
Abstract
Natural Killer (NK) cells have long been known for their potent cytokine producing and cytotoxic capacities against transformed or infected cells. In particular, they are recognized to play an early role in immune surveillance (detection and elimination) of developing tumors (especially hematological cancers) and metastasis to distant organs. NK cells are also the first identified protagonists of a new emerging family of innate lymphoid cells (ILCs) that regulate several physiological processes such as immunity, tissue remodeling, and metabolism to maintain body integrity. NK cells express an array of receptors allowing them to sense malignant cells. NK cells exert their anti-tumor function mainly through the direct killing of the cancer cells and the secretion of the anti-tumor cytokine IFN-γ. Recently, it has been suggested that ILC subsets other than NK cells could also respond to cancer cells and play a role in anti-tumor immunity. In this chapter, we will detail the various mechanisms by which NK cells recognize and eliminate cancer cells. Moreover, we will revisit the role of NK cells in cancer immunosurveillance in light of the recent findings on the other ILC family members.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Natural killer cells
- Innate lymphoid cells
- Cancer
- Immunosurveillance
- Tumor
- Immunoediting
- Innate receptors
- Cytotoxicity
1 Introduction
T cells screen the body for antigens presented on Major Histocompatibility Complex (MHC) molecules and have gained a central position in cancer immunotherapy for their ability to mount a response specifically directed against the tumor [1]. Indeed, tumor-infiltrating cytotoxic CD8+ T cells and Th1-polarized CD4+ T cells are usually associated with better prognosis [2]. However, conventional T cells are not the sole mediators of anti-cancer immunity. Several immune cells are able to detect the earliest signs of malignant transformation despite their lack of receptors specific for antigenic peptides. Innate immune cells provide immediate protection and are therefore likely to play a crucial role in the early stages of tumorigenesis [3, 4]. Innate immune surveillance of cancers involves myeloid cells such as macrophages [5] and neutrophils [6], unconventional T cells bearing invariant or semi-invariant T cell receptors (TCR) such as Natural Killer T (NKT) cells [7] and γδ T cells [8], and a growing family of lymphocytes called innate lymphoid cells (ILCs) [9].
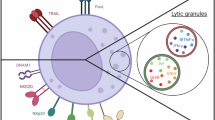
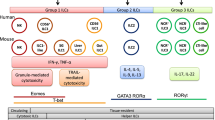
Innate lymphoid cells are characterized by a lymphoid morphology, the lack of RAG-rearranged antigen-specific receptors, and the absence of surface markers of the dendritic or myeloid lineages [10]. ILCs constitute a heterogeneous population of cells that share a common origin [11]. They have been divided between killer and helper-like ILCs [12] (Fig. 9.1). Natural Killer (NK) cells are the best-known cytotoxic members of the ILC family. These killer ILCs may be seen as the innate equivalents of CD8+ cytotoxic T cells, housing similar cytotoxic granules and contents, whereas helper-like ILCs mirror the activity of CD4+ helper T cells. Moreover, ILCs have been split into 3 categories based on their cytokine expression profiles and their specific transcription factor requirement. Group 1 ILCs are defined by their ability to produce IFNγ and depend on the Th1 cell-associated transcription factor T-bet. This group encompasses NK cells and helper-like type 1 ILCs (ILC1s). NK cells and ILC1s are developmentally distinct [15]. However, these two innate cell populations are not always easy to discriminate because they share many phenotypic markers [16]. Importantly, expression of the transcription factor Eomes is considered a hallmark of NK cells that distinguishes them from other group 1 ILCs [17], with the exception of salivary gland ILC1s [18]. Group 2 ILCs rely on the transcription factors GATA3 and RORα for their development and function and produce type 2 cytokines, mainly IL-5 and IL-13. Finally, group 3 ILCs are defined by their ability to produce IL-17 and/or IL-22 and their dependency on the transcription factor RORγt for their development and function.

The ILC family members and their putative role in cancer. The ILC family comprises killer blood-circulating ILCs and helper-like tissue-resident ILCs. ILCs are further divided into three groups according their signature cytokines and their transcription factor requirement. The main characteristics of the three classes of ILCs are represented here, as well as their anti-tumor activity. Question marks (?) indicate possible roles that have not been demonstrated yet. Direct killing of tumor cells is well established for NK cells but only few studies have described ILC1-mediated cytotoxicity through the lytic granule pathway [13] or through TRAIL in the case of liver ILC1s [14]. Of note, only the protective roles of ILCs against tumors are depicted here
NK cells constitute the first ILC subset to be discovered and were initially identified for their spontaneous cytotoxic activity [19]. NK cells are often characterized as CD3 negative cells expressing CD56 in humans, NK1.1 in mice, and NKp46 in both species [20]. Of note, such phenotypic definition may include other ILC subsets. Moreover, NK cells are not a homogeneous population and can be divided into different subtypes [21]. In humans, the two main NK cell subsets are CD56brightCD16− NK cells which are important cytokine producers and are abundant in lymph nodes, and CD56dimCD16+ NK cells which represent the main NK cell population in the blood and are highly cytotoxic [22]. NK cell-depleting antibodies as well as mouse models displaying NK cell deficiencies have been fundamental for the demonstration of NK cell anti-cancer activity (Table 9.1). NK cells harbor many surface receptors allowing them to distinguish malignant-transformed cells from healthy cells. Moreover, NK cells are endowed with potent cytotoxic functions and are a major source of the anti-tumor cytokine, IFN-γ. These characteristics make NK cells key protagonists of innate immunosurveillance of cancers [29]. In neat opposition to the numerous reports on NK cells in cancer, the possible pro- or anti-tumor functions of helper-like ILCs remain largely unexplored [30]. Nonetheless, recent work suggested an involvement of tissue-resident type 1-like ILCs in the immune surveillance of spontaneous tumors [13]. Furthermore, various reports described tumor-suppressive activities of ILC2s [31, 32] and ILC3s [33, 34].
In this chapter, we will review the different mechanisms by which ILCs detect malignant cells and prevent cancer development. Most of our current knowledge is restricted to NK cells which constitute the prototypical anti-cancer ILC subset. Therefore, we will predominantly focus on NK cells while introducing emerging data on helper-like ILCs. It should be noted that both pro- and anti-tumor activities of helper-like ILCs have been described [35] but herein we will only discuss their potential host protective functions.
2 Surface Receptors Involved in Tumor Recognition by ILCs
Innate cells express a fixed set of germline-encoded receptors that allow the recognition of foreign, aged and damaged cells [36]. Activating and inhibitory surface receptors are crucial for the regulation of NK cell functions and some of these receptors are also expressed on helper-like ILCs subsets (Fig. 9.2). Furthermore, interactions with accessory cells, generally monocytes or dendritic cells (DCs), also stimulate NK cells to produce pro-inflammatory cytokines and potentiate their killing functions [37]. Historically, NK cells were described for their ability to kill tumor cells having down-regulated MHC class I molecules (MHC-I), a concept termed “missing-self” recognition [38]. Most cell types express self-peptide-MHC-I complexes on their surface but partial or complete loss of MHC-I expression is a common feature of cancer cells [39]. This phenomenon is often caused by CD8+ T cell-mediated immune pressure and renders tumor cells susceptible to NK cell-mediated cytotoxicity. Conversely, MHC-I molecules expressed at the surface of healthy autologous cells bind to inhibitory NK cell receptors and deliver negative signals thereby avoiding NK cell autoreactivity. Besides the “missing-self” recognition, the detection of stress-induced self-ligands expressed at the surface of damaged cells also promotes NK cell killing capacities. In fact, the outcome of NK cell interaction with a target cell is determined by the balance between inhibitory signals transmitted by NK cell receptor binding to self MHC-I and activating signals transmitted upon recognition of stress ligands at the surface of the target cell [40]. Activating NK cell receptors involved in the immunosurveillance of cancers include the natural cytotoxicity receptors (NCRs), NKG2D (also known as CD314 and KLRK1) and DNAM-1 (also known as CD226) [36]. Other receptors such as the low-affinity activating receptor FcγRIIIa (CD16) or the co-stimulatory molecules CD137, OX40, and GITR are promising clinical targets for their ability to mediate potent NK cell activation [41]. Nonetheless, these receptors have not been reported to play any role in the early detection of nascent tumor and therefore will not be discussed here.

