
Abstract
Targeted immunotherapy in cancer is a rapidly expanding and evolving field with a developmental history spanning at least three decades. Beginning with the identification and characterisation of tumour-specific antigens (TSA)—protein molecules which are exclusively present on the surface of cancer cells—and very recently the dawn of neoantigen-specific immune-cell reactivity—championed by immune checkpoint blockade therapy—demonstrates that immune-based interventions will shape the future of cancer therapy. Neoantigens arise from naturally processed host protein molecules—eventually presented as immunogenic peptides to the immune system. However, a deeper understanding concerning the generation and recognition of neoantigens is indispensable in order to better understand the immunological and biological underpinnings in diagnostics and therapeutic applications in to enhance healthcare for patients with cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Tumour-infiltrating lymphocytes
- Immunotherapy
- Neoepitopes
- Glioblastoma
- Interferon gamma
- Immune response
- Personalised medicine
- Mutant epitope
- TAA
- TSA
- Pancreatic cancer
- T-cells
- TCR
1 Introduction
Targeted immunotherapy in cancer is a rapidly expanding and evolving field with a developmental history spanning at least three decades. Beginning with the identification and characterisation of tumor-specific antigens (TSA)—protein molecules which are exclusively associated with transformed cells—and very recently the dawn of neoantigen-specific immune-cell reactivity—championed by immune checkpoint blockade therapy—demonstrates that immune-based interventions will substantially shape the future of cancer therapy. Neoantigens arise from naturally processed mutated host protein molecules—eventually presented as immunogenic peptides to the immune system. However, a deeper understanding concerning the generation and recognition of neoantigens is indispensable in order to better understand the immunological and biological underpinnings in diagnostics and therapeutic applications to enhance healthcare for patients with cancer. We briefly introduce the reader to the antigen processing and presenting machinery in cancer, and provide a condensed history of cancer antigen discovery, touching upon seminal findings. Last but not least, we discuss the latest development in cancer immunotherapy—with a strong focus on neoantigen-directed strategies, which may be improved for the time to come in the context of clinical translation and therapy. We limit the focus in this chapter to active cellular therapy (ACT) for patients with cancer and the potential of using mutant epitopes in combination witih cellular therapy.
Harnessing the potential of neoepitope-specific T-cell subsets is highly attractive, due to their ability to recognize and respond to tumor cells with limited off-target toxicity, superior efficiency, and with the capacity to provide durable and clinically meaningful outcome in patients with cancer [1]. This anti-cancer reactivity directed against transformed cells is in essence a targeted but productive autoimmune response and dependent on the presence of a T-cell receptor T-cell receptor (TCR) repertoire capable of recognising mutant targets. Some cancer antigens have been identified as ‘cancer antigens’ due to their selective tissue expression or overexpression in malignant/transformed cells, i.e. mesothelin, or cancer testis antigens (discussed later in this chapter). In other cases, mutations that occur in otherwise normally expressed and functional proteins may cause them to become cancer-inducing agents. These mutated host molecules may be involved in cancer initiation (oncogenesis), disease maintenance, or in metastasis. Since some mutations are crucial for malignant transformation and for tumor cell survival, they may also be instrumental in immune escape mechanisms, either by selecting tumor-promoting T-cell responses, or - not mutually exclusive, ‘blinding’ anti-cancer immune responses by inducing loss of immune - ‘fitness’.
Recent findings in cancer research show that thesuccess of immune - based therapies requires a T-cell receptor repertoire capable of recognizing mutant targets along with anti-cancer directed cellular immune responses (e.g. cytotoxicity, Th1 - type immune responses, see Fig. 4.1). In line with this, T-cell-based cancer immunotherapy is gaining momentum since the most successful novel interventions against solid tumorsrely on cancer-specific T-cell activity and their mobilisation to sites of disease, i.e. immune checkpoint inhibitors, chimeric antigen receptors (CARs) and T-cell receptor (TCR)-modified T-cell products [2, 3]. Local activation of antigen-specific tumour-infiltrating T lymphocytes, known as TILs, allows for re-circulation of cells, robust killing of tumour cells, reduction in tumour mass and orchestration of anti-tumour responses in tissue. Monoclonal antibodies targeting PD-1 and CTLA-4 have thus revolutionised cancer therapy, with signs of potential use in treating chronic infectious diseases such as viral hepatitis, human immunodeficiency virus (HIV) infection, malaria and tuberculosis [4,5,6,7]. In particular, anti-PD-1 therapy has been shown to activate CD8 T cells specific for mutated antigens (neoantigens) associated with cancer progression in metastatic melanoma [3]. Patients showing durable responses following immunotherapy had increased numbers of neoantigen-specific T-cells in their blood, signifying the underlying mechanism of anti-PD-1 therapy.

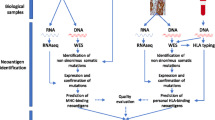
Strategies to identify mutant epitopes from transformed cells, CSF: cerebrospinal fluid
2 Antigen Processing and Presentation in Cancer
In order to gain an understanding of the dynamics driving the generation and ‘visibility’ of antigens to the immune system, it is advantageous to provide an overview about antigen processing and presentation to immune effector cells. Antigens can be generally viewed as being either intrinsic or extrinsic in nature; they are biochemically processed within cells and presented to various T-cell subsets, B cells as well as natural killer (NK) cells [8]. The essential molecule associated with presenting antigens to the immune system is termed as the major histocompatibility complex (MHC), or specifically in humans, the human leukocyte antigen (HLA) [8]. The function of MHC/HLA system was discovered and first described by Rolf Zinkernagel and Peter Doherty in the early 1970s, the seminal work for which they were awarded the Nobel prize in Medicine and Physiology in 1996 [9,10,11,12].
Antigens can derive from whole pathogens, i.e. bacteria, viruses and parasitic organisms, or by non-mutant, or mutant proteins associated by transformed cells. Intrinsic antigens, also called ‘endogenous’ antigens, are processed and presented to the immune system in the form of specific peptides called epitopes. This pathway is termed the MHC/HLA class I pathway (hereafter referred to as the ‘HLA class I pathway’), and plays a crucial role in eliciting immune responses to viruses (viral components synthesised within the host cell), intracellular bacteria as well as to cancer - associated antigens - which relies on the immune system’s capacity to recognized ‘self’ or ‘mutant self’ antigens [8]. All cells of the body (with the exception of erythrocytes) are capable of processing and presenting antigens via the HLA class I pathway. The processing of antigens in this pathway involves a crucial step, where the immunoproteasome (occurring in the cytosol) cuts up denatured (unfolded) protein structures into small peptide sequences between 8 and 10 amino acids long. The amino acid junctions at which the proteasome enzymatically cuts a protein decides on which peptide or epitopes are naturally presented to immune cells. Epitopes presented by HLA class I molecules are recognised by CD8+ T-cells, which can respond by i) proliferation, ii) cytokine production and / or iii) production of cytotoxic molecules, capable of killing transformed cells [8]. CD8+ T-cells may produce perforin, granzymes, and granulolysin (that can be easily measured using an CD107a induction assay), or - not mutually exclusive - IFN-gamma in response to transformed cells [13]. If (cancer) target epitopes are identified using the ‘reverse immunology strategy’, i.e. that epitopes are selected based on their predicted capacity to bind to MHC class I or class II molecules, it cannot be assumed with a very high degree of certainty that T-cells will also recognise the naturally processed and presented epitopes on tumour cells—a scenario which was described more than two decades ago [14]: T-cells that were shown to be peptide specific were not able to react against naturally processed and presented peptides on tumor cells. One of the reasons driving this phenomenon is that the specialised, or ‘skewed’ antigen processing and presentation machinery in transformed cells may be different compared to professional and non-professional antigen presenting cells [15] that are responsible for activating and expanding antigen-reactive T-cells. Alternatively, epitopes may have been created via post-translational modifications (such as phosphorylation) that could not be predicted from the primary structure of the wildtype and/or the mutant protein [16].
Antigens that are taken up from the external environment by professional antigen presenting cells or APCs (i.e. dendritic cells, macrophages), including B-cells, that have also professional APC functions, are usually processed and presented to the immune system via the HLA class II pathway. Whole pathogens, as well as proteins, e.g. generated via destruction of cancer cells by antibody-mediated mechanism, NK or CD8 T-cells, are actively taken up by APCs in endocytic vesicles called phagosomes, after which proteolytic enzymes contained within lysosomal compartments fuse with the phagosome to digest the antigen to yield smaller peptide sequences, usually 13–17 amino acids in length. These epitopes are then presented to CD4+ T-cells, which are also termed as helper T-cells (Th), and have an indispensable role in orchestrating immune responses mainly by producing effector cytokines, i.e. IFN-γ, tumor necrosis factor alpha (TNF-α), interleukin (IL)-2 (Th1 cells), IL-4, IL-10 (Th2 cells) and in some cases, IL-17 (Th17 cells). Cytotoxic activity is not exclusively attributed to CD8+ CTLs; cytotoxic CD4+ T-cells have also been reported to mediate biologically relevant immune responses in cancer as well as in viral infections [17,18,19].
