production period of over 7 days, an increased respiration and starch accumulation and a smaller decrease in functional PSII [PSII was only reduced by 20% instead of 80% as in sulphur deprivation (Volgusheva et al. 2013)], resulting in a higher electron availability to the hydrogenase (Volgusheva et al. 2015).
5 Targets for Improved Microalgal H2 Production
The major problem with nutrient deprivation is that H2 production cannot be sustained for more than a few days. This is because the algae eventually die due to the lack of the respective nutrient. Therefore, the prolongation of photobiological H2 production via other strategies has been the focus of much research. Significantly higher H2 production efficiencies are required in order for biological H2 production to be commercially viable. Microalgae systems currently only have photon conversion efficiencies of 3%, while the theoretical maximum is ∼12–14% (Scoma et al. 2012; Volgusheva et al. 2013). The highest efficiency strains will therefore require a combination of strategies and involve significant re-engineering of the H2 production process. The ultimate goal is continuous H2 production where O2 production and consumption are in balance and the water-splitting reaction is active at the same time as the hydrogenase.
However, there are a few major factors currently limiting sustained H2 production. These include proton and electron supply to the hydrogenase and the extreme oxygen sensitivity of the hydrogenase enzyme. Factors limiting the growth of the algae and photosynthetic efficiency, such as light capture, also limit H2 production. In attempts to overcome these bottlenecks, extensive genetic engineering has been performed. A number of high H2-producing mutants with improvements in one or more of these bottlenecks have been generated and are detailed below. A tabular summary of the various mutants and their H2 production efficiencies can also be found in the recent review by Dubini and Ghirardi (2015).
5.1 Electron Supply to the Hydrogenase
Electron flow to the hydrogenase is one of the main bottlenecks for sustainable H2 production. This is due to the large number of other pathways competing for electrons from PETF (Winkler et al. 2011). For example, ferredoxin-NADP+ reductase (FNR), sulphite reductase, nitrate reductase, glutamate synthase and fatty acid desaturases all compete with HYDA1 for electrons (Hemschemeier and Happe 2011). To improve electron flow to the hydrogenase, PETF, FNR and the hydrogenase itself have all been engineered (Long et al. 2009; Yacoby et al. 2011; Wittenberg et al. 2013; Lubner et al. 2011; Rumpel et al. 2014; Sun et al. 2013). For example, a PETF variant with a reduced affinity for FNR has been developed (Rumpel et al. 2014), and PETF and PSI have both been fused to the hydrogenase (Yacoby et al. 2011; Lubner et al. 2011). However, all of the above work has so far only been performed in vitro and remains to be tested in vivo.
Engineering has also focused on various indirect targets. For example, small and large subunit Rubisco mutants (Pinto et al. 2013; Hemschemeier et al. 2008b), cyclic electron flow (CEF) mutants (Kruse et al. 2005; Johnson et al. 2014; Steinbeck et al. 2015; Tolleter et al. 2011), starch degradation mutant strains (Chochois et al. 2009) and respiration mutants (Ruehle et al. 2008) have all been reported to display increased H2 production levels. In fact, the cyclic electron flow mutants state transition 6 (Stm6) (Kruse et al. 2005) and the proton gradient regulation like 1 (pgrl1) (Tolleter et al. 2011) and pgr5 mutants (Steinbeck et al. 2015) have been reported to have the highest H2 production rates, producing between 540 and 840 mL of H2 per litre of culture, which is ∼10× that produced by wild-type C. reinhardtii. Interestingly, these high H2-producing mutants all display similar phenotypes: they have an enhanced oxygen consumption capacity and thus increased PSII stability and electron supply to the hydrogenase. The mutant cultures also become anaerobic earlier than the wild type following sulphur deprivation, which is thought to result in an increase in PSII stability due to a reduction in photo-oxidative damage (Steinbeck et al. 2015; Volgusheva et al. 2013). It is important to note that Stm6 also has increased starch reserves compared to the wild type and thus more available substrate (Kruse et al. 2005).
The effect of adding extra components to the photosynthetic electron transfer pathway has also been tested. The expression of plastid-expressed NAD(P)H dehydrogenase (Baltz et al. 2014) and native and exogenous hydrogenases (Reifschneider-Wegner et al. 2014; Chien et al. 2012) has all been reported.
5.2 Proton Supply to the Hydrogenase
Proton supply to the hydrogenase is another bottleneck for H2 production. This is because the ATP requirement drops during H2 production (Das et al. 2014), resulting in a reduced electron transport at cytochrome b6f (Burgess et al. 2011; Antal et al. 2009) and an impaired dissipation of the proton gradient and therefore decreased proton availability for the hydrogenase. One strategy to improve H2 production is to artificially dissipate the proton gradient to increase H2 production transiently in the presence of the chemical uncoupler carbonyl cyanide m-chlorophenyl hydrazine (CCCP), which causes an efflux of protons from the thylakoid lumen into the stroma (Lee 2013; Kruse et al. 2005; Lee and Greenbaum 2003). This suggests that the integration of a proton channel into the thylakoid membrane could more permanently restore proton and electron flow to the hydrogenase. Such a proton channel however would need to be inducibly expressed, as the addition of the uncoupler prior to anaerobiosis was found to abolish hydrogenase activity, suggesting that the proton gradient is important for initial hydrogenase expression (Lee 2013) and aerobic growth. A similar strategy involves the development of a leaky ATPase to increase proton flow and reduce ATP production (Das et al. 2014; Robertson et al. 1990). Reduced ATP production caused by the introduction of a proton channel or mutated ATPase may additionally reduce reactions competing for reducing equivalents and therefore increase electron supply to the hydrogenase (Kumar and Das 2013).
5.3 Oxygen Sensitivity of the Hydrogenase
Sustained H2 production under standard growth conditions remains a major challenge. The O2 sensitivity of the hydrogenase is a multifaceted problem due to the fact that O2 not only inhibits the activity of the hydrogenase enzyme, but also transcription and protein maturation (Cohen et al. 2005). However, to produce H2 from water using photosynthesis, O2 will be released; hence O2 production (photosynthesis) needs to be balanced with O2 consumption (respiration). Approaches have focused on developing O2-tolerant hydrogenases or changing the balance between O2 production and O2 consumption. As detailed in Sect. 5.1, the highest reported H2-producing mutants all display increased respiration rates and reach anaerobiosis earlier than the wild type following sulphur deprivation (Kruse et al. 2005; Steinbeck et al. 2015; Schonfeld et al. 2004), thus obtaining the correct balance between oxygen evolution and oxygen consumption appears to be of vital importance for further improvements in H2 production efficiencies.