Cell surface receptors involved in tumor cell recognition by ILCs. NK cells express an array of activating (green) or inhibitory (red) cell surface receptors. Some receptors are only expressed on activated NK cells (underline). The outcome of NK cell interactions with a target cell is determined by the balance between activating and inhibitory signals. Healthy cells express MHC-I molecules that engage NK cell inhibitory receptors whereas cancer cells down-regulate MHC-I molecules and/or express stress ligands recognized by NK cell activating receptors. An excess of activating signals over inhibitory signals leads to NK cell activation and the cytotoxicity of the target cell. Helper-like ILCs also express receptors that may regulate the sensing of tumor cells and some of these receptors have been found to regulate cytokine production by helper-like ILCs. DNAM-1 and PD-1 have recently been observed on mouse ILCs but their expression on human ILCs has not been investigated yet
There are three members of the NCR family: NKp46 (NCR1; CD335) is expressed in both mice and humans, whereas NKp44 (NCR2, CD336) and NKp30 (NCR3; CD337) are restricted to human NK cells [42]. Unlike NKp46 and NKp30, NKp44 is not detected on resting NK cells but is up-regulated after activation. NCR engagement triggers NK cell-mediated cytotoxicity and secretion of IFNγ [36]. Importantly, NCR expression is not restricted to NK cells and is shared with helper ILC1s [16], a subset of human ILC2s [43] as well as a subset of ILC3s named NCR+ ILC3s [10]. Engagement of NKp44 by tumor cells and tumor-associated fibroblast stimulates NCR+ ILC3 to release IL-8 and TNF [33]. Similarly, NKp30-mediated recognition of human tumor cell lines induces the NF-κB signaling pathway in ILC2s, leading to the production of IL-13 and other type 2 cytokines [43]. Whether NCRs also govern helper-like ILC1 recognition of malignant cells is yet to be demonstrated. NCR ligands on tumor cells have only been partially defined and those reported include NKp44L, HLA-B associated transcript 3 (BAT3), B7-H6, heparan sulfates, and proliferating cell nuclear antigen (PCNA) [42]. Curiously, PCNA differs from other NCR ligands as it does not stimulate but rather inhibits NK cell functions [44]. Interestingly, NCR genes encode different splice variants, some of them being immunosuppressive. A recent report suggested that the cytokine-defined microenvironment may influence NKp30 and NKp44 isoform expression profile in NK cells and that alternative splicing gives rise to inhibitory isoforms that dampen NK cell functions [45]. Furthermore, in gastrointestinal stromal tumors, predominant expression of the immunosuppressive NKp30c isoform over the immunostimulatory NKp30a and NKp30b isoforms is associated with reduced survival [46]. Remarkably, mice lacking NKp46 have been useful to define NKp46 involvement in the control of initial tumor growth [47] and the prevention of tumor metastasis [48] in vivo. Unfortunately, in vivo investigation of the other NCRs in tumor immunosurveillance is limited by the lack of mouse orthologs for NKp30 and NKp44.
NKG2D is a major NK cell activating receptor also expressed by some T cell subsets [36]. NKG2D recognizes several MHC-related ligands that are poorly expressed at the surface of healthy cells but are frequently up-regulated with the process of malignant transformation [49]. For instance, in non-transformed mouse or human cells, activation of the DNA-damage response induces the up-regulation of NKG2D ligands and enhances cellular sensitivity to NK cell killing [50]. NKG2D ligands are RAE-1α-ɛ, MULT1 and H60a-c in mice; and MICA-B and ULBP1–6 in humans. NKG2D engagement stimulates signaling cascades leading to cell activation, killing, and cytokine production. A pioneer study demonstrated that mouse tumor cell lines engineered to express high levels of NKG2D ligands and injected subcutaneously into syngeneic mice are rapidly rejected by conventional NK cells without a requirement for T and B cells [51]. Notably, the pivotal role of NKG2D in tumor immunosurveillance has been evidenced in mouse models of de novo tumorigenesis [52] and carcinogenesis [53]. In a model where expression of the Epstein–Barr virus transforming protein LPM1 in mouse B cells led to the development of B cell lymphomas, the arising lymphoma cells expressed ligands for NKG2D and were killed in vitro by NK cells [54]. However, in this model, T cells were the major effectors of immunosurveillance. It is possible that arising lymphomas may have developed escape mechanisms to circumvent NK cell anti-tumor activity in vivo. In fact, tumor progression is usually associated with an immunoediting process resulting in the emergence of malignant clones that are resistant to NK cell activity [55]. This has been illustrated in multiple myeloma where the transition from a pre-malignant to a malignant stage of the disease is associated with shedding of MICA from the surface of the tumor cells [56]. In addition to reducing NKG2D ligand surface density on tumor cells, this shedding process generates soluble ligands that down-regulate NKG2D expression on immune cells and promote tumor immune evasion [57]. Actually, chronic exposure to low-affinity surface-attached NKG2D ligands also leads to NK cell desensitization to both NKG2D-dependent and -independent pathways [58]. Surprisingly, instead of blocking tumor cell recognition, shedding of the high affinity NKG2D ligand MULT1 promotes tumor rejection by boosting NK cell effector functions [59]. It was suggested that MULT1 prevents immunosuppressive interaction with low-affinity ligands such as RAE-1 expressed in the tumor microenvironment and restores NK cell responsiveness.
DNAM-1 is an adhesion molecule expressed on NK cells and T cells that associates with the integrin LFA-1 and participates to the stabilization of the cytolytic synapse [60]. DNAM-1 recognizes a the family of nectin and nectin-like molecules initially identified for their role in cell-cell adhesion [61]. Nectin and nectin-like molecules have been involved in a wide range of biological processes and they notably regulate the immune functions of T cells, NK cells, and antigen presenting cells. In addition to enhancing NK cell adhesion and cytotoxicity, DNAM-1 promotes the secretion of IFN-γ. Moreover, DNAM-1 expression distinguishes two functional NK cell subsets in mouse [62]. Compared with DNAM-1− NK cells, DNAM-1+ NK cells have enhanced IL-15 signaling and produce higher levels of pro-inflammatory cytokines. Intriguingly, DNAM-1 is expressed at high levels on mouse liver ILC1s [17] and is also detected on early ILC precursors as well as ILC2 progenitors in the mouse bone marrow [63]. Still, the putative role of DNAM-1 in the regulation of helper-like ILC function remains to be investigated. DNAM-1 ligands, CD155 (also known as PVR) and CD112 (also known as nectin-2, PRR2, or PVRL2) are often over-expressed by solid and hematological malignancies [60]. Similarly to what has been described for NKG2D ligands, DNA-damage resulting from replication stress seems responsible for tumor cell expression of the DNAM-1 ligand CD155, and this pathway is dependent on ATM, an enzyme that senses double strand DNA breaks [64]. An interesting study performed in the Eμ-Myc mouse lymphoma model established that the DNA-damage response induces CD155 expression on early stage transformed B cells and thereby leads to spontaneous tumor regression that is partially DNAM-1-dependent [65]. Further strong evidence of DNAM-1 contribution to immunosurveillance comes from the observation of enhanced development of carcinogen-induced fibrosarcomas [66] as well as accelerated growth of spontaneous and transplantable tumors in DNAM-1-deficient mice [67, 68].