The T-cell receptor (TCR) on the surface of T-cells binds to the HLA-epitope complex, along with co-receptors CD8 or CD4, to initiate an immune synapse. Interactions between T-cells and tumor cells are governed by HLA-restriction—the alleles encoding a person’s HLA repertoire and matching TCRs available in the tissue microenvironment and/or in blood, which dictates the nature and strength of the immune response. HLA allele-restriction of epitopes and immune cross-reactivity thereof plays an indispensable role in dictating the nature of immune responses. For example, HLA-DQ variants have been associated with increased susceptibility to certain infectious diseases; mutations in the β57 subunit of HLA-DQ may perpetrate progression to pulmonary disease [20]. Interestingly, mutations in HLA-DQ alleles have been attributed to susceptibility to contract type 1 diabetes mellitus (T1DM). While HLA-DQ is highly prevalent among Caucasians in the Americas as well as Europe, East Asians and Africans are much less likely to express these alleles [21]. Indeed, individual HLA alleles may also favour certain immune-recognition profiles, independent of the peptide repertoire displayed by the nominal restricting MHC element, i.e. HLA-DQ0602 favours IL-17 production independent of binding peptides, as shown in the transgenic murine model of multiple sclerosis [22]. This IL-17-centric reactivity represents a double-edged sword; it may more effectively contain certain bacterial infections [23, 24] and IL-17 may be beneficial to attract immune cells to the tumor site [25] while the chronic exposure to IL-17 may rather promote malignant transformation [26,27,28]. Therefore, the nature, quality and quantity of immune responses following vaccination appear to greatly depend on an individual’s HLA profile, which shapes the quality and quantity of ensuing cellular immune responses, including increased or decreased risk for infections, autoimmune responses or the ability to present (neo) epitopes to T-cells dependent on the restrictions imposed by the MHC–peptide complex and the responding TCR repertoire. For instance, even if neoepitopes are generated during malignant transformation, they may not be visible to the cellular immune system, if they are not processed and ultimately complexed to the respective HLA molecule and presented to responding T-cells.
Th1–Th2 Responses and MHC Restriction
Most studies use IFN-gamma as the readout of T-cells responding to wildtype and mutant epitopes provided from cancer cells, yet Th2 responses, with the signature cytokines IL-4, IL-5 and IL-13 may also be present, either as an ‘original’ Th2 response or as a result of partial agonist peptides, imposed by the mutational event (see below) that may turn Th1 T-cells into Th2 cytokine-producing T-cells [29]. Th2-type T-cell responses may not per se signify an unproductive and potentially ‘tolerizing’ immune response; more recent reports indicate that Th2-type immune responses may also be able to mediate clinically relevant anti-cancer immune reactivities [30]. In a preclinical model, antigen-specific Th2 cells eradicated myelomas without the help of CD8 T-cells, leading to massive inflammation at the tumor site [30]. Th2-mediated tumour destruction has been shown to be associated with IL-1, TNF-alpha (Th1) and Th2 cytokine (IL-4, IL-5, IL-13) production in situ, while passively transferred Th2 cells were able to confer long-lasting cellular anti-cancer directed immune responses. CD8-independent and antigen-specific T-cells in Th2-mediated immune responses were shown to be eotaxin- and STAT6-dependent [31,32,33,34,35,36]. In general, Th2 infiltrates in human cancers have not been studied extensively and some studies even suggested a better outcome with Th2-type cytokines [36]. The nature of Th2 responses in recognising mutant epitopes is not well explored at this time. The more detailed association of CD4 Th2 responses may also benefit from closer association of T-cells with the restricting MHC class II elements. For instance, previous studies reported Th1/Th2 CD4+ T-cell responses against NY-ESO-1 in DPB1*0401/0402-positive patients with ovarian cancer [37]. Much more information is available concerning the nature of the cellular immune response directed against peptides presented by the rather less variant (as compared to HLA-DR) HLA-DP molecules from infectious pathogens, e.g. Hepatitis B or MHC class II molecules that pre-dispose humans to certain autoimmune diseases (e.g. gluten-associated colitis) [37,38,39,40,41,42,43,44,45]. The impact of variant epitopes in association with certain MHC alleles that are associated with certain cytokine production patterns (IL-17, Th1, Th2) is unexplored up to now. Table 4.1 provides an overview of wildtype and mutant target epitopes recognised in TIL from patients with glioma, demonstrating that Th2 responses exist in the TCR repertoire from individual patients directed against mutant epitopes.
Processing and presentation of neoantigens may yield mutant epitopes (neoepitopes) that are shared as well as patient-specific (‘private’). This of course depends on the location of the mutation, i.e. point mutation which might disrupt the naturally occurring cleavage site and the nature of the mutation itself i.e. point mutation vs. chromosomal deletion vs. premature stop codons. A comprehensive analysis of somatic mutations in the HLA class I pathway, using DNA isolated from tumour and non-tumour tissue from patients representing 20 different cancer types, revealed a high likelihood for loss-of-function mutations occurring in the N-terminus of the HLA class I molecule, which abrogates transport of the peptide-HLA complex to the cell surface [46]. Furthermore, in all cancers tested, the most frequent mutations were found to occur in the α3 region of the HLA class I molecule, which is required for binding of the CD8 co-receptor on T-cells during an immune synapse for subsequent activation of the CD8- TCR complex [8].
3 Cancer Antigens and Epitopes: From Discovery to Therapeutic Application
Preclinical studies in the mouse model of human cancer, in particular melanoma, provided the first insights into cancer antigen discovery and functional characterisation, in the context of tumour rejection. Thierry Boon and colleagues had shown in the late 1980s that the tumor- antigen P19A, heterologously expressed in mouse P815 tumour cells (isolated from DBA/2 mice bearing methylcholanthrene-induced sarcoma), contains an HLA class I epitope (within a 13-mer sequence harbouring a point mutation) capable of eliciting potent CTL responses and lysis of target cells [47].
Epitope mining in the human cancer setting was first performed using tumour tissue derived from human melanoma lesions, spearheaded by groups in Europe and the United States. Thierry Boon, Pierre Coulie and colleagues at the Ludwig Institute in Brussels, Belgium discovered the first tumour-associated antigen (TAA) in 1991, after in vitro characterisation of CTL responses using melanoma cell lines derived from an anonymous patient MZ2 who had metastatic disease [48]. This TAA, first annotated as MZ2-E and later renamed as melanoma-associated antigen 1 (MAGE-1, cancer testis antigen), was recognised by an autologous CTL line and induced lysis of the tumour cell line expressing the MAGE-1 DNA and restricted by HLA-A1 [48]. Further work with a cell line from the same patient led to the discovery of MZ2-F, or as it is known today, G antigen 1 (GAGE-1) [49]. Much of the ongoing work at the time focussed on discovering novel immunogenic HLA class I-restricted antigens that mediated CTL reactivity and lysis of melanoma cells from patients, with a strong interest to first understand and then to develop immune-based interventions; Melan-A (HLA-A2+ epitope) [50]; MAGE-3 (HLA-A1+ epitope)-specific CTL response in a patient vaccinated with MAGE-3.A1 peptide [51].
Simultaneous efforts by researchers in Europe and the United States revealed another important cancer antigen, the cancer testis antigen NY-ESO-1, which was discovered by serological analysis of expression cDNA libraries (SEREX) (indicating the presence of antibody responses), using cDNA prepared from human oesophageal squamous carcinoma cells [52]. NY-ESO-1 was later shown by Elke Jäger and co-workers (Frankfurt) to contain biologically functional CD8+ (HLA-A2/B51) and CD4+ (HLA-DRB*1) T-cell epitopes, based on seminal studies performed on human melanoma cells as well as transfected T2 cells as a model [53,54,55,56]. The afore-mentioned T2 cells harbour a defect in the transporter associated with antigen processing (TAP), which in turn inhibits them to present endogenous cytosolic cytosolic peptides (except for some leader peptide sequences loaded onto HLA-A2 molecules), but accommodates the introduction of exogenously added HLA class I epitopes for CTL recognition assays [57].
Steven Rosenberg and colleagues at the Surgery Branch, National Cancer Institute (NCI, National Institutes of Health (NIH), Bethesda, MD) made pivotal contributions to antigen discovery in human melanoma, in particular those that induce reactivity among TILs: the tyrosine related protein 1 (TRP-1) or gp75 restricted by the HLA-A31 molecule in 1995 [58]; HLA-A31-restricted TRP-2 peptide LLPGGRPYR, which was a major target of TILs infused into a patient with metastatic melanoma who thereafter showed disease regression [58]; epitopes from TRP-1 and TRP-2 (TRP197–205) restricted by HLA-A31 as well as HLA-A33 [59]; a mutated epitope derived from triosephosphate isomerase restricted by HLA-DR1 and recognised by CD4+ TIL and cell division cycle protein 27 homolog (CDC27) epitope restricted by HLA-DR4 [60, 61]. Collectively, these early efforts (over a span of 15 years, from the late 1980s to early 2000s) provided an excellent foundation which lead to the expansion of the field of targeted cancer immunotherapy.
A whole series of other molecules were identified to be associated with transformed cells. For instance, mesothelin was discovered as a marker of several important solid cancers, i.e. mesothelioma, ovarian cancer, pancreatic ductal adenocarcinoma based on serological (a murine ‘Ki antibody’ recognising human mesothelin) and genetic analyses [62,63,64]. Further exploration of the clinical significance of this molecule in ovarian cancer, mesothelioma and squamous cell carcinomas, and in conjunction with measurable mesothelin as well as antibody responses in sera of patients, indicated the immunogenic potential of mesothelin and its designation as a legitimate cancer antigen [65, 66]. An experimental immunotoxin developed based on the mesothelin-binding region of the K1 antibody was among the earliest attempted targeted immune-based interventions, with preclinical studies performed in a murine model of human carcinoma xenografts [67].
Work implemented in the later part of the 1990s placed a greater focus on studying mutated proteins in human cancer cells, and the possibility of discovering mutated antigenic determinants (neoepitopes) presented by HLA restricting elements, with biological and clinical relevance in therapy. An early example is a neoepitope derived from melanoma ubiquitous mutated 1 protein (MUM-1, initially named LB33-B, after the patient from whom the melanoma tumour was obtained, LB33 [68]), which is restricted by the HLA-B*44*02 allele. This 9-mer neoepitope was identified following in vitro cytotoxicity studies directed against the autologous melanoma cell line LB33-MEL.A-1; the same cytolytic activity was not seen with the wildtype peptide sequence [69]. A 10-mer neoepitope (amino acids 23–32) from mutated cyclin-dependent kinase 4 (CDK4R24C) protein, restricted by HLA-A*0201, was also shown to mediate cytolytic activity by autologous CTLs in a dose-dependent manner, when exposed to T2 cells transfected with the CDK4R24C cDNA [70]. A caspase 8-derived mutated peptide restricted by HLA-B*3503, which showed potent cytolytic activity against the autologous head and neck cancer cells as well as tumour cDNA-transfected B-cell lines [71] further strengthened the field of neoepitope mining from human cancer cells.
