
Abstract
Rare facial cleft has a broad spectrum of clinical presentation as soft tissue and bony structures can be malformed, clefting or absent. The face is a complete three-dimensional structure, composed by regions with different topography, skin thickness, texture, and color. In patients with rare facial cleft, hair follicles may accompany the cleft, especially those above the orbit or that are completely absent causing alopecia, absence of eyebrow and eyelash.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Rare facial cleft has a broad spectrum of clinical presentation as soft tissue and bony structures can be malformed, clefting or absent. The face is a complete three-dimensional structure, composed by regions with different topography, skin thickness, texture, and color. In patients with rare facial cleft, hair follicles may accompany the cleft, especially those above the orbit or that are completely absent causing alopecia, absence of eyebrow and eyelash.
Numerous classification systems for the rare craniofacial clefts have been proposed, including those by American Association of Cleft Palate Rehabilitation (AACPR). Boo-Chai 1969, Karfik 1966. Strengths and drawbacks have been observed in these classification systems. Paul Tessier, the father of craniofacial surgery, described his system for classifying rare facial clefts, offering to the surgical community a comprehensive analysis of the wide spectrum of this disease, including a concise way to categorize not only the obvious soft-tissue deformities of the various clefts but also a description of how the soft-tissue defects related to the underlying aberrant bony anatomy associated with the clefts (Tessier 1976). In other words, he added a practical component to the descriptive narrative that generates clinical significance to the classification system, facilitates diagnosis, and helps guide therapy. Moreover, Tessier’s original description was based on the embryological origins of cleft development. For these reasons the rare craniofacial clefts have been come to be commonly known as the Tessier rare craniofacial clefts.
Tessier divided the face into various “time zones” centered on an imaginary equatorial axis running horizontally through the orbits and vertically through the facial midline (Kawamoto 1976). Clefts occurring below the orbits are numbered 0 through 8 and those above 9 through 14, including both soft-tissue and bone defects (Tessier 1976; Kawamoto 1976) (Fig. 21.1a). This numbering system encourages the clinician to evaluate the entire height of the face and more readily identify instances where caudal and cranial facial clefts are coincident. When upper and lower median/paramedian or oblique clefts occur simultaneously, the numbers of the individual upper and lower clefts add up to 14 (e.g., 0–14, 2–12, 4–10 clefts). Deviating from this overall scheme, Tessier defined a cleft of the lower lip, mandible, and associated midline soft-tissue and skeletal structures of the lower face and neck system as a number 30 cleft.

Tessier diagram of rare facial cleft. In the original diagram, Tessier correlates the bony cleft to its associated soft-tissue deformity
The preoperative surgical planning should be tailored to each individual deformity and it has been impossible to delineate an algorithm or a protocol to approach each specific condition. It has been a consensus among craniofacial plastic surgeons that the craniofacial skeleton should be reconstructed first; however in some major craniofacial cleft treated at 3–4 months of age, the bony reconstruction is postponed at later age and soft tissue has been a priority to offer satisfactory aesthetic and functional outcomes. The treatment of rare facial cleft demands a complete understanding of the concept of the aesthetic units of the face described by Gonzalez-Ulloa (1956, 1958).
Burget and Menick applied this concept to develop a state-of-the-art philosophy for nasal reconstruction (Burget and Menick 1986). We have been using similar principles to avoid multiple scars crossing the face that can be apparent and evident by reflected highlights and cast shadows. Interestingly, a plastic surgeon approaching a rare facial cleft should have in mind that there is a continuous need to construct the congenitally absent structures. Thus, although these terms construct and reconstruct are used interchangeably, in most scenarios one has to create an absent structure using local tissues that may or may not have similar characteristics. In this chapter we aimed to emphasize some of the modern principles and surgical strategies to approach soft-tissue and bony reconstruction in patients with rare facial cleft. Orthodontics and speech pathology are critical fields in the rehabilitation process. However, technical details of both fields are beyond the scope of this chapter. A detailed description of each cleft type will be followed.
21.1 Rare Facial Cleft Number 0
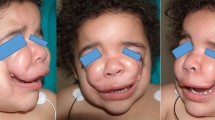
The rare facial cleft number 0 is a midline defect that may involve soft tissue and bone from the central incisors up through the nasal cavity to the perpendicular plate of the ethmoid. These clefts may be characterized by severe tissue deficiency to tissue excess. On one end of the spectrum are deformities that are defined by lack of development of midline facial structures. Mild manifestation may include isolated midline cleft of the lip. The palate may be high and arched or completely cleft, with absent premaxilla and philtrum. A central cleft of the nose, absent columella and nasal septum, hypoplasia of the nasoethmoidal complex, or arrhinia with or without proboscis may contribute to hypotelorism. Associated central nervous system defects can result in holoprosencephaly. Alternatively, midline tissue excess may result in enlarged and broadened nasal bones, widened nasal septum, and enlarged and laterally/superiorly displaced nasal cartilages. Nasofrontal skeletal deficiency may paradoxically present as hypertelorism due to herniation of intracranial contents in the form of frontonasal encephalocele (Pittet et al. 2004; da Silva et al. 2008; Nam and Kim 2014) (Fig. 21.2).

Patient with number 0 Tessier cleft. This cleft crosses the nose and divided it into two equal halves, but not beyond the orbital level. Wide and bifid tip and wide collumela is found in this particular cleft (left). Other type of clinical features can be also found (center, right). Isolated number 0 is very rare as it is usually associated with number 14 Tessier cleft
21.2 Rare Facial Cleft Number 1
The rare facial cleft number 1 is a vertical paramedian cleft characterized by soft-tissue notching through the dome of the nose that extends toward the medial canthus and medial brow. A cleft of the lip may occur in the region of Cupid’s bow, where the “traditional” cleft lip manifests. The nasal defect may range from completely missing upper and lower lateral cartilages to paramedian soft-tissue fissures or contour irregularities over the nasal dorsum. A bony cleft between the central and lateral incisors may extend into the pyriform aperture lateral to the anterior nasal spine. The ethmoid sinuses may be involved and the cleft may extend between the nasal bone (which may be notched or absent) and frontal process of the maxilla, resulting in hypertelorism. Heminasal atrophy or a proboscis may be seen in severe forms. The nasolacrimal system is spared in this facial cleft (Kawamoto 1976; Agrawal et al. 1998) (Figs. 21.3–21.6).