Several genetic engineering approaches have been utilised to reduce the O2 sensitivity of the hydrogenase (reviewed in Ghirardi 2015), including random mutagenesis (Flynn et al. 2002) and targeted mutagenesis of the catalytic site to restrict O2 access (Stiebritz and Reiher 2012). While engineering approaches have been successful for a bacterial [NiFe]-hydrogenase (Dementin et al. 2009), there has so far been no success for the microalgal [FeFe]-hydrogenase. However, two algae strains were recently reported to be able to express the hydrogenase in the presence of more than 21% O2 and to produce low levels of H2 at 15% atmospheric O2 (Hwang et al. 2014). One problem with an O2-tolerant hydrogenase however, is that H2 would be produced alongside O2, which would make gas separation difficult and potentially lead to a dangerous gas mixture.
Balancing O2 production and consumption is perhaps a better, more feasible approach and has already been demonstrated in a number of studies. Photosystem II itself has been the target of engineering. For example, mutant strains with downregulated PSII subunits (Surzycki et al. 2007; Lin et al. 2013) and a mutant containing amino acid substitutions in the D1 protein (Scoma et al. 2012; Torzillo et al. 2009) all displayed increased H2 production efficiencies. The repression of psbD translation (Surzycki et al. 2007) and the downregulation of psbO in Chlorella sp. DT (Lin et al. 2013) both resulted in a lower O2 evolution. A double amino acid substitution in the D1 protein resulted in an increase in the quantum yield of photosynthesis, a higher respiration rate, a higher carbohydrate accumulation following sulphur deprivation and a higher synthesis of xanthophyll cycle pigments, which was proposed to result in an improved photoprotection (Torzillo et al. 2009, 2015). A mutant with an inactivated Calvin-Benson cycle also displayed a higher respiration rate and higher H2 evolution compared to the sulphur-deprived wild type (Ruehle et al. 2008).
Alternative approaches have also been tested to control the balance between oxygen evolution and consumption. Leghemoglobins, which are able to sequester O2, have been expressed in C. reinhardtii (Wu et al. 2010, 2011), as has a pyruvate oxidase from E. coli, which was found to lower O2 evolution (Xu et al. 2011). A sulphate permease mutant was also developed, which allowed a greater control over sulphur deprivation (Chen et al. 2005). Another approach targeting controlled sulphur deprivation implemented sulphur microdosing (Kosourov et al. 2005), whereby the sulphur in a sulphur-deprived culture was replaced in small amounts, allowing the repair of PSII and renewed production of protons and electrons to drive H2 production. Finally, cocultivation of algae and bacteria was reported to result in algal anaerobiosis due to an increased bacterial respiration (Lakatos et al. 2014).
5.4 Indirect Targets
Hydrogen production can be improved further by optimising the general culture conditions. For example, light capture can be optimised so that the photosystems function to their maximal capacities. The LHC antennae systems function to capture photons and dissipate excess light energy to provide photoprotection (Takahashi et al. 2006; Niyogi 1999; Pascal et al. 2005; Oey et al. 2013). Biomass production efficiency, at least in the laboratory, can be improved by reducing LHC antenna size, as this enhances light distribution through the bioreactors and enables the use of increased operational cell concentrations, resulting in improved overall photosynthetic efficiencies (Melis et al. 1999; Mussgnug et al. 2007; Polle et al. 2003; Oey et al. 2013; Beckmann et al. 2009). A number of C. reinhardtii antenna mutants have been developed, including Stm6Glc4T7, a mutant expressing a permanently active LHC translational repressor NAB1, which resulted in a 10–17% reduction in LHC antenna size and a ∼50% increase in photosynthetic efficiency (Beckmann et al. 2009); tla1 (truncated light-harvesting Chl antenna), an insertional mutant with a reduced LHCI and LHCII antenna complex (50% and 65% of the wild type, respectively) (Polle et al. 2003; Kosourov et al. 2011); and LO1, an RNAi knockdown of LHCBM1, 2 and 3 (21%, 81% and 41% expression of LHCBM1, 2 and 3, respectively) (Oey et al. 2013).
6 Conclusion
Microalgal H2 production is not only a fascinating biological phenomenon but is also of enormous biotechnological significance, as it offers a carbon neutral or even carbon negative approach for renewable energy production. In the more than 20 years since HYDA1 was isolated, remarkable progress has been made in elucidating this protein's structure, catalytic mechanism, induction and regulation. This in vitro groundwork has provided us with a detailed understanding of the enzyme and has paved the way for its in vivo engineering. Considerable work has also been carried out at the physiological level, and a large number of mutants displaying enhanced H2 productions have now been created. To date, the mutants with the highest reported H2 production rates have been the CEF mutants, which share the phenotype of an increased oxygen consumption, earlier onset of anaerobiosis and more stable PSII in common. However, further improvements are needed if microalgal H2 production is to become a commercial reality, particularly in regards to limitations in electron transport, competition for reductants from ferredoxin and the O2 sensitivity of the enzyme. The most successful approach is likely the combination of a number of different strategies. Solar conversion efficiency also needs to be improved. However, unresolved questions regarding, for example, the underlying regulation of HYDA1 in algae, need to be understood before such improvements can be made. Despite this, we are in a good position to solve these questions, and hopefully a H2 economy is no longer a distant dream but a future reality.
References
Adamska A, Silakov A, Lambertz C, Rudiger O, Happe T, Reijerse E, Lubitz W (2012) Identification and characterization of the “super-reduced” state of the H-cluster in [FeFe] hydrogenase: a new building block for the catalytic cycle? Angew Chem Int Ed Engl 51(46):11458–11462. doi:10.1002/anie.201204800
Adamska-Venkatesh A, Krawietz D, Siebel J, Weber K, Happe T, Reijerse E, Lubitz W (2014) New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster. J Am Chem Soc 136(32):11339–11346. doi:10.1021/ja503390c
Antal TK, Volgusheva AA, Kukarskih GP, Krendeleva TE, Rubin AB (2009) Relationships between H2 photoproduction and different electron transport pathways in sulfur-deprived Chlamydomonas reinhardtii. Int J Hydrog Energy 34(22):9087–9094. doi:10.1016/j.ijhydene.2009.09.011
Antal TK, Krendeleva TE, Tyystjarvi E (2015) Multiple regulatory mechanisms in the chloroplast of green algae: relation to hydrogen production. Photosynth Res 125(3):357–381. doi:10.1007/s11120-015-0157-2
Atteia A, van Lis R, Tielens AGM, Martin WF (2013) Anaerobic energy metabolism in unicellular photosynthetic eukaryotes. Biochim Biophys Acta Bioenerg 1827(2):210–223. doi:10.1016/j.bbabio.2012.08.002
Bachmeier A, Esselborn J, Hexter SV, Kramer T, Klein K, Happe T, McGrady JE, Myers WK, Armstrong FA (2015) How formaldehyde inhibits hydrogen evolution by [FeFe]-hydrogenases: determination by (1)(3)C ENDOR of direct Fe-C coordination and order of electron and proton transfers. J Am Chem Soc 137(16):5381–5389. doi:10.1021/ja513074m
Balk J, Schaedler TA (2014) Iron cofactor assembly in plants. Annu Rev Plant Biol 65:125–153. doi:10.1146/annurev-arplant-050213-035759
Ball SG, Dirick L, Decq A, Martiat JC, Matagne RF (1990) Physiology of starch storage in the monocellular alga Chlamydomonas reinhardtii. Plant Sci 66(1):1–9. doi:10.1016/0168-9452(90)90162-h
Baltz A, Kieu-Van D, Beyly A, Auroy P, Richaud P, Cournac L, Peltier G (2014) Plastidial expression of type II NAD(P)H dehydrogenase increases the reducing state of Plastoquinones and hydrogen photoproduction rate by the indirect pathway in Chlamydomonas reinhardtii. Plant Physiol 165(3):1344–1352. doi:10.1104/pp.114.240432
Batyrova KA, Tsygankov AA, Kosourov SN (2012) Sustained hydrogen photoproduction by phosphorus-deprived Chlamydomonas reinhardtii cultures. Int J Hydrog Energy 37(10):8834–8839. doi:10.1016/j.ijhydene.2012.01.068
Beckmann J, Lehr F, Finazzi G, Hankamer B, Posten C, Wobbe L, Kruse O (2009) Improvement of light to biomass conversion by de-regulation of light-harvesting protein translation in Chlamydomonas reinhardtii. J Biotechnol 142(1):70–77. doi:10.1016/j.jbiotec.2009.02.015
Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca JM, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M (2013) Biomimetic assembly and activation of FeFe-hydrogenases. Nature 499(7456):66–69. doi:10.1038/nature12239
Betz JN, Boswell NW, Fugate CJ, Holliday GL, Akiva E, Scott AG, Babbitt PC, Peters JW, Shepard EM, Broderick JB (2015) [FeFe]-hydrogenase maturation: insights into the role HydE plays in dithiomethylamine biosynthesis. Biochemistry 54(9):1807–1818. doi:10.1021/bi501205e
Broderick JB, Byer AS, Duschene KS, Duffus BR, Betz JN, Shepard EM, Peters JW (2014) H-cluster assembly during maturation of the [FeFe]-hydrogenase. J Biol Inorg Chem 19(6):747–757. doi:10.1007/s00775-014-1168-8
Burgess SJ, Tamburic B, Zemichael F, Hellgardt K, Nixon PJ (2011) Solar-driven hydrogen production in green algae. In: Laskin AI, Sariaslani S, Gadd GM (eds) Advances in applied microbiology, vol 75, pp 71–110. doi:10.1016/b978-0-12-387046-9.00004-9
Castruita M, Casero D, Karpowicz SJ, Kropat J, Vieler A, Hsieh SI, Yan W, Cokus S, Loo JA, Benning C, Pellegrini M, Merchant SS (2011) Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23(4):1273–1292. doi:10.1105/tpc.111.084400
Chen HC, Newton AJ, Melis A (2005) Role of SulP, a nuclear-encoded chloroplast sulfate permease, in sulfate transport and H2 evolution in Chlamydomonas reinhardtii. Photosynth Res 84(1–3):289–296. doi:10.1007/s11120-004-7157-y
Chien LF, Kuo TT, Liu BH, Lin HD, Feng TY, Huang CC (2012) Solar-to-bio H2 production enhanced by homologous overexpression of hydrogenase in green alga Chlorella sp. DT. Int J Hydrog Energy 37(23):17738–17748. doi:10.1016/j.ijhydene.2012.09.068
Chochois V, Dauvillee D, Beyly A, Tolleter D, Cuine S, Timpano H, Ball S, Cournac L, Peltier G (2009) Hydrogen production in Chlamydomonas: photosystem II-dependent and -independent pathways differ in their requirement for starch metabolism. Plant Physiol 151(2):631–640. doi:10.1104/pp.109.144576
Cohen J, Kim K, Posewitz M, Ghirardi ML, Schulten K, Seibert M, King P (2005) Molecular dynamics and experimental investigation of H2 and O2 diffusion in Fe-hydrogenase. Biochem Soc Trans 33:80–82
Cornish AJ, Gaertner K, Yang H, Peters JW, Hegg EL (2011) Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum. J Biol Chem 286(44):38341–38347. doi:10.1074/jbc.M111.254664
Das D, Khanna N, Dasgupta CN (2014) Biohydrogen production: fundamentals and technology advances. CRC Press, London