The variety of activating receptors expressed by NK cells is complemented by numerous inhibitory receptors that prevent the killing of healthy tissue. Receptors binding to self-MHC-I are responsible for the “missing-self” recognition. NK cell receptors of the KIR family in humans and of the Ly49 family in mice directly recognize MHC-Ia molecules [36]. Moreover, the CD94/NKG2A heterodimeric receptor is expressed in both species and binds to a peptide presented by the non-classical MHC molecule HLA-E in humans and Qa-1 in mice. Successful engagement of MHC-I by these receptors transmits an inhibitory signal that disrupts activating pathways. For instance, engagement of the inhibitory receptor KIR2DL2 blocks activating receptor clustering and induces actin remodeling and concomitant retraction from the target cell [69]. The importance of the missing-self recognition in NK cell-mediated immunosurveillance has been highlighted by the report of accelerated onset of carcinogen-induced sarcomas and spontaneous B cell lymphomas in mice expressing reduced levels of Ly49 inhibitory receptors [70]. So far, NK cells are considered the sole ILC subset mediator of the “missing-self” recognition. Notwithstanding, Ly49 receptors have been detected on the surface of other ILCs subsets [13, 16] but their function needs to be assessed.
Additional receptors may regulate ILC functions in the tumor microenvironment. The immune checkpoint molecules CTLA-4 and PD-1 have been found to hinder NK cell activity and constitute important clinical targets [41] but little is known about their role in innate immunosurveillance of cancer. Interestingly, mouse precursors for helper-like ILCs have been characterized by high expression levels of PD-1 [63]. Furthermore, few mature ILCs express PD-1, but they up-regulate this immune checkpoint molecule upon activation [71]. More investigation would be required to assess the role of PD-1 on ILCs within tumors. Besides, two receptors interacting with the nectin and nectin-like molecule family, TIGIT and CD96 (also known as TACTILE), have recently gained clinical interest for their potent inhibition of NK cell- and T cell-functions [72]. TIGIT and CD96 bind to CD155 and counterbalance DNAM-1-mediated activation of NK cells [60]. TIGIT inhibits mouse and human NK cell-mediated cytotoxicity [73, 74] and CD96 was shown to reduce mouse NK cell production of IFN-γ [75]. CD96-deficient mice display robust resistance to experimental lung metastasis and carcinogenesis [75], but the role of TIGIT in NK cell-mediated surveillance of cancers remains to be established.
3 Cytokines and Soluble Factors that Activate ILCs
Besides cell-to-cell interactions, ILCs integrate multiple signals provided by soluble mediators such as cytokines, alarmins, lipids, or hormones produced by epithelial, stromal, or myeloid cells [76] (Fig. 9.3). Tumor growth is likely to disturb the homeostasis of the surrounding tissue, leading to the release of cytokines and danger signals that might shape ILC-mediated immunosurveillance. Both NK cells and helper-like ILC1s are responsive to IL-12, IL-15, and IL-18, while ILC2s mainly respond to IL-25, IL-33, and TSLP; and ILC3s are activated by IL-23 and IL-1β. There is little direct evidence of the role of physiologically secreted cytokines on ILCs in cancer and most studies have rather investigated the effect of exogenous cytokine administration or used genetically manipulated tumors or mice. For example, a report described the resistance of transgenic mice over-expressing IL-15 to subcutaneously injected B16 melanoma cells lacking MHC-I [77]. In this study, the anti-tumor activity was maintained in the absence of CD4+ or CD8+ T cells, but protection was lost upon NK cell depletion with anti-asialo-GM1 antibodies. Moreover, IL-2, IL-12, and IL-18 promote NK cell-mediated control of experimental metastasis in mice [78, 79] and NK cells pre-activated with IL-12, IL-15, and IL-18 display sustained effector functions and delay the growth of MHC-I-deficient tumors in mice [80]. Intriguingly, a more recent study established that the IL-12-induced suppression of subcutaneous B16 melanoma tumors was mediated by a NKp46+NK1.1− ILC3 population identified using a fate-mapping reporter mouse strain for the transcription factor RORγt [34]. Finally, the expression of Toll-like receptors by NK cells and ILC3s [81, 82] might enable these cells to detect danger-associated molecular patterns (DAMPs) present in the tumor microenvironment. DAMP may induce NK cell activation either directly, or indirectly through the stimulation of accessory cells [37]. Interestingly, a recent study established that NK cells from TLR3-deficient mice are hyporesponsive to cytokine stimulation, a defect that is associated with increased experimental lung metastasis in these mice upon challenge with B16F10 melanoma cells [83].

Cytokines and membrane-bound ligands contribute to ILC activation. Cytokines such as IL-12, IL-15, and IL-18 produced by DCs and macrophages prime NK cells increase their capacities upon engagement of activating receptors. IL-12 and IL-18 also stimulate IFN-γ production by ILC1s. ILC2s respond to IL-25, IL-33, and TSLP. In addition, human ILC2s are activated upon NKp30 engagement, while human ILC3s respond to NKp44 stimulation. Moreover, ILC3s are activated by the cytokines IL-1β and IL-23
Notably, helper-like ILCs are mainly activated via soluble mediators while NK cell receptors represent a predominant pathway for NK cell activation. Nevertheless, cytokine-activated NK cells are more responsive to NK cell receptor signaling than resting NK cells. NK cell-activating cytokines are generally secreted by myeloid cells or dendritic cells (DCs) [84]. In fact, it has been suggested that naïve NK cells only acquire their full killing capacity following an interaction with DCs or macrophages termed “NK cell priming” [85], a process that is influenced by the commensal microbiota [86]. Of particular interest, similar synergy between cytokine-mediated activation and NCR signaling has recently been described for NCR+ ILC3s [87].
4 Direct Clearance of Cancer Cells by ILCs
The release of cytotoxic granules containing perforin and granzymes constitutes the main pathway by which NK cells exert their killing activity [88]. Perforin is a pore-forming protein that allows granzymes to enter into target cells, thereby triggering apoptosis [89]. Perforin protects mice against spontaneous lymphomas [90] and mouse studies have established the major contribution of perforin to NK cell-mediated rejection of tumor lacking MHC-I [91, 92], as well as NK cell-mediated control of metastasis [23] and protection against carcinogen-induced fibrosarcomas [93]. Granule-dependent cytotoxicity was originally thought to be a characteristic of NK cells distinguishing them from ILC1s [12]. However, it was recently demonstrated that a population of ILC1-like cells clearly distinct from conventional NK cells could also kill tumor cells in a perforin-dependent fashion [13]. Notably, this study used a spontaneous mouse mammary cancer model to suggest that ILC1-like cells, but not conventional NK cells, contribute to reduce tumor growth.
Fas ligand (FasL or CD95L) and TNF-related apoptosis inducing ligand (TRAIL) belong to the death receptor pathway and represent alternative mechanisms by which NK cells eliminate target cells [14, 94, 95]. The binding of Fas or TRAIL to their receptors (Fas and DR5 or DR4, respectively) triggers the activation of the common death signaling molecules FADD, caspase 8, and caspase 3, and leads to apoptosis [96]. The relevance of targeting Fas- and TRAIL-death receptor pathways to bypass the refractory nature of cancer stem cells to conventional therapy has been demonstrated in mice [97]. Initially, TRAIL-positive NK cells were described in the liver of naïve mice; and in vitro killing activity of hepatic but not splenic NK cells was found to be TRAIL-dependent [14]. Accordingly, TRAIL appears necessary for the control of experimental liver metastasis in mice [14]. Interestingly, TRAIL is largely expressed by immature NK cells in new-born mice and is required for fetal NK cell killing activity of TRAIL-sensitive targets in vitro [98]. Similarly, TRAIL is required for the in vitro killing activity of human cord blood NK cells whereas the cytotoxicity of mature human NK cells mostly relies on the perforin and FasL pathways [99]. While the origin of TRAIL+ fetal NK cells remains unclear, mouse liver TRAIL+ NKp46+ cells have now been assigned to the ILC1 lineage [100]. In the healthy human liver, NK cells do not express TRAIL but its expression can be induced by pro-inflammatory cytokines [101]. It was recently shown that TRAIL up-regulation is confined to a specific population of human intra-hepatic NK cells that express CXCR6 and are absent from the periphery [102]. Thus, in the absence of inflammatory stimuli, TRAIL expression might be restricted to tissue-resident subsets of the group 1 ILCs. As a matter of fact, in mice, TRAIL has also been detected on salivary gland ILC1s [18] and tumor-infiltrating ILC1-like cells [13]. However, the observation that cytokine stimulation induces TRAIL expression on CD3−NK1.1+ cells in the murine spleen [103] and CD3−CD56+ cells in the human blood [104] suggests that TRAIL also contributes to conventional NK cell functions under some circumstances. In fact, membrane-bound TRAIL supplements perforin-mediated killing of neuroblastoma and multiple myeloma cell lines by activated NK cells isolated from human peripheral blood [105, 106]. Additional investigation should shed light on the respective roles played by blood-circulating conventional NK cells and tissue-resident ILC1s in the TRAIL-mediated control of nascent tumors. In opposition to TRAIL, there is very limiting data supporting a role of FasL in NK cell-mediated-control of tumors in vivo [107]. Noteworthy, a recent study elegantly demonstrated that IL-18 induces a rapid expression of FasL on the surface of mouse NK cells and NK cell-mediated FasL-dependent cytotoxicity was found to control MC38 liver metastases in mice [108].
An important characteristic of group 1 ILCs is the secretion of IFN-γ and TNF [109]. These two cytokines play a major role in tumorigenesis. Not only do they modulate immune responses, but they also directly impact on tumor cell biology. Actually, enhanced in vivo growth of various mouse cell lines has been observed upon ablation of tumor-responsiveness to IFN-γ [110]. Responses to IFN-γ are induced by the JAK-STAT signaling pathway. In cancer cells, this pathway has been shown to inhibit cellular proliferation and to promote apoptosis [110]. The central role played by endogenously produced IFN-γ in promoting the immune-mediated elimination of nascent tumor cells has been demonstrated by Schreiber and colleagues [111]. An early study indicated that the combination of perforin and IFN-γ pathways fully accounts for NK cell anti-metastatic activity in mice [112]. Nonetheless, since IFN-γ can be produced by many different innate and adaptive immune cell types, the formal proof of ILC contribution to IFN-γ-mediated immunosurveillance is still lacking. As for TNF, the role of this cytokine in cancer biology is rather ambiguous [113]. The two receptors to TNF are TNFR1, which is expressed on all cell types and TNFR2, which expression is restricted to immune and endothelial cells. Paradoxically, TNFR1 can transmit both pro-survival and pro-apoptosis signals. As a result, some reports described cytostatic or cytotoxic effects on tumor cells while others observed an enhancement of malignant cell proliferation (for a review, see [113]). The observation of reduced in vitro killing capacity of NK1.1+ splenocytes from TNF-deficient mice against YAC-1 suggested that TNF contributes to NK cell-mediated killing [114]. Furthermore, NK cell cytotoxicity against chemotherapeutic-sensitized mouse MC38 tumors was found to be TNF-dependent [115]. But overall, to date there is no convincing evidence of a major contribution of TNF to innate cell-mediated cytotoxic activity. In fact, spleen cells from TNF-deficient mice are perfectly able to lyse MHC-I deficient RMA-S tumor cells in vitro [116]. However, defective elimination of RMA-S cells was observed following intraperitoneal injection in TNF-deficient mice [116]. This phenomenon was explained by reduced NK cell accumulation in the peritoneum in the absence of TNF. Moreover, TNF-neutralization inhibits NK cell activation and thus reduces human NK cell-mediated cytotoxic activity against myeloma cells in the presence of anti-CD319 mAbs (Elotuzumab) [117]. Collectively, these data indicate that albeit not directly cytotoxic, TNF secreted by NK cells might act to regulate the tumor microenvironment.
Of note, although direct cytotoxic activity toward tumor cells is considered as a specificity of group 1 ILCs, ILC2s were recently found to induce tumor cell apoptosis via the CXCR2 pathway [32].
5 Cross-Talk between ILCs and Other Immune Cells Resulting in Anti-Cancer Immunity
NK cell functions expend far beyond the simple killing of cancer cells [118]. In addition to IFN-γ and TNF, activated NK cells release a broad range of cytokines, including GM-CSF, IL-6, and IL-10, and they may facilitate the recruitment of other immune cells by secreting chemokines such as MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5), IL-8 (CXCL8), and IP-10 (CXCL10) [119]. IFN-γ production by NK cells has been shown to promote macrophage-mediated immunoediting of carcinogen-induced tumors in mice [120]. Moreover, NK cells influence the outcome of developing T cell responses in many different ways [121]. For instance, NK cells enhance the priming cytotoxic CD8+ T cell responses by eliminating myeloid-derived suppressor cells [122] and activating DCs [123, 124]. Tumor cell killing by NK cells and the subsequent release of antigen further contributes to T cell priming [125]. Besides, NK cells recruited to inflamed lymph nodes provide an early source of IFN-γ necessary for Th1 polarization of CD4+ T cells [126]. The direct cytotoxicity of activated regulatory T cells (Tregs) represents another mechanism by which NK cell could potentiate effector T cell responses [127]. Importantly, in some mouse tumor models, NK cells were found to contribute to the priming of tumor-specific memory T cells required for the long-term control of cancer [123, 128].
Our understanding of helper-like ILC mediated-regulation of anti-tumor responses is still in its infancy. Given the important role of IFN-γ in NK cell-mediated tuning of adaptive immune responses, it is tempting to hypothesize that IFN-γ production by ILC1s would contribute in a similar manner. However, there is currently no study supporting this statement. In fact, the evaluation of the relative contribution of NK cells and ILC1s to anti-tumor immunity is complicated by the high phenotypic resemblance of these cells. Of note, TRAIL+ members of the group 1 ILC might also dampen T cell responses [129]. Regarding ILC2s, the type 2 cytokines produced by these cells usually inhibit type 1 anti-tumor responses and foster tumor progression [9]. Nevertheless, in the mouse B16F10 model of experimental metastasis, IL-5 production by lung ILC2s cells has been shown to promote eosinophil recruitment and clearance of lung tumors [31]. A positive role of ILC3s in anti-tumor immunity was suggested in a study where combined treatment of chemotherapy with tumor-targeting antibodies resulted in delayed growth of B16 subcutaneous tumors [130]. In this setting, tumor clearance was found to be dependent on CD90+NK1.1−RORγt+ innate lymphocytes and was associated with increased infiltration of macrophages within the tumor tissue. Another report demonstrated that NKp46+ ILC3s suppress the growth of subcutaneously injected B16F10 tumor cells engineered to secrete IL-12 [34]. IL-12-secreting tumors were still repressed in the absence of T cells or of conventional NK cells. It was suggested that NKp46+ ILC3s mediated their anti-tumor functions by up-regulating adhesion molecules on the tumor endothelium. Similarly, the production of soluble factors by NCR+ ILC3s present in human non-small cell lung cancer (NSCLC) tissues was found to activate mesenchymal stem cells and endothelial cells [33]. In this study, ILC3 numbers within the tumor tissue were found to correlate with the density of TLS tertiary lymphoid structures (TLS) which are ectopic lymphoid organs associated with a favorable prognosis in NSCLC patients [131]. Very interestingly, lower frequencies of tumor-infiltrating NCR+ ILC3s were observed in advanced tumors, suggesting that NCR+ ILC3s might be associated with a better prognosis for NSCLC patients [33].
Conclusions
ILCs act as sentinels that react promptly upon disturbance of host homeostasis [76]. Their rapid and robust response allows the temporary control of the danger and alerts other immune cells to provide long-term protection (Fig. 9.4). NK cells have long been known as the most powerful innate guardians against cancer development. However, the recent expansion of the ILC family may challenge this idea and raises the question of the relative contribution of the different ILC subsets, in particular NK cells and ILC1s. Noteworthy, despite considerable transcriptomic overlap with ILC1s, NK cells express higher transcripts encoding proteins of the cytotoxic machinery as well as cell surface receptors involved in the detection of transformed cells [100]. Moreover, helper-like ILCs are tissue-resident cells [132] that would only sense alterations of the specific organ where they are located whereas conventional NK cells circulate in the blood and can scan the whole body for the presence of damaged cells. Thus, NK cells are probably the most efficient ILC subset in tumor clearance.

Innate and adaptive lymphocytes contribute to cancer immunosurveillance. Left panel: ILCs directly induce tumor cell apoptosis and/or growth arrest and they also stimulate other innate cells to ensure the fast clearance of cancer cells. Group 1 ILCs exert a direct cytotoxic activity against tumor cells either via the cytotoxic granule pathway (perforin/granzymes) or the death ligand pathway (TRAIL). Of note, such cytotoxic activity has been described for both conventional NK cells and ILC1s but NK cells are currently considered more potent killers. IFN-γ production by group 1 ILCs also directly inhibits tumor growth and may stimulate macrophage tumoricidal activity. ILC2s might directly induce the apoptosis of CXCR2-expressing tumor cells via the secretion of CXCL1 and CXCL2. ILC2s also secrete IL-5 and thereby recruit eosinophils. ILC3s may increase macrophage infiltration into the tumor. Right panel: ILCs stimulate T cell responses and thereby ensure long-term protection against cancer cells. NK cells activate DCs, enhance anti-tumor T cell responses, and also kill immunosuppressive cells such as Tregs and myeloid-derived suppressor cells (MDSC). ILC3s activate stroma and endothelial cells and thereby facilitate immune cell infiltration into the tumor bed
The importance of ILCs in the presence of functional adaptive immunity was recently questioned as ILC deficiency occurring in a cohort of SCID patients appeared to have no major clinical consequences [133]. This study provided a 7–39 year follow-up of 18 patients with mutation of IL2RG or JAK3 treated with hematopoietic stem cell transplantation in the absence of myeloablation. However, this cohort was too small to address the role of ILCs in tumor immunosurveillance and such investigation may also require a longer follow-up. Perhaps the most convincing evidence of the importance of cytotoxic ILCs for human cancer immunosurveillance comes a prospective study demonstrating that individuals with high spontaneous cytotoxic activity of peripheral blood lymphocytes were at significantly lower risk of developing cancers [134]. In gastrointestinal sarcoma patients, NK cell infiltration correlates with the absence of metastasis at diagnosis [46] and in renal cell carcinoma, high densities of NK cells in lung metastases are associated with prolonged survival [135]. Conversely, a study in NSCLC reported no correlation between NK cell numbers and clinical outcome [136]. An absence of NK cell activity against advanced cancers may be caused by (1) tumor escape from NK-cell immunosurveillance due the selection of resistant variant clones through the immunoediting process [120] and/or (2) the exhaustion of NK cells within the tumor microenvironment where a variety of mechanisms contribute to hinder NK cell functions [55]. Adenosine and TGF-β are two examples of soluble factors proven to affect NK cell activity against tumors [137, 138]. Furthermore, poor NK cell infiltration in human cancer tissues could explain their limited impact on solid tumor progression. However, NK cells seem particularly efficient against metastatic disease [27] and hematological malignancies [139].
Evidence for helper-like ILC subsets involvement in human cancers is limited. ILC3 infiltration has been observed in human colorectal cancer [140], primary tumors of breast cancer patients [141], and NSCLC tissues [33]. Importantly, the function of helper-like ILCs in malignant diseases remains unclear and may depend on the cancer type and stage. This chapter is focused on the protective role of ILCs in the detection and elimination of nascent tumors and the reader is invited to refer to other reviews for a complete discussion of the opposing abilities of helper-like ILCs to either promote or repress tumor growth [9, 30, 35]. It is possible that the tumor microenvironment hijacks ILCs, either by dampening ILC anti-tumor activity such as IFN-γ release, or by influencing ILC plasticity. Indeed, some ILC subsets are not stable and depending on the cytokine microenvironment, NK cells can acquire an ILC1 phenotype [18], ILC2s can convert into ILC1s [142] or ILC3s [143], and ILC3s can convert into ILC1s [144]. The hypothesis that tumor may escape immunosurveillance by modulating ILCs is supported by the report that both ILC functions and subtype composition are dysregulated in the blood of acute myeloid leukemia patients [145].
ILCs represent an interesting clinical target since they react immediately to stimulation and their responses are not antigen-driven. Their production of large amounts of cytokines could shift the tumor microenvironment and awaken the anti-cancer capacities of myeloid cells and adaptive lymphocytes. However, a better understanding of helper-like ILC functions, biology, and plasticity is definitely needed before these cells could be efficiently exploited in the clinic. On the other hand, manipulation of NK cells has emerged as a very promising therapeutic option for cancer patients. The multiple strategies employed to take advantage of NK cell anti-tumor activity have been reviewed elsewhere [41] and are the subject of another chapter of this volume. Importantly, some strategies currently developed to target T cells or NK cells, such as anti-PD1 mAbs [146], NKG2D-bispecific engagers [147], or cytokine infusions [148] might also influence ILC functions. As our knowledge of the ILC family increases, the interest of the cancer immunotherapy field in these cells is likely to rise.
References
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–46.
Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011;71(17):5601–5.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71.
Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010;31(6):212–9.
Blaisdell A, Crequer A, Columbus D, Daikoku T, Mittal K, Dey SK, Erlebacher A. Neutrophils oppose uterine epithelial carcinogenesis via debridement of hypoxic tumor cells. Cancer Cell. 2015;28(6):785–99.
Altman JB, Benavides AD, Das R, Bassiri H. Antitumor responses of invariant natural killer T cells. J Immunol Res. 2015;2015:652875.
Girardi M. Immunosurveillance and immunoregulation by gammadelta T cells. J Invest Dermatol. 2006;126(1):25–31.
van Beek JJP, Martens AWJ, Bakdash G, de Vries IJM. Innate lymphoid cells in tumor immunity. Biomedicine. 2016;4(7) pii E7. doi: 10.3390/biomedicines4010007.
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–9.
Zook EC, Kee BL. Development of innate lymphoid cells. Nat Immunol. 2016;17(7):775–82.
Eberl G, Di Santo JP, Vivier E. The brave new world of innate lymphoid cells. Nat Immunol. 2015;16(1):1–5.
Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, Li MO. Cancer immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell. 2016;164(3):365–77.
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7(1):94–100.
Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell. 2014;157(2):340–56.
Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. 2016;17(7):758–64.
Seillet C, Belz GT, Huntington ND. Development, homeostasis, and heterogeneity of NK cells and ILC1. Curr Top Microbiol Immunol. 2016;395:37–61.
Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, Gilfillan S, Fuchs A, Vivier E, Sun JC, Cella M, Colonna M. Transforming growth factor-beta signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. 2016;44(5):1127–39.
Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5(2):112–7.
Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci U S A. 2007;104(9):3384–9.
Hayakawa Y, Huntington ND, Nutt SL, Smyth MJ. Functional subsets of mouse natural killer cells. Immunol Rev. 2006;214:47–55.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–40.
Smyth MJ, Thia KY, Cretney E, Kelly JM, Snook MB, Forbes CA, Scalzo AA. Perforin is a major contributor to NK cell control of tumor metastasis. J Immunol. 1999;162(11):6658–62.
Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13(4):459–63.
Talmadge JE, Meyers KM, Prieur DJ, Starkey JR. Role of NK cells in tumour growth and metastasis in beige mice. Nature. 1980;284(5757):622–4.
Vosshenrich CA, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, Di Santo JP. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol. 2005;174(3):1213–21.
Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, Rankin LC, Mielke LA, Vikstrom I, Kolesnik TB, Nicholson SE, Vivier E, Smyth MJ, Nutt SL, Glaser SP, Strasser A, Belz GT, Carotta S, Huntington ND. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun. 2014;5:4539.
Merzoug LB, Marie S, Satoh-Takayama N, Lesjean S, Albanesi M, Luche H, Fehling HJ, Di Santo JP, Vosshenrich CA. Conditional ablation of NKp46+ cells using a novel Ncr1(greenCre) mouse strain: NK cells are essential for protection against pulmonary B16 metastases. Eur J Immunol. 2014;44(11):3380–91.
Guillerey C, Smyth MJ. NK cells and cancer immunoediting. Curr Top Microbiol Immunol. 2016;395:115–45.
Vallentin B, Barlogis V, Piperoglou C, Cypowyj S, Zucchini N, Chene M, Navarro F, Farnarier C, Vivier E, Vely F. Innate lymphoid cells in cancer. Cancer Immunol Res. 2015;3(10):1109–14.
Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, Kouro T, Itakura A, Nagai Y, Takaki S, Takatsu K. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol. 2012;188(2):703–13.
Kim J, Kim W, Moon UJ, Kim HJ, Choi HJ, Sin JI, Park NH, Cho HR, Kwon B. Intratumorally establishing type 2 innate lymphoid cells blocks tumor growth. J Immunol. 2016;196(5):2410–23.
Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S, Bonaccorsi I, Morandi B, Truini M, Mingari MC, Moretta L, Ferlazzo G. NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun. 2015;6:8280.
Eisenring M, Vom Berg J, Kristiansen G, Saller E, Becher B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol. 2010;11(11):1030–8.
Mattner J, Wirtz S. Friend or foe? The ambiguous role of innate lymphoid cells in cancer development. Trends Immunol. 2017;38(1):29–38.
Marcus A, Gowen BG, Thompson TW, Iannello A, Ardolino M, Deng W, Wang L, Shifrin N, Raulet DH. Recognition of tumors by the innate immune system and natural killer cells. Adv Immunol. 2014;122:91–128.
Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat Rev Immunol. 2007;7(4):279–91.
Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11(7):237–44.
Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195(3):346–55.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–36.
Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol. 2014;92(3):221–9.
Salimi M, Xue L, Jolin H, Hardman C, Cousins DJ, McKenzie AN, Ogg GS. Group 2 innate lymphoid cells express functional NKp30 receptor inducing type 2 cytokine production. J Immunol. 2016;196(1):45–54.
Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F, Yossef R, Rosenberg LA, Aharoni A, Cerwenka A, Campbell KS, Braiman A, Porgador A. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J Immunol. 2011;187(11):5693–702.
Siewiera J, Gouilly J, Hocine HR, Cartron G, Levy C, Al-Daccak R, Jabrane-Ferrat N. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat Commun. 2015;6:10183.
Delahaye NF, Rusakiewicz S, Martins I, Menard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, Minard-Colin V, Poirier-Colame V, Chaba K, Flament C, Baud V, Authier H, Kerdine-Romer S, Pallardy M, Cremer I, Peaudecerf L, Rocha B, Valteau-Couanet D, Gutierrez JC, Nunes JA, Commo F, Bonvalot S, Ibrahim N, Terrier P, Opolon P, Bottino C, Moretta A, Tavernier J, Rihet P, Coindre JM, Blay JY, Isambert N, Emile JF, Vivier E, Lecesne A, Kroemer G, Zitvogel L. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. 2011;17(6):700–7.
Halfteck GG, Elboim M, Gur C, Achdout H, Ghadially H, Mandelboim O. Enhanced in vivo growth of lymphoma tumors in the absence of the NK-activating receptor NKp46/NCR1. J Immunol. 2009;182(4):2221–30.
Glasner A, Ghadially H, Gur C, Stanietsky N, Tsukerman P, Enk J, Mandelboim O. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J Immunol. 2012;188(6):2509–15.
Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3(10):781–90.
Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436(7054):1186–90.
Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165–71.
Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28(4):571–80.
Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med. 2005;202(5):583–8.
Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, Wang Z, Derudder E, Li S, Chakraborty T, Cotter SE, Koyama S, Currie T, Freeman GJ, Kutok JL, Rodig SJ, Dranoff G, Rajewsky K. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell. 2012;148(4):739–51.
Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. 2014;44(6):1582–92.
Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, Neuberg D, Anderson KC, Carrasco DR, Dranoff G. MHC class I chain-related protein a antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A. 2008;105(4):1285–90.
Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–8.
Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 2008;111(7):3571–8.
Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348(6230):136–9.
Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15(4):243–54.
Samanta D, Almo SC. Nectin family of cell-adhesion molecules: structural and molecular aspects of function and specificity. Cell Mol Life Sci. 2015;72(4):645–58.
Martinet L, Ferrari De Andrade L, Guillerey C, Lee JS, Liu J, Souza-Fonseca-Guimaraes F, Hutchinson DS, Kolesnik TB, Nicholson SE, Huntington ND, Smyth MJ. DNAM-1 expression marks an alternative program of NK cell maturation. Cell Rep. 2015;11(1):85–97.
Seillet C, Mielke LA, Amann-Zalcenstein DB, Su S, Gao J, Almeida FF, Shi W, Ritchie ME, Naik SH, Huntington ND, Carotta S, Belz GT. Deciphering the innate lymphoid cell transcriptional program. Cell Rep. 2016;17(2):436–47.
Tang ML, Gasser S. ATM activation mediates anticancer immunosurveillance by natural killer and T cells. Oncoimmunology. 2013;2(6):e24438.
Croxford JL, Tang ML, Pan MF, Huang CW, Kamran N, Phua CM, Chng WJ, Ng SB, Raulet DH, Gasser S. ATM-dependent spontaneous regression of early Emu-myc-induced murine B-cell leukemia depends on natural killer and T cells. Blood. 2013;121(13):2512–21.
Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya K, Shibuya A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med. 2008;205(13):2959–64.
Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Med. 2008;205(13):2965–73.
Guillerey C, Ferrari de Andrade L, Vuckovic S, Miles K, Ngiow SF, Yong MC, Teng MW, Colonna M, Ritchie DS, Chesi M, Bergsagel PL, Hill GR, Smyth MJ, Martinet L. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J Clin Invest. 2015;125(5):2077–89.
Abeyweera TP, Merino E, Huse M. Inhibitory signaling blocks activating receptor clustering and induces cytoskeletal retraction in natural killer cells. J Cell Biol. 2011;192(4):675–90.
Tu MM, Mahmoud AB, Wight A, Mottashed A, Belanger S, Rahim MM, Abou-Samra E, Makrigiannis AP. Ly49 family receptors are required for cancer immunosurveillance mediated by natural killer cells. Cancer Res. 2014;74(14):3684–94.
Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, Brandt C, Kane L, Campos LS, Lu L, Belz GT, McKenzie AN, Teichmann SA, Dougan G, Liu P. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature. 2016;539(7627):102–6.
Blake SJ, Dougall WC, Miles JJ, Teng MW, Smyth MJ. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183–8.
Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, Enk J, Jonjic S, Mandelboim O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur J Immunol. 2013;43(8):2138–50.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(42):17858–63.
Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15(5):431–8.
Bando JK, Colonna M. Innate lymphoid cell function in the context of adaptive immunity. Nat Immunol. 2016;17(7):783–9.
Yajima T, Nishimura H, Wajjwalku W, Harada M, Kuwano H, Yoshikai Y. Overexpression of interleukin-15 in vivo enhances antitumor activity against MHC class I-negative and -positive malignant melanoma through augmented NK activity and cytotoxic T-cell response. Int J Cancer. 2002;99(4):573–8.
Smyth MJ, Swann J, Kelly JM, Cretney E, Yokoyama WM, Diefenbach A, Sayers TJ, Hayakawa Y. NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J Exp Med. 2004;200(10):1325–35.
Smyth MJ, Taniguchi M, Street SE. The anti-tumor activity of IL-12: mechanisms of innate immunity that are model and dose dependent. J Immunol. 2000;165(5):2665–70.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–65.
Crellin NK, Trifari S, Kaplan CD, Satoh-Takayama N, Di Santo JP, Spits H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity. 2010;33(5):752–64.
Sivori S, Carlomagno S, Pesce S, Moretta A, Vitale M, Marcenaro E. TLR/NCR/KIR: which one to use and when? Front Immunol. 2014;5:105.
Guillerey C, Chow MT, Miles K, Olver S, Sceneay J, Takeda K, Moller A, Smyth MJ. Toll-like receptor 3 regulates NK cell responses to cytokines and controls experimental metastasis. Oncoimmunology. 2015;4(9):e1027468.
Degli-Esposti MA, Smyth MJ. Close encounters of different kinds: dendritic cells and NK cells take centre stage. Nat Rev Immunol. 2005;5(2):112–24.
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26(4):503–17.
Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, Lienenklaus S, Weiss S, Staeheli P, Aichele P, Diefenbach A. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37(1):171–86.
Glatzer T, Killig M, Meisig J, Ommert I, Luetke-Eversloh M, Babic M, Paclik D, Bluthgen N, Seidl R, Seifarth C, Grone J, Lenarz M, Stolzel K, Fugmann D, Porgador A, Hauser A, Karlas A, Romagnani C. RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity. 2013;38(6):1223–35.
Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369(6475):31–7.
Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6(12):940–52.
Street SE, Hayakawa Y, Zhan Y, Lew AM, MacGregor D, Jamieson AM, Diefenbach A, Yagita H, Godfrey DI, Smyth MJ. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and gammadelta T cells. J Exp Med. 2004;199(6):879–84.
Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, Kawano T, Pelikan SB, Crowe NY, Godfrey DI. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191(4):661–8.
van den Broek MF, Kagi D, Zinkernagel RM, Hengartner H. Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol. 1995;25(12):3514–6.
van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, Lutz WK, Melief CJ, Zinkernagel RM, Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J Exp Med. 1996;184(5):1781–90.
Screpanti V, Wallin RP, Grandien A, Ljunggren HG. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol. 2005;42(4):495–9.
Smyth MJ, Cretney E, Takeda K, Wiltrout RH, Sedger LM, Kayagaki N, Yagita H, Okumura K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193(6):661–70.
Vujanovic NL. Role of TNF superfamily ligands in innate immunity. Immunol Res. 2011;50(2–3):159–74.
Li M, Knight DA, Smyth MJ, Stewart TJ. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol Immunother. 2012;61(8):1255–68.
Takeda K, Cretney E, Hayakawa Y, Ota T, Akiba H, Ogasawara K, Yagita H, Kinoshita K, Okumura K, Smyth MJ. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood. 2005;105(5):2082–9.
Zamai L, Ahmad M, Bennett IM, Azzoni L, Alnemri ES, Perussia B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J Exp Med. 1998;188(12):2375–80.
Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, Immunological Genome C. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol. 2015;16(3):306–17.
Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, Das A, Lopes AR, Borrow P, Williams K, Humphreys E, Afford S, Adams DH, Bertoletti A, Maini MK. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204(3):667–80.
Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, Christophides T, Pallett LJ, Peppa D, Dunn C, Fusai G, Male V, Davidson BR, Kennedy P, Maini MK. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci Rep. 2016;6:26157.
Kayagaki N, Yamaguchi N, Nakayama M, Takeda K, Akiba H, Tsutsui H, Okamura H, Nakanishi K, Okumura K, Yagita H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J Immunol. 1999;163(4):1906–13.
Johnsen AC, Haux J, Steinkjer B, Nonstad U, Egeberg K, Sundan A, Ashkenazi A, Espevik T. Regulation of APO-2 ligand/trail expression in NK cells-involvement in NK cell-mediated cytotoxicity. Cytokine. 1999;11(9):664–72.
Jungkunz-Stier I, Zekl M, Stuhmer T, Einsele H, Seggewiss-Bernhardt R. Modulation of natural killer cell effector functions through lenalidomide/dasatinib and their combined effects against multiple myeloma cells. Leuk Lymphoma. 2014;55(1):168–76.
Sheard MA, Asgharzadeh S, Liu Y, Lin TY, Wu HW, Ji L, Groshen S, Lee DA, Seeger RC. Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J Immunother. 2013;36(5):319–29.
Screpanti V, Wallin RP, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol. 2001;167(4):2068–73.
Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, Rodrigue-Gervais IG, Breton V, Colpitts SL, Beauchemin N, Saleh M. The Nlrp3 Inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell Tumoricidal activity. Immunity. 2015;43(4):751–63.
Cortez VS, Robinette ML, Colonna M. Innate lymphoid cells: new insights into function and development. Curr Opin Immunol. 2015;32:71–7.
Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13(2):95–109.
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95(13):7556–61.
Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood. 2001;97(1):192–7.
Bertazza L, Mocellin S. The dual role of tumor necrosis factor (TNF) in cancer biology. Curr Med Chem. 2010;17(29):3337–52.
Baxevanis CN, Voutsas IF, Tsitsilonis OE, Tsiatas ML, Gritzapis AD, Papamichail M. Compromised anti-tumor responses in tumor necrosis factor-alpha knockout mice. Eur J Immunol. 2000;30(7):1957–66.
Nagasaki E, Takahara A, Koido S, Sagawa Y, Aiba K, Tajiri H, Yagita H, Homma S. Combined treatment with dendritic cells and 5-fluorouracil elicits augmented NK cell-mediated antitumor activity through the tumor necrosis factor-alpha pathway. J Immunother. 2010;33(5):467–74.
Smyth MJ, Kelly JM, Baxter AG, Korner H, Sedgwick JD. An essential role for tumor necrosis factor in natural killer cell-mediated tumor rejection in the peritoneum. J Exp Med. 1998;188(9):1611–9.
Balasa B, Yun R, Belmar NA, Fox M, Chao DT, Robbins MD, Starling GC, Rice AG. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-alpha pathways. Cancer Immunol Immunother. 2015;64(1):61–73.
Andoniou CE, Coudert JD, Degli-Esposti MA. Killers and beyond: NK-cell-mediated control of immune responses. Eur J Immunol. 2008;38(11):2938–42.
Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–76.
O'Sullivan T, Saddawi-Konefka R, Vermi W, Koebel CM, Arthur C, White JM, Uppaluri R, Andrews DM, Ngiow SF, Teng MW, Smyth MJ, Schreiber RD, Bui JD. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med. 2012;209(10):1869–82.
Crouse J, Xu HC, Lang PA, Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. 2015;36(1):49–58.
Geldhof AB, van Ginderachter JA, Liu Y, Noel W, de Baetselier P. Ablation of NK cell function during tumor growth favors type 2-associated macrophages, leading to suppressed CTL generation. Clin Dev Immunol. 2003;10(2–4):71–81.
Adam C, King S, Allgeier T, Braumuller H, Luking C, Mysliwietz J, Kriegeskorte A, Busch DH, Rocken M, Mocikat R. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood. 2005;106(1):338–44.
Vitale M, Della Chiesa M, Carlomagno S, Pende D, Arico M, Moretta L, Moretta A. NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood. 2005;106(2):566–71.
Krebs P, Barnes MJ, Lampe K, Whitley K, Bahjat KS, Beutler B, Janssen E, Hoebe K. NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood. 2009;113(26):6593–602.
Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5(12):1260–5.
Roy S, Barnes PF, Garg A, Wu S, Cosman D, Vankayalapati R. NK cells lyse T regulatory cells that expand in response to an intracellular pathogen. J Immunol. 2008;180(3):1729–36.
Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, Smyth MJ. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol. 2002;3(1):83–90.
Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA. “Natural regulators”: NK cells as modulators of T cell immunity. Front Immunol. 2016;7:235.
Moskalenko M, Pan M, Fu Y, de Moll EH, Hashimoto D, Mortha A, Leboeuf M, Jayaraman P, Bernardo S, Sikora AG, Wolchok J, Bhardwaj N, Merad M, Saenger Y. Requirement for innate immunity and CD90(+) NK1.1(−) lymphocytes to treat established melanoma with chemo-immunotherapy. Cancer Immunol Res. 2015;3(3):296–304.
Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, Rabbe N, Laurans L, Tartour E, de Chaisemartin L, Lebecque S, Fridman WH, Cadranel J. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. 2008;26(27):4410–7.
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350(6263):981–5.
Vely F, Barlogis V, Vallentin B, Neven B, Piperoglou C, Ebbo M, Perchet T, Petit M, Yessaad N, Touzot F, Bruneau J, Mahlaoui N, Zucchini N, Farnarier C, Michel G, Moshous D, Blanche S, Dujardin A, Spits H, Distler JH, Ramming A, Picard C, Golub R, Fischer A, Vivier E. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17(11):1291–9.
Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356(9244):1795–9.
Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, Crozet L, Ouakrim H, Goc J, Cazes A, Flejou JF, Gibault L, Verkarre V, Regnard JF, Pages ON, Oudard S, Mlecnik B, Sautes-Fridman C, Fridman WH, Damotte D. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res. 2013;19(15):4079–91.
Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, Andre P, Dieu-Nosjean MC, Alifano M, Regnard JF, Fridman WH, Sautes-Fridman C, Cremer I. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71(16):5412–22.
Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A. 2013;110(36):14711–6.
Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, Bienvenu J, Marie JC, Caux C, Marvel J, Town L, Huntington ND, Bartholin L, Finlay D, Smyth MJ, Walzer T. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9(415):ra19.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–100.
Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210(5):917–31.
Irshad S, Flores-Borja F, Lawler K, Monypenny J, Evans R, Male V, Gordon P, Cheung A, Gazinska P, Noor F, Wong F, Grigoriadis A, Fruhwirth G, Barber PR, Woodman N, Patel D, Rodriguez-Justo M, Owen J, Martin SG, Pinder S, Gillett C, Poland SP, Ameer-Beg S, McCaughan F, Carlin L, Hasan U, Withers DR, Lane P, Vojnovic B, Quezada SA, Ellis P, Tutt AN, Ng T. RORgammat+ innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res. 2017;(77):1083–96.
Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, van Drunen CM, Lutter R, Jonkers RE, Hombrink P, Bruchard M, Villaudy J, Munneke JM, Fokkens W, Erjefalt JS, Spits H, Ros XR. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17(6):636–45.
Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, Williamson PR, Urban JF Jr, Paul WE. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential 'inflammatory' type 2 innate lymphoid cells. Nat Immunol. 2015;16(2):161–9.
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14(3):221–9.
Trabanelli S, Curti A, Lecciso M, Salome B, Riether C, Ochsenbein A, Romero P, Jandus C. CD127+ innate lymphoid cells are dysregulated in treatment naive acute myeloid leukemia patients at diagnosis. Haematologica. 2015;100(7):e257–60.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Kellner C, Hallack D, Glorius P, Staudinger M, Mohseni Nodehi S, de Weers M, van de Winkel JG, Parren PW, Stauch M, Valerius T, Repp R, Humpe A, Gramatzki M, Peipp M. Fusion proteins between ligands for NKG2D and CD20-directed single-chain variable fragments sensitize lymphoma cells for natural killer cell-mediated lysis and enhance antibody-dependent cellular cytotoxicity. Leukemia. 2012;26(4):830–4.
Romee R, Leong JW, Fehniger TA. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica (Cairo). 2014;2014:205796.
Acknowledgements
The authors wish to thank past and present members of the Immunology in Cancer and Infection Laboratory.
C.G. is supported by a National Health and Medical Research Council (NH&MRC) of Australia Early Career Fellowship (1107417) and a Project grant from Cure Cancer Australia (1122183). M. J. S. is supported by a NH&MRC Senior Principal Research Fellowship (1078671) and Project Grant (1098960).
Conflict of Interest
M.J. S. has contract research agreements with Bristol Myers Squibb, Aduro Biotech, Corvus Pharmaceuticals, and Medimmune. M. J. S. is an advisory board member for Arcus Biosciences, Astrazeneca, Kymab, and F-star. C.G. declares no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Guillerey, C., Smyth, M.J. (2018). Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells. In: Zitvogel, L., Kroemer, G. (eds) Oncoimmunology. Springer, Cham. https://doi.org/10.1007/978-3-319-62431-0_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-62431-0_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62430-3
Online ISBN: 978-3-319-62431-0
eBook Packages: MedicineMedicine (R0)