A high-throughput analysis of whole genomic as well as exomic DNA from clinical tumor samples representing thirty different human cancers revealed the unique mutational burden in each cancer type, in addition to specific mutational signatures characterising these cancers [72]. Although this provides an elegant view of the general landscape of mutational burden in human cancers, the mutational signature in each patient varies—thus giving rise to a ‘compendium’ of private mutational signatures involved not only in driving and maintaining malignant transformation, but also in the activation and expansion of immune effector cells.
The mutated form of the V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog, or known as KRAS in short, is a well-established neoantigen implicated in the pathogenesis of pancreatic, colorectal and lung cancers [73,74,75,76]. Native KRAS was discovered in 1982 following gene sequencing of human lung adenocarcinomas, and is a guanine triphosphatase involved in cellular signal transduction [77]; however, mutations at positions 12, 13 and 16 are associated with oncogenesis, thus making it a proto-oncogene in humans.
Steven Rosenberg and colleagues at the Surgery Branch, National Institutes of Health (Bethesda, MD) recently developed a cutting-edge approach to screen for neoepitope-specific T-cell responses for individual patients. This method has been termed the ‘tandem minigene (TMG)’ approach, which first requires whole-exome sequencing data of genomic DNA isolated from patients’ tumor tissue samples. The sequencing data then yield all non-somatic mutations contained within gene-coding DNA of the patient. This allows for constructing a personalised library of the patient’s ‘private’ mutations that potentially code for neoepitopes. These short gene sequences are then put together, an artificial construct, and inserted into an expression plasmid, which is subsequently transfected into a lentiviral vector for infection of APCs, i.e. dendritic cells (DCs) from a patient. Autologous TILs (from the same patient) are then co-cultured with the TMG-bearing DCs to allow induction of immune-reactivity. A positive response, represented by IFN-γ production by the TILs, would signal that the co-cultured DCs harbour a TMG that includes a neoepitope-encoding sequence(s) that is/are naturally processed and presented to the immune system [78].
Mutations may lead to different, not mutually exclusive effects on the responding T-cell population based on the prerequisite that a mutant epitope is indeed processed and presented to T-cells: (1) a T-cell may be newly recruited that would exclusively recognize the mutant sequence, (2) potential T-cell receptor (TCR) cross-reactivity between wildtype and mutant epitope sequences (if both, the wildtype and the mutant epitopes are being processed and presented to T-cells). It is biologically relevant whether there is already a T-cell population expanded that recognizes the wildtype epitope and then, following malignant transformation, recognizes the mtuant target, since this situation may lead to differential TCR triggering and subsequently to differential T-cell effector functions.
Mutant Epitopes as the ‘Biological Scalpel’ Against Cancer Cells
Increasing immune effector functions by recruiting T-cells that recognise mutant epitope sequences is a clinically attractive attempt to improve and broaden the TCR repertoire directed against mutations that exclusively exist in malignancies and would therefore represent the ‘ideal’ cancer-associated antigen - a ‘biological scalpel’ that would only target cancer cells and not harm non-transformed cells. One method is to modify peptides at residues that do not interact with the nominal MHC restricting molecule, yet with the TCR contact residues: these variants are called ‘heteroclitic’ analogues and are able to trigger the nominal TCR with differential T-cell effector functions, e.g. cytotoxicity, quality and quantity of cytokine production, as well as proliferation [79]. This approach has been used to induce T-cells that react to wildtype peptides, e.g. to p53, yet are elicited with a variant peptide. If single mutations occur in epitopes, it could yield peptides that are naturally processed and presented by tumour cells to TCRs. What could potentially happen if a T-cell response, directed against a wildtype target, will also be able to able to react to the corresponding mutant target epitope, presented by the identical, nominal HLA-restricting element?. The following scenarios may occur, which have been described already in the early 1990s from several groups: In general, a single ligand specificity for each individual TCR appears to be rare. In contrast, the TCR recognition has been shown to be flexible, induced by altered peptide ligands, grouped into antagonists, partial agonists and superagonists. Mutations within peptide targets can induce differential phosphorylation of the TCR/CD3 complex with a differential downstream signalling pathway configuration [80]. Mutations may therefore—in case the wildtype peptide is also recognized—lead to abrogation of T-cell recognition simply because the ligand is not processed and presented. Alternatively, the T-cell ligand may well be processed and presented, but the T-cell signal may be abrogated potentially due to cellular anergy [29]. Partial agonists, i.e. by inducing a single amino acid residue, will still be able to stimulate the T-cells directed against the wildtype peptide, even across a similar dose range of the nominal epitopes: as the T-cells with the wildtype TCR ligand react with proliferation, cytokine production and cytotoxicity, peptide variants may either induce cytotoxicity and/or cytokine production, in the absence of T-cell proliferation [81, 82]. Of note, a similar observation may be true for TCRs directed against the mutant epitope that would react with a qualitatively and quantitatively differential T-cell reactivity pattern. Single amino acid exchanges may also turn T-cell clones from a Th1 into a Th2 cytokine production pattern, or lead to T-cell clones with abrogated cytokine production, yet strong cytotoxic T-cell responses as shown for viral pathogens [81,82,83]. Mutations in nominal targets, associated with differential signalling events, may also be crucial for the differentiation status of T-cells reacting to wildtype as well as to mutant targets, first described in preclinical models of thymocyte differentiation and maturation [84, 85]. Differential triggering of the nominal TCR is associated with T-cell maturation and differentiation—a quality that is important for long-term immune memory, access to (tumor) tissue as well as for clinical efficacy of T-cell therapy. The passive transfer of immune cells directed to TAAs has been shown to be clinically relevant as the transfer of the T-cell product leads to the generation of central memory T-cells [86]. The biochemical signals that govern T-cell memory rely not only on the cytokine environment, yet also on the signal strength delivered by the TCRζ chain complex; the quality and quantity of T-cell responses including T-cell memory is strikingly shaped by the strength of the MHC/peptide–TCR interaction [87], which may in part be relevant for CD8+ T-cells. A decreasing potential model has been proposed, gauging the signal strength delivered by the target epitope to the corresponding TCR that is dictating whether the T-cell most likely enters the T-cell memory pool [88, 89]. This ‘signal strength’ model will need to take into account the locally produced cytokines and pro-inflammatory signals associated with moving T-cells into the diversity of the memory T-cell pool. In general, weaker TCR signals are sufficient in order to move T-cells into a memory T-cell program [90, 91]. Not mutually exclusive, the length of the TCR signalling (i.e. shortening the TCR stimulation) will also decide whether T-cells enter the memory T-cell pool [92, 93]. The observation that point mutations within peptides affect the contact with the nominal TCR also impairs CD8+ T-cell memory development, mediated in part by TCR-dependent NFκB signalling [94]. This may partly explain why T-cell clones targeting the identical (mutant) tumor epitope exist in heterogeneous differentiation states (see Fig. 4.2).

Different T-cell clones (A: VB5.1, B-D: VB9) recognize a naturally processed and presented (mutant) target on autologous pancreatic cancer cells. Note that the cancer - directed T-cell clones expresses different T-cell homing and differentiation markers, defined by CD45RA and CCR7 expression, i.e. CD45RA+CCR7+ T-cells are precursor T-cells, CD45RA-CCR7+ central memory T-cells, CD45RA-CCR7- T-cells memory effector T-cells and CD45RA+CCR7- T-cells represent terminally differentiated effector T-cells. Note that the majority of cells derived from clone B reside in the central memory T-cell subset that has been shown to be associated with increased responsiveness in the cellular therapy of cancer
The role of gamma-delta (γδ) T-cells in cancer is now also being revisited, due to their non-classical recognition of antigens. γδ T-cells recognise non-peptide structures, i.e. phosphoantigens such as derivatives of the eukaryotic isoprenoid (mevalonate) pathway presented by the CD1d molecule [95]. The Vγ9Vδ2 (Vδ2+) subset of γδ T-cells, which are found in peripheral blood. They have also been described to express the CXCR3 surface marker which is crucial for tissue penetration, an important feature in accessing transformed cells or tissue-residing pathogens. An interesting feature of Vδ2+ γδ T-cells is that they express the CD16 co-receptor, which can bind to FcγRIII present on tumour cells in addition to the killer receptor NKG2A [96]. Thus, like NK cells, γδ T-cells can also orchestrate antibody-dependent cellular cytotoxicity (ADCC), which is implicated in the therapeutic activity of several monoclonal antibody-based cancer drugs, i.e. rituximab, trastuzumab, ofatumumab and alemtuzumab [97,98,99]. An intermediate of the isoprenoid pathway, isopentenyl pyrophosphate (IPP), is strongly recognised by Vγ9Vδ2 T-cells, as shown in the context of zoledronic acid-treated human cancer cells [100]. Zoledronic acid induces accumulation of IPP in cancer cells, thus stimulating the activity of Vγ9Vδ2 T-cells, subsequently promoting the production of IFN-γ as well as cytotoxic molecules. This effect can be further enhanced in the presence of IL-2 and/or IL-15 conditioning. Although altered/mutated forms of IPP are yet to be reported, the significance of γδ T-cells in targeted cellular therapy should be explored further. There have also been reports of the recognition and killing of overexpressed human heat shock protein 60/70 on cancer cells by γδ T-cells, indicating that the overall T-cell repertoire in human which recognizes tumor antigens is rather generous [101, 102].
4 Clinical Significance of Neoepitope-specific Immune Responses
The clinical value of neoantigen-specific responses is most evident in immune checkpoint blockade therapy. Case reports of patients with melanoma or non-small cell lung cancer treated with anti-PD-1 and anti-CTLA-4 monoclonal antibodies showed that the repertoire of neoantigen-directed CD8+ T-cell responses (based on the diversity of TCRs recognizing mutated peptides) is is associated with clinical responses [103,104,105,106,107,108]. The most relevant examples are the T-cell responses from patients with metastatic melanoma or non-small cell lung cancer NSCLC, whereby the number of PD-1+ circulating T-cells directed against neoepitopes (visualised by flow cytometry) associates with clinical outcome in patients [105, 109, 110]. Furthermore, more recent clinical observations indicate that neoepitope-specific immune responses in peripheral blood can be used as a prognostic marker for several solid cancers [107, 110, 111].