The clinical characteristic of rare facial cleft number 0–14 may broadly vary from a bifid nose from a complete absence of the midline structures with encephalocele

The cleft number 1 crosses the lateral incisor, nasal cavity, and nasal bone. An alternative root is through the region between nasal bone and frontal process of the maxilla. The cranial extension of number 1 is the number 13 Tessier cleft

Patient photograph of cleft number 1

Photograph of patients with number 1 Tessier clefts and its cranial extension number 13
21.3 Rare Facial Cleft Number 2
The rare facial cleft number 2 may be characterized as a transitional or an incomplete form of the number 1 and 3 clefts. It is very rare to identify an isolated number 2 cleft. Similarly to number 1 cleft, the cleft of the lip occurs in the area of Cupid’s bow. The medial third of the nasal nostril is flat that can be either apparent without true notching or absent. The nasal dorsum may be widened and the septum may be deviated. The medial canthi and medial brow are intact but may exhibit inferior displacement or epicanthal folding. The bony cleft begins in the region of the lateral incisor and the skeletal fissure extends cephalad into pyriform aperture. The frontal process of maxilla is broad and flat and can be notched. Ethmoid enlargement contributes to hypertelorism, specially in its cranial extension (Kawamoto 1976; Ozek et al. 2001; Tiwari et al. 1991) (Figs. 21.7 and 21.8).

Patient photograph of cleft number 2, more laterally than number 1 with flat nasal nostril and nasal ala clefting

Different clinical presentations of 2–12 Tessier cleft. The hypertelorbitism occurs as a consequence of ethmoidal bone enlargement
21.4 Rare Facial Cleft Number 3
The rare facial cleft number 3 is more frequent than the rare facial cleft number 1 and 2. It has also been referred as oro-naso-ocular cleft or oblique cleft as it involves these anatomical structures leading to oblique facial cavities. The cleft lies in the area of union of the embryologic median nasal, lateral nasal, and maxillary processes and may therefore affect any of the structures that arise from normal fusion of these processes. In addition, failure of the embryonic naso-optic groove to invaginate and form the nasolacrimal drainage system at this confluence of advancing processes may explain the nasolacrimal defects associated with this cleft.
Similarly to rare facial cleft number 1 and 2, the cleft begins at the cleft lip and continues through the alar base toward the medial canthi. The soft-tissue cheek deficiency decreases the distance between the nasal ala to the medial canthi, leading to inferior displacement of the medial canthi, resulting in either vertical orbital dystopia or telecanthus. A lower eyelid coloboma, medial to lacrimal punctum, may also be present, as with agenesis of part or all of nasolacrimal system. Orbital and ocular malformations may include bony defects of the orbital rim and floor, dystopia, microphthalmia, anophthalmia, and epibulbar dermoids. The inferior orbital rim may be incomplete or grooving toward the pyriform aperture, generating a bony communication between the the orbit and nose. The cleft in the alveolus occurs between the lateral incisor and canine and extends through the lateral border of the pyriform into the nasal cavity. The osseous defect can involve the nasal cavity along the nasomaxillary process to the level of the lacrimal bone leading to a compromise of the nasolacrimal system function. The orbital, maxillary, nasal, and oral cavities may be confluent and difficult to distinguish (Allam et al. 2014; Gawrych et al. 2010; Wenbin et al. 2007; Bodin et al. 2006) (Figs. 21.9 and 21.10).

Schematic drawing of number 3 Tessier cleft. The lip cleft is usually located in cupid bow. The base of the nasal ala is involved and the cleft runs toward the medial canthi. In the complete forms the cleft can be seen in the lateral incisors

Photographs of patients with Tessier number 3 cleft. All clinical characteristics can be seen in these patients
21.5 Rare Facial Cleft Number 4
The rare facial cleft number 4 is characterized by the soft-tissue involvement laterally to labial philtrum, usually between the philtral ridge and commissure. It differs from the common cleft lip and palate characteristics, as the cleft moves laterally to the common affected structures, affecting the check and lateral nasal aesthetic subunits, sparing the nasal alar base, but shortening the distance between the alar base and medial canthi. The medial canthus are not involved, as the cleft continues from the cheek onto the lower lid lateral to the punctum. The lacrimal drainage structures are therefore intact, but often dysfunctional. The globe can show anophthalmia, microphthalmia, or normal anatomy. Similarly to the rare facial cleft number 3, the cleft alveolus occurs between the lateral incisor and canine, sparing the pyriform aperture. The infraorbital foramen is an anatomical landmark for separation of cleft numbers 4 and 5, as number 4 moves medially to the foramen and then to the orbital rim and floor. Orbital contents may be herniated into the maxillary sinus and the bony orbit may be hypoplastic and dystopic as the eyeball can also be smaller. In the complete form of the number 4 cleft, the oral, maxillary, orbital cavities are confluent, for which this cleft has been referred to as the oro-ocular oblique cleft (Abdollahifakhim et al. 2013; Laure et al. 2010; Portier-Marret et al. 2008; Tokioka et al. 2005; Kale and Pakhmode 2000; Akoz et al. 1996) (Figs. 21.11–21.13).

Schematic drawing of Tessier number 4 cleft. (a) Patient’s photograph of number 4 Tessier cleft. The cleft crosses laterally to the cupid bow. The nose may be distorted and the cleft crosses the medial canthi ligament. (b) Patient’s photographs of severe forms of number 4 Tessier cleft (left, center) and unusual presentation of number 4 Tessier cleft (right)

Patients photograph of number 4 Tessier cleft. The cleft crosses laterally to the cupid bow. The nose may be distorted and the cleft crosses the medial canthi ligament

Patient’s photograph of number 4 Tessier cleft. The cleft crosses laterally to the cupid bow. The nose may be distorted and the cleft crosses the medial canthi ligament. Patient’s photographs of severe forms of number 4 Tessier cleft (left, center) and unusual presentation of number 4 Tessier cleft (right)
21.6 Rare Facial Cleft Number 5
The rare facial cleft number 5 is the rarest of the oblique facial clefts and is rarely seen in isolation. The cleft of the lip occurs medial to the oral commissure and continues cephalad with a curvilinear trajectory on the lateral cheek toward the lower lid at the junction of its median and lateral thirds. Oblique clefts and abnormal muscle attachments of the soft palate may be identified. The lateral oro-ocular height is shortened. The skeletal defect is characterized by an alveolar cleft that occurs distal to canine and extends superiorly along the anterior maxillary wall lateral to the infraorbital foramen and then across the rim and onto the lateral orbital floor. Similar to orbital floor defects seen in the number 3 and 4 facial cleft, herniation of orbital contents into the maxilla can occur, along with orbital hypoplasia and dystopia with associated microphthalmia or anophthalmia (Abdollahifakhim et al. 2013; Garg and Goyal 2009; Galante and Dado 1991) (Figs. 21.14–21.16).