Dementin S, Leroux F, Cournac L, de Lacey AL, Volbeda A, Leger C, Burlat B, Martinez N, Champ S, Martin L, Sanganas O, Haumann M, Fernandez VM, Guigliarelli B, Fontecilla-Camps JC, Rousset M (2009) Introduction of methionines in the gas channel makes NiFe hydrogenase aero-tolerant. J Am Chem Soc 131(29):10156–10164. doi:10.1021/ja9018258
Desplats C, Mus F, Cuine S, Billon E, Cournac L, Peltier G (2009) Characterization of Nda2, a plastoquinone-reducing type II NAD(P)H dehydrogenase in Chlamydomonas chloroplasts. J Biol Chem 284(7):4148–4157. doi:10.1074/jbc.M804546200
Dubini A, Ghirardi ML (2015) Engineering photosynthetic organisms for the production of biohydrogen. Photosynth Res 123(3):241–253. doi:10.1007/s11120-014-9991-x
Erbes DL, King D, Gibbs M (1979) Inactivation of hydrogenase in cell-free extracts and whole cells of Chlamydomonas reinhardi by oxygen. Plant Physiol 63(6):1138–1142
Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Nothl J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T (2013) Spontaneous activation of FeFe-hydrogenases by an inorganic 2Fe active site mimic. Nat Chem Biol 9(10):607–609. doi:10.1038/nchembio.1311
Esselborn J, Muraki N, Klein K, Engelbrecht V, Metzler-Nolte N, Apfel UP, Hofmann E, Kurisu G, Happe T (2016) A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem Sci 7(2):959–968. doi:10.1039/c5sc03397g
Flynn T, Ghirardi ML, Seibert M (2002) Accumulation of O2-tolerant phenotypes in H2-producing strains of Chlamydomonas reinhardtii by sequential applications of chemical mutagenesis and selection. Int J Hydrog Energy 27(11–12):1421–1430. doi:10.1016/s0360-3199(02)00117-9
Forestier M, King P, Zhang LP, Posewitz M, Schwarzer S, Happe T, Ghirardi ML, Seibert M (2003) Expression of two [Fe]-hydrogenases in Chlamydomonas reinhardtii under anaerobic conditions. Eur J Biochem 270(13):2750–2758. doi:10.1046/j.1432-1033.2003.03656
Fouchard S, Hemschemeier A, Caruana A, Pruvost K, Legrand J, Happe T, Peltier G, Cournac L (2005) Autotrophic and mixotrophic hydrogen photoproduction in sulfur-deprived Chlamydomonas cells. Appl Environ Microbiol 71(10):6199–6205. doi:10.1128/aem.71.10.6199-6205.2005
Gaffron H, Rubin J (1942) Fermentative and photochemical production of hydrogen in algae. J Gen Physiol 26(2):219–240. doi:10.1085/jgp.26.2.219
Ghirardi ML (2015) Implementation of photobiological H2 production: the O2 sensitivity of hydrogenases. Photosynth Res 125(3):383–393. doi:10.1007/s11120-015-0158-1
Ghirardi ML, Posewitz MC, Maness P-C, Dubini A, Yu J, Seibert M (2007) Hydrogenases and hydrogen photoproduction in oxygenic photosynthetic organisms. Annu Rev Plant Biol 58:71–91. doi:10.1146/annurev.arplant.58.032806.103848
Godde D, Trebst A (1980) NADH as electron donor for the photosynthetic membrane of Chlamydomonas reinhardtii. Arch Microbiol 127(3):245–252. doi:10.1007/bf00427200
Goldet G, Brandmayr C, Stripp ST, Happe T, Cavazza C, Fontecilla-Camps JC, Armstrong FA (2009) Electrochemical kinetic investigations of the reactions of FeFe-hydrogenases with carbon monoxide and oxygen: comparing the importance of gas tunnels and active-site electronic/redox effects. J Am Chem Soc 131(41):14979–14989. doi:10.1021/ja905388j
Gonzalez-Ballester D, Luis Jurado-Oller J, Fernandez E (2015) Relevance of nutrient media composition for hydrogen production in Chlamydomonas. Photosynth Res 125(3):395–406. doi:10.1007/s11120-015-0152-7
Grossman AR, Gonzalez-Ballester D, Shibagaki N, Pootakham W, Moseley J, Pootakham W (2010) Responses to macronutrient deprivation. In: Abiotic stress adaptation in plants: physiological, molecular and genomic foundation. Springer, Dordrecht. doi:10.1007/978-90-481-3112-9_15
Grossman AR, Catalanotti C, Yang W, Dubini A, Magneschi L, Subramanian V, Posewitz MC, Seibert M (2011) Multiple facets of anoxic metabolism and hydrogen production in the unicellular green alga Chlamydomonas reinhardtii. New Phytol 190(2):279–288. doi:10.1111/j.1469-8137.2010.03534.x
Gupta SK, Kumari S, Reddy K, Bux F (2013) Trends in biohydrogen production: major challenges and state-of-the-art developments. Environ Technol 34(13–14):1653–1670. doi:10.1080/09593330.2013.822022
Happe T, Kaminski A (2002) Differential regulation of the Fe-hydrogenase during anaerobic adaptation in the green alga Chlamydomonas reinhardtii. Eur J Biochem 269(3):1022–1032. doi:10.1046/j.0014-2956.2001.02743.x
Happe T, Naber JD (1993) Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii. Eur J Biochem 214(2):475–481. doi:10.1111/j.1432-1033.1993.tb17944.x
Happe T, Mosler B, Naber JD (1994) Induction, localization and metal content of hydrogenase in the green alga Chlamydomonas reinhardtii. Eur J Biochem 222(3):769–774. doi:10.1111/j.1432-1033.1994.tb18923.x
Hemschemeier A, Happe T (2011) Alternative photosynthetic electron transport pathways during anaerobiosis in the green alga Chlamydomonas reinhardtii. BBA-Bioenergetics 1807(8):919–926. doi:10.1016/j.bbabio.2011.02.010
Hemschemeier A, Fouchard S, Cournac L, Peltier G, Happe T (2008a) Hydrogen production by Chlamydomonas reinhardtii: an elaborate interplay of electron sources and sinks. Planta 227(2):397–407. doi:10.1007/s00425-007-0626-8
Hemschemeier A, Jacobs J, Happe T (2008b) Biochemical and physiological characterization of the pyruvate Formate-Lyase Pfl1 of Chlamydomonas reinhardtii, a typically bacterial enzyme in a eukaryotic alga. Eukaryot Cell 7(3):518–526. doi:10.1128/ec.00368-07
Hemschemeier A, Casero D, Liu BS, Benning C, Pellegrini M, Happe T, Merchant SS (2013) Copper response regulator1-dependent and -independent responses of the Chlamydomonas reinhardtii Transcriptome to dark anoxia. Plant Cell 25(9):3186–3211. doi:10.1105/tpc.113.115741
Hexter SV, Grey F, Happe T, Climent V, Armstrong FA (2012) Electrocatalytic mechanism of reversible hydrogen cycling by enzymes and distinctions between the major classes of hydrogenases. Proc Natl Acad Sci U S A 109(29):11516–11521. doi:10.1073/pnas.1204770109
Hong G, Pachter R (2012) Inhibition of biocatalysis in [Fe–Fe] hydrogenase by oxygen: molecular dynamics and density functional theory calculations. ACS Chem Biol 7(7):1268–1275. doi:10.1021/cb3001149
Hwang J-H, Kim H-C, Choi J-A, Abou-Shanab RAI, Dempsey BA, Regan JM, Kim JR, Song H, Nam I-H, Kim S-N, Lee W, Park D, Kim Y, Choi J, Ji M-K, Jung W, Jeon B-H (2014) Photoautotrophic hydrogen production by eukaryotic microalgae under aerobic conditions. Nat Commun 5. doi:10.1038/ncomms4234
Jans F, Mignolet E, Houyoux P-A, Cardol P, Ghysels B, Cuine S, Cournac L, Peltier G, Remacle C, Franck F (2008) A type II NAD(P) H dehydrogenase mediates light-independent plastoquinone reduction in the chloroplast of Chlamydomonas. Proc Natl Acad Sci U S A 105(51):20546–20551. doi:10.1073/pnas.0806896105
Johnson X, Steinbeck J, Dent RM, Takahashi H, Richaud P, Ozawa SI, Houille-Vernes L, Petroutsos D, Rappaport F, Grossman AR, Niyogi KK, Hippler M, Alric J (2014) Proton gradient regulation 5-mediated cyclic electron flow under ATP- or redox-limited conditions: a study of ΔATPase pgr5 and ΔrbcL pgr5 mutants in the green alga Chlamydomonas reinhardtii. Plant Physiol 165(1):438–452. doi:10.1104/pp.113.233593
Knörzer P, Silakov A, Foster CE, Armstrong FA, Lubitz W, Happe T (2012) Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J Biol Chem 287(2):1489–1499. doi:10.1074/jbc.M111.305797
Kosourov S, Makarova V, Fedorov AS, Tsygankov A, Seibert M, Ghirardi ML (2005) The effect of sulfur re-addition on H2 photoproduction by sulfur-deprived green algae. Photosynth Res 85(3):295–305. doi:10.1007/s11120-005-5105-0
Kosourov SN, Ghirardi ML, Seibert M (2011) A truncated antenna mutant of Chlamydomonas reinhardtii can produce more hydrogen than the parental strain. Int J Hydrog Energy 36(3):2044–2048. doi:10.1016/j.ijhydene.2010.10.041
Kropat J, Tottey S, Birkenbihl RP, Depege N, Huijser P, Merchant S (2005) A regulator of nutritional copper signaling in Chlamydomonas is an SBP domain protein that recognizes the GTAC core of copper response element. Proc Natl Acad Sci U S A 102(51):18730–18735. doi:10.1073/pnas.0507693102
Kruse O, Rupprecht J, Bader KP, Thomas-Hall S, Schenk PM, Finazzi G, Hankamer B (2005) Improved photobiological H2 production in engineered green algal cells. J Biol Chem 280(40):34170–34177. doi:10.1074/jbc.M503840200
Kumar K, Das D (2013) CO2 sequestration and hydrogen production using cyanobacteria and green algae. In: Natural and artificial photosynthesis. Wiley, New York, pp 173–215. doi:10.1002/9781118659892.ch6
Lakatos G, Deak Z, Vass I, Retfalvi T, Rozgonyi S, Rakhely G, Oerdoeg V, Kondorosi E, Maroti G (2014) Bacterial symbionts enhance photo-fermentative hydrogen evolution of Chlamydomonas algae. Green Chem 16(11):4716–4727. doi:10.1039/c4gc00745j
Lambertz C, Leidel N, Havelius KGV, Noth J, Chernev P, Winkler M, Happe T, Haumann M (2011) O2 reactions at the six-iron active site (H-cluster) in [FeFe]-hydrogenase. J Biol Chem 286(47):40614–40623. doi:10.1074/jbc.M111.283648
Lee J (2013) Designer transgenic algae for photobiological production of hydrogen from water. In: Lee JW (ed) Advanced biofuels and bioproducts. Springer, New York, pp 371–404. doi:10.1007/978-1-4614-3348-4_20
Lee JW, Greenbaum E (2003) A new oxygen sensitivity and its potential application in photosynthetic H2 production. Appl Biochem Biotechnol 105:303–313
Lin H-D, Liu B-H, Kuo T-T, Tsai H-C, Feng T-Y, Huang C-C, Chien L-F (2013) Knockdown of PsbO leads to induction of HydA and production of photobiological H2 in the green alga Chlorella sp. DT. Bioresour Technol 143:154–162. doi:10.1016/j.biortech.2013.05.101
Long H, King PW, Ghirardi ML, Kim K (2009) Hydrogenase/Ferredoxin charge-transfer complexes: effect of hydrogenase mutations on the complex association. J Phys Chem A 113(16):4060–4067. doi:10.1021/jp810409z
Lubitz W, Ogata H, Rudiger O, Reijerse E (2014) Hydrogenases. Chem Rev 114(8):4081–4148. doi:10.1021/cr4005814
Lubner CE, Applegate AM, Knoerzer P, Ganago A, Bryant DA, Happe T, Golbeck JH (2011) Solar hydrogen-producing bionanodevice outperforms natural photosynthesis. Proc Natl Acad Sci U S A 108(52):20988–20991. doi:10.1073/pnas.1114660108
Ludwig M, Schulz-Friedrich R, Appel J (2006) Occurrence of hydrogenases in cyanobacteria and anoxygenic photosynthetic bacteria: implications for the phylogenetic origin of cyanobacterial and algal hydrogenases. J Mol Evol 63(6):758–768. doi:10.1007/s00239-006-0001-6
Maione TE, Gibbs M (1986) Association of the chloroplastic respiratory and photosynthetic electron transport chains of Chlamydomonas reinhardtii with photoreduction and the oxyhydrogen reaction. Plant Physiol 80(2):364–368. doi:10.1104/pp.80.2.364
Melis A, Happe T (2001) Hydrogen production. Green algae as a source of energy. Plant Physiol 127(3):740–748
Melis A, Neidhardt J, Benemann J (1999) Dunaliella salina (Chlorophyta) with small chlorophyll antenna sizes exhibit higher photosynthetic productivities and photon use efficiencies than normally pigmented cells. J Appl Phycol 10(6):515–525. doi:10.1023/a:1008076231267
Melis A, Zhang LP, Forestier M, Ghirardi ML, Seibert M (2000) Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol 122(1):127–135. doi:10.1104/pp.122.1.127
Meuser JE, D’Adamo S, Jinkerson RE, Mus F, Yang WQ, Ghirardi ML, Seibert M, Grossman AR, Posewitz MC (2012) Genetic disruption of both Chlamydomonas reinhardtii [FeFe]-hydrogenases: insight into the role of HYDA2 in H2 production. Biochem Biophys Res Commun 417(2):704–709. doi:10.1016/j.bbrc.2011.12.002
Meyer J (2007) [FeFe] hydrogenases and their evolution: a genomic perspective. Cell Mol Life Sci 64(9):1063–1084. doi:10.1007/s00018-007-6477-4
Mignolet E, Lecler R, Ghysels B, Remacle C, Franck F (2012) Function of the chloroplastic NAD(P)H dehydrogenase Nda2 for H2 photoproduction in sulphur-deprived Chlamydomonas reinhardtii. J Biotechnol 162(1):81–88. doi:10.1016/j.jbiotec.2012.07.002
Morra S, Giraudo A, Di Nardo G, King PW, Gilardi G, Valetti F (2012) Site saturation mutagenesis demonstrates a central role for cysteine 298 as proton donor to the catalytic site in CaHydA [FeFe]-Hydrogenase. Plos One 7(10):e48400. doi:10.1371/journal.Pone.0048400
Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW (2010) Stepwise [FeFe]-hydrogenase H-cluster assembly revealed in the structure of HydADEFG. Nature 465(7295):248–251. doi:10.1038/nature08993
Mulder DW, Shepard EM, Meuser JE, Joshi N, King PW, Posewitz MC, Broderick JB, Peters JW (2011) Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure 19(8):1038–1052. doi:10.1016/j.str.2011.06.008
Mulder DW, Ratzloff MW, Bruschi M, Greco C, Koonce E, Peters JW, King PW (2014) Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase. J Am Chem Soc 136(43):15394–15402. doi:10.1021/ja508629m
Mus F, Cournac L, Cardettini W, Caruana A, Peltier G (2005) Inhibitor studies on non-photochemical plastoquinone reduction and H2 photoproduction in Chlamydomonas reinhardtii. Biochim Biophys Acta Bioenerg 1708(3):322–332. doi:10.1016/j.bbabio.2005.05.003
Mus F, Dubini A, Seibert M, Posewitz MC, Grossman AR (2007) Anaerobic acclimation in Chlamydomonas reinhardtii – anoxic gene expression, hydrogenase induction, and metabolic pathways. J Biol Chem 282(35):25475–25486. doi:10.1074/jbc.M701415200
Mussgnug JH, Thomas-Hall SR, Rupprecht J, Foo A, Klassen V, McDowall A, Schenk PM, Kruse O, Hankamer B (2007) Engineering photosynthetic light capture: impacts on improved solar energy to biomass conversion. Plant Biotechnol J 5(6):802–814. doi:10.1111/j.1467-7652.2007.00285.x
Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC (1999) Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Struct Fold Des 7(1):13–23. doi:10.1016/s0969-2126(99)80005-7
Niyogi KK (1999) Photoprotection revisited: genetic and molecular approaches. Annu Rev Plant Physiol Plant Mol Biol 50(1):333–359. doi:10.1146/annurev.Arplant.50.1.333
Noth J, Krawietz D, Hemschemeier A, Happe T (2013) Pyruvate: ferredoxin oxidoreductase is coupled to light-independent hydrogen production in Chlamydomonas reinhardtii. J Biol Chem 288(6):4368–4377. doi:10.1074/jbc.M112.429985
Noth J, Kositzki R, Klein K, Winkler M, Haumann M, Happe T (2015) Lyophilization protects [FeFe]-hydrogenases against O2-induced H-cluster degradation. Sci Rep 5:13978. doi:10.1038/srep13978
Oey M, Ross IL, Stephens E, Steinbeck J, Wolf J, Radzun KA, Kuegler J, Ringsmuth AK, Kruse O, Hankamer B (2013) RNAi knock-down of LHCBM1, 2 and 3 increases photosynthetic H2 production efficiency of the green alga Chlamydomonas reinhardtii. Plos One 8(4):e61375. doi:10.1371/journal.Pone.0061375
Oey M, Sawyer AL, Ross IL, Hankamer B (2016) Challenges and opportunities for hydrogen production from microalgae. Plant Biotechnol J 14(7):1487–1499. doi:10.1111/pbi.12516
Pagnier A, Martin L, Zeppieri L, Nicolet Y, Fontecilla-Camps JC (2016) CO and CN- syntheses by [FeFe]-hydrogenase maturase HydG are catalytically differentiated events. Proc Natl Acad Sci U S A 113(1):104–109. doi:10.1073/pnas.1515842113
Paila YD, Richardson LGL, Schnell DJ (2015) New insights into the mechanism of chloroplast protein import and its integration with protein quality control, organelle biogenesis and development. J Mol Biol 427(5):1038–1060. doi:10.1016/j.jmb.2014.08.016
Pape M, Lambertz C, Happe T, Hemschemeier A (2012) Differential expression of the Chlamydomonas [FeFe]-hydrogenase-encoding HYDA1 gene is regulated by the copper response regulator1. Plant Physiol 159(4):1700–1712. doi:10.1104/pp.112.200162
Pascal AA, Liu Z, Broess K, van Oort B, van Amerongen H, Wang C, Horton P, Robert B, Chang W, Ruban A (2005) Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436(7047):134–137
Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC (1998) X-ray crystal structure of the Fe-only hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282(5395):1853–1858. doi:10.1126/science.282.5395.1853
Peters JW, Schut GJ, Boyd ES, Mulder DW, Shepard EM, Broderick JB, King PW, Adams MWW (2015) [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim Biophys Acta Mol Cell Res 1853(6):1350–1369. doi:10.1016/j.bbamcr.2014.11.021
Philipps G, Happe T, Hemschemeier A (2012) Nitrogen deprivation results in photosynthetic hydrogen production in Chlamydomonas reinhardtii. Planta 235(4):729–745. doi:10.1007/s00425-011-1537-2
Pinto TS, Malcata FX, Arrabaça JD, Silva JM, Spreitzer RJ, Esquível MG (2013) Rubisco mutants of Chlamydomonas reinhardtii enhance photosynthetic hydrogen production. Appl Microbiol Biotechnol 97(12):5635–5643. doi:10.1007/s00253-013-4920-z
Polle JEW, Kanakagiri SD, Melis A (2003) tla1, a DNA insertional transformant of the green alga Chlamydomonas reinhardtii with a truncated light-harvesting chlorophyll antenna size. Planta 217(1):49–59. doi:10.1007/s00425-002-0968-1
Posewitz MC, King PW, Smolinski SL, Zhang LP, Seibert M, Ghirardi ML (2004) Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active Fe hydrogenase. J Biol Chem 279(24):25711–25720. doi:10.1074/jbc.M403206200
Reifschneider-Wegner K, Kanygin A, Redding KE (2014) Expression of the [FeFe] hydrogenase in the chloroplast of Chlamydomonas reinhardtii. Int J Hydrog Energy 39(8):3657–3665. doi:10.1016/j.ijhydene.2013.12.157
Robertson D, Boynton JE, Gillham NW (1990) Cotranscription of the wild-type chloroplast atpE gene encoding the CF1/CF0 epsilon subunit with the 3′ half of the rps7 gene in Chlamydomonas reinhardtii and characterization of frameshift mutations in atpE. Mol Gen Genet 221(2):155–163
Roessler PG, Lien S (1984) Purification of hydrogenase from Chlamydomonas reinhardtii. Plant Physiol 75(3):705–709. doi:10.1104/pp.75.3.705
Roseboom W, De Lacey AL, Fernandez VM, Hatchikian EC, Albracht SP (2006) The active site of the [FeFe]-hydrogenase from desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy. J Biol Inorg Chem 11(1):102–118
Ruehle T, Hemschemeier A, Melis A, Happe T (2008) A novel screening protocol for the isolation of hydrogen producing Chlamydomonas reinhardtii strains. BMC Plant Biol 8. doi:10.1186/1471-2229-8-107
Rumpel S, Siebel JF, Fares C, Duan J, Reijerse E, Happe T, Lubitz W, Winkler M (2014) Enhancing hydrogen production of microalgae by redirecting electrons from photosystem I to hydrogenase. Energy Environ Sci 7(10):3296–3301. doi:10.1039/c4ee01444h
Schonfeld C, Wobbe L, Borgstadt R, Kienast A, Nixon PJ, Kruse O (2004) The nucleus-encoded protein MOC1 is essential for mitochondrial light acclimation in Chlamydomonas reinhardtii. J Biol Chem 279(48):50366–50374. doi:10.1074/jbc.M408477200
Scoma A, Krawietz D, Faraloni C, Giannelli L, Happe T, Torzillo G (2012) Sustained H2 production in a Chlamydomonas reinhardtii D1 protein mutant. J Biotechnol 157(4):613–619. doi:10.1016/j.jbiotec.2011.06.019
Shepard EM, McGlynn SE, Bueling AL, Grady-Smith CS, George SJ, Winslow MA, Cramer SP, Peters JW, Broderick JB (2010) Synthesis of the 2Fe subcluster of the [FeFe]-hydrogenase H cluster on the HydF scaffold. Proc Natl Acad Sci U S A 107(23):10448–10453. doi:10.1073/pnas.1001937107
Simmons TR, Berggren G, Bacchi M, Fontecave M, Artero V (2014) Mimicking hydrogenases: from biomimetics to artificial enzymes. Coord Chem Rev 270:127–150. doi:10.1016/j.ccr.2013.12.018
Steinbeck J, Nikolova D, Weingarten R, Johnson X, Richaud P, Peltier G, Hermann M, Magneschi L, Hippler M (2015) Deletion of proton gradient regulation 5 (PGR5) and PGR5-like 1 (PGRL1) proteins promote sustainable light-driven hydrogen production in Chlamydomonas reinhardtii due to increased PSII activity under sulfur deprivation. Front Plant Sci 6:892. doi:10.3389/fpls.2015.00892
Stiebritz MT, Reiher M (2012) Hydrogenases and oxygen. Chem Sci 3(6):1739–1751. doi:10.1039/c2sc01112c
Stripp ST, Goldet G, Brandmayr C, Sanganas O, Vincent KA, Haumann M, Armstrong FA, Happe T (2009) How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc Natl Acad Sci U S A 106(41):17331–17336. doi:10.1073/pnas.0905343106
Stuart TS, Gaffron H (1972) The mechanism of hydrogen photoproduction by several algae: 2. The contribution of photosystem II. Planta 106(2):101–112. doi:10.1007/bf00383990
Suess DLM, Pham CC, Buerstel I, Swartz JR, Cramer SP, Britt RD (2016) The radical SAM enzyme HydG requires cysteine and a dangler iron for generating an organometallic precursor to the [FeFe]-hydrogenase H-cluster. J Am Chem Soc 138(4):1146–1149. doi:10.1021/jacs.5b12512
Sun YL, Chen M, Yang HM, Zhang J, Kuang TY, Huang F (2013) Enhanced H2 photoproduction by down-regulation of ferredoxin-NADP+ reductase (FNR) in the green alga Chlamydomonas reinhardtii. Int J Hydrog Energy 38(36):16029–16037. doi:10.1016/j.ijhydene.2013.10.011
Surzycki R, Cournac L, Peltiert G, Rochaix J-D (2007) Potential for hydrogen production with inducible chloroplast gene expression in Chlamydomonas. Proc Natl Acad Sci U S A 104(44):17548–17553. doi:10.1073/pnas.0704205104
Swanson KD, Ratzloff MW, Mulder DW, Artz JH, Ghose S, Hoffman A, White S, Zadvornyy OA, Broderick JB, Bothner B, King PW, Peters JW (2015) [FeFe]-hydrogenase oxygen inactivation is initiated at the H cluster 2Fe subcluster. J Am Chem Soc 137(5):1809–1816. doi:10.1021/ja510169s
Takahashi H, Iwai M, Takahashi Y, Minagawa J (2006) Identification of the mobile light-harvesting complex II polypeptides for state transitions in Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A 103(2):477–482. doi:10.1073/pnas.0509952103
Terashima M, Specht M, Naumann B, Hippler M (2010) Characterizing the anaerobic response of Chlamydomonas reinhardtii by quantitative proteomics. Mol Cell Proteomics 9(7):1514–1532. doi:10.1074/mcp.M900421-MCP200
Tolleter D, Ghysels B, Alric J, Petroutsos D, Tolstygina I, Krawietz D, Happe T, Auroy P, Adriano JM, Beyly A, Cuine S, Plet J, Reiter IM, Genty B, Cournac L, Hippler M, Peltier G (2011) Control of hydrogen photoproduction by the proton gradient generated by cyclic electron flow in Chlamydomonas reinhardtii. Plant Cell 23(7):2619–2630. doi:10.1105/tpc.111.086876
Torzillo G, Scoma A, Faraloni C, Ena A, Johanningmeier U (2009) Increased hydrogen photoproduction by means of a sulfur-deprived Chlamydomonas reinhardtii D1 protein mutant. Int J Hydrog Energy 34(10):4529–4536. doi:10.1016/j.ijhydene.2008.07.093
Torzillo G, Scoma A, Faraloni C, Giannelli L (2015) Advances in the biotechnology of hydrogen production with the microalga Chlamydomonas reinhardtii. Crit Rev Biotechnol 35(4):485–496. doi:10.3109/07388551.2014.900734
van Lis R, Baffert C, Coute Y, Nitschke W, Atteia A (2013) Chlamydomonas reinhardtii chloroplasts contain a homodimeric pyruvate: ferredoxin oxidoreductase that functions with FDX1. Plant Physiol 161(1):57–71. doi:10.1104/pp.112.208181
Vignais PM, Billoud B, Meyer J (2001) Classification and phylogeny of hydrogenases. FEMS Microbiol Rev 25(4):455–501. doi:10.1111/j.1574-6976.2001.tb00587.x
Volgusheva A, Styring S, Mamedov F (2013) Increased photosystem II stability promotes H2 production in sulfur-deprived Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A 110(18):7223–7228. doi:10.1073/pnas.1220645110
Volgusheva A, Kukarskikh G, Krendeleva T, Rubin A, Mamedov F (2015) Hydrogen photoproduction in green algae Chlamydomonas reinhardtii under magnesium deprivation. RSC Adv 5(8):5633–5637. doi:10.1039/c4ra12710b
Winkler M, Kawelke S, Happe T (2011) Light driven hydrogen production in protein based semi-artificial systems. Bioresour Technol 102(18):8493–8500. doi:10.1016/j.biortech.2011.05.019
Winkler M, Esselborn J, Happe T (2013) Molecular basis of [FeFe]-hydrogenase function: an insight into the complex interplay between protein and catalytic cofactor. Biochim Biophys Acta. doi:10.1016/j.bbabio.2013.03.004
Wittenberg G, Sheffler W, Darchi D, Baker D, Noy D (2013) Accelerated electron transport from photosystem I to redox partners by covalently linked ferredoxin. Phys Chem Chem Phys 15(45):19608–19614. doi:10.1039/C3CP53264J
Wu SX, Huang R, Xu LL, Yan GY, Wang QX (2010) Improved hydrogen production with expression of hemH and lba genes in chloroplast of Chlamydomonas reinhardtii. J Biotechnol 146(3):120–125. doi:10.1016/j.jbiotec.2010.01.023
Wu S, Xu L, Huang R, Wang Q (2011) Improved biohydrogen production with an expression of codon-optimized hemH and lba genes in the chloroplast of Chlamydomonas reinhardtii. Bioresour Technol 102(3):2610–2616. doi:10.1016/j.biortech.2010.09.123
Wykoff DD, Davies JP, Melis A, Grossman AR (1998) The regulation of photosynthetic electron transport during nutrient deprivation in Chlamydomonas reinhardtii. Plant Physiol 117(1):129–139. doi:10.1104/pp.117.1.129
Xu F-Q, Ma W-M, Zhu X-G (2011) Introducing pyruvate oxidase into the chloroplast of Chlamydomonas reinhardtii increases oxygen consumption and promotes hydrogen production. Int J Hydrog Energy 36(17):10648–10654. doi:10.1016/j.ijhydene.2011.05.130
Yacoby I, Pochekailov S, Toporik H, Ghirardi ML, King PW, Zhang S (2011) Photosynthetic electron partitioning between FeFe-hydrogenase and ferredoxin:NADP(+)-oxidoreductase (FNR) enzymes in vitro. Proc Natl Acad Sci U S A 108(23):9396–9401. doi:10.1073/pnas.1103659108
Zhang LP, Happe T, Melis A (2002) Biochemical and morphological characterization of sulfur-deprived and H2-producing Chlamydomonas reinhardtii (green alga). Planta 214(4):552–561. doi:10.1007/s004250100660
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Sawyer, A., Esselborn, J., Winkler, M., Happe, T. (2017). Chlamydomonas: Hydrogenase and Hydrogen Production. In: Hippler, M. (eds) Chlamydomonas: Biotechnology and Biomedicine. Microbiology Monographs, vol 31. Springer, Cham. https://doi.org/10.1007/978-3-319-66360-9_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-66360-9_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66359-3
Online ISBN: 978-3-319-66360-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)