Tissue scarring, arising from inflammatory processes processes, associated with infection(s), may lead to to genetic aberrations, which in time may perpetrate oncogenesis. Observations in patients with lung adenocarcinomas who had previously contracted M. tuberculosis infection in the lung showed that immune responses to mycobacterial antigens (‘old’ tuberculosis (TB) lesions) caused mutational changes to the gene encoding epidermal growth factor receptor (EGFR), in association with cancer development [112]. More strikingly, these patients had a worse 1-year survival prognosis compared to those who did not have ‘old’ TB lesions in the lung at cancer diagnosis. Patients presenting with ‘old TB lesions’ and adenocarcinomas in the same lung did not harbour the EGFRL858R mutation (occurring in exon 21 of the EGFR gene, which encodes the intracellular tyrosine kinase domain of the receptor), which is implicated in positive clinical outcomes in patients with lung cancer who are treated with the tyrosine kinase inhibitor gefitinib [113]. The EGFRL858R mutation has been shown to give rise to neoepitopes that induce antibody responses in patients with NSCLC who received gefitinib therapy [114]. Another EGFR-associated mutation, EGFRT790M, which is found in approximately 60% of patients with NSCLC, yields HLA-A2-restricted neoepitopes that are linked to favourable anti-tumor immune responsesthat could be implemented for designing better immunotherapies [115, 116].
The agonistic activity of peptides, namely their ability to stimulate T-cell activation can in fact shape the cellular immune response milieu due to mutational changes in their molecular structure. Paul Allen and co-workers had elegantly shown in the mid-1990s that peptide analogues of staphylococcal enterotoxin A, derived from haemoglobin, can abrogate the effector functions while inhibiting the proliferation of T helper cells (CD4+ T-cells with a Th1 or Th2 phenotype) [117]. While some mutations in the haemoglobin peptides inhibited T-cell proliferation, other mutations did not have a deleterious effect on the T-cell. Further research showed that partial phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM), which forms an indispensable component of the intracellular TCR zeta (ζ) chain, can either totally abrogate or even lead to T-cell death during an immune synapse [118]. Importantly, this phenomenon can be due to the binding of TCR with HLA molecules presenting mutated peptides, and more importantly, the nature of the mutation itself and the very position of the mutation within the epitope sequence. It is undeniable that the local inflammatory milieu in cancer lesions (such as those described in chronic infections [119]) may also contribute to chromosomal aberrations resulting in strong downregulation or loss of the TCRζ chain. These seminal findings were first reported in a preclinical murine model of colon carcinoma and later in TILs from patients with renal cell carcinoma and peripheral blood T-cells from patients with non-Hodgkin’s lymphoma [120,121,122].
Preclinical studies of infectious disease models may provide an insight into TCR repertoire shaping in relation to neoepitope-specific immune responses. Analyses of splenic and bronchoalveolar lavage fluid-derived T-cells from mice primed with a wildtype strain of influenza A virus (HK/PR8) by intraperitoneal infection showed that animals’ CD8 TCRVβ repertoire was shaped by primary viral challenge to efficiently recognize and respond to a secondary challenge with another wildtype strain but not a mutated version of either virus (HK/PR8-NPN3A) [123]. Also, while challenge with a wildtype virus strain provided a broader TCRVβ repertoire, the mutant strain of the virus induces a more focussed and narrow antigen-specific T-cell compartment, with subtle TCR re-arrangement patterns. Furthermore, an immunogenic epitope from the wildtype virus (NP366, ASNENMETM) induced a measurable CD8+ T-cell response among mice primed and re-challenged with a mutated viral strain. Conversely, the mutated version of the NP366 epitope, harbouring only a single amino acid change (NPN3A366, ASAENMETM), did not promote strong binding between MHC and TCR among T-cells from mice challenged with a wildtype virus, exhibiting a high ‘off-rate’ (large percentage of mutated epitope-bearing tetramers dissociating from the TCR within minimal time), requiring greater dependence on the CD8 co-receptor binding to MHC to elicit an immune response. This is of relevance to immune responses in cancer; T-cell reactivity to neoepitopes may be subdued owing to poor binding kinetics between the HLA-restricting element and the TCR. However, vaccination with a broader array of personalised neoepitopes may help prime the immune system to either re-awaken the smaller populations of central memory T-cell that are tumor-reactive, or not mutually exclusive, generate a fresh pool of (as yet not activated) antigen-specific T-cells. [124].
5 Harnessing Basic Immunology to Improve Clinical Immunotherapeutic Approaches
The effect of gut commensal bacteria on shaping (and re-shaping) immune responses in health and disease has been at the heart of current immunological research. It was recently shown that induction of T-cell responses to select, ‘immunogenic’ intestinal bacteria (Bacteroides thetaiotaomicron and B. fragilis) driven by anti-CTLA-4 therapy correlates with clinically beneficial outcomes in patients with metastatic melanoma [125]. In a murine model highly susceptible to tumors, the introduction of Bifidobacterium sp. notably improved cytotoxic lymphocyte-dependent control of tumor burden [126]. Combination of Bifidobacterium inoculation and anti-PD-L1 monoclonal antibody administration further enhanced tumor control in these animals, thereby underlining the critical role of gut microbiota in dictating anti-cancer immune responses. Thus, supplementing biologically active material from intestinal bacteria with immune blockade therapy or T-cell immunotherapy may potentially improve neoantigen-specific immune responses in patients with advanced cancer.
Small molecules and cytokines that target the activation of fatty acid oxidation (FAO) in CD8+ T-cells and promote maintenance of cellular memory can be used as an adjunct to mainstream therapeutic regimens in cancer and infectious diseases. For example, the antidiabetic drug metformin activates 5′ adenosine monophosphate-activated protein kinase (AMPK) and improves FAO in memory CD8+ TILs, as shown in a proof-of-concept study in a murine model of chemically induced skin cancer [127]. IL-15 also promotes lipid metabolism by upregulating mitochondrial biogenesis and inducing the expression of carnitine palmitoyl transferase, an enzyme that is critical for mitochondrial beta-oxidation [128]. This process has been shown to be upregulated in memory CD8+ T-cells in mice, and enhances their survival. In cancer therapy, IL-15 has already been evaluated as an instrumental adjuvant with pronounced effects on proliferation of TIL and enhanced cytotoxic activity of tumour-antigen specific T-cells [129,130,131,132,133,134].
Although immunological tolerance of T-cells is necessary to prevent overt pathology, increased numbers of regulatory T-cells (Tregs) have significant implications for the success of cell-based immunotherapies. While infusion of mesenchymal stromal cells for downregulation of severe inflammation requires subsequent TGF-β production and Treg activation [135], T-cell products reinfused into patients with cancer are allowed to contain only minimal Treg populations in order to optimise anti-tumor activity mediated by cancer-specific T-cells [136]. In addition, IL-17 production in response to chronic inflammation in the tumor microenvironment can induce TGF-β production and suppression of CD8+ T-cell responses [27]. Tregs could also be stimulated by TAAs, i.e. NY-ESO-1157–170-specific Treg responses in patients with melanoma given the NY-ESO-1/ISCOMATRIXTM therapeutic vaccine [137]; NY-ESO-1119–143 and TRAG-334–48 (derived from another cancer testis antigen, Cancer/Testis Antigen Family 24) can induce the expansion of both Th1 cells and FoxP3+ Tregs in patients with melanoma [138], and therefore contribute to immune evasion. More research is needed to better understand whether certain mutations would represent the nominal epitopes for Tregs directed specifically against cancer mutations.
New information arising from basic research needs to be considered for inclusion into preclinical (pre-GMP) evaluation of T-cell products. For example, analysis of BTB Domain and CNC Homolog 2 (BACH2), a transcription factor that promotes the generation and maintenance of regulatory as well as central memory T-cells in the host while repressing immune effector mechanisms could be a useful tool in characterising the T-cell populations which may persist in the patient to fight transformed cells [139]. Mice lacking BACH2 were able to mount a strong T-cell response in the tumor microenvironment (marked by CD4+ and CD8+ T-cell proliferation and IFN-γ production), concomitant with reduced numbers of FoxP3+ regulatory T-cells, which subsequently allowed forallowed for improved tumor control. BACH2 deficiency also increased gene transcription of cytotoxic molecules, i.e. granzymes and perforin. From a translational viewpoint, this finding has direct implications for enhancing the T-cell-mediated anti-tumor effect in targeted cellular immunotherapy. Regulating the expression of BACH2 in neoantigen-reactive T-cells in diseased tissue, i.e. TILs as well as peripheral blood T-cells may improve the quality and efficacy of immune cells for exploitation in clinical therapy [140].