Schematic drawing of severe forms of number 5 Tessier cleft. The cleft of the lip occurs medial to the oral commissure and continues cephalad with a curvilinear trajectory on the lateral cheek toward the lower lid at the junction of its median and lateral thirds. Oblique clefts and abnormal muscle attachments of the soft palate may be identified. The lateral oro-ocular height is shortened

Photographs of patients with number 5 Tessier cleft showing main clinical characteristics of this very rare cleft

Photographs of a very rare forms of number 5 Tessier cleft on the right side and number 4 on the left side
21.7 Rare Facial Cleft Number 6
The association of the number 6, 7, and 8 rare craniofacial clefts is known as Treacher Collins syndrome (TCS), characterized by the absence of the zygoma. Tessier defines the TCS as a confluence of clefts at the maxillary zygomatic, temporozygomatic, and frontozygomatic regions.
The isolated form of number 6 cleft is described as an incomplete form of TCS but authors have identified very rare forms of complete number 6.
Macrostomia and soft palate are almost always present. An isolate number 6 can be seen as uni- or bilateral when not associated to TCS. There is a fusion between the corner of the mouth and the soft palate. The cleft bone in the retro molar area is responsible for the abnormal insertion of the palate muscles. Palatopharyngeal muscle is not inserted in the central part of the soft palate. Velopharyngeal incompetence is very difficult to solve because the soft palate is too short and the muscles are inserted in a wrong direction. When the cleft continues until the orbit facial, mimic muscles can be damaged. It is unusual to observe facial palsy. Lower eye lid retraction is not related just to lack of fusion of facial soft tissue but also for cleft or absence of zygomatic bone. The frontal process of the zygomatic bone is absent but sometimes is present; for this reason there is no antimongoloid slant in few cases. The number 6 is always parallel to number 5 but more laterally displaced. The infraorbital foramen is in a medial position. Tessier cleft number 6 could be present in Treacher Collins syndrome associated with number 7 and 8 cleft. In this association the phenotype is a little bit different from the isolated ones. Maxillary duplication is seen in bilateral number 6 (Fig. 21.17).

Patient with number 6 Tessier cleft. Those are the incomplete form of Treacher Collins syndrome. Lower eyelid coloboma is seen, but the lateral canthi is less severely dystopic in comparison to Treacher Collins syndrome. These patients are characterized by a tight soft-tissue envelope in the zygomatic region and hypoplastic or clefting zygomatic bone is seen
In contrast to the complete TCS, characterized by absence of the zygoma and its arch, the isolated number 6 cleft is defined by a present but hypoplastic zygomatic body and an intact zygomatic arch. The bony cleft occurs at the junction of the malar bone and maxilla, the zygomaticomaxillary suture. Soft-tissue manifestations are similar to some of those seen in TCS patients, but more mild in nature. These can include external ear deformities, lower lid colobomas at the junction of the median and lateral thirds of the lower eyelids, and hypoplastic or absent lateral canthal tendons. The resultant midface hypoplasia, negative palpebral cant, and inferior displacement of the lateral canthi are reminiscent of the syndromic presentation (Ligh et al. 2015; Nguyen et al. 2016; Plomp et al. 2016).
21.8 Rare Facial Cleft Number 7
The number 7 cleft is the most common isolated rare craniofacial cleft. It has a variable clinical presentation, with a diversity of anatomical structures that may be involved in the clefting. This might explain the numerous ways the number 7 cleft is referred to, including craniofacial microsomia, microtia, otomandibular dysostosis, first and second branchial arch syndrome, and oromandibular-auricular syndrome. Macrostomia is the main feature of the cleft, but soft-tissue involvement can affect all tissue layers of the face. Presentation may range from small preauricular skin tags to mild facial asymmetry to significant soft-tissue hypoplasia that includes the cheek, tongue, soft palate, parotid gland, trigeminal and facial nerves, and muscles they innervate. External ear deformities include the entire spectrum from mild microtia to complete anotia. The cardinal bony defects are centered around the zygomaticotemporal suture, with resultant absence or hypoplasia of the body and arch of the zygoma. As with the soft-tissue deficiency, the mandibular hypoplasia also occurs on a gradient, ranging from a structurally normal but small mandible to complete absence of the mandibular condyle and ramus. Zygomatic arch may be absent and zygoma may terminate in a stump (Bodin et al. 2006; Presti et al. 2004; Woods et al. 2008; Horgan et al. 1995; Poon et al. 2003) (Figs. 21.18–21.20).

Photographs of patient with number 7 Tessier cleft

Photograph of a patient with hemifacial microsomia

Frontal photograph of a patient with an unusual presentation of number 6 and 7 Tessier cleft
21.9 Rare Facial Cleft Number 8
While commonly associated with the number 6 and 7 rare facial clefts in Treacher Collins syndrome, an isolated number 8 cleft is a rare entity. Centered on the frontozygomatic suture, the cleft begins at the lateral palpebral fissure and extends to temporal region. The bony defect manifests as bone loss of the lateral orbital rim at the level of the suture. More severe forms of bony involvement are likely representative of Treacher Collins syndrome; when these are present defects in the areas of the number 6 and 7 clefts should be sought out. Soft-tissue deficits affect the lateral canthal tendon and its insertion, ranging from a furrow in the lateral palpebral area with discontinuity of the orbicularis oculi ring to a coloboma of the lateral canthus, to a dermatocele (Kawamoto 1976; Fuente-del-Campo 1990) (Figs. 21.21–21.23).

Photograph of a patient with an unusual presentation of number 8 Tessier cleft with hemifacial microsomia and cleft lip. This cleft affects the lateral palpebrum commissure that can be occupied by a dermatocele

Schematic drawing of number 6, 7, 8 Tessier cleft known as Treacher Collins syndrome

This particular patient shows a complete form of Treacher Collins syndrome
21.10 Rare Facial Cleft Number 9
The isolated number 9 cleft is extremely rare. The cleft is located at the superolateral angle of orbit. As a result, the upper eyelid may exhibit full-thickness tissue loss between its median and lateral thirds. This defect may extend through the brow and obliquely toward the hairline and involve the underlying orbit and forehead (David et al. 1989; Dumortier et al. 1999) (Fig. 21.24).

Frontal photograph of a patient with number 9 Tessier cleft, a very rare condition
21.11 Rare Facial Cleft Number 10
This cleft represents the cranial extension of the number 4 cleft. A coloboma of middle third of eyelid with disruption of middle third of brow and continuation to the hairline is possible. A whorl of hair-bearing scalp may be seen in the frontal region. The underlying skeletal defect can include absence of the middle orbital rim and roof as well as adjacent frontal bone, resulting in a fronto-orbital encephalocele of varying size. With herniation of intracranial contents through the bony defect, the orbit is rotated laterally and inferiorly (Kawamoto 1976; Lee et al. 2012) (Figs. 21.25 and 21.26).

Schematic drawing of number 10 Tessier cleft. Note the presence of the encephalocele

Photographs of patients with number 10 Tessier cleft. Encephalocele and upper-lower eyelid distortion and absent eyebrow are seen
21.12 Rare Facial Cleft Number 11
The number 11 cleft usually occurs along with the number 3 facial cleft, and rarely occurs alone. It passes through the medial third of the upper eyelid and eyebrow, extending to the frontal hairline. At the level of the orbit, it can pass through the orbit itself, causing a defect of the medial orbital rim. Alternatively it can pass through the ethmoid complex, leading to hypertelorism (Kawamoto 1976; Bodin et al. 2006) (Fig. 21.27).

Photograph of patient with number 11 Tessier cleft. This cleft crosses the medial portion of eyelid and eyebrow bilaterally toward the frontal hair line
21.13 Rare Facial Cleft Number 12
The number 12 cleft represents the cranial extension of the number 2 cleft. It passes either through the frontal process of the maxilla or between this process and the nasal bone, with continuation of the bony defect seen in its facial counterpart. Due to increase in the width of the ethmoid sinuses, hypertelorism is a hallmark characteristic. The olfactory organs and cribriform plate are spared. The overlying soft tissue may include brow irregularities immediately lateral to its medial edge (Fig. 21.28).

Photograph of a patient with isolated number 12
21.14 Rare Facial Cleft Number 13:
Displacement of an otherwise uninterrupted medial brow is a characteristic of a number 13 cleft. The underlying skeletal anomaly is consistent with the facial number 1 cleft: the cleft may extend between the nasal bone and frontal process of the maxilla. Cranially, the cleft passes through the cribriform plate, widening the olfactory grooves. The concurrent presence of a frontal encephalocele can cause inferior displacement of the cribriform plate as well. The ethmoids may also be increased in their transverse dimension. Collectively, these skeletal components result in hypertelorism, and in the bilateral form the hypertelorism can be remarkable (Kawamoto 1976; Gargano et al. 2015) (Fig. 21.29 and 21.30).

Photograph of a patient with number 1–13 Tessier cleft

Photographs of patients with 2–12 and 1–13 Tessier clefts. This combination brings the most severe forms of hypertelorism
21.15 Rare Facial Cleft Number 14
The zero cleft continues onto the central forehead as the number 14 cleft, which has a variety of presentations. When tissue agenesis is pathogenetically implicated, clinical manifestations can include hypotelorism that is defined by severe underdevelopment of midline frontal and orbital structures, such as cyclopia, ethmocephaly, cebocephaly, and a single central orbit. In these circumstances, the brain can be significantly hypoplastic in its development, resulting in holoprosencephaly and microcephaly. As Kawamoto describes, the more severe the brain deformity, the more severe the facial appearance, and the more limited life expectancy and meaningful neurologic function (Kawamoto 1976). Paradoxically, the 14 cleft may also be characterized by hypertelorism. Improper migration of the frontonasal process, a bifid cranium, or the presence of a frontal and ethmoid encephalocele may contribute to arrested medial migration of the embryological orbits and an ultimate lateralized position. An enlarged or a duplicated crista galli, widened olfactory grooves, inferiorly displaced cribriform plates, and enlarged ethmoids are also characteristic. The cranial sequelae are wide bossed forehead with midline furrow, depressed nasal root, protrusion of the lateral forehead, or encephalocele (Nam and Kim 2014; Pidgeon et al. 2014; Raposo-Amaral et al. 2011) (Fig. 21.27).
21.16 Rare Facial Cleft Number 30
Cleft of the lower lip and mandible may fall on the same axis as 0 cleft and involve the tongue, hyoid bone, and midline structure of the neck down to the sternum but mandibular involvement classified as distinct 30 cleft by Tessier (Kececi et al. 1994; Morioka et al. 2003; Bhattacharyya et al. 2012) (Fig. 21.31).

Patients with number 30 Tessier cleft
21.17 Treatment
21.17.1 Nonoperative Management
Definitive management of rare craniofacial clefts is surgical in nature. Nonsurgical care of patients with rare craniofacial clefts is generally supportive in nature and helps facilitate successful surgical outcomes. Under ideal circumstances, patients are treated in a multidisciplinary craniofacial clinic with close coordination among all involved caregivers.
21.17.1.1 Feeding and Nutrition
In the immediate postnatal period, rare facial clefts that involve the upper lip and/or palate may pose challenges to feeding and potentially contribute to dehydration and malnutrition. Parents must be provided with education and guidance on techniques and materials that can facilitate adequate oral feeds. Consultation with an occupational therapist who is experienced in the management of children with facial clefts may be indicated. This support should coincide with a larger more holistic approach to providing longitudinal psychosocial support to patients and their families.
21.17.1.2 Neurological Assessment
Children with rare craniofacial clefts with any suggestion of encephalocele, holoprosencephaly, or other structural cranial deformity should undergo neurological and neurosurgical assessment. These evaluations should include radiographic examination of the craniofacial skeleton via CT scan and of the brain via MRI.
21.17.1.3 Dentistry/Orthodontia
Virtually all patients with rare craniofacial clefts will require attention from a pediatric dentist and an orthodontist during their growth and development. Orthodontic treatment should be tailored to each individual’s needs, as hypodontia, alveolar misalignment, malocclusion, and even orthodontic sequelae of surgical interventions can have aesthetic and functional consequences upon occlusal relationships.
21.17.1.4 Speech
Cleft patients may have difficulties with speech stemming from structural abnormalities or from learned misarticulations. The speech pathologist plays a critical and ongoing role in identifying undesirable speech patterns early in language development, providing speech therapy, and monitoring speech patterns throughout childhood and young adulthood.
21.17.2 Operative Management
Due to the wide breadth of clinical diversity that is inherent to the rare craniofacial clefts, it is impossible to define specific algorithms or protocols for any one specific cleft entity. Tessier’s ordered classification system, however, allows us to draw upon well-established basic principles of cleft, craniofacial, and general plastic surgery to address the complex defects we encounter in these patients. Below is first a summary of general approaches to surgical management, followed by some cleft-specific considerations. While by no means comprehensive, this overview provides a template for developing surgical treatment plans.
The rare facial cleft number 1 and number 2 require a nasal reconstruction with local flaps. We have been using a nasal reconstruction in two or three stages using a paramedian flap. Nasal lining is usually required with folded paramedian flap or local transpositioning flap. Costal and conchal cartilages are used for adding nasal support to the reconstructed nose (Tessier 1976; Kawamoto 1976; Ozek et al. 2001; Tiwari et al. 1991; da Silva et al. 2010).
The rare facial cleft number 3 is one of the most challenging owing to magnitude of soft-tissue distortion. We aim to construct the lower eyelid and lacrimal system offering ocular protection. The alar base and the lip should be repositioned by enhancing the vertical dimension of the paranasal region. Tessier described a Z-plasty in the medial canthi, positioning one arm of the flap to correct the medial canthi and the other one to fill the vertical dimension of the nose (Tessier 1976). The medial canthi should be elevated by using the contralateral side as a template and a myocutaneous flap can be needed to fill the defect created by the elevation of the medial canthi. The cleft lip can be approached as a common unilateral cleft deformity. The mobilization of the distorted alar base is technically challenging. Glabelar or paramedian forehead flaps can be used to accomplish this task. Kawamoto has used a Z-plasty to gain length of vertical paranasal dimension, and prefer a cheek rotation flap in more severe cases. Elongation of the ala was done by complete degloving of the nose and mucosal lining on that side through an oral vestibular incision. The authors described a detailed approach for each specific spectrum of Tessier number 3 (Allam et al. 2014). We have proposing the unilateral orbital box osteotomy to lift the affected orbit, and completely release the periorbital contents and herniation into the cleft and bone graft in the cleft region and orbital rim. In addition, we also advocate a complete undermining of the medial canthi ligament in the affect side associated with a medial canthopexy with wires. This operation corrects the vertical orbital dystopia commonly found in these patients.
21.18 The Rare Facial Cleft Number 4
Multiple surgical stages are necessary to accomplish a successful clinical rehabilitation in a rare facial cleft number 4. As the oro-ocular distance is deficient, one should aim to construct this dimension by establishing its normal length. Corneal protection is urgent as corneal exposure may jeopardize visual acuity. The patient is usually treated early in life. Tessier, in the late 1970s, described a multiple Z-plasty to approach and reallocate tissue in the clefting region (Tessier 1976). This technique was used by many surgeons as a protocol to approach the number 4. We discard tissue between the philtrum and the cleft edge, offering an aesthetic pleasing lip reconstruction. Instead of sparing tissue one has to discard tissue to obtain better scar positioning into the aesthetic lip unit. This maneuver “discarding tissue where the tissue is already missing” may sound odd. However it is a necessary trade-off to obtain a long-term satisfactory outcome by avoiding the scars crossing the delineations of facial subunits. Alonso has described an approach respecting the aesthetic subunits of the face by using a medially based upper eyelid myocutaneous flap to construct the lower eyelid and lift the medial canthi to the same level (or slightly higher) using the contralateral side as a template. Bone graft has been used primarily or secondarily (Alonso et al. 2008). Millard rotation-advancement principle is used to repair the lip by discarding tissue and placing the scars in the facial subunits (Tessier 1976; Kawamoto 1976; Allam et al. 2014; Kale and Pakhmode 2000; Akoz et al. 1996).
21.19 The Rare Facial Cleft Number 5
Similarly to other oblique facial cleft, corneal exposure should be carefully managed. Radiological imaging is necessary to rule out encephalocele and to define orbital and cranial base anatomy; however soft tissue is approached first, early in life. Z-plasty is usually chosen along the cleft margins and in close proximity to the lower eyelid. Upper eyelid to lower eyelid transpositioning flap can be performed associated to lateral canthopexy as the lateral canthi is usually off. Bone graft can be used in the orbital floor, but in cases of microphthalmia it is extremely difficult to mobilize forward the eyeball as most of the cases there is a certain degree of enophthalmia (Tessier 1976; Kawamoto 1976; Galante and Dado 1991; da Silva et al. 2009).
21.20 The Rare Facial Cleft Number 6
Patients of isolate number 6 present a face that is quite similar to patients with Treacher Collins syndrome. The key point on number 6 is to deal with coloboma, and expand the midface that is tight in all cases. Correction of the coloboma can be accomplished with a combination of Z-plasty and upper to lower eyelid transpositioning flaps. The expansion of craniofacial skeleton is usually performed with parietal bone grating to the zygoma and zygomatic arch. In a second stage, soft-tissue expansion is accomplished using free fat grafting from abdomen, gluteal or thigh regions. Low quantity of free fat, less than 10 cc, is harvested in these regions and injected carefully in the zygomatic region using a 1 cc syringe. The systematization of the technique was previously described in Parry Romberg Syndrome and Hemifacial Microsomia by our group Denadai et al. 2016, 2017, Raposo-Amaral et al. 2013, Plomp et al. 2016.
21.21 The Rare Facial Cleft Number 7
Initial macrostomia repair should not only match the transverse length and wet/dry vermilion orientation of the lip on the uninvolved side, but also reorient the zygomaticus major, risorius, and depressor angulioris muscles to create the absent modiolus. Straight-line and z-plasty skin closure are both acceptable. Microtia repair using either autologous rib cartilage, porous polyethylene implants, or osseointegrated prostheses are all well described.
The technique and timing of skeletal reconstruction of the deficient mandible depend on the degree of hypoplasia, mandibular growth, and secondary effects on the maxilla. Options include rib grafting for the congenitally absent mandible, distraction osteogenesis for the significantly shortened mandibular body or ramus, and conventional orthognathic correction for skeletally mature patients with mandibular asymmetry and malocclusion. Free vascularized bone grafts have also been described for mandibular agenesis (Tessier 1976; Kawamoto 1976; Woods et al. 2008).
21.22 The Rare Facial Cleft Number 8
Soft-tissue reconstruction involves excision of the cleft tissue and repair with tarsopalpebral flaps and laterally based cutaneous flaps. A lateral canthoplasty is performed and covered with adjacent tissue.
21.23 Median and Paramedian Clefts: 0–14, 1–13, 2–12
The rare facial clefts from 8 to 14 are those whose cleft affects all structures above the orbit leading to symmetric and asymmetric hypertelorbitism. Surgical treatment depends on the extent of the deformity. Treatment of the facial and cranial components should be staged, starting with reconstruction of the cleft lip deformity at approximately 3 months of age according to standard cleft lip repair principles. Subsequent repair of the nose can be accomplished with a combination of local and regional tissue advancement, rotation, and transposition flaps in combination with composite or cartilaginous auricular grafts for support. More complex reconstructions are necessary in clefts with cranial extension that affect the orbits and forehead, following the guidelines for management of hypertelorbitism with or without encephalocele as described above. When there is significant deformity of the nose requiring an extensive amount of soft tissue for coverage, initial correction of the hypertelorbitism may provide additional mobile tissue to recruit for nasal reconstruction.
Once the patient presents a mild-to-severe interorbital distance according to Tessier classification, the key point is to medialize the orbit to allow nasal and further soft-tissue reconstruction and refinements.
The current techniques of hypertelorbitism are well described in the literature as well as the best age to perform the surgery to avoid long-term relapse of the orbit positioning (Raposo-Amaral et al. 2011).
In general, adult patients with adequate occlusion underwent an orbital box osteotomy procedure. Skeletally immature patients and patients with rare craniofacial clefts characterized by an inverted-V maxillary morphology underwent a facial bipartition procedure.
21.24 Box Osteotomy
Intracranial and extracranial approaches were used to make orbital box osteotomies. Coronal and gingivobuccal sulcus incisions were used to gain subperiosteal exposure of the frontal bones, orbits, and midface. A bifrontal craniotomy was performed. The interdacyron distance was measured with calipers. Circumferential orbital osteotomies were made with great care to preserve the integrity of the medial canthal tendons. The zygomaticomaxillary and nasomaxillary buttresses were also cut. The planned central frontoethmoidal segment osteotomy was marked and cut with a reciprocating saw, following adequate intracranial midline dissection and retraction. After removal of the median segment, the orbits were translocated medially. The nasofrontal processes of the maxilla were fixed with wires, and the vertical buttresses were rigidly fixed with titanium plates and screws. A pericranial flap was raised and sutured down to the cranial base in the midline before closure (Raposo-Amaral et al. 2011) (Fig. 21.32).

Schematic drawing of a box osteotomy to medialize the orbits and corrects hypertelorism
21.25 Facial Bipartition
For the facial bipartition, pterygomaxillary, septal, and median palatal osteotomies were added to the bone cuts described above to allow complete midface mobilization. Rowe disimpaction forceps were used to downfracture the midface. A wedge of central nasal, frontal, and ethmoid bone was removed, and the hemifacial segments were rotated toward the midline. Preoperative vertical orbital discrepancies were corrected with asymmetric wedge removal. The nasofrontal processes and the lateral aspects of the bipartition halves were fixed to the zygomatic processes with wire. The inferomedial aspects of the bipartition halves were rigidly fixed to one another with plates and screws. Autologous bone grafts were placed at the advanced portions of the lateral orbital rims and zygomatic arches. Where indicated, a cantilever autologous nasal bone graft was placed and rigidly secured (Raposo-Amaral et al. 2011) (Fig. 21.33).

Schematic drawing of facial bipartition osteotomy
21.26 The Rare Facial Cleft Number 10
Patients with gigantic encephalocele, feature commonly found in the number 10 cleft, can be corrected by exenteration of abnormal nonfunctioning brain and autologous bone graft from the parietal region. These procedures can be previously or simultaneously performed to the orbital medialization. After correcting the hypertelorbitism with an asymmetric medial wedge resection, soft-tissue reconstruction using local flaps to lengthen the medial paranasal dimension and to allow medial canthi upward mobilization is fundamental. This can be accomplished with transpositioning flaps. The challenge in this treatment relies on those number 10 patients with complete absence of the eyebrow and inverted eyelash (the upper eyelash with a downward rotation and lower eyelash with an upward rotation). This characteristic can cause corneal irritation in some patients. These distorted eyelash orientations are very difficult to correct.
Similar line of thinking should be used when treating the Tessier 1–13 cleft as similar soft-tissue malformations may be found.
The Tessier number 0–14 is the most common and encephalocele may be accompanied. Patients with complete nasal medial separation and encephalocele as a consequence of a wide hypertelorbitism should be carefully planned. The nose is usually constructed with converse scalping flaps; thus if the encephalocele is needed to be correct at very early age, the incisions should be carefully planned to not jeopardize the vascular pedicle of the scalping flaps based on the superficial and deep temporal arteries. All technical details of bony orbital medialization were extensively described in the literature. Local transpositioning flaps and Z-plastys can be used to adjust the nasal morphology and in most cases to descend the alar base unilaterally or bilaterally to a more gracious positioning (Raposo-Amaral et al. 2017).
21.27 Complications
There are a wide range of complications that may arise from the treatment of these patient populations. Complications may be related to either bone operation or soft-tissue operation. The magnitude of bony movements especially those to treat patients with rare facial cleft above the orbit requires an intracranial route to offer access for the orbital roof cuts and craniofacial disjunction when needed. As a consequence, several complications can be shown that generally occur as a result of severe blood loss or related to the communication between the oral nasal cavity and the anterior cranial fossa. As a result, cerebrospinal fluid leak may occur leading to ascending infection and postoperative meningitis that usually correlated with high morbidity/mortality rates. However, these complications that can be frequently seen in the treatment of syndromic craniosynostosis (Dunaway et al. 2012) are less frequent in the treatment of rare facial cleft mainly because of the normal intracranial pressure seen in these patients and no requirement to bring the face forward. Partial bone flap devascularization may also occur and require second operation for removal of the necrotic bone. Accidental fractures can be seen in patients whose bone cuts are not completely performed especially at the level of the pterygomaxillary junction. During the craniofacial disjunction the fracture can run toward the cranial base causing a CFS or an encephalocele. Orbital translocation carries the additional risk of optic nerve injury, as well as inadvertent medial canthal avulsion. Frontoethmoidal dissection and resection carry the risk of olfactory sensory loss. Soft-tissue complications are more frequently seen than bony complication. Local flaps can be necrotic compromising the postoperative result. Suboptimal result in management of nasal reconstruction and lower eyelid reconstruction and innumerous scars crossing the face can also be seen and this may be a challenge to correct in a later postoperative period.
21.28 Arhinia and Hemi-Arhinia
Congenital absence of nose termed total arhinia have possible genetic component. Gene candidates have been tested; however extended genetic analysis has not been performed in a large series of patients to determine common sporadic mutations. Thus, total arhinia has not been associated to any syndrome and most of the patients described in the literature are sporadic reports of isolated cases in different regions of the world. These patients present with different clinical characteristics and all scientific efforts to classify and grade this striking craniofacial deformity are of paramount importance.
Allam et al. (2011) classified the craniofacial dysplasia into three main division and total arhinia is identified as median craniofacial hypoplasia, described as tissue deficiency or agenesis. A subclassification also proposed by the authors divided the median craniofacial hypoplasia into four subcategories: (1) holoprosencephalic spectrum; (2) median cerebrofacial hypoplasia (lobar brain); (3) median facial hypoplasia; and (4) microforms of median facial hypoplasia. Interestingly, Binder syndrome known as maxillo-nasal dysplasia was included in the last division. Thus, a wide spectrum of bony and soft-tissue deformity makes a surgical algorithm difficult to be developed and proposed (Fig. 21.34).

Photographs of patients with facial agenesis, hemi-ahrinia and total ahrinia
21.29 Summary
This chapter highlighted some of very rare clinical examples of Tessier rare facial clefts. Some cases are so unique that similar clinical features may not be seen again in a lifetime.
References
Abdollahifakhim S, Shahidi N, Bayazian G. A bilateral tessier number 4 and 5 facial cleft and surgical strategy: a case report. Iran J Otorhinolaryngol. 2013;25(73):259–62.
Agrawal K, Panda KN, Prasad S. Isolated Tessier no. 1 cleft of the nose. Ann Plast Surg. 1998;41(3):311–3.
Akoz T, Erdogan B, Gorgu M, Kutlay R, Dag F. Bilaterally involved Tessier no. 4 cleft: case report. Cleft Palate Craniofac J. 1996;33(3):252–4.
Allam KA, Wan DC, Kawamoto HK, Bradley JP, Sedano HO, Saied S. The spectrum of median craniofacial dysplasia. Plast Reconstr Surg. 2011 Feb;127(2):812–21.
Allam KA, Lim AA, Elsherbiny A, Kawamoto HK. The Tessier number 3 cleft: a report of 10 cases and review of literature. J Plast Reconstr Aesthet Surg. 2014;67(8):1055–62.
Alonso N, Freitas RS, de Oliveira e Cruz GA, Goldenberg D, DallOglio Tolazzi AR. Plast Reconstr Surg. 2008;122(5):1505–13.
Bhattacharyya NC, Kalita K, Gogoi M, Deuri PK. Tessier 30 facial cleft. J Indian Assoc Pediatr Surg. 2012;17(2):75–7.
Bodin F, Salazard B, Bardot J, Magalon G. Craniofacial cleft: a case of Tessier no. 3, 7 and 11 cleft. J Plast Reconstr Aesthet Surg. 2006;59(12):1388–90.
Boo-Chai K. The transverse facial cleft: its repair. Br J Plast Surg. 1969;22:119–24.
Burget GC, Menick FJ. Nasal reconstruction: seeking a fourth dimension. Plast Reconstr Surg. 1986;78(2):145–57.
David DJ, Moore MH, Cooter RD, Chow SK. The Tessier number 9 cleft. Plast Reconstr Surg. 1989;83(3):520–7.
Denadai R, Raposo-Amaral CA, Pinho AS, et al. Predictors of autologous free fat graft retention in the management of craniofacial contour deformities. Plast Reconstr Surg. 2017;140:50e–61e.
Denadai R, Raposo-Amaral CA, Buzzo CL, et al. Isolated autologous free fat grafting for management of facial contour asymmetry in a subset of growing patients with craniofacial microsomia. Ann Plast Surg. 2016;76:288–94.
Dumortier R, Delhemmes P, Pellerin P. Bilateral Tessier no. 9 cleft. J Craniofac Surg. 1999;10(6):523–5.
Dunaway DJ, Britto JA, Abela C, Evans RD, Jeelani NU. Complications of frontofacial advancement. Childs Nerv Syst. 2012;28(9):1571–6.
Fuente-del-Campo A. Surgical correction of Tessier number 8 cleft. Plast Reconstr Surg. 1990;86(4):658–61. discussion 62-3
Galante G, Dado DV. The Tessier number 5 cleft: a report of two cases and a review of the literature. Plast Reconstr Surg. 1991;88(1):131–5.
Garg A, Goyal S. Tessier number 5 cleft. Indian Pediatr. 2009;46(10):907.
Gargano F, Szymanski K, Bosman M, Podda S. Tessier 1-13 atypical craniofacial cleft. Eplasty. 2015;15:ic32.
Gawrych E, Janiszewska-Olszowska J, Chojnacka H. Tessier type 3 oblique facial cleft with a contralateral complete cleft lip and palate. Int J Oral Maxillofac Surg. 2010;39(11):1133–6.
Gonzalez-Ulloa M. Restoration of the face covering by means of selected skin in regional aesthetic units. Br J Plast Surg. 1956;9(3):212–21.
Gonzalez-Ulloa M. Selective regional restoration by means of esthetic units. Prensa Med Mex. 1958;23(2):68–73.
Horgan JE, Padwa BL, LaBrie RA, Mulliken JB. OMENS-plus: analysis of craniofacial and extracraniofacial anomalies in hemifacial microsomia. Cleft Palate Craniofac J. 1995;32(5):405–12.
Karfik V. [Proposed classification of rare congenital cleft defects of the face]. Rozhl Chir. 1966;45:518–22.
Kale SM, Pakhmode VK. Bilateral Tessier no. 4 facial cleft with left eye anophthalmos: a case report. J Indian Soc Pedod Prev Dent. 2000;18(3):87–9.
Kawamoto HK Jr. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg. 1976;3(4):529–72.
Kececi Y, Gencosmanoglu R, Gorken C, Cagdas A. Facial cleft no. 30. J Craniofac Surg. 1994;5(4):263–4.
Laure B, Picard A, Bonin-Goga B, Letouze A, Petraud A, Goga D. Tessier number 4 bilateral orbito-facial cleft: a 26-year follow-up. J Craniomaxillofac Surg. 2010;38(4):245–7.
Lee HM, Noh TK, Yoo HW, Kim SB, Won CH, Chang SE, et al. A wedge-shaped anterior hairline extension associated with a tessier number 10 cleft. Ann Dermatol. 2012;24(4):464–7.
Ligh CA, Swanson J, Yu J, Samra F, Bartlett SP, Taylor JA. A morphological classification scheme for the mandibular hypoplasia in Treacher Collins syndrome. Plast Reconstr Surg. 2015;136(4 Suppl):46.
Morioka D, Simic R, Vlahovic A, Kravljanac D. Tessier 30 median mandibular cleft associated with lower lip hemangioma. Plast Reconstr Surg. 2003;112(3):935.
Nam SM, Kim YB. The Tessier number 14 facial cleft: a 20 years follow-up. J Craniomaxillofac Surg. 2014;42(7):1397–401.
Nguyen PD, Caro MC, Smith DM, Tompson B, Forrest CR, Phillips JH. Long-term orthognathic surgical outcomes in Treacher Collins patients. J Plast Reconstr Aesthet Surg. 2016;69(3):402–8.
Ozek C, Gundogan H, Bilkay U, Cankayali R, Guner U, Gurler T, et al. Rare craniofacial anomaly: Tessier no. 2 cleft. J Craniofac Surg. 2001;12(4):355–61.
Pidgeon TE, Flapper WJ, David DJ, Anderson PJ. From birth to maturity: midline tessier 0-14 craniofacial cleft patients who have completed protocol management at a single craniofacial unit. Cleft Palate Craniofac J. 2014;51(4):e70–9.
Pittet B, Jaquinet A, Rilliet B, Montandon D. Simultaneous correction of major hypertelorism, frontal bone defect, nasal aplasia, and cleft of the upper lip (Tessier 0-14). Plast Reconstr Surg. 2004;113(1):299–303.
Plomp RG, van Lieshout MJ, Joosten KF, Wolvius EB, van der Schroeff MP, Versnel SL, et al. Treacher Collins syndrome: a systematic review of evidence-based treatment and recommendations. Plast Reconstr Surg. 2016;137(1):191–204.
Poon CC, Meara JG, Heggie AA. Hemifacial microsomia: use of the OMENS-plus classification at the Royal Children's Hospital of Melbourne. Plast Reconstr Surg. 2003;111(3):1011–8.
Portier-Marret N, Hohlfeld J, Hamedani M, de Buys Roessingh AS. Complete bilateral facial cleft (Tessier 4) with corneal staphyloma: a rare association. J Pediatr Surg. 2008;43(10):e15–8.
Presti F, Celentano C, Marcazzo L, Dolcetta G, Prefumo F. Ultrasound prenatal diagnosis of a lateral facial cleft (Tessier number 7). Ultrasound Obstet Gynecol. 2004;23(6):606–8.
Raposo-Amaral CE, Raposo-Amaral CM, Raposo-Amaral CA, Chahal H, Bradley JP, Jarrahy R. Age at surgery significantly impacts the amount of orbital relapse following hypertelorbitism correction: a 30-year longitudinal study. Plast Reconstr Surg. 2011;127(4):1620–30.
Raposo-Amaral CE, Denadai R, Camargo DN, et al. Parry-Romberg syndrome: severity of the deformity does not correlate with quality of life. Aesthetic Plast Surg. 2013;37:792–801.
Raposo-Amaral CE, Denadai R, Ghizoni E, et al. Surgical strategies for soft tissue management in hypertelorbitism. Ann Plast Surg. 2017;78:421–27.
da Silva FR, Alonso N, Shin JH, Busato L, Ono MC, Cruz GA. Surgical correction of Tessier number 0 cleft. J Craniofac Surg. 2008;19(5):1348–52.
da Silva FR, Alonso N, Shin JH, Busato L, DallOglio Tolazzi AR, de Oliveira e Cruz GA. The Tessier number 5 facial cleft: surgical strategies and outcomes in six patients. Cleft Palate Craniofac J. 2009;46(2):179–86.
da Silva FR, Alonso N, Busato L, Ueda WK, Hota T, Medeiros GH, Kunz RT. Oral-nasal-ocular cleft: the greatest challeng among the rare clefts. J Craniofac Surg. 2010;21(2):390–5.
Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg. 1976;4(2):69–92.
Tiwari P, Bhatnagar SK, Kalra GS. Tessier number 2 cleft, a variation. Case report. J Craniomaxillofac Surg. 1991;19(8):346–7.
Tokioka K, Nakatsuka T, Park S, Okouchi M, Aiba E. Two cases of Tessier no. 4 cleft with anophthalmia. Cleft Palate Craniofac J. 2005;42(4):448–52.
Wenbin Z, Hanjiang W, Xiaoli C, Zhonglin L. Tessier 3 cleft with clinical anophthalmia: two case reports and a review of the literature. Cleft Palate Craniofac J. 2007;44(1):102–5.
Woods RH, Varma S, David DJ. Tessier no. 7 cleft: a new subclassification and management protocol. Plast Reconstr Surg. 2008;122(3):898–905.
Acknowledgments
The authors thank the plastic surgeon and artist Jorge Vitale for the drawings given to Prof. Dr. Cassio Raposo do Amaral.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Raposo-Amaral, C.E., Jarrahy, R., Lim, R., Alonso, N. (2018). The Rare Facial Cleft. In: Alonso, N., Raposo-Amaral, C. (eds) Cleft Lip and Palate Treatment. Springer, Cham. https://doi.org/10.1007/978-3-319-63290-2_21
Download citation
DOI: https://doi.org/10.1007/978-3-319-63290-2_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-63289-6
Online ISBN: 978-3-319-63290-2
eBook Packages: MedicineMedicine (R0)