
Abstract
Haploidentical hematopoietic stem cell transplantation (haplo-HSCT) is increasingly used as potentially curative option for pediatric patients affected by leukemia or other malignancies, as well as by several non malignant disorders such as primary immunodeficiencies, constitutional and acquired bone marrow failure syndromes, hemoglobinopathies and others.
Different techniques have been developed in order to overcome the HLA barrier and make HSCT feasible in virtually all patients. These approaches are mainly based on the in vitro physical removal of alloreactive T cells capable of mediating graft-versus-host disease (GVHD) or on in vivo pharmacological treatment with immunosuppressive drugs or immunomodulating agents.
The possibility to receive a transplant from a haploidentical donor (usually a parent) is particularly relevant both for children needing to have rapid access to this procedure, and for patients belonging to ethnic minorities or with rare HLA haplotypes that are underrepresented in the international voluntary donor registries.
The delayed immune reconstitution after haplo-HSCT may cause impaired graft-versus-leukemia (GVL) effect and defective control of potentially life-threatening opportunistic infections, thus increasing the risk of relapse and of transplant-related mortality (TRM). For this reason, several strategies of post-transplant adoptive cell therapy have been developed over time, from the use of donor lymphocyte infusions (DLI), to the administration of selected immunocompetent cell populations or genetically modified T lymphocytes, or the production of leukemia or pathogen-specific cytotoxic T cells (CTL) to be infused after the transplant.
Even if prospective clinical trials are still needed in order to define the optimal strategy to treat each different disease, the results of haplo-HSCT in children have significantly improved over time and, nowadays, the probability of disease cure is often similar to that of transplants performed from matched unrelated donors, thus making this approach extremely appealing in the pediatric population.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

11.1 Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is a potentially curative treatment for children and adolescents with malignant and non-malignant diseases. Recent progress in HSCT contributed to the improvement of outcomes for patients with diseases curable by HSCT. Human leukocyte antigen (HLA)-identical sibling is the preferred donor choice, however the probability of having such a donor correlates to the number of siblings within the family, and is approximately 25%. HLA-matched unrelated volunteer donor (MUD) is also a good option for successful HSCT, but the probability to find a HLA-matched donor correlates with race and ethnicity, and, in the current multiethnic context, identifying a MUD in a timely manner remains a challenge. Indeed, the search for a HLA-matched volunteer donor may result in unacceptable delay in certain diseases, such as very high risk acute leukemias, or severe combined immunodeficiency (SCID), for which the goal is to proceed to transplantation as early as possible after diagnosis. For those without a HLA matched donor, alternative hematopoietic progenitor cell sources include mismatched unrelated donors, umbilical cord blood, and haploidentical related donors.
Transplantation from a full HLA-haplotype mismatched family member (haplo-HSCT), in addition to ensuring a donor for the large majority of patients, offers several other advantages, including prompt availability of the stem cell source, the possibility to select the best donor from a pool of family candidates, and immediate access to donor-derived cellular therapies either for the prevention of relapse or the treatment of infections after HSCT. Despite these advantages, widespread use of haplo-HSCT has been limited for many years by relevant complications mediated by bidirectional alloreactivity responsible for unacceptably high rates of graft rejection and severe graft-versus-host disease (GvHD).
The continuous development of graft engineering and pharmacologic GVHD prevention strategies, better supportive care, and optimal conditioning regimens have significantly improved the outcomes of haploidentical HSCT, and this progress has led to establishment of haplo-HSCT as a standard therapeutic option for patients with both malignant and non malignant disease needing a HSCT procedure and lacking a HLA-identical or compatible donor.
11.2 Donor Selection
Most patients requiring HSCT have more than one haploidentical donor. In the majority of pediatric patients, however, the best donor will have to be selected from one of the parents. Ideally, the goal is to employ a graft that will enable complete and permanent engraftment of donor hematopoiesis, ensure rapid immune reconstitution, and exert effective graft-versus-leukemia (GVL) effect in the absence of GvHD. Several studies investigated donor/recipient characteristics that influenced haplo-HSCT outcome.
The role of HLA disparity degree was evaluated in large clinical trials, mainly in the T-cell replete context [85, 146], and it did not influence the incidence of acute GVHD and treatment related mortality (TRM). In the study by Wang et al., conducted on 1210 pediatric and adult patients transplanted with G-CSF mobilized T-cell replete bone marrow and peripheral stem cells, donor age less than 30 years was associated with a lower incidence of acute GVHD compared to older donor age, this observation being also confirmed in the pediatric T-cell depleted setting [67]; younger donor age and male gender were also associated with less TRM and better overall survival (OS). Moreover, having a maternal donor was associated with a higher GVHD incidence and TRM than having a paternal donor. These findings are in contrast to observations from other studies demonstrating a lower risk of relapse and survival advantage for grafts from maternal donors [136], in which the anti-leukemic effect of maternal donor HSCT had been explained by maternal immune system exposure to fetal antigens during pregnancy. When analyzing the role of non-inherited maternal antigen (NIMA) disparities, also Wang et al. found that NIMA mismatched sibling haplo donors conferred a lower incidence of acute GVHD compared to non-inherited paternal antigen (NIPA) mismatched donors [146].
The use of highly purified CD34+ stem cells in haploidentical HSCT has allowed deep insights into the biology of NK cells and into the understanding of NK alloreactivity [110, 129]. Among adult patients affected by AML, a subgroup of patients given T cell depleted HSCT from an HLA-disparate relative having alloreactive NK cells showed a low risk of leukemia relapse [9]. Cytotoxic activity of NK cells is under the negative feedback control from inhibitory killer immunoglobulin-like receptors (KIRs), that recognize epitopes shared by HLA class I alleles. KIR-KIR ligand mismatches in the donor-recipient direction lead to loss of the inhibitory feedback and activation of donor NK cells targeting recipient hematopoietic cells and leukemic cells [130]. In contrast to alloreactive T-lymphocytes, NK cells are thought to be capable of inducing GVL effect without promoting GvHD, through elimination of residual recipient antigen-presenting cells in addition to leukemia blasts [130]. Accordingly, KIR analysis to identify donor/recipient KIR mismatch has been proposed as a tool for donor selection prior to T-cell depleted haplo-HSCT [131].
Notably, other factors may contribute to NK alloreactivity. In particular, killing of target cells may also depend on the surface density of activating receptors on NK cells and on the expression of their ligands on target cells [47]; indeed, activating KIRs (in particular KIR2DS1) were shown to play a substantial role in mediating alloreactivity [36, 123]. Thus, phenotypic identification of the alloreactive NK cell subset and assessment of the NK cytolytic activity against leukemic cells represent important criteria in donor selection [123]. Moreover, recent reports have proposed a novel approach for optimal donor selection based on the KIR genotype analysis. Among these, a study in a haplo-HSCT setting provided evidence that the selection of donors with KIR B haplotypes was associated with significant improvements in both overall and relapse free survival [114, 140].
KIR mismatch between recipient and donor has been associated with improved outcomes in both T-cell deplete and T-cell replete haplo-HSCTs [103, 123, 129, 130, 140]; however, some studies failed to find an association between the presence of donor NK alloreactivity and a favorable clinical outcome of transplanted patients [75, 113, 143]. The apparent discrepancies are likely due to the different clinical settings, in particular the type of graft (manipulated vs unmanipulated), the conditioning regimen, stem cell source and number (i.e. CD34+ HSC megadose), the type of GVHD prophylaxis, and the disease status of the patient at the time of HSCT, and these variables have to be taken into account in the donor selection algorithm.
Finally, another factor that may have a negative influence on haplo-HSCT outcome is the presence of recipient antibodies directed to donor HLA antigens (DSAs), as DSAs have been associated to graft rejection [40]. Thus, pre-transplant DSA analysis may help donor selection, and, in case DSAs to antigens of all available donors are present, may guide antibody removal by anti-B cell monoclonal antibodies or plasma exchange.
11.3 Haploidentical HSCT Strategies
11.3.1 T-Cell Depletet Graft
HLA-haploidentical HSCT had been experimented with varying success since mid-seventies [42], at first in the setting of acute leukemia and aplastic anemia. The greatest challenge to performing haplo-HSCT had been high rates of graft failure and severe GVHD. Notable success came with the demonstration that in children with SCID it was possible to reconstitute a functional immune system with HSCT without inducing life-threatening acute GVHD by performing a T-cell depletion using soybean agglutinin and sheep red blood cell rosette formation technique [31, 61, 62, 126]. Primary immune deficiencies were an ideal setting for haplo-HSCT, as, in contrast to other disorders, the profound immunodeficiency in SCID minimized the risk of immunologic graft rejection, conceptually eliminating the need for immunosuppressive conditioning before HSCT and, consequently, reducing the toxicity of the procedure.
In contrast to SCID, haplo-HSCT was less successful in the setting of acute leukemia owing to a high rate of graft failure, attributed to host derived T-lymphocytes that survived the conditioning regimen [87]. A major breakthrough intervened when preclinical studies in murine models demonstrated that infusion of large numbers of donor hematopoietic stem cells (HSC “mega dose”) could overcome the major histocompatibility complex (MHC) barrier and promote engraftment [14]. Seminal clinical studies showed that transplantation of mega doses of stem cells (>10 × 106 CD34+ cells/kg), obtained by supplementing T cell-depleted bone marrow transplants with granulocyte colony-stimulating factor (G-CSF) mobilized peripheral blood stem cells (PBSC), after a conditioning regimen consisting of single fraction total body irradiation (TBI), thiotepa, cyclophosphamide (CY) and rabbit antithymocyte globulin (rATG), allowed for successful primary engraftment and relatively low incidence of acute and chronic GVHD despite the use of T-cell depletion as the only GVHD prophylaxis [10, 11]. The initial protocol was implemented with time, as highly purified CD34+ cells selected from mobilized PBSC by magnetic sorting replaced soybean agglutination and E-rosetting T cell depleted inoculum, and fludarabine replaced CY in the conditioning regimen [12]. In the pediatric setting, different versions of the “Perugia protocol” were applied successfully to treat both malignant and non-malignant hematologic disorders [6, 44, 72, 104].
Despite acceptable rates of engraftment and GVHD, TRM due to infectious complications and malignancy relapse remained a major problem after CD34+ HSC-selected haplo-HSCT. In the attempt to ameliorate immune reconstitution, attempts were made to switch from positive CD34 HSC selection to negative T and B cell depletion, in order retain, besides the CD34+ stem cells, large numbers of other cells including γδ and NK cells, monocytes, and dendritic cells. With introduction of automated devices, simultaneous depletion of CD3+ T cells and CD19+ B cells allowed to prevent GVHD and posttransplant lymphoproliferative disease (PTLD) [18, 35]. In order to reduce toxicity, a non-myeloablative conditioning regimen based on the use of fludarabine, thiotepa, melphalan and the anti-CD3 T monoclonal antibody OKT3 was employed. T-cell reduction did not reach the 4.5–5 log depletion obtained with CD34+ selection, and despite post-HSCT GVHD prophylaxis, the rate of acute and chronic GVHD were higher than those observed in the CD34+ T cell-depleted haplo-HSCT; moreover, notwithstanding a faster immune recovery, leukemia relapse remained a major problem.
A more effective approach to negative depletion of T cells is the more recently described negative depletion of T-cell receptor (TcR) αβ + T lymphocytes from mobilized peripheral stem cell grafts, coupled with B cell depletion [34]. With this technique, Bertaina et al. reported high OS and disease-free survival (DFS) (91%) coupled with a low incidence of acute GvHD (13%) and chronic GvHD in 23 children with a variety of non-malignant disorders, including SCID, Fanconi anemia (FA), severe aplastic anemia (SAA), osteopetrosis, and primary immunodeficiencies (PIDs) [22]. Recently, a multicenter Italian study comparing the outcome of Tαβ/B cell depleted haplo-HSCT vs UD-HSCT in children with acute leukemia transplanted with a myeloablative regimen reported primary engraftment in 95 of 97 patients receving haplo-HSCT and, with the only pharmacologic GVHD prophylaxis of pretransplant ATG in the haplo-HSCT setting, 16% and 0% grade II–IV and III–IV acute GVHD, respectively, as compared to 39% and 12% in UD-HSCT recipients [23]. After a median follow-up of 3.3 years, the 3-year leukemia-free survival was 63% vs 62% in the UD-HSCT setting, with chronic GVHD rates of 6% vs 20%, respectively. Encouraging results in the setting of ALL and AML were also reported from other groups [94, 106].
Different means to deplete alloreactive T cells within the graft have been experimented in the setting of haplo-HSCT. Triggering of alloreactivity in vitro through a mixed lymphocyte reaction (MLR) obtained by co-culturing donor T cells with recipient antigen-presenting cells has been generally followed by depletion of the activated donor T cells through surface activation markers or photoactive dyes. Cavazzana-Calvo et al. designed a protocol to allo-activate donor T cells responsible for GVHD and eliminate them with an immunotoxin that reacted with the cell surface activation antigen CD25, and demonstrated in a group of pediatric patients receiving haplo-HSCT the ability of an allodepleted T-cell “add-back” to exert an anti-viral infection effect without causing GVHD [5, 108]. Using a similar approach, Amrolia et al. infused donor allodepleted lymphocytes in 16 pediatric recipients of T-cell depleted haplo-HSCT, showing a low rate of GVHD, but inability to prevent posttransplant leukemia relapse and viral infections [4]. A method to deplete alloactivated T cells, based on the use TH9402, a phototoxic dye that accumulates in activated T cells due to their inability to efflux rhodamidelike drugs, was also developed and employed in the setting of haplo-HSCT [19].
An alternative approach to prevent GVHD while preserving anti-leukemia and anti-infectious immunity is to functionally inactivate alloreactive T cells by inducing alloantigen-specific anergy. Several groups demonstrated how blockade with antibodies directed to costimulatory molecules during allostimulation in MLR could induce anergy directed to the specific alloantigen, while preserving other immune responses [29, 45]. In a pilot trial conducted in 12 pediatric haplo-HSCT recipients, Guinan et al. incubated donor marrow cells with CTLA-4–Ig, an agent that inhibits B7-CD28 costimulatory signal, in the presence of irradiated mononuclear cells from the recipient. Primary engraftment after myeloablative conditioning was demonstrated in 10 of the 11 evaluable patients, and acute GVHD of the gastrointestinal tract developed in three patients, despite posttransplant prophylaxis with cyclosporine and short-course methotrexate. Five of the 12 patients were alive and in remission 4.5–29 months after transplantation [70]. In a follow-up study, a 50% rate of TRM was observed, and 8 of 24 reported patients developed acute GVHD [49].
Finally, studies conducted in animal models had suggested that coinfusion of CD4 + CD25+ regulatory T cells (Tregs) and conventional donor T cells could inhibit lethal GVHD after allogeneic HSCT across MHC, while preserving GVL surveillance [56].
Di Ianni et al. were able to prevent GVHD, improve immune reconstitution, and induce a strong GVL effect after infusion of donor-derived Tregs, followed by conventional T cells, in 28 recipients of CD34+ selected haplo-HSCT, in the absence of any post-transplant immunosuppression [52]. Despite prompt immune reconstitution, however, the rate of opportunistic infections, and thus TRM, remained high, perhaps due to Tregs hampering immunity to infectious agents.
11.3.2 T-Cell Replete Graft
Haplo-HSCT with ex-vivo T cell depletion ensures the best mean to prevent GVHD. However, it requires specific cell processing expertise, a myeloablative conditioning regimen to eradicate residual recipient immune cells and support engraftment, and posttransplant immunologic interventions to boost immune recovery. Being costly and technically demanding, its application has been generally confined to highly experienced centers, consequently limiting widespread application of haplo-HSCT. Recently, this scenario has dramatically changed thanks to the impressive results obtained with unmanipulated haplo-HSCT strategies in patients affected by malignant diseases.
Historical attempts at using unmanipulated haploidentical allografts were associated with an unacceptably high rate of GVHD. To overcome this hurdle, efforts have been made to increase the intensity of posttransplant GVHD prophylaxis regimen. The first unmanipulated approach, pioneered by the Johns Hopkins group, relied on the use of post-transplantation cyclophosphamide (PTCY) [102]. The concept behind this strategy relied on the observation that CY could induce skin graft tolerance [20]. It was hypothesized that in vivo donor and recipient alloreactive T cell depletion could be obtained by exposing to PTCY donor/recipient lymphocytes proliferating to reciprocal alloantigens within the first posttransplant days. This procedure could ensure both engraftment and GVHD prevention, while sparing quiescent T cells, that are less sensitive to CY due to high levels of aldehyde dehydrogenase, an enzyme responsible for CY metabolism [81]. Using a reduced-intensity conditioning regimen including fludarabine, CY and 200 Gy TBI, and PTCY, Luznik et al. obtained a 87% rate of engraftment, with 6% grade III–IV acute GVHD, and demonstrated a significantly higher rate of chronic GVHD with the use of PTCY on day +3 vs day +3 and +4 (25% vs 5%, respectively). The 2-year overall survival and event-free survival (EFS) rates were as low as 36% and 26%, due to high incidence of relapse (58% at 2 years) [102]. Similar results of acceptable incidence of GVHD (32% acute grade II–IV and 13% chronic GVHD) and very low TRM were obtained in a large multicenter trial conducted in patients with malignancies, in which relapse was a major cause of mortality and was primarily attributed to the use of nonmyeloablative conditioning for patients with acute leukemias [30]. To address this obstacle, the use of myeloablative preparative regimens was explored and found successful. Raiola et al. showed a 4-year disease-free survival of 43% in 92 adult patients transplanted for hematological malignancies [125].
Data on the feasibility of this approach in children are scarce, although the procedure is increasingly employed [21, 78].
A different unmaninuplated haplo-HSCT strategy was applied in 250 pediatric and adult patients with acute leukemia, based on the use of myeloablative haplo-HSCT with non-T-cell depleted, cytokine-primed marrow and peripheral blood grafts, associated with in vivo T-cell depletion by ATG and GVHD prophylaxis consisting of cyclosporine, short-course methotrexate and mycophenolate mofetil (MMF) [74]. All but one patients engrafted. The cumulative incidence of grade II-IV acute GVHD was 46%, grade III-IV was 13%, and incidence of chronic GVHD was 54%. At 3 years after HSCT, the cumulative incidence of opportunistic infections was 49%, and 141 of 250 patients were alive and disease free. An Italian study reported a significantly lower incidence of acute and chronic GVHD with a similar strategy, but with bone marrow as the only source of HSC [51].
A multicenter Italian trial explored the feasibility of a haplo-HSCT protocol consisting of unmanipulated PBSC infusion after a treosulfan and fludarabine conditioning regimen, and GVHD prophylaxis based on antithymocyte globulin Fresenius (ATG-F), rituximab and oral administration of sirolimus and mycophenolate [121]. Incidence of acute GvHD grade II-IV was 35%, and correlated negatively with Treg frequency, while that of chronic GvHD was 47%. At 3 years after HSCT, TRM was 31%, with 48% relapse incidence and 25% OS. The high rate of chronic GVHD might have been partly due to the use of PBSC rather than BM as stem cell source.
11.4 Indications for Haplo-HSCT in Children
11.4.1 Haplo-HSCT in Childhood Malignancies
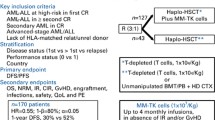
At present, allogeneic HSCT in children with ALL is reserved to patients who experience an early or very early marrow relapse after first line chemotherapy, or to the subpopulation of high-risk ALL in first complete remission (CR1), i.e. those with known molecular biological markers or chromosomal abnormalities, and clinical factors such as poor prednisone response and resistance to initial chemotherapy including persistence of minimal residual disease (MRD) [138] (Table 11.1). Likewise, in the setting of pediatric AML, indications for allo-HSCT are high/very high risk disease (infant AML and children with unfavorable karyotype, or FAB M0, M6 or M7), or patients in CR2 [138]. Conversely, childhood myelodysplastic syndromes (MDS), including juvenile myelomonocytic leukemia (JMML) without germ line PTPN11 and CBL mutations, myelodysplasia-related AML, advanced MDS and refractory cytopenia of childhood (RCC) with high risk of disease progression (monosomy 7 or >2 chromosomal abnormalities), have an indication to HSCT [84, 99, 137] (Table 11.1). In JMML and in children ≤12 years with advanced MDS, a myeloablative conditioning regimen of busulfan (16 g/kg over 4 days), cyclophosphamide (120 mg/kg over 2 days) and melphalan (140 mg/m2 in single dose) and rATG is indicated. In older patients with advanced MDS, who experience a higher TRM, and in high risk RCC children with normo/hypercellular bone marrow, a myeloablative conditioning based on thiotepa (4–5 mg/kg/day for 2 days), treosulfan (14 g/m2/day for 3 days), fludarabine (40 mg/m2/day for 4 days,) and rATG may be employed. Finally, RCC children with hypocellular bone marrow may receive a reduced-intensity conditioning with fludarabine (40 mg/m2/day for 4 days), thiotepa (5 mg/kg/day for 2 days) and rATG.
In the setting of Hodgkin disease (HD) or non-hodgkin lymphoma (NHL), allogeneic HSCT may be considered in patients with relapsed/refractory disease or disease relapsed after autologous HSCT [3, 32, 69, 132].
In all cases, haplo-HSCT can be considered when a matched sibling donor or a well-matched unrelated donor are not available. However, with haplo-HSCT results constantly improving [23], it is now debatable if UD-HSCT is more indicated than haplo-HSCT in refractory malignancy, a setting where haplo-HSCT (especially Tαβ/B cell depleted haplo-HSCT without posttransplant GVHD prophylaxis), may be an ideal platform for strategies to boost immune surveillance and control disease outgrowth.
In childhood acute leukemia, early studies of haplo-HSCT were heterogeneous and carried out on small cohorts. Moreover, as the procedure was considered experimental, the patients enrolled were mostly very high risk patients or refractory ALL with dismal outcome. With the development of the T-cell depleted, CD34+ megadose haplo-HSCT, the Perugia group obtained for the first time encouraging results [10, 11]. With a strategy based on fractionated TBI, thiotepa, fludatabine and rATG conditioning and no posttransplant GVHD prophylaxis, in their larger cohort that included also pediatric patients, they obtained 95% primary engraftment, with 8% and 7% acute and chronic GVHD; in children, a 15% TRM was observed, with a DFS probability of 38% in ALL and 62% in AML in any CR. For patients transplanted in relapse, DFS was 5% in ALL and 38% in AML [12]. In 47 pediatric patients transplanted with the same strategy, 5% graft failure was observed, with grade III-IV acute and extensive chronic GVHD of 6% and 3%, and 25% TRM (10% in patients transplanted after 2005). The 5-year estimate of DFS for the whole cohort was 50%; interestingly, 70% DFS was observed in children with ALL, 75% in MDS, while only 20% in AML patients. The DFS of the 18 patients with ALL transplanted from an NK-alloreactive donor was 81% [100, 123]. In 2006, Chen et al. published data providing evidence that a CD3+/CD19+ depletion strategy using PBSCs, in combination with a reduced intensity conditioning based on fludarabine, melphalan, thiotepa and OKT-3 monoclonal antibody, was a feasible option for children with hematological malignancy [35]. Although higher rates of acute GVHD were seen in comparison to CD34+ selected grafts, due to lower T cell depletion, TRM was low. However, the 2-year disease-free survival was 25%. A retraspective study by the European Blood and Marrow Transplant group on the outcome of haplo-HSCT in children transplanted for very high risk ALL, showed five-year TRM, relapse incidence, and DFS of 37%, 36%, and 27%, respectively. The study highlighted the importance of performing the transplant in remission, using CD34+ cell megadose, and indicated a significant impact of center experience (centers performing large numbers of allo-HSCT: DFS of 39% vs 15% in small centers) [90]. Recently, a multicenter Italian study comparing the outcome of Tαβ/B cell depleted haplo-HSCT vs UD-HSCT in children with acute leukemia transplanted with a myeloablative regimen reported primary engraftment in 95 of 97 patients receving haplo-HSCT and, with the only pharmacologic GVHD prophylaxis of pretransplant ATG in the haplo-HSCT setting, 16% and 0% grade II-IV and III–IV acute GVHD, respectively, as compared to 39% and 12% in UD-HSCT recipients [23]. After a median follow-up of 3.3 years, the 3-year leukemia-free survival was 63% vs 62% in the UD-HSCT setting, with chronic GVHD rates of 6% vs 20%, respectively. Encouraging results in the setting of ALL and AML were also reported from other groups. Lang et al. reported on 41 children with acute leukemia, MDS and nonmalignant diseases receiving Tαβ/B cell depleted haplo-HSCT with conditioning regimens consisting of fludarabine or clofarabine, thiotepa, melphalan and serotherapy with OKT3 or ATG-Fresenius [94]. Primary engraftment occurred in 88%, acute GvHD grades II and III-IV occurred in 10% and 15%, respectively. The 1.6 year survival rate was 51%, with 41% relapse being the major cause of death. With the same manipulation approach, and a conditioning based on treosulfan, melphalan, fludarabine and ATG, Maschan et al. reported 100% primary engraftment and 39% acute GVHD II-IV in 33 children transplanted with UD-HSCT or haplo-HSCT for AML. At 2 years, the cumulative incidence of relapse was 40% in the haplo group, with a DSF of 59%, whereas TRM was 0% [106].
Regarding the T-cell replete approach, a recent pilot study of PTCY-based haplo-HSCT, showed a cumulative incidence of disease progression of 26% in children with acute leukemia, with 24% NRM and 40% aGVHD. Out of a total of ten grades II–IV acute GVHD cases, severe GVHD occurred exclusively in children below the age of 10 years, and the authors hypothesize defective clearance of alloreactive T cells due to altered CY metabolism in the young age group [78]. A multicenter Italian study of T-cell replete haplo-HSCT based on myeloablative or reduced-intensity conditioning, and GVHD prophylaxis with PTCY, MMF and calcineurin inhibitor, conducted in 33 children with high-risk hematologic malignancies and lacking a match-related or -unrelated donor, showed aGVHD and cGVHD rates of 22% and 4%, respectively, with a DFS rate of 61%, 24% cumulative incidence of relapse and 9% TRM [21].
Regarding lymphoma, the experience in children is limited. Broader use has been hampered for a long time mainly by high TRM, offsetting the advantage of a GVL effect. However, since the use of nonmyeloablative conditioning and T-cell replete haplo-HSCT with PTCY, results in adult patients have dramatically improved. In a cohort that included also adolescents, Burroughs and colleague first observed an advantage of haplo-HSCT on matched related and unrelated donor HSCT in HD, as, due to a lower TRM with comparable disease control, the haplo group showed a 51% DFS compared to <30% in the other two groups [33]. Recent data from two large multicenter, retrospective, registry studies showed similar results in adult patients with HD and NHL transplanted with PTCY haplo-HSCT. The analysis by Kanate et al. registered the same relapse rate of 36% compared with UD HSCT, despite haplo cohort having higher disease risk index scores, but less acute and chronic GVHD, with a OS of 60% vs 50%, respectively [82]. Gosh et al. compared haplo-HSCT with matched sibling donor HSCT in adults, finding superimposable TRM and PFS rates, but significantly less chronic GVHD in the haplo-HSCT cohort [65]. As relapse remains the major cause of treatment failure, it will be important to use the haplo-HSCT platform as a basis for GVL effect, by integrating transplant with novel immunological therapies. An example in the pediatric setting is the use of DLIs modified by insertion of the inducible caspase 9 suicide gene, that proved of efficacy in two patients with lymphoma enrolled in a phase I trial [155].
11.4.2 Haplo-HSCT in Severe Aplastic Anemia
Currently, haplo-HSCT in acquired severe aplastic anemia (SAA) is reserved to children who have failed previous immunosuppressive treatment with ATG and cyclosporine-A and who do not have a suitable matched family or unrelated donor or cord blood unit, or to patients who have rejected a previous unrelated donor transplant [15, 16].
In the recent years, several series of children [59, 76, 147, 148, 150, 151, 153] and adults [41, 50, 57, 97] with SAA and given haplo-HSCT have been reported. However, the number of pediatric patients in each study is often relatively small, and the preparative regimens and GVHD prophylaxis are different. Nevertheless, the reported average 1-year EFS is good and in the order of 75% or more. Both T-cell depletion and unmanipulated bone marrow or peripheral blood stem cells have been successfully used. Because of the high risk of rejection, unmanipulated bone marrow or peripheral blood stem cells have been preferred in several cases, usually in combination with high-dose PTCY or with monoclonal antibodies or ATG as GVHD prophylaxis.
In conclusion, even if it is still in the experimental stage, haplo-HSCT should be considered in patients with SAA failing first-line immune suppressive therapy and lacking an HLA-matched related or unrelated donor. Unfortunately, the data available so far do not allow to make strong recommendations regarding the best conditioning regimen, the optimal composition of the graft, and the best GVHD prophylaxis strategy. Both ex vivo T-cell-depleted and unmanipulated graft strategies have been explored; results show comparable efficacy and acceptable toxicities of both these approaches [15, 39, 64].
11.4.3 Haplo-HSCT in Constitutional Cytopenias
Constitutional bone marrow failure syndromes represent a group of rare genetically and phenotypically heterogeneous disorders characterized by the variable presence of multiple congenital somatic abnormalities, the gradual onset of bone marrow failure involving one or more hematopoietic cell lineages, and the predisposizion to develop clonal hematopoietic disorders as well as, in some cases, solid tumors [24]. The bone marrow insufficiency can be uni-linear, such as usually in Diamond-Blackfan anemia or in congenital amegakaryocytic thrombocytopenia, or it can involve all the three lineages, such as in Fanconi anemia, diskeratosis congenita or Shwackmann-Diamond syndrome. Also the degree of cytopenia is variable among the different disease and can worsen over time: in Fanconi anemia the cytopenia is typically absent at birth and usually appears during childhood, while in Diamond-Blackfan anemia the hyporegenerative anemia appears in infancy [24].
Optimized supportive care, including red blood cell and platelet transfusions, and prevention of infectious complications, are critical for the conservative management of these patients [139]. Some children, namely those affected by Fanconi anemia and dyskeratosis congenital, can benefit from treatment with androgens, while those with Diamond-Blackfan anemia can improve anemia with steroid treatment. Allogeneic HSCT is currently the only curative treatment able to restore normal hematopoiesis. Nevertheless, the underlying defect in DNA repair, typical for example of Fanconi anemia, is responsible of the hypersensitivity to the treatment with irradiation and alkylating agents as cyclophosphamide, leading to excessive regimen-related toxicity and severe acute GVHD [66]. Furthermore, a strong association between chronic GVHD and the development of secondary malignancies (squamous cell carcinoma) has been demonstrated [26, 122], thus increasing the risk of late mortality notwithstanding the cure of bone marrow insufficiency.
Current evidence in the medical literature on the use of haplo-HSCT in this particular setting is often limited to case reports and small retrospective case series [2, 54, 101, 141]. Recently, Zecca et al. described 12 children with Fanconi anemia treated with haplo-HSCT, who received T cell-depleted, CD34+ positively selected stem cells after a conditioning regimen including fludarabine (30 mg/m2/day for 4 days), cyclophosphamide (300 mg/m2/day for 4 days), rATG (10 mg/kg/day for 4 days) and single dose TBI (200 cGy). Survival and DFS were 83%, while the cumulative incidence of TRM was 17%, with no fatal regimen-related toxicity. The incidence of acute and chronic GVHD was limited. Low infused CD34+ cell dose seemed to correlate with graft rejection [152]. Bertaina et al. described four further patients who received the same conditioning regimen, and were successfully transplanted using T-cell depleted PBSC after T α/β + and B CD19+ negative selection [22]. Furthermore, Bonfim et al. reported 30 children with Fanconi anemia given haplo-HSCT with unmanipulated bone marrow and post-transplant cyclophosphamide (25 mg/kg/day on day +3 and +4) [27]. The conditioning regimen included fludarabine (150 mg/m2), cyclophosphamide (10 mg/kg) and single dose TBI (200 cGy). Pre-transplant rATG (4–5 mg/kg) was added to the conditioning regimen after the first 12 transplants, because of the high incidence of severe acute and chronic GVHD. Hemorrhagic cystitis occurred in 50% of the patients, but overall survival was 73% with all surviving patients achieving full donor chimerism.
Taken together, these results demonstrate the feasibility of haplo-HSCT also for Fanconi anemia patients and, more in general, for children with constitutional bone marrow failure syndromes. However, because of the peculiar frailty of this heterogeneous patient population, particular attention must be payed to the choice of the conditioning regimen, because of the high regimen-related toxicity. Furthermore, a very effective GVHD prophylaxis should be adopted, in view of the strong association between chronic GVHD and secondary malignancies in otherwise cured long-term survivors.
11.4.4 Haplo-HSCT in PID
Primary immunodeficiencies (PID) are a group of heterogeneous diseases, many of which are caused by monogenic defects, resulting in susceptibility to life threatening infections, uncontrolled inflammation, or autoimmunity. Historically, allogeneic HSCT has been a curative option for several primary PID, including severe combined immunodeficiency (SCID), Wiskott-Aldrich syndrome (WAS), chronic granulomatous disease (CGD) hemophagocytic lymphohistiocytosis (HLH) and many others [13, 63]. This field has rapidly expanded over the last years. Currently, more than 300 PIDs have been genetically defined and 34 new genetic disorders have been added to the International Union of Immunological Societies (PIDTC) PID classification in the last 2 years [124]. Many of these diseases can be cured by allogeneic HSCT even if, given the heterogeneity and rarity of some diseases, in some cases the indication to HSCT can be controversial. Table 11.2 summarizes the most important, established or still debated indications [71].
HSCT in PID can be a challenge. Comorbidities such as chronic infections and severe pulmonary dysfunction, that could make patients ineligible to the procedure, are common. Myeloablation may be avoided in order to reduce excessive toxicity, but reduced-intensity regimens could lead to higher rejection rate or to increased mixed chimerism. Also the degree of donor engraftment necessary for disease cure is yet not completely understood. In children with SCID, HSCT is considered an urgent and life-saving procedure, while in other forms of PID, where the immune defect does not result in an imminent risk, the transplant could be delayed until a properly matched donor is found. Indeed, a causative molecular defect can be identified in many patients with PID, leading to formulation of a definitive diagnosis. In this case, the decisional process is relatively simple and allows to rapidly proceed to the transplant on the basis of the existing knowledge about the underlying disease. Unfortunately, in other cases, in which a genetic diagnosis cannot be achieved, the decision to transplant is often delayed until the susceptibility to severe recurrent infections or autoimmunity are clearly demonstrated.
The major advantage of using a haplo-HSCT is that a healthy donor, usually a parent, is immediately available so that the transplant can be performed very quickly. In the study of the Primary Immunodeficiency Treatment Consortium (PIDTC), reporting 240 infants with SCID transplanted between 2009 and 2009, more than 50% of the patients (138/240) received a transplant from a partially matched related donor [118]. Children who received a T-cell depleted graft from partially matched related donors and did not received any conditioning regimen had a survival probability of 79% while those receiving any type of conditioning had a survival probability of 66%. However, the use of a reduced intensity or myeloablative conditioning was associated with improved T-cell count and better B-cell function. Older age (>3.5 years) and active infection at time of HSCT were associated with lower survival rate, while children transplanted in early infancy (<3.5 months) had an excellent outcome, similar to that of patients transplanted from a matched sibling, even if grafted from an alternative donor.
Also some patients with WAS and given haplo-HSCT, either with or without T-cell depletion, have been reported in different series of patients usually including also transplants from matched family or unrelated donors [17, 77, 88, 93, 109, 112, 133]. These studies show that haplo-HSCT can be an effective form of treatment. However, it must be noted that in the setting of WAS a mixed chimerism appeared to have a strong detrimental effect on EFS because of an increased incidence of autoimmunity [117]. For this reason, a stable multilineage donor engraftment is required to fully correct the disease [109] and this consideration supports the use of fully myeloablative conditioning regimens, in order to minimize the chance of autologous reconstitution and recurrence or persistence of the WAS phenotype.
Also rare cases of haplo-HSCT in children with CGD have been recently reported [73, 111, 120, 154]. However, the experience with CGD is still too limited to give specific recommendations and haplo-HSCT in CGD should sill be considered experimental.
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory clinical syndrome with uncontrolled immune response which results in hypercytokinemia due to an underlying primary or secondary immune defect. HCT is recommended in patients with documented familial HLH, recurrent or progressive HLH despite chemoimmunotherapy, and CNS involvement [80]. The selection of the optimal stem cell donor and source, as well as of the conditioning regimen, is important for HLH patients undergoing HSCT, because of the high risk of rejection and unstable mixed chimerism reported in this disease [116]. HSCT using haploidentical donors for HLH patients who do not have matched donor was demonstrated to be feasible, and the outcome has improved over time [55, 96, 98, 115, 116].
Conventional myeloablative conditioning regimens, mostly including busulfan, cyclophosphamide and etoposide with or without ATG have been usually adopted. However, it has been reported that the outcome of myeloablative conditioning for HLH can be impaired by high early TRM. The increased TRM has prompted the use of less toxic approaches adopting reduced intensity regimens. The combination of alemtuzumab, fludarabine, and melphalan has demonstrated good efficacy [46, 105]. Also melphalan or treosulfan are promising alternatives. Fludarabine, treosulfan, alemtuzumab, and thiotepa could represent a conditioning regimen with a high rate of disease-free survival and low toxicity [96]. Reduced intensity conditioning before haplo-HSCT was reported to sufficiently restore immune regulation in infants with FHL, while decreasing TRM and long-term sequelae [115]. However, the high incidences of mixed chimerism and graft loss still remains a significant concern in HLH patients.
11.4.5 Haplo-HSCT in Hemoglobinopathies
Allogeneic HSCT offers a potentially curative treatment for patients with hemoglobinopathies, mainly thalassemia major (TM) and severe sickle cell disease (SCD). Nevertheless, so far the applicability of HSCT has been limited mainly by donor availability, with a less than 20% of eligible patients having a HLA-matched sibling donor [1]. Furthermore, the probability of finding a potential MUD is largely dependent on the ethnic and racial background [68].
In TM, transfusion-dependency is an indication for HSCT, especially in younger patients before development of iron-related tissue damage. In SCD, allogeneic HSCT is currently limited to patients with a severe phenotype, with frequent vaso-occlusive crises and acute chest syndrome that are unresponsive to hydroxyurea, with end organ damage such as stroke or osteonecrosis of multiple joints, and with severe disease that has been associated with an increased risk of early mortality and/or requiring regular transfusion therapy [7, 89].
Haplo-HSCT could significantly increase the donor pool for patients with SCD, who historically have limited donor options. In adult patients with SCD (age 15–46 years), a single-center experience using a novel non myeloablative haploidentical or HLA-mismatched donor regimen employing PTCY showed the feasibility of this approach: more than 80% of eligible patients had suitable related haploidentical donors, and in the cohort of 14 haploidentical recipients with SCD, 60% of the patients engrafted with no deaths or significant GVHD; however, the graft failure rate was 43%. So, the experience using this approach is still relatively limited and remains under clinical investigation [25]. In 2013, Dallas et al. reported eight children with SCD given unmanipulated haploidentical HSCT after a myeloablative conditioning regimen and a GVHD prophylaxis based on the use of an anti-CD3 monoclonal antibody. However, only three of the eight patients achieved a sustained engraftment and are alive and disease-free, while graft failure and SCD recurrence were observed in 38% of the patients [48]. A further study was published in 2017 by Foell et al., describing nine children or adolescents with SCD and given haplo-HSCT [60]. The conditioning regimen included thiotepa, treosulfan, fludarabine and ATG, and GVHD prophylaxis consisted of CD3+/CD19+ depletion followed by the administration of cyclosporine-A and mycophenolate until day +120 post-HSCT. All nine children achieved stable engraftment with only one patient dying of transplant-related complications.
Moving to TM patients, two studies evaluated the role of haplo-HSCT in children. In 2010, Sodani et al. presented 22 patients with TM given HSCT from their haploidentical mother after an intensive conditioning regimen and a GVHD prophylaxis based on T-cell depletion. The reported survival probability was 90%, the thalassemia-free survival was 61% and the cumulative incidence of the rejection was 29% [134]. More recently, in 2016, Anurathapan et al. described 31 patients (median age 10 years, range 2–20) transplanted after a myeloablative conditioning regimen (busulfan + fludarabine + ATG) and with a GVHD prophylaxis including PTCY, tacrolimus and mycophenolate mophetil [8]. The authors reported a survival probability of 95%, an EFS probability of 94%, with only 2 out of the 31 patients rejecting the transplant. In both studies, all patient received an intensive pre-conditioning treatment, including high-dose hydroxyurea, azathioprine, hematopoietic growth factors, hypertransfusion regimen and intensive iron chelation [134] or 2 cycles of fludarabine (40 mg/m2/day for 5 days) and dexamethasone (25 mg/m2/day for 5 days) [8]. The pre-conditioning treatment was administered in order to decrease marrow cellularity and to suppress erythropoiesis, with the aim of reducing the risk of rejection and graft failure. The pre-conditioning immunosuppressive and myelo- and erithrosuppressive treatment could play a relevant role in the success of the transplant in this otherwise difficult disease, and deserves further investigation.
Overall, despite the higher incidence of graft rejection as compared to other diseases, these recent results are encouraging because of the low toxicity of the procedure. Only few patients with TM and, still less with SCD, often belonging to ethnic minorities with rare HLA phenotype [68], are able to receive a HLA compatible donor transplant. The use of haploidentical donors could extend the use of HSCT in a setting where this procedure is still largely underutilized. Prospective trials are needed to determine the risk-benefit ratio of this approach, and many such studies are currently ongoing.
11.5 Strategies to Enhance Immune Reconstitution and GVL After Haplo-HSCT
As we have seen in previous sections, transplant-related mortality (in the pediatric setting mostly ascribable to opportunistic infections), and relapse in patients transplanted for malignancy, are the major causes of failure after T-cell depleted haplo-HSCT. These complications are likely related to the delayed immune reconstitution, and, in order to overcome their development, different means to boost immune surveillance have been implemented.
Proof of principle studies had demonstrated the feasibility to administer unmanipulated donor lymphocytes (DLI) to treat viral complications or leukemia relapse after T-cell depleted HSCT [91, 119]. The rate of acute GVHD developing after the procedure, however, prompted manipulation of donor lymphocytes to reduce alloreactivity while maintaining immune surveillance potency. Two strategies have been explored to reduce the risks derived from alloreactivity associated with DLI. The first approach was based on transduction of nonspecific T cells with a retroviral construct containing suicide genes, to induce susceptibility to drug-mediated lysis in case of development of alloreactive response [28]. Infusion of HSV-thymidine kinase gene-marked lymphocytes has proved safe and devoid of adverse effects [38]. However, its mechanism of action requires interference with DNA synthesis so that cell killing may take several days and be incomplete, resulting in a delay in clinical benefit. Recently, an alternative strategy that relies on inducible caspase proteins (iCasp9) to exploit the mitochondrial apoptotic pathway has been explored. The use of DLI modified by iCasp9 cell-suicide system in a small cohort of children transplanted for acute leukemia demonstrated the potential advantages in terms of rapid and consistent cell removal in case of GVHD development [53]. Escalating doses of iCasp9-modified DLI have been employed in 20 pediatric patients receiving Tα/β depleted haplo-HSCT for PID, and proved safe (25% cumulative incidence of aGVHD, no TRM) and able to provide prompt immune reconstitution [83].
An alternate strategy consists in delivering infectious/leukemia antigen-specific T cells selected by cell culture or by sorting. A major breakthrough was achieved by the adoptive transfer of virus-specific cytotoxic T lymphocytes reactivated from the peripheral blood of HSCT donors as prophylaxis/treatment against CMV disease or EBV-positive post-transplant lymphoproliferative disease in patients given T-cell depleted, HLA-disparate, unrelated HSCT [127, 145]. This approach has been successful in preventing and treating infectious complications after T-cell depleted haplo-HSCT, both in the pediatric and adult setting, while limiting the risk of inducing GVHD [44].
In the setting of leukemia, attempts have been made to boost tumor-specific responses and control leukemia relapse by post-transplant add-backs of donor cytotoxic T cells (CTLs) directed towards patients blasts [58], minor histocompatibility antigens [149], or leukemia-related antigens [43]. One of the main limitations is that CTL antigen recognition is major histocompatibility complex (MHC)- restricted. Moreover, in many cases, tumor-specific antigens able to elicit protective immune responses have not been identified.
To extend the recognition specificity of T lymphocytes beyond their classical MHC-peptide complexes, a gene-therapeutic strategy has been developed that allows redirecting T cells to defined tumor cell surface antigens, by the transfer of an antigen-binding moiety, most commonly a single chain variable fragment derived from a monoclonal antibody, together with an activating T-cell receptor (chimeric antigen receptors, CARs). Recently, CARs directed to the CD19 molecule, expressed on B-cell malignancies, have been employed in pediatric and adult patients with refractory ALL and proven highly efficient, with CR rates of 70–90% [92, 95, 107, 142]. These studies included patients with a prior history of allogeneic HSCT, and no GVHD was recorded. A phase I study of CD19 CAR T cell infusion after autologous and allogeneic HSCT included also 8 haplo-HSCT recipients, and the OS and DFS at 12 months for the haplo group were 100% and 75%, respectively. In the allogeneic setting, CAR T cell doses up to 108/m2 were safe and did not exacerbate GVHD [86].
It has been shown that leukemia blasts may escape immune control mediated by T cells and cause relapse by losing HLA mismatched alleles after HSCT, due to an acquired uniparental disomy, with consecutive total loss of the HLA-mismatched haplotype [144]. In this case, infusion of selected and/or activated NK cells may help control leukemia relapse. In addition, NK cells mostly target hematopoietic cells sparing solid organs, suggesting that an NK-mediated antitumor effect can be achieved in the absence of GVHD.
Studies have shown that infusion of haploidentical NK cells to exploit KIR/HLA alloreactivity is safe and can mediate impressive clinical activity in some patients with AML [128], and donor NK cells have been infused after haplo-HSCT with some evidence of efficacy [37, 79, 135]. Despite reports of clinical efficacy, a number of factors limit the application of NK cell immunotherapy for the treatment of cancer, such as the failure of infused NK cells to expand and persist in vivo. Therefore, means to maximize NK persistence and efficacy are currently being implemented.
11.6 Conclusions
Dramatic progress in the outcomes of haplo-HSCT in pediatric patients has been registered over the past decade, providing a chance to cure the children and adolescents in need of a HSCT.
Although the optimal strategy to overcome the HLA–histoincompatibility barrier is still debated, results in the pediatric populations appear equally encouraging with both T-cell depleted and T-replete HSCT approaches. In order to evaluate which strategy may be more appropriate in the different disease settings, multicentre controlled/randomized trials will have to be eventually conducted. Haplo-HSCT with PTCY has the potential to be the preferred transplant option for patients without HLA-matched donors in developing countries, where cell processing laboratories with specialized expertise and unrelated donor registries may be difficult to establish and maintain.
The excellent results obtained with Tαβ/B cell depleted haplo-HSCT, as well as with T-replete HSCT with PTCY, could challenge, in the near future, the current hierarchical algorithm in which MUD and unrelated cord blood are preferred to haploidentical donors, also in view of the possibility to exploit postransplant immune interventions in malignancy. Recent studies comparing haplo-HSCT to other types of allo-HSCT in both adult and children suggest that such a step may not be far to come.
References
Alfraih F, Aljurf M, Fitzhugh CD, Kassim AA. Alternative donor allogeneic hematopoietic cell transplantation for hemoglobinopathies. Semin Hematol. 2016;53(2):120–8. doi:10.1053/j.seminhematol.2016.01.001.
Algeri M, Comoli P, Strocchio L, Perotti C, Corbella F, Del Fante C, et al. Successful T-cell-depleted haploidentical hematopoietic stem cell transplantation in a child with dyskeratosis congenita after a fludarabine-based conditioning regimen. J Pediatr Hematol Oncol. 2015;37(4):322–6. doi:10.1097/MPH.0000000000000283.
Alinari L, Blum KA. How I treat relapsed classical Hodgkin lymphoma after autologous stem cell transplant. Blood. 2016;127(3):287–95. doi:10.1182/blood-2015-10-671826.
Amrolia PJ, Muccioli-Casadei G, Huls H, Adams S, Durett A, Gee A, et al. Adoptive immunotherapy with allodepleted donor T-cells improves immune reconstitution after haploidentical stem cell transplantation. Blood. 2006;108(6):1797–808. doi:10.1182/blood-2006-02-001909.
Andre-Schmutz I, Le Deist F, Hacein-Bey-Abina S, Vitetta E, Schindler J, Chedeville G, et al. Immune reconstitution without graft-versus-host disease after haemopoietic stem-cell transplantation: a phase 1/2 study. Lancet. 2002b;360(9327):130–7. doi:10.1016/S0140-6736 (02)09413-8.
Andre-Schmutz I, Le Deist F, Hacein-Bey S, Hamel Y, Vitetta E, Schindler J, et al. Donor T lymphocyte infusion following ex vivo depletion of donor anti-host reactivity by a specific anti-interleukin-2 receptor P55 chain immunotoxin. Transplant Proc. 2002a;34(7):2927–8.
Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–20. doi:10.3324/haematol.2013.099747.
Anurathapan U, Hongeng S, Pakakasama S, Sirachainan N, Songdej D, Chuansumrit A, et al. Hematopoietic stem cell transplantation for homozygous beta-thalassemia and beta-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant. 2016;51(6):813–8. doi:10.1038/bmt.2016.7.
Aversa F, Reisner Y, Martelli MF. The haploidentical option for high-risk haematological malignancies. Blood Cells Mol Dis. 2008;40(1):8–12. doi:10.1016/j.bcmd.2007.07.004.
Aversa F, Tabilio A, Terenzi A, Velardi A, Falzetti F, Giannoni C, et al. Successful engraftment of T-cell-depleted haploidentical "three-loci" incompatible transplants in leukemia patients by addition of recombinant human granulocyte colony-stimulating factor-mobilized peripheral blood progenitor cells to bone marrow inoculum. Blood. 1994;84(11):3948–55.
Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F, et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med. 1998;339(17):1186–93. doi:10.1056/NEJM199810223391702.
Aversa F, Terenzi A, Tabilio A, Falzetti F, Carotti A, Ballanti S, et al. Full haplotype-mismatched hematopoietic stem-cell transplantation: a phase II study in patients with acute leukemia at high risk of relapse. J Clin Oncol. 2005;23(15):3447–54. doi:10.1200/JCO.2005.09.117.
Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet. 1968;2(7583):1364–6.
Bachar-Lustig E, Rachamim N, Li HW, Lan F, Reisner Y. Megadose of T cell-depleted bone marrow overcomes MHC barriers in sublethally irradiated mice. Nat Med. 1995;1(12):1268–73.
Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129(11):1428–36. doi:10.1182/blood-2016-08-693481.
Bacigalupo A, Sica S. Alternative donor transplants for severe aplastic anemia: current experience. Semin Hematol. 2016;53(2):115–9. doi:10.1053/j.seminhematol.2016.01.002.
Balashov D, Shcherbina A, Maschan M, Trakhtman P, Skvortsova Y, Shelikhova L, et al. Single-center experience of unrelated and Haploidentical stem cell transplantation with TCRalphabeta and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015;21(11):1955–62. doi:10.1016/j.bbmt.2015.07.008.
Barfield RC, Otto M, Houston J, Holladay M, Geiger T, Martin J, et al. A one-step large-scale method for T- and B-cell depletion of mobilized PBSC for allogeneic transplantation. Cytotherapy. 2004;6(1):1–6. doi:10.1080/14653240310004411.
Bastien JP, Roy J, Roy DC. Selective T-cell depletion for haplotype-mismatched allogeneic stem cell transplantation. Semin Oncol. 2012;39(6):674–82. doi:10.1053/j.seminoncol.2012.09.004.
Berenbaum MC, Brown IN. Prolongation of homograft survival in mice with single doses of cyclophosphamide. Nature. 1963;200:84.
Berger M, Lanino E, Cesaro S, Zecca M, Vassallo E, Faraci M, et al. Feasibility and outcome of Haploidentical hematopoietic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescents with hematologic malignancies: an AIEOP-GITMO retrospective multicenter study. Biol Blood Marrow Transplant. 2016;22(5):902–9. doi:10.1016/j.bbmt.2016.02.002.
Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA-haploidentical stem cell transplantation after removal of alphabeta+ T and B cells in children with nonmalignant disorders. Blood. 2014;124(5):822–6. doi:10.1182/blood-2014-03-563817.
Bertaina A, Zecca M, Algeri M, Saglio F, Perotti C, Rovelli A et al. Outcome of children with acute leukemia given allogeneic HSCT either from an unrelated donor (UD-HSCT) or from an HLA-partially matched relative after αβ-T cell/B-cell depletion (αβhaplo-HSCT). Presented at the 2017 EBMT Annual Meeting, 27/03/2017, 2017.
Bessler M, Mason PJ, Link DC, Wilson DB. Inherited bone marrow failure syndromes. In: Saunders, editor. Nathan and Oski's hematology of infancy and childhood. Philadelphia: Saunders; 2009. p. 307–97.
Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285–91. doi:10.1182/blood-2012-07-438408.
Bonfim C, Ribeiro L, Nichele S, Bitencourt M, Loth G, Koliski A, et al. Long-term survival, organ function, and malignancy after hematopoietic stem cell transplantation for Fanconi anemia. Biol Blood Marrow Transplant. 2016;22(7):1257–63. doi:10.1016/j.bbmt.2016.03.007.
Bonfim C, Ribeiro L, Nichele S, Loth G, Bitencourt M, Koliski A, et al. Haploidentical bone marrow transplantation with post-transplant cyclophosphamide for children and adolescents with Fanconi anemia. Biol Blood Marrow Transplant. 2017;23(2):310–7. doi:10.1016/j.bbmt.2016.11.006.
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276(5319):1719–24.
Boussiotis VA, Freeman GJ, Gray G, Gribben J, Nadler LM. B7 but not intercellular adhesion molecule-1 costimulation prevents the induction of human alloantigen-specific tolerance. J Exp Med. 1993;178(5):1753–63.
Brunstein CG, Fuchs EJ, Carter SL, Karanes C, Costa LJ, Wu J, et al. Alternative donor transplantation after reduced intensity conditioning: results of parallel phase 2 trials using partially HLA-mismatched related bone marrow or unrelated double umbilical cord blood grafts. Blood. 2011;118(2):282–8. doi:10.1182/blood-2011-03-344853.
Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999;340(7):508–16. doi:10.1056/NEJM199902183400703.
Burkhardt B, Reiter A, Landmann E, Lang P, Lassay L, Dickerhoff R, et al. Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the berlin-frankfurt-muenster group. J Clin Oncol. 2009;27(20):3363–9. doi:10.1200/JCO.2008.19.3367.
Burroughs LM, O'Donnell PV, Sandmaier BM, Storer BE, Luznik L, Symons HJ, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant. 2008;14(11):1279–87. doi:10.1016/j.bbmt.2008.08.014.
Chaleff S, Otto M, Barfield RC, Leimig T, Iyengar R, Martin J, et al. A large-scale method for the selective depletion of alphabeta T lymphocytes from PBSC for allogeneic transplantation. Cytotherapy. 2007;9(8):746–54. doi:10.1080/14653240701644000.
Chen X, Hale GA, Barfield R, Benaim E, Leung WH, Knowles J, et al. Rapid immune reconstitution after a reduced-intensity conditioning regimen and a CD3-depleted haploidentical stem cell graft for paediatric refractory haematological malignancies. Br J Haematol. 2006;135(4):524–32. doi:10.1111/j.1365-2141.2006.06330.x.
Chewning JH, Gudme CN, Hsu KC, Selvakumar A, Dupont B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J Immunol. 2007;179(2):854–68.
Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Jang YJ, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: a dose-escalation study. Biol Blood Marrow Transplant. 2014;20(5):696–704. doi:10.1016/j.bbmt.2014.01.031.
Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10(5):489–500. doi:10.1016/S1470-2045(09)70074-9.
Ciceri F, Lupo-Stanghellini MT, Korthof ET. Haploidentical transplantation in patients with acquired aplastic anemia. Bone Marrow Transplant. 2013;48(2):183–5. doi:10.1038/bmt.2012.231.
Ciurea SO, de Lima M, Cano P, Korbling M, Giralt S, Shpall EJ, et al. High risk of graft failure in patients with anti-HLA antibodies undergoing haploidentical stem-cell transplantation. Transplantation. 2009;88(8):1019–24. doi:10.1097/TP.0b013e3181b9d710.
Clay J, Kulasekararaj AG, Potter V, Grimaldi F, McLornan D, Raj K, et al. Nonmyeloablative peripheral blood haploidentical stem cell transplantation for refractory severe aplastic anemia. Biol Blood Marrow Transplant. 2014;20(11):1711–6. doi:10.1016/j.bbmt.2014.06.028.
Clift RA, Hansen JA, Thomas ED, Buckner CD, Sanders JE, Mickelson EM, et al. Marrow transplantation from donors other than HLA-identical siblings. Transplantation. 1979;28(3):235–42.
Comoli P, Basso S, Riva G, Barozzi P, Guido I, Gurrado A, et al. BCR-ABL-specific T-cell therapy in Ph+ ALL patients on tyrosine-kinase inhibitors. Blood. 2017;129(5):582–6. doi:10.1182/blood-2016-07-731091.
Comoli P, Basso S, Zecca M, Pagliara D, Baldanti F, Bernardo ME, et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant. 2007;7(6):1648–55. doi:10.1111/j.1600-6143.2007.01823.x.
Comoli P, Montagna D, Moretta A, Zecca M, Locatelli F, Maccario R. Alloantigen-induced human lymphocytes rendered nonresponsive by a combination of anti-CD80 monoclonal antibodies and cyclosporin-A suppress mixed lymphocyte reaction in vitro. J Immunol. 1995;155(12):5506–11.
Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood. 2006;107(3):1233–6. doi:10.1182/blood-2005-05-1819.
Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, et al. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood. 2002;99(10):3661–7.
Dallas MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820–30. doi:10.1016/j.bbmt.2013.02.010.
Davies JK, Gribben JG, Brennan LL, Yuk D, Nadler LM, Guinan EC. Outcome of alloanergized haploidentical bone marrow transplantation after ex vivo costimulatory blockade: results of 2 phase 1 studies. Blood. 2008;112(6):2232–41. doi:10.1182/blood-2008-03-143636.
Dezern AE, Luznik L, Fuchs EJ, Jones RJ, Brodsky RA. Post-transplantation cyclophosphamide for GVHD prophylaxis in severe aplastic anemia. Bone Marrow Transplant. 2011;46(7):1012–3. doi:10.1038/bmt.2010.213.
Di Bartolomeo P, Santarone S, De Angelis G, Picardi A, Cudillo L, Cerretti R, et al. Haploidentical, unmanipulated, G-CSF-primed bone marrow transplantation for patients with high-risk hematologic malignancies. Blood. 2013;121(5):849–57. doi:10.1182/blood-2012-08-453399.
Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117(14):3921–8. doi:10.1182/blood-2010-10-311894.
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83. doi:10.1056/NEJMoa1106152.
Dufort G, Pisano S, Incoronato A, Castiglioni M, Carracedo M, Pages C, et al. Feasibility and outcome of haploidentical SCT in pediatric high-risk hematologic malignancies and Fanconi anemia in Uruguay. Bone Marrow Transplant. 2012;47(5):663–8. doi:10.1038/bmt.2011.148.
Durken M, Finckenstein FG, Janka GE. Bone marrow transplantation in hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2001;41(1–2):89–95. doi:10.3109/10428190109057957.
Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9(9):1144–50. doi:10.1038/nm915.
Esteves I, Bonfim C, Pasquini R, Funke V, Pereira NF, Rocha V, et al. Haploidentical BMT and post-transplant Cy for severe aplastic anemia: a multicenter retrospective study. Bone Marrow Transplant. 2015;50(5):685–9. doi:10.1038/bmt.2015.20.
Falkenburg JH, Wafelman AR, Joosten P, Smit WM, van Bergen CA, Bongaerts R, et al. Complete remission of accelerated phase chronic myeloid leukemia by treatment with leukemia-reactive cytotoxic T lymphocytes. Blood. 1999;94(4):1201–8.
Fang B, Li N, Song Y, Li J, Zhao RC, Ma Y. Cotransplantation of haploidentical mesenchymal stem cells to enhance engraftment of hematopoietic stem cells and to reduce the risk of graft failure in two children with severe aplastic anemia. Pediatr Transplant. 2009;13(4):499–502. doi:10.1111/j.1399-3046.2008.01002.x.
Foell J, Pfirstinger B, Rehe K, Wolff D, Holler E, Corbacioglu S. Haploidentical stem cell transplantation with CD3+−/CD19+− depleted peripheral stem cells for patients with advanced stage sickle cell disease and no alternative donor: results of a pilot study. Bone Marrow Transplant. 2017. doi:10.1038/bmt.2017.49.
Friedrich W, Goldmann SF, Ebell W, Blutters-Sawatzki R, Gaedicke G, Raghavachar A, et al. Severe combined immunodeficiency: treatment by bone marrow transplantation in 15 infants using HLA-haploidentical donors. Eur J Pediatr. 1985;144(2):125–30.
Friedrich W, Goldmann SF, Vetter U, Fliedner TM, Heymer B, Peter HH, et al. Immunoreconstitution in severe combined immunodeficiency after transplantation of HLA-haploidentical, T-cell-depleted bone marrow. Lancet. 1984;1(8380):761–4.
Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968;2(7583):1366–9.
Georges GE, Storb R. Hematopoietic stem cell transplantation for acquired aplastic anemia. Curr Opin Hematol. 2016;23(6):495–500. doi:10.1097/MOH.0000000000000281.
Ghosh N, Karmali R, Rocha V, Ahn KW, DiGilio A, Hari PN, et al. Reduced-intensity transplantation for lymphomas using Haploidentical related donors versus HLA-matched sibling donors: a Center for International Blood and Marrow Transplant Research Analysis. J Clin Oncol. 2016;34(26):3141–9. doi:10.1200/JCO.2015.66.3476.
Gluckman E, Devergie A, Dutreix J. Radiosensitivity in Fanconi anaemia: application to the conditioning regimen for bone marrow transplantation. Br J Haematol. 1983;54(3):431–40.
Gonzalez-Vicent M, Molina B, Deltoro N, Sevilla J, Vicario JL, Castillo A, et al. Donor age matters in T-cell depleted haploidentical hematopoietic stem cell transplantation in pediatric patients: faster immune reconstitution using younger donors. Leuk Res. 2017;57:60–4. doi:10.1016/j.leukres.2017.03.001.
Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371(4):339–48. doi:10.1056/NEJMsa1311707.
Gross TG, Hale GA, He W, Camitta BM, Sanders JE, Cairo MS, et al. Hematopoietic stem cell transplantation for refractory or recurrent non-Hodgkin lymphoma in children and adolescents. Biol Blood Marrow Transplant. 2010;16(2):223–30. doi:10.1016/j.bbmt.2009.09.021.
Guinan EC, Boussiotis VA, Neuberg D, Brennan LL, Hirano N, Nadler LM, et al. Transplantation of anergic histoincompatible bone marrow allografts. N Engl J Med. 1999;340(22):1704–14. doi:10.1056/NEJM199906033402202.
Hagin D, Burroughs L, Torgerson TR. Hematopoietic stem cell transplant for immune deficiency and immune dysregulation disorders. Immunol Allergy Clin N Am. 2015;35(4):695–711. doi:10.1016/j.iac.2015.07.010.
Handgretinger R, Schumm M, Lang P, Greil J, Reiter A, Bader P, et al. Transplantation of megadoses of purified haploidentical stem cells. Ann N Y Acad Sci. 1999;872:351–61. discussion 61-2
Hoenig M, Niehues T, Siepermann K, Jacobsen EM, Schutz C, Furlan I, et al. Successful HLA haploidentical hematopoietic SCT in chronic granulomatous disease. Bone Marrow Transplant. 2014;49(10):1337–8. doi:10.1038/bmt.2014.125.
Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W, et al. Treatment of acute leukemia with unmanipulated HLA-mismatched/haploidentical blood and bone marrow transplantation. Biol Blood Marrow Transplant. 2009;15(2):257–65. doi:10.1016/j.bbmt.2008.11.025.
Huang XJ, Zhao XY, Liu DH, Liu KY, Xu LP. Deleterious effects of KIR ligand incompatibility on clinical outcomes in haploidentical hematopoietic stem cell transplantation without in vitro T-cell depletion. Leukemia. 2007;21(4):848–51. doi:10.1038/sj.leu.2404566.
Im HJ, Koh KN, Choi ES, Jang S, Kwon SW, Park CJ, et al. Excellent outcome of haploidentical hematopoietic stem cell transplantation in children and adolescents with acquired severe aplastic anemia. Biol Blood Marrow Transplant. 2013;19(5):754–9. doi:10.1016/j.bbmt.2013.01.023.
Inagaki J, Park YD, Kishimoto T, Yoshioka A. Successful unmanipulated haploidentical bone marrow transplantation from an HLA 2-locus-mismatched mother for Wiskott-Aldrich syndrome after unrelated cord blood stem cell transplantation. J Pediatr Hematol Oncol. 2005;27(4):229–31.
Jaiswal SR, Chakrabarti A, Chatterjee S, Bhargava S, Ray K, O'Donnell P, et al. Haploidentical peripheral blood stem cell transplantation with post-transplantation cyclophosphamide in children with advanced acute leukemia with fludarabine-, busulfan-, and melphalan-based conditioning. Biol Blood Marrow Transplant. 2016;22(3):499–504. doi:10.1016/j.bbmt.2015.11.010.
Jaiswal SR, Zaman S, Nedunchezhian M, Chakrabarti A, Bhakuni P, Ahmed M, et al. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced incidence of acute graft versus host disease: a pilot study. Cytotherapy. 2017;19(4):531–42. doi:10.1016/j.jcyt.2016.12.006.
Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–52. doi:10.1182/blood-2011-03-278127.
Kanakry CG, Ganguly S, Zahurak M, Bolanos-Meade J, Thoburn C, Perkins B, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. 2013;5(211):211ra157. doi:10.1126/scitranslmed.3006960.
Kanate AS, Mussetti A, Kharfan-Dabaja MA, Ahn KW, DiGilio A, Beitinjaneh A, et al. Reduced-intensity transplantation for lymphomas using haploidentical related donors vs HLA-matched unrelated donors. Blood. 2016;127(7):938–47. doi:10.1182/blood-2015-09-671834.
Kapoor N, Bertaina A, Merli P, Naik S, Quasim W, Slatter M, et al. Outcome of Children with Primary Immune-Deficiencies (PIDs) enrolled in a phase I-II trial based on the infusion of BPX-501 donor t cells genetically modified with a novel suicide gene (inducible Caspase 9, iC9) after t-cell depleted HLA-haploidentical allogeneic stem cell transplantation (haplo-HSCT). Blood. 2016;128:4683. Presented at the 2016 ASH Annual Meeting, 12/2016. Blood. 2016;128:4683. Abstract presented at the 2016 ASH Annual Meeting.
Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR, et al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood. 2003;102(6):1997–2003. doi:10.1182/blood-2002-11-3444.
Kasamon YL, Luznik L, Leffell MS, Kowalski J, Tsai HL, Bolanos-Meade J, et al. Nonmyeloablative HLA-haploidentical bone marrow transplantation with high-dose posttransplantation cyclophosphamide: effect of HLA disparity on outcome. Biol Blood Marrow Transplant. 2010;16(4):482–9. doi:10.1016/j.bbmt.2009.11.011.
Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using sleeping beauty to generate CD19-specific CAR T cells. J Clin Invest. 2016;126(9):3363–76. doi:10.1172/JCI86721.
Kernan NA, Flomenberg N, Dupont B, O'Reilly RJ. Graft rejection in recipients of T-cell-depleted HLA-nonidentical marrow transplants for leukemia. Identification of host-derived antidonor allocytotoxic T lymphocytes. Transplantation. 1987;43(6):842–7.
Kharya G, Nademi Z, Leahy TR, Dunn J, Barge D, Schulz A, et al. Haploidentical T-cell alpha beta receptor and CD19-depleted stem cell transplant for Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2014;134(5):1199–201. doi:10.1016/j.jaci.2014.04.041.
King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood. 2014;123(20):3089–3094.; quiz 210. doi:10.1182/blood-2013-01-435776.
Klingebiel T, Cornish J, Labopin M, Locatelli F, Darbyshire P, Handgretinger R, et al. Results and factors influencing outcome after fully haploidentical hematopoietic stem cell transplantation in children with very high-risk acute lymphoblastic leukemia: impact of center size: an analysis on behalf of the Acute Leukemia and Pediatric Disease Working Parties of the European Blood and Marrow Transplant group. Blood. 2010;115(17):3437–46. doi:10.1182/blood-2009-03-207001.
Klingebiel T, Handgretinger R, Lang P, Bader P, Niethammer D. Haploidentical transplantation for acute lymphoblastic leukemia in childhood. Blood Rev. 2004;18(3):181–92. doi:10.1016/S0268-960X(03)00063-8.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129–39. doi:10.1182/blood-2013-08-519413.
Kreetapirom P, Hongeng S, Manuyakorn W, Anurathapan U, Pakakasama S, Sirachainan N, et al. Successful HLA haploidentical HSCT with post-transplant cyclophosphamide in Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2017. doi:10.1038/bmt.2017.25.
Lang P, Feuchtinger T, Teltschik HM, Schwinger W, Schlegel P, Pfeiffer M, et al. Improved immune recovery after transplantation of TCRalphabeta/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant. 2015;50(Suppl 2):S6–10. doi:10.1038/bmt.2015.87.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. doi:10.1016/S0140-6736(14)61403-3.
Lehmberg K, Albert MH, Beier R, Beutel K, Gruhn B, Kroger N, et al. Treosulfan-based conditioning regimen for children and adolescents with hemophagocytic lymphohistiocytosis. Haematologica. 2014;99(1):180–4. doi:10.3324/haematol.2013.094730.
Li XH, Gao CJ, Da WM, Cao YB, Wang ZH, Xu LX, et al. Reduced intensity conditioning, combined transplantation of haploidentical hematopoietic stem cells and mesenchymal stem cells in patients with severe aplastic anemia. PLoS One. 2014;9(3):e89666. doi:10.1371/journal.pone.0089666.
Li Z, Wang Y, Wang J, Zhang J, Wang Z. Successful haploidentical stem cell transplantation for three adults with primary hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2017;52(2):330–3. doi:10.1038/bmt.2016.284.
Locatelli F, Nollke P, Zecca M, Korthof E, Lanino E, Peters C, et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105(1):410–9. doi:10.1182/blood-2004-05-1944.
Locatelli F, Pende D, Comoli P, Zecca M, Giorgiani G, Perotti C, et al. Transplantation of T-cell depleted peripheral blood haematopoietic stem cells from an HLA-disparate family donor for children with hematological malignancies. Blood. 2007a;110:3071.
Locatelli F, Zecca M, Pession A, Morreale G, Longoni D, Di Bartolomeo P, et al. The outcome of children with Fanconi anemia given hematopoietic stem cell transplantation and the influence of fludarabine in the conditioning regimen: a report from the Italian pediatric group. Haematologica. 2007b;92(10):1381–8. doi:10.3324/haematol.11436.
Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641–50. doi:10.1016/j.bbmt.2008.03.005.
Mancusi A, Ruggeri L, Urbani E, Pierini A, Massei MS, Carotti A, et al. Haploidentical hematopoietic transplantation from KIR ligand-mismatched donors with activating KIRs reduces nonrelapse mortality. Blood. 2015;125(20):3173–82. doi:10.1182/blood-2014-09-599993.
Marks DI, Khattry N, Cummins M, Goulden N, Green A, Harvey J, et al. Haploidentical stem cell transplantation for children with acute leukaemia. Br J Haematol. 2006;134(2):196–201. doi:10.1111/j.1365-2141.2006.06140.x.
Marsh RA, Jordan MB, Filipovich AH. Reduced-intensity conditioning haematopoietic cell transplantation for haemophagocytic lymphohistiocytosis: an important step forward. Br J Haematol. 2011;154(5):556–63. doi:10.1111/j.1365-2141.2011.08785.x.
Maschan M, Shelikhova L, Ilushina M, Kurnikova E, Boyakova E, Balashov D, et al. TCR-alpha/beta and CD19 depletion and treosulfan-based conditioning regimen in unrelated and haploidentical transplantation in children with acute myeloid leukemia. Bone Marrow Transplant. 2016;51(5):668–74. doi:10.1038/bmt.2015.343.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. doi:10.1056/NEJMoa1407222.
Montagna D, Yvon E, Calcaterra V, Comoli P, Locatelli F, Maccario R, et al. Depletion of alloreactive T cells by a specific anti-interleukin-2 receptor p55 chain immunotoxin does not impair in vitro antileukemia and antiviral activity. Blood. 1999;93(10):3550–7.
Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood. 2011;118(6):1675–84. doi:10.1182/blood-2010-11-319376.
Moretta A, Bottino C, Pende D, Tripodi G, Tambussi G, Viale O, et al. Identification of four subsets of human CD3-CD16+ natural killer (NK) cells by the expression of clonally distributed functional surface molecules: correlation between subset assignment of NK clones and ability to mediate specific alloantigen recognition. J Exp Med. 1990;172(6):1589–98.
Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins AM, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. 2016;128(3):440–8. doi:10.1182/blood-2016-03-704015.
Munoz A, Olive T, Martinez A, Bureo E, Maldonado MS. Diaz de Heredia C et al. allogeneic hemopoietic stem cell transplantation (HSCT) for Wiskott-Aldrich syndrome: a report of the Spanish working Party for Blood and Marrow Transplantation in children (GETMON). Pediatr Hematol Oncol. 2007;24(6):393–402. doi:10.1080/08880010701454404.
Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al Jijakli A, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood. 2005;105(10):4135–42. doi:10.1182/blood-2004-10-4113.
Oevermann L, Michaelis SU, Mezger M, Lang P, Toporski J, Bertaina A, et al. KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood. 2014;124(17):2744–7. doi:10.1182/blood-2014-03-565069.
Ohta H, Miyashita E, Hirata I, Matsumura R, Yoshida H, Hashii Y, et al. Hematopoietic stem cell transplantation with reduced intensity conditioning from a family haploidentical donor in an infant with familial hemophagocytic lymphohistocytosis. Int J Hematol. 2011;94(3):285–90. doi:10.1007/s12185-011-0916-6.
Ouachee-Chardin M, Elie C, de Saint BG, Le Deist F, Mahlaoui N, Picard C, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117(4):e743–50. doi:10.1542/peds.2005-1789.
Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111(1):439–45. doi:10.1182/blood-2007-03-076679.
Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014;371(5):434–46. doi:10.1056/NEJMoa1401177.
Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330(17):1185–91. doi:10.1056/NEJM199404283301703.
Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, Malech H, et al. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: a first report. J Clin Immunol. 2015;35(7):675–80. doi:10.1007/s10875-015-0204-y.
Peccatori J, Forcina A, Clerici D, Crocchiolo R, Vago L, Stanghellini MT, et al. Sirolimus-based graft-versus-host disease prophylaxis promotes the in vivo expansion of regulatory T cells and permits peripheral blood stem cell transplantation from haploidentical donors. Leukemia. 2015;29(2):396–405. doi:10.1038/leu.2014.180.
Peffault de Latour R, Porcher R, Dalle JH, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122(26):4279–86. doi:10.1182/blood-2013-01-479733.
Pende D, Marcenaro S, Falco M, Martini S, Bernardo ME, Montagna D, et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood. 2009;113(13):3119–29. doi:10.1182/blood-2008-06-164103.
Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for primary immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726. doi:10.1007/s10875-015-0201-1.
Raiola AM, Dominietto A, Ghiso A, Di Grazia C, Lamparelli T, Gualandi F, et al. Unmanipulated haploidentical bone marrow transplantation and posttransplantation cyclophosphamide for hematologic malignancies after myeloablative conditioning. Biol Blood Marrow Transplant. 2013;19(1):117–22. doi:10.1016/j.bbmt.2012.08.014.
Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Cunningham-Rundles S, Dupont B, et al. Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood. 1983;61(2):341–8.
Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345(8941):9–13.
Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28(6):955–9. doi:10.1200/JCO.2009.24.4590.
Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333–9.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100. doi:10.1126/science.1068440.
Ruggeri L, Mancusi A, Capanni M, Urbani E, Carotti A, Aloisi T, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. 2007;110(1):433–40. doi:10.1182/blood-2006-07-038687.
Satwani P, Jin Z, Martin PL, Bhatia M, Garvin JH, George D, et al. Sequential myeloablative autologous stem cell transplantation and reduced intensity allogeneic hematopoietic cell transplantation is safe and feasible in children, adolescents and young adults with poor-risk refractory or recurrent Hodgkin and non-Hodgkin lymphoma. Leukemia. 2015;29(2):448–55. doi:10.1038/leu.2014.194.
Shin CR, Kim MO, Li D, Bleesing JJ, Harris R, Mehta P, et al. Outcomes following hematopoietic cell transplantation for Wiskott-Aldrich syndrome. Bone Marrow Transplant. 2012;47(11):1428–35. doi:10.1038/bmt.2012.31.
Sodani P, Isgro A, Gaziev J, Polchi P, Paciaroni K, Marziali M, et al. Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010;115(6):1296–302. doi:10.1182/blood-2009-05-218982.
Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant. 2013;48(3):433–8. doi:10.1038/bmt.2012.162.
Stern M, Ruggeri L, Mancusi A, Bernardo ME, de Angelis C, Bucher C, et al. Survival after T cell-depleted haploidentical stem cell transplantation is improved using the mother as donor. Blood. 2008;112(7):2990–5. doi:10.1182/blood-2008-01-135285.
Strahm B, Nollke P, Zecca M, Korthof ET, Bierings M, Furlan I, et al. Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia. 2011;25(3):455–62. doi:10.1038/leu.2010.297.
Sureda A, Bader P, Cesaro S, Dreger P, Duarte RF, Dufour C, et al. Indications for allo- and auto-SCT for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2015. Bone Marrow Transplant. 2015;50(8):1037–56. doi:10.1038/bmt.2015.6.
Svahn J, Bagnasco F, Cappelli E, Onofrillo D, Caruso S, Corsolini F, et al. Somatic, hematologic phenotype, long-term outcome, and effect of hematopoietic stem cell transplantation. An analysis of 97 Fanconi anemia patients from the Italian national database on behalf of the Marrow Failure Study Group of the AIEOP (Italian Association of Pediatric Hematology-Oncology). Am J Hematol. 2016;91(7):666–71. doi:10.1002/ajh.24373.
Symons HJ, Leffell MS, Rossiter ND, Zahurak M, Jones RJ, Fuchs EJ. Improved survival with inhibitory killer immunoglobulin receptor (KIR) gene mismatches and KIR haplotype B donors after nonmyeloablative, HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant. 2010;16(4):533–42. doi:10.1016/j.bbmt.2009.11.022.
Thakar MS, Bonfim C, Sandmaier BM, O'Donnell P, Ribeiro L, Gooley T, et al. Cyclophosphamide-based in vivo T-cell depletion for HLA-haploidentical transplantation in Fanconi anemia. Pediatr Hematol Oncol. 2012;29(6):568–78. doi:10.3109/08880018.2012.708708.
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38. doi:10.1172/JCI85309.
Vago L, Forno B, Sormani MP, Crocchiolo R, Zino E, Di Terlizzi S, et al. Temporal, quantitative, and functional characteristics of single-KIR-positive alloreactive natural killer cell recovery account for impaired graft-versus-leukemia activity after haploidentical hematopoietic stem cell transplantation. Blood. 2008;112(8):3488–99. doi:10.1182/blood-2007-07-103325.
Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MT, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361(5):478–88. doi:10.1056/NEJMoa0811036.
Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333(16):1038–44. doi:10.1056/NEJM199510193331603.
Wang Y, Chang YJ, Xu LP, Liu KY, Liu DH, Zhang XH, et al. Who is the best donor for a related HLA haplotype-mismatched transplant? Blood. 2014a;124(6):843–50. doi:10.1182/blood-2014-03-563130.
Wang Z, Yan H, Zhu L, Wang H. Successful unmanipulated stem cell transplantation from HLA-haploidentical 3-loci-mismatched parents in two children with severe aplastic anemia not responding to immunosuppressive therapy. Am J Hematol. 2010;85(5):389–90. doi:10.1002/ajh.21693.
Wang Z, Zheng X, Yan H, Li D, Wang H. Good outcome of haploidentical hematopoietic SCT as a salvage therapy in children and adolescents with acquired severe aplastic anemia. Bone Marrow Transplant. 2014b;49(12):1481–5. doi:10.1038/bmt.2014.187.
Warren EH, Fujii N, Akatsuka Y, Chaney CN, Mito JK, Loeb KR, et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood. 2010;115(19):3869–78. doi:10.1182/blood-2009-10-248997.
Woodard P, Cunningham JM, Benaim E, Chen X, Hale G, Horwitz E, et al. Effective donor lymphohematopoietic reconstitution after haploidentical CD34+−selected hematopoietic stem cell transplantation in children with refractory severe aplastic anemia. Bone Marrow Transplant. 2004;33(4):411–8. doi:10.1038/sj.bmt.1704358.
Xu LP, Zhang XH, Wang FR, Mo XD, Han TT, Han W, et al. Haploidentical transplantation for pediatric patients with acquired severe aplastic anemia. Bone Marrow Transplant. 2017;52(3):381–7. doi:10.1038/bmt.2016.281.
Zecca M, Strocchio L, Pagliara D, Comoli P, Bertaina A, Giorgiani G, et al. HLA-haploidentical T cell-depleted allogeneic hematopoietic stem cell transplantation in children with Fanconi anemia. Biol Blood Marrow Transplant. 2014;20(4):571–6. doi:10.1016/j.bbmt.2014.01.015.
Zhang Y, Guo Z, Liu XD, He XP, Yang K, Chen P, et al. Comparison of Haploidentical hematopoietic stem cell transplantation and immunosuppressive therapy for the treatment of acquired severe aplastic anemia in pediatric patients. Am J Ther. 2017;24(2):e196–201. doi:10.1097/MJT.0000000000000366.
Zhou L, Dong LJ, Gao ZY, Yu XJ, Lu DP. Haploidentical hematopoietic stem cell transplantation for a case with X-linked chronic granulomatous disease. Pediatr Transplant. 2017;21(1) doi:10.1111/petr.12861.
Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123(25):3895–905. doi:10.1182/blood-2014-01-551671.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zecca, M., Comoli, P. (2017). Applications of Haploidentical SCT in Pediatric Patients. In: Demirer, T. (eds) Haploidentical Stem Cell Transplantation. Stem Cell Biology and Regenerative Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-65319-8_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-65319-8_11
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-65318-1
Online ISBN: 978-3-319-65319-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)