Exploiting novel technology platforms to screen for TCR specificities in diseased tissue, i.e. deep (TCR) sequencing, peptide microarrays, cellular microarrays and TCR-epitope docking studies are contributing substantially to our current knowledge of disease mechanisms and immune dynamics. This may allow to characterise in greater detail the immune repertoires crucial for orchestrating long-term immunological protection against cancer. Combining mathematical knowledge and biological understanding of cancer dynamics and tumor development algorithms will benefit this field greatly. Furthermore, implementing comparative studies using these techniques with clinical samples from various anatomical sites of healthy individuals and patients will deliver new information for the development of next-generation biotherapeutics. The differential TCR repertoires in TIL versus PBMCs, in addition to the mutational load in a patient with cancer relate to the success of checkpoint inhibitors. This is evident in patients with metastatic melanoma and NSCLC, who have among the highest mutational burden and respond well to anti-PD-1 and/or anti-CTLA-4 therapy [3, 103,104,105, 107]. ‘Mining’ biologically and clinically relevant TCRs targeting cancer mutations may lead to the generation of T-cell products for therapy by cloning and transferring specific TCRs to PBMCs; at present a viable and pursuable platform, as already shown in a patient with metastatic colorectal cancer [86, 141, 142]. These examples underline the importance of a topic that has been discussed for decades, e.g. in the field of cellulary immune responses directed against HIV: How much focus and how much diversity should a tailored immune response directed against mutant epitopes afford? How much diversity, with regard to focus on single epitopes, is biologically and clinically relevant taking into account the (1) similarity of mutations in primary tumors versus relapse, (2) the mutational diversity displayed by the primary tumour and distant metastasis as well as (3) the ‘local’ imprint of gene expression (bearing in mind that not all gene-encoding DNA may at all, or at some points be translated into RNA and then subsequently into protein), associated with the tissue environment (e.g. lung versus liver-metastases). These questions will represent a matter of clinically relevant research with impact on the design of biologically and clinically relevant studies. For instance, an educated decision will take into account the similarity and dissimilarity of the primary tumor versus the corresponding relapsed malignancy, if TIL would be immediately available from the primary tumor upon clinical detection of a relapse.
The humoral immune response to cancer antigens, and thus its significance in mediating clinically relevant and beneficial anti-tumour responses in patients calls for greater emphasis [114, 143,144,145]. Along these lines, peptide microarray studies possess the sensitivity and specificity to discover naturally presented epitopes recognised by circulating antibodies in serum as well as those derived from patients’ B cells in culture. In serum derived from patients with cancer, disease-associated epitopes may include those belonging to neoantigens, and can be screened for using the high-content peptide microarray (HCPM) platform. The HCPM is a novel technology used for profiling antibodies in many research areas, which has been more recently been developed, including our research group, in order to visualize an unbiased view of serum reactivity to a wide range of epitopes. This sophisticated technology allows to display on each individual microarray slide 2.9 million peptide sequences (spots), corresponding to unique epitopes. Using only a small sample volume (i.e. 4 μL of biological fluid per slide), it is possible to identify immune-recognition patterns associated with relevant endpoints on a HCPM microarray chip containing the whole human proteome, at the highly detailed level of 16-mer peptides. Well-documented experience in the use of the HCPM platform with regard to chip design, pre-processing and methods of analysis as well as techniques [146,147,148,149,150,151], and different applications of HCPM in various clinical settings, i.e. bacterial infections [149, 152, 153], viral infections [154,155,156], sarcoidosis [157] and pertussis [151] (further references for readers: 146,147,148,149,150,151,152,153,154,155,156,157) strongly suggests that this platform is also able to pick up very specific serum reactivities directed against mutant versus wildtype target molecules. This technique can also be used to detect humoral immune responses to ‘private’ neoepitopes in the peripheral blood of patients with cancer [158]. Results from HCPM studies can contribute to developing novel antibody-based therapies including the identification of novel,clinically relevant cancer - associated targets for CARs, or augment cellular immune responses directed against (intracellular) mutant antigens via ADCC. On the other hand, B cell-dependent immune responses in disease may aid to modulate a T-cell driven ‘immunopathological’ milieu, such as that observed in patients with post-transplantation lymphoproliferative disease (PTLD) [159]. The early studies and identification of cancer-associated antigens supported the hypothesis that strong B-cell responses point to the existence of strong anti-cancer T-cell responses in patients with cancer. For instance, the cancer testis antigens MAGE or NY-ESO-1, which are clinically relevant T-cell targets, were identified via B-cell responses and are currently used in clinical protocols to induce disease-modifying T-cell responses targeting NY-ESO-1+ cancer lesions [132, 160,161,162,163,164].
Where Could Anti-Mutation-Reactive T-cells be Harvested?
Immune cells from peripheral blood express tissue-specific homing markers of their surface (e.g. VLA-4 for the central nervous system, CXCR3 for the lung, CCR6 for the gut) that can be instrumental in gauging circulating T-cells among PBMCs that are travelling either from or to the respective target organ [165]. Enrichment, e.g. for VLA-4+ T-cells, will result in selecting T-cells trafficking to and from the tumor lesions in the patient, which can then be tested for their recognition of neoepitopes and potential immunoreactivity targeting transformed cells. Furthermore, some of the epitopes recognised in target organs overlap with the recognition patterns observed in PBMCs, while others do not and are either exclusively recognised in PBMCs or TILs [109]. In line with this, 15 out of 20 HLA-A2+ patients with breast cancer whose tumor and blood samples were analyzed were shown to harbor 18 TCR specificities shared between TILs and PBMC-derived T-cells [166]. Sim et al. reported in 2016 that the complementarity determining region (CDR) 3 of the TCR, the portion of the complex which binds to the HLA-peptide complex on targets cells, is greatly diverse between PBMCs and TILs among patients with glioma [167]. Importantly, this research consortium also found a unique TCR signature present in peripheral blood of the patients exhibiting a minimally divergent TIL TCR repertoire concomitant with low-grade glioma, while patients with glioblastoma showed a wider selection of TCRs. We have also noticed this among patients with glioblastoma, where some of the somatic mutations are recognized by PBMCs but not TILs, and vice versa (Liu et al., unpublished data). Thus, information arising from such studies is already translated into clinical products for patients with advanced cancer, i.e. genetically transferring the TCR repertoire associated with better prognosis into PBMCs, for re-infusion as adjunctive therapy, given the feasibility of using advanced gene transfer technologies [141, 142, 168]. Dr. Rosenberg’s group at the NIH has in fact treated patients with advanced cancer harbouring particular mutations using autologous T-cell products expressing specific TCRs directed against neoepitopes. Pivotal examples include the treatment of a patient with metastatic cholangiocarcinoma with CD4+ IFN-γ+ TILs reognizing an HLA-DQ*06-restricted neoepitope derived from the receptor tyrosine-protein kinase erbB-2 (ERBB2), or HER-2 interacting protein (ERBB2IP) [169], and a patient with metastatic colorectal cancer, who received a TCR-transferred T-cell product specific for the KRASG12D mutation (HLA-C*08*02-restricted) driver mutation, with subsequent regression of metastases expressing the KRASG12D mutation [86]. The latter strategy was initiated by a TMG screen performed on TILs isolated from tumour tissue samples obtained from 10 patients with metastatic gastrointestinal cancers, in the quest to detect neoepitope-specific reactivity [170]. Patients with the HLA-C*0802 allele had TIL responses to their tumor cells (directed to KRAS) underlining that the restricting HLA element in the patient may limit the therapeutic targeting of KRAS, as other (KRAS) mutations may not be visible to the cellular immune system, since these mutations may not be naturally processed and ultimately be presented to the patients cellular immune repertoire.
Identification and verification of clinically and biologically relevant neoepitopes remains a challenge, i.e. whether the epitopes are expressed in a representative fashion, whether they are processed and presented on tumour cells, and whether a ‘fit’ TCR repertoire is available and capable of reacting to it—leading to immune effector functions that will most likely facilitate a strong and long-lasting anti-tumor immune response. A possible way is to design and generate potential neoepitopes based on whole genome sequences from patients with cancer. Once these neoepitopes are chemically synthesised, they could be submitted for large-scale in vitro screening of T-cell cultures to carefully select for high quality, mutation-specific T-cells that could be expanded and reinfused into patients. In a non-mutually exclusive fashion, the TCRs exclusively targeting mutant TAAs, and not the wildtype peptide sequence, may be cloned and subsequently transferred into recipient target cells (T-cells) for the active cellular therapy of patients with cancer [86, 103]. PBMCs may serve as a very good starting point to screen for populations of neoepitope-specific T-cells circulating (and re-circulating) in the patient. The fact that peripheral T-cells are able to recognise mutant epitopes/neoepitopes has been heralded by the Rosenberg group, pointing to PD-1+ T-cell populations that are enriched for tumor neoantigen-specific T-cells that rather reflect the repertoire of antigen-experienced and not only ‘exhausted’ T-cells [109, 171]. This echoes earlier findings in TIL from patients with metastatic melanoma: PD1+ TIL recognize ‘private’ mutations presented by tumor cells. However, the heterogeneity of cancer lesions needs to be further investigated concerning the anatomy of the ‘diversity of mutanomes’ and the diversity of the corresponding immune effector cells that could be harvested from individual cancer lesions [140]. An essential point for TCR transfer targeting commonly shared or private mutations is that the HLA-restriction element presenting cancer epitopes differs from individual to individual. Thus, not all patients would benefit from a single TCR-HLA-matched T-cell product/TCR transfer but rather a more personalized approach, taking into account the neoepitope structure, the corresponding TCR sequence(s) as well as the HLA-restricting element. An illustrative example in this regard is the occurrence of PTLD associated with Epstein–Barr virus (EBV)-induced inflammatory T-cell responses [159]. Individuals with an HLA- A2, A11, B5, B18, B21, Bw22 and B35 background suffer a greater risk of PTLD onset following solid organ transplantation compared to those an HLA-A03 or HLA-DR7 (CD4+ T helper cell response) genetic background, while individuals with an HLA-A1, B8 or DR8 appeared to be protected against PTLD [172,173,174]. Careful selection of specific TCRs and HLA restriction is also being pursued in targeted immunotherapy of patients with hepatitis B virus (HBV)-induced hepatocellular carcinoma, with a focus of HBV-specific epitopes and potentially, neoepitopes [175]. Ton Schumacher’s research group at the Netherlands Cancer Institute in Amsterdam recently showed that since naturally occurring neoepitope-specific T-cell responses may be silenced in patients with cancer and that T-cells isolated from peripheral blood from healthy individuals may contain neoepitope-recognising TCRs [176], the latter option may represent the starting point of generating anti-mutant epitope reactive T-cells. Five different HLA-A2-restricted neoepitopes were recognized by CTL lines, established from PBMCs obtained from different healthy donors, and produced IFN-γ and/or CD107a in response to patient-derived melanoma cell lines that expresses the cognate (mutant) T-cell epitope. Also, T-cells which recognized the neoepitopes as well as the tumor cells expressed epitopes which exhibited strong peptide-HLA binding kinetics (half-life of MHC-class I–β2 microglobulin–epitope interaction) Specific TCRs could then be cloned and heterologously expressed in T-cells from an individual who is in need of an ‘improved’ TCR repertoire tailored to target mutation-specific T-cells. This has been shown in PBMCs from patients with cancer using T-cells from MHC-matched donors [176] and has been shown to be clinically feasible in the context of infectious diseases, i.e. by transferring CMV-specific T-cells from an allogeneic-donor matched for the HLA class I-restricting allele, to patients with CMV infection (and a non-functional immune system) after allogeneic hematopoietic stem cell transplantation [177, 178]. Similar observations have been generated in our laboratory using HLA class I-matched TIL (see Table 4.2), that have been shown to react—i.e. without restimulation and expansion—to mutant target epitopes that are reognized on autologous tumor cells from an HLA-B*2705 matched patient, suggesting that precursor T-cells exist, even in TIL, following exposure to epitope arising from ‘driver’ mutations, i.e. KRAS, SMAD4, p53 or even commonly shared mutations among individuals [1, 86, 179].
Preparing the Ground: Enhancing the TCR Repertoire to Mutant Targets
The activation of the innate immune compartment using standard anti-cancer drugs, i.e. gemcitabine, decitabine, cisplatin and doxorubicin can prompt the activation of APCs such as macrophages and dendritic cells, and facilitate antigen-processing and orchestrating pro-inflammatory immune response, i.e. IL-12 production [180]. In addition, chemoimmunotherapy with decitabine has also been shown to induce potent anti-cancer cytotoxic responses mediated by CD8+ T-cells [181], while its use in patients with pancreatic cancer along with cytokine-activated killer cells leads to improved progression-free survival compared to chemotherapy alone [182]. Increase in neoantigen-specific cellular immune responses following adjuvant cancer therapy has not been explored in depth, yet could be the focus of future evaluations - directed towards enhancing the ‘visibility’ of the patients’ mutanome to their immune repertoire. Several approaches could be explored, which is beyond the scope of this overview. A strong argument that anti-cancer immune responses can be increased with ‘standard therapeutic manipulations’ is the observation that in patients with brain metastases who underwent whole-brain radiation therapy (WBRT), immune responses were activated that resulted in tumor regression at distant lesions [183]. Patients with advanced melanoma-related brain metastases appear to have a greater survival advantage when they are treated with ipilimumab (anti-CTLA-4) in addition to WBRT [184]. Along these lines, preclinical evaluation of radiotherapy-induced CTL responses have shown that the mutational landscape shifts alongside modifications in HLA class I antigen processing and presentation, subsequently concomitant with enhanced control of the tumor burden [185]. In general terms, ionising radiation promotes immune stimulatory events such as (1) increased HLA molecule expression, MHC-I expression, (2) generation of specific peptides involved in cytotoxic T-cell recognition and (3) promotion of cytotoxic T-cell activity by the release of tumor associated antigens [186, 187]. The latter processes lead to the activation and trafficking of effector cells promoting in situ cellular immune responses, although distant (abscopal) responses may also occur [186,187,188,189,190]. In this particular context, dose and fractionation seem to play a determining role for eliciting anti-tumor immunological effects. The preclinical studies of the Demaria group on breast- and colon cancer models showed a clear indications of anti-tumor T-cell responses when combining local fractionated radiation schedules (8gy × 3, 6gy × 5) in addition to CTLA-4 blockade [186, 187, 191, 192]. Other preclinical/clinical studies lend support to the relevance of such combined approaches [186, 187, 193]. Considering the inhibitory effects of monoclonal antibodies on CTLA-4 and PD-1-modulated immune suppressive actions taking place in metastatic non-small cell lung cancer, melanoma and renal cancer, further studies aiming to identify the potential adjunctive effects of radiation on local and distant sites are certainly necessary [186]. Postow et al. reported a case of a patient diagnosed with metastatic melanoma treated with ipilimumab; at a stage, the patient underwent hypofractionated radiotherapy to treat a paraspinal metastasis; the patient was treated with 28.5 Gy delivered in three fractions over a period of 7 days. Four to five months later, the paraspinal mass and a group of previously identified distant hilar lymphadenopathies and splenic lesions (not targeted by radiation) had considerably diminished. CT-imaging 10 months after treatment still showed a stable condition [194]. Reproducing these latter described effects in a larger group of patients requires consideratino of many biological and clinical variables, among others mathematical models able to provide qualitative and quantitative predictive data on radiation-induced immune responses. Image analysis that may even reflect the ‘mutational load’ and T-cell infiltration may also provide a prognostic tool, yet also an instrument to gauge how ‘focused’ and narrow or broad a T-cell response directed against mutated antigens should be (see Fig. 4.3) in order to provide increased survival.

MRI scans of 4 patients with glioma (grade 4, WHO classification) with (i) survival time in days after surgery and (ii) the number of mutant epitopes recognized by the patients’ TIL defined by IFN-gamma production in relation to the total number of mutations recognized. A focused TIL response appears to be associated with increased survival, an observation that needs to be followed up in larger studies
Although (limited) data suggests that a very focused immune recognition is clinically more favourable (as defined by survival), more basic and clinical research needs to be undertaken to visualize a link between mutational load, the number of targets recognized, the possibility to tailor the T-cell graft targeting mutations and, subsequently, clinical responsiveness. Thus, it is plausible that activation of neoepitope-specific T-cells in patients after a combination of radiotherapy and immune checkpoint blockade therapy is key to improved clinical outcomes, and thus warrants further exploration in well-controlled clinical settings. Full-scale analyses of surface tissue-homing markers as well as memory markers on neoantigen-reactive T-cells, the relationship between these readouts and the anatomical locations of metastasis in the patient as well as the functionality of the T-cells are of paramount importance to enrich our understanding of targeted cellular immunotherapies in cancer [195]. These immunological analyses can be performed on clinical samples obtained from patients undergoing therapy, i.e. peripheral blood drawn at various time points during ‘standard’ or immunological treatment strategies. Another major conundrum in further optimising targeted T-cell-based therapies lies in the lack of our understanding of antigen processing and presentation, i.e. which epitopes are naturally presented, what is their respective tissue expression pattern, their corresponding HLA restriction and their capacity to drive antigen-specific T-cell responses, based on the TCR repertoire capable of reacting to individual mutant target epitopes. Further research into the ‘immunological fitness’ of antigen-specific T-cell populations may impact on the quality of cell-based therapies and further aid to tailor T-cell products. For instance, recent advances in T-cell therapy for cancer, viral infections and autoimmune diseases highlight the broad therapeutic potential of T-cell engineering. Even as site-specific genetic manipulation in primary human T-cells remains challenging, they hold great clinical promise to tailor T-cell products, e.g. genome editing in T-cells using the CRISPR and TALEN approaches [196] along with the detailed analysis of asymmetric T-cell division in order to better understand and define the quality of mutational epitopes that would give rise to immediate immune effector cells, as well as to long-term memory T-cells [197]. New and clinically relevant insights as to how exposures to pathogens and ‘environmental factors’ may impact on TCR repertoires and ultimately disease susceptibility [198] will aid to decipher the molecular ‘decision-making process’ in adaptive cellular immune responses targeting mutant epitopes in patients with cancer [199, 200] and provide the necessary tools to enhance treatment decisions to offer more effective, multi-layered and long-term cellular immune responses for patients with malignancies.
References
Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4):209–22.
Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337–50.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81.
Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013;8(5):e63818.
Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12(10):1198–202.
Singh A, Dey AB, Mohan A, Mitra DK. Programmed death-1 receptor suppresses gamma-IFN producing NKT cells in human tuberculosis. Tuberculosis. 2014;94(3):197–206.
Wykes MN, Horne-Debets JM, Leow CY, Karunarathne DS. Malaria drives T cells to exhaustion. Front Microbiol. 2014;5:249.
Abbas A, Lichtman A, Pilai S. Basic immunology: functions and disorders of the immune system. Philadelphia: Elsevier Saunders; 2014.
Hammerling GJ. The 1996 Nobel Prize to Rolf Zinkernagel and Peter Doherty. Cell Tissue Res. 1997;287(1):1–2.
Zinkernagel RM, Doherty PC. Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248(5450):701–2.
Zinkernagel RM, Doherty PC. Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature. 1974;251(5475):547–8.
Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18(1):14–7.
Brighenti S, Andersson J. Induction and regulation of CD8+ cytolytic T cells in human tuberculosis and HIV infection. Biochem Biophys Res Commun. 2010;396(1):50–7.
Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369-377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res. 1998;58(21):4902–8.
Valmori D, Fonteneau JF, Valitutti S, Gervois N, Dunbar R, Lienard D, et al. Optimal activation of tumor-reactive T cells by selected antigenic peptide analogues. Int Immunol. 1999;11(12):1971–80.
Zarling AL, Ficarro SB, White FM, Shabanowitz J, Hunt DF, Engelhard VH. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J Exp Med. 2000;192(12):1755–62.
Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–50.
Tian Y, Sette A, Weiskopf D. Cytotoxic CD4 T cells: differentiation, function, and application to dengue virus infection. Front Immunol. 2016;7:531.
Gjertsen MK, Bjorheim J, Saeterdal I, Myklebust J, Gaudernack G. Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12Val) peptide vaccination of a patient, recognize 12Val-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int J Cancer. 1997;72(5):784–90.
Delgado JC, Baena A, Thim S, Goldfeld AE. Aspartic acid homozygosity at codon 57 of HLA-DQ beta is associated with susceptibility to pulmonary tuberculosis in Cambodia. J Immunol. 2006;176(2):1090–7.
Dorman JS, Bunker CH. HLA-DQ locus of the human leukocyte antigen complex and type 1 diabetes mellitus: a HuGE review. Epidemiol Rev. 2000;22(2):218–27.
Kaushansky N, Altmann DM, David CS, Lassmann H, Ben-Nun A. DQB1*0602 rather than DRB1*1501 confers susceptibility to multiple sclerosis-like disease induced by proteolipid protein (PLP). J Neuroinflammation. 2012;9:29.
Bandaru A, Devalraju KP, Paidipally P, Dhiman R, Venkatasubramanian S, Barnes PF, et al. Phosphorylated STAT3 and PD-1 regulate IL-17 production and IL-23 receptor expression in Mycobacterium tuberculosis infection. Eur J Immunol. 2014;44(7):2013–24.
Perreau M, Rozot V, Welles HC, Belluti-Enders F, Vigano S, Maillard M, et al. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur J Immunol. 2013;43(4):939–48.
Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99(6):2114–21.
Fabre J, Giustiniani J, Garbar C, Antonicelli F, Merrouche Y, Bensussan A, et al. Targeting the tumor microenvironment: the protumor effects of IL-17 related to cancer type. Int J Mol Sci. 2016;17(9):E1433.
Liang H, Yi L, Wang X, Zhou C, Xu L. Interleukin-17 facilitates the immune suppressor capacity of high-grade glioma-derived CD4 (+) CD25 (+) Foxp3 (+) T cells via releasing transforming growth factor beta. Scand J Immunol. 2014;80(2):144–50.
Xiang T, Long H, He L, Han X, Lin K, Liang Z, et al. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene. 2015;34(2):165–76.
Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 1994;79(5):913–22.
Lorvik KB, Hammarstrom C, Fauskanger M, Haabeth OA, Zangani M, Haraldsen G, et al. Adoptive transfer of tumor-specific Th2 cells eradicates tumors by triggering an in situ inflammatory immune response. Cancer Res. 2016;76(23):6864–76.
Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. 1999;190(5):617–27.
Zhang L, Ge C, Zhao F, Zhang Y, Wang X, Yao M, et al. NRBP2 overexpression increases the chemosensitivity of hepatocellular carcinoma cells via Akt signaling. Cancer Res. 2016;76(23):7059–71.
Shen Y, Fujimoto S. A tumor-specific Th2 clone initiating tumor rejection via primed CD8+ cytotoxic T-lymphocyte activation in mice. Cancer Res. 1996;56(21):5005–11.
Mattes J, Hulett M, Xie W, Hogan S, Rothenberg ME, Foster P, et al. Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med. 2003;197(3):387–93.
Haabeth OA, Bogen B, Corthay A. A model for cancer-suppressive inflammation. Oncoimmunology. 2012;1(7):1146–55.
Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306.
Qian F, Gnjatic S, Jager E, Santiago D, Jungbluth A, Grande C, et al. Th1/Th2 CD4+ T cell responses against NY-ESO-1 in HLA-DPB1*0401/0402 patients with epithelial ovarian cancer. Cancer Immun. 2004;4:12.
Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41(5):591–5.
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010;11:568.
Mandic M, Castelli F, Janjic B, Almunia C, Andrade P, Gillet D, et al. One NY-ESO-1-derived epitope that promiscuously binds to multiple HLA-DR and HLA-DP4 molecules and stimulates autologous CD4+ T cells from patients with NY-ESO-1-expressing melanoma. J Immunol. 2005;174(3):1751–9.
Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10(10):1096–101.
Sidney J, Steen A, Moore C, Ngo S, Chung J, Peters B, et al. Five HLA-DP molecules frequently expressed in the worldwide human population share a common HLA supertypic binding specificity. J Immunol. 2010;184(5):2492–503.
Hovhannisyan Z, Weiss A, Martin A, Wiesner M, Tollefsen S, Yoshida K, et al. The role of HLA-DQ8 beta57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456(7221):534–8.
Siebold C, Hansen BE, Wyer JR, Harlos K, Esnouf RE, Svejgaard A, et al. Crystal structure of HLA-DQ0602 that protects against type 1 diabetes and confers strong susceptibility to narcolepsy. Proc Natl Acad Sci U S A. 2004;101(7):1999–2004.
Ettinger RA, Kwok WW. A peptide binding motif for HLA-DQA1*0102/DQB1*0602, the class II MHC molecule associated with dominant protection in insulin-dependent diabetes mellitus. J Immunol. 1998;160(5):2365–73.
Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152–8.
Lurquin C, Van Pel A, Mariame B, De Plaen E, Szikora JP, Janssens C, et al. Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell. 1989;58(2):293–303.
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–7.
Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, Boon T. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med. 1995;182(3):689–98.
Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. 1994;180(1):35–42.
Coulie PG, Karanikas V, Colau D, Lurquin C, Landry C, Marchand M, et al. A monoclonal cytolytic T-lymphocyte response observed in a melanoma patient vaccinated with a tumor-specific antigenic peptide encoded by gene MAGE-3. Proc Natl Acad Sci U S A. 2001;98(18):10290–5.
Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A. 1997;94(5):1914–8.
Jager E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jager D, et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. 1998;187(2):265–70.
Neumann F, Wagner C, Kubuschok B, Stevanovic S, Rammensee HG, Pfreundschuh M. Identification of an antigenic peptide derived from the cancer-testis antigen NY-ESO-1 binding to a broad range of HLA-DR subtypes. Cancer Immunol Immunother. 2004;53(7):589–99.
Jager E, Karbach J, Gnjatic S, Jager D, Maeurer M, Atmaca A, et al. Identification of a naturally processed NY-ESO-1 peptide recognized by CD8+ T cells in the context of HLA-B51. Cancer Immun. 2002;2:12.
Gnjatic S, Nishikawa H, Jungbluth AA, Gure AO, Ritter G, Jager E, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30.
Hosken NA, Bevan MJ. Defective presentation of endogenous antigen by a cell line expressing class I molecules. Science. 1990;248(4953):367–70.
Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med. 1995;181(2):799–804.
Wang RF, Johnston SL, Southwood S, Sette A, Rosenberg SA. Recognition of an antigenic peptide derived from tyrosinase-related protein-2 by CTL in the context of HLA-A31 and -A33. J Immunol. 1998;160(2):890–7.
Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 1999;284(5418):1351–4.
Wang RF, Wang X, Rosenberg SA. Identification of a novel major histocompatibility complex class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4(+) T cells. J Exp Med. 1999;189(10):1659–68.
Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A. 1996;93(1):136–40.
Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862–8.
Chang K, Pastan I. Molecular cloning and expression of a cDNA encoding a protein detected by the K1 antibody from an ovarian carcinoma (OVCAR-3) cell line. Int J Cancer. 1994;57(1):90–7.
Suarez-Alvarez B, Garcia Suarez MM, Arguelles ME, Sampedro A, Alvarez Marcos C, Mira E, et al. Circulating IgG response to stromelysin-3, collagenase-3, galectin-3 and mesothelin in patients with pharynx/larynx squamous cell carcinoma. Anticancer Res. 2001;21(5):3677–84.
Scholler N, Fu N, Yang Y, Ye Z, Goodman GE, Hellstrom KE, et al. Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc Natl Acad Sci U S A. 1999;96(20):11531–6.
Hassan R, Viner JL, Wang QC, Margulies I, Kreitman RJ, Pastan I. Anti-tumor activity of K1-LysPE38QQR, an immunotoxin targeting mesothelin, a cell-surface antigen overexpressed in ovarian cancer and malignant mesothelioma. J Immunother. 2000;23(4):473–9.
Lehmann F, Marchand M, Hainaut P, Pouillart P, Sastre X, Ikeda H, et al. Differences in the antigens recognized by cytolytic T cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur J Immunol. 1995;25(2):340–7.
Coulie PG, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci U S A. 1995;92(17):7976–80.
Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269(5228):1281–4.
Mandruzzato S, Brasseur F, Andry G, Boon T, van der Bruggen P. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med. 1997;186(5):785–93.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Burmer GC, Loeb LA. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proc Natl Acad Sci U S A. 1989;86(7):2403–7.
Chiosea SI, Sherer CK, Jelic T, Dacic S. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod Pathol. 2011;24(12):1571–7.
Hartman DJ, Davison JM, Foxwell TJ, Nikiforova MN, Chiosea SI. Mutant allele-specific imbalance modulates prognostic impact of KRAS mutations in colorectal adenocarcinoma and is associated with worse overall survival. Int J Cancer. 2012;131(8):1810–7.
Krasinskas AM, Moser AJ, Saka B, Adsay NV, Chiosea SI. KRAS mutant allele-specific imbalance is associated with worse prognosis in pancreatic cancer and progression to undifferentiated carcinoma of the pancreas. Mod Pathol. 2013;26(10):1346–54.
Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756(2):81–2.
YC L, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):3401–10.
Hoffmann TK, Loftus DJ, Nakano K, Maeurer MJ, Chikamatsu K, Appella E, et al. The ability of variant peptides to reverse the nonresponsiveness of T lymphocytes to the wild-type sequence p53(264-272) epitope. J Immunol. 2002;168(3):1338–47.
Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annu Rev Immunol. 1995;13:93–126.
Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252(5010):1308–10.
Evavold BD, Sloan-Lancaster J, Hsu BL, Allen PM. Separation of T helper 1 clone cytolysis from proliferation and lymphokine production using analog peptides. J Immunol. 1993;150(8 Pt 1):3131–40.
Bertoletti A, Sette A, Chisari FV, Penna A, Levrero M, De Carli M, et al. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 1994;369(6479):407–10.
Spain LM, Jorgensen JL, Davis MM, Berg LJ. A peptide antigen antagonist prevents the differentiation of T cell receptor transgenic thymocytes. J Immunol. 1994;152(4):1709–17.
Sprent J, Tough DF. Lymphocyte life-span and memory. Science. 1994;265(5177):1395–400.
Tran E, Robbins PF, YC L, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–62.
Daniels MA, Teixeiro E. TCR signaling in T cell memory. Front Immunol. 2015;6:617.
Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12(11):749–61.
Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. 2013;25(5):556–63.
Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458(7235):211–4.
Sabatino JJ Jr, Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med. 2011;208(1):81–90.
D’Souza WN, Hedrick SM. Cutting edge: latecomer CD8 T cells are imprinted with a unique differentiation program. J Immunol. 2006;177(2):777–81.
Knudson KM, Goplen NP, Cunningham CA, Daniels MA, Teixeiro E. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep. 2013;4(3):554–65.
Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, et al. Different T cell receptor signals determine CD8+ memory versus effector development. Science. 2009;323(5913):502–5.
Morita CT, Jin C, Sarikonda G, Wang H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol Rev. 2007;215:59–76.
Gogoi D, Chiplunkar SV. Targeting gamma delta T cells for cancer immunotherapy: bench to bedside. Indian J Med Res. 2013;138(5):755–61.
Gertner-Dardenne J, Bonnafous C, Bezombes C, Capietto AH, Scaglione V, Ingoure S, et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood. 2009;113(20):4875–84.
Todaro M, D’Asaro M, Caccamo N, Iovino F, Francipane MG, Meraviglia S, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol. 2009;182(11):7287–96.
Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med. 2003;197(2):163–8.
Nada MH, Wang H, Workalemahu G, Tanaka Y, Morita CT. Enhancing adoptive cancer immunotherapy with Vgamma2Vdelta2 T cells through pulse zoledronate stimulation. J Immunother Cancer. 2017;5:9.
Zhang H, Hu H, Jiang X, He H, Cui L, He W. Membrane HSP70: the molecule triggering gammadelta T cells in the early stage of tumorigenesis. Immunol Investig. 2005;34(4):453–68.
Laad AD, Thomas ML, Fakih AR, Chiplunkar SV. Human gamma delta T cells recognize heat shock protein-60 on oral tumor cells. Int J Cancer. 1999;80(5):709–14.
Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18(3):255–62.
Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. 2014;6(254):254ra128.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22:433.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9.
Kowalewski DJ, Stevanovic S, Rammensee HG, Stickel JS. Antileukemia T-cell responses in CLL—we don’t need no aberration. Oncoimmunology. 2015;4(7):e1011527.
Luo YH, CH W, WS W, Huang CY, WJ S, Tsai CM, et al. Association between tumor epidermal growth factor receptor mutation and pulmonary tuberculosis in patients with adenocarcinoma of the lungs. J Thorac Oncol. 2012;7(2):299–305.
Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015;16(9):e447–59.
Azuma K, Komatsu N, Hattori S, Matsueda S, Kawahara A, Sasada T, et al. Humoral immune responses to EGFR-derived peptides predict progression-free and overall survival of non-small cell lung cancer patients receiving gefitinib. PLoS One. 2014;9(1):e86667.
Yamada T, Azuma K, Muta E, Kim J, Sugawara S, Zhang GL, et al. EGFR T790M mutation as a possible target for immunotherapy; identification of HLA-A*0201-restricted T cell epitopes derived from the EGFR T790M mutation. PLoS One. 2013;8(11):e78389.
Sasada T, Azuma K, Ohtake J, Fujimoto Y. Immune responses to epidermal growth factor receptor (EGFR) and their application for cancer treatment. Front Pharmacol. 2016;7:405.
Evavold BD, Sloan-Lancaster J, Allen PM. Antagonism of superantigen-stimulated helper T-cell clones and hybridomas by altered peptide ligand. Proc Natl Acad Sci U S A. 1994;91(6):2300–4.
Kersh EN, Kersh GJ, Allen PM. Partially phosphorylated T cell receptor zeta molecules can inhibit T cell activation. J Exp Med. 1999;190(11):1627–36.
Seitzer U, Kayser K, Hohn H, Entzian P, Wacker HH, Ploetz S, et al. Reduced T-cell receptor CD3zeta-chain protein and sustained CD3epsilon expression at the site of mycobacterial infection. Immunology. 2001;104(3):269–77.
Mizoguchi H, O'Shea JJ, Longo DL, Loeffler CM, McVicar DW, Ochoa AC. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992;258(5089):1795–8.
Finke JH, Zea AH, Stanley J, Longo DL, Mizoguchi H, Tubbs RR, et al. Loss of T-cell receptor zeta chain and p56lck in T-cells infiltrating human renal cell carcinoma. Cancer Res. 1993;53(23):5613–6.
Frydecka I, Kaczmarek P, Bocko D, Kosmaczewska A, Morilla R, Catovsky D. Expression of signal-transducing zeta chain in peripheral blood T cells and natural killer cells in patients with Hodgkin’s disease in different phases of the disease. Leuk Lymphoma. 1999;35(5-6):545–54.
Valkenburg SA, Gras S, Guillonneau C, La Gruta NL, Thomas PG, Purcell AW, et al. Protective efficacy of cross-reactive CD8+ T cells recognising mutant viral epitopes depends on peptide-MHC-I structural interactions and T cell activation threshold. PLoS Pathog. 2010;6(8):e1001039.
Tureci O, Vormehr M, Diken M, Kreiter S, Huber C, Sahin U. Targeting the heterogeneity of cancer with individualized neoepitope vaccines. Clin Cancer Res. 2016;22(8):1885–96.
Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9.
Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):1809–14.
van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78.
Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, et al. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res. 2013;19(1):106–17.
Waldmann TA. Interleukin-15 in the treatment of cancer. Expert Rev Clin Immunol. 2014;10(12):1689–701.
Floros T, Tarhini AA. Anticancer cytokines: biology and clinical effects of interferon-alpha2, interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol. 2015;42(4):539–48.
Meng Q, Liu Z, Rangelova E, Poiret T, Ambati A, Rane L, et al. Expansion of tumor-reactive T cells from patients with pancreatic cancer. J Immunother. 2016;39(2):81–9.
Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, et al. The IL-15-based ALT-803 complex enhances FcgammaRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin Cancer Res. 2016;22(3):596–608.
Weng J, Moriarty KE, Baio FE, Chu F, Kim S-D, He J, et al. IL-15 enhances the anti-tumor effect of human antigen-specific CD8 T cells by cellular senescence delay. Oncoimmunology. 2016. 5(12):e1237327.
English K, Ryan JM, Tobin L, Murphy MJ, Barry FP, Mahon BP. Cell contact, prostaglandin E(2) and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4+CD25(High) forkhead box P3+ regulatory T cells. Clin Exp Immunol. 2009;156(1):149–60.
Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71.
Ebert LM, MacRaild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS One. 2012;7(10):e48424.
Fourcade J, Sun Z, Kudela P, Janjic B, Kirkwood JM, El-Hafnawy T, et al. Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J Immunol. 2010;184(12):6709–18.
Roychoudhuri R, Eil RL, Clever D, Klebanoff CA, Sukumar M, Grant FM, et al. The transcription factor BACH2 promotes tumor immunosuppression. J Clin Invest. 2016;126(2):599–604.
Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–59.
de Witte MA, Coccoris M, Wolkers MC, van den Boom MD, Mesman EM, Song JY, et al. Targeting self-antigens through allogeneic TCR gene transfer. Blood. 2006;108(3):870–7.
Linnemann C, Heemskerk B, Kvistborg P, Kluin RJ, Bolotin DA, Chen X, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med. 2013;19(11):1534–41.
Winograd EK, Ciesielski MJ, Fenstermaker RA. Novel vaccines for glioblastoma: clinical update and perspective. Immunotherapy. 2016;8(11):1293–308.
Fremd C, Schuetz F, Sohn C, Beckhove P, Domschke C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology. 2013;2(7):e25443.
Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63(2):81–99.
Nahtman T, Jernberg A, Mahdavifar S, Zerweck J, Schutkowski M, Maeurer M, et al. Validation of peptide epitope microarray experiments and extraction of quality data. J Immunol Methods. 2007;328(1-2):1–13.
Ngo Y, Advani R, Valentini D, Gaseitsiwe S, Mahdavifar S, Maeurer M, et al. Identification and testing of control peptides for antigen microarrays. J Immunol Methods. 2009;343(2):68–78.
Reilly M, Valentini D. Visualisation and pre-processing of peptide microarray data. Methods Mol Biol. 2009;570:373–89.
Valentini D, Rao M, Ferrara G, Perkins M, Dodoo E, Zumla A, et al. Immune recognition surface construction of Mycobacterium tuberculosis epitope-specific antibody responses in tuberculosis patients identified by peptide microarrays. Int J Infect Dis. 2017;56:155–66.
Valentini D, Gaseitsiwe S, Maeurer M. Humoral ‘reactome’ profiles using peptide microarray chips. Trends Immunol. 2010;31(11):399–400.
Valentini D, Ferrara G, Advani R, Hallander HO, Maeurer MJ. Serum reactome induced by Bordetella pertussis infection and pertussis vaccines: qualitative differences in serum antibody recognition patterns revealed by peptide microarray analysis. BMC Immunol. 2015;16:40.
Gaseitsiwe S, Valentini D, Mahdavifar S, Magalhaes I, Hoft DF, Zerweck J, et al. Pattern recognition in pulmonary tuberculosis defined by high content peptide microarray chip analysis representing 61 proteins from M. tuberculosis. PLoS One. 2008;3(12):e3840.
Gaseitsiwe S, Valentini D, Mahdavifar S, Reilly M, Ehrnst A, Maeurer M. Peptide microarray-based identification of Mycobacterium tuberculosis epitope binding to HLA-DRB1*0101, DRB1*1501, and DRB1*0401. Clin Vaccine Immunol. 2010;17(1):168–75.
Ambati A, Valentini D, Montomoli E, Lapini G, Biuso F, Wenschuh H, et al. H1N1 viral proteome peptide microarray predicts individuals at risk for H1N1 infection and segregates infection versus Pandemrix vaccination. Immunology. 2015;145(3):357–66.
Perez-Bercoff L, Valentini D, Gaseitsiwe S, Mahdavifar S, Schutkowski M, Poiret T, et al. Whole CMV proteome pattern recognition analysis after HSCT identifies unique epitope targets associated with the CMV status. PLoS One. 2014;9(4):e89648.
Gaseitsiwe S, Valentini D, Ahmed R, Mahdavifar S, Magalhaes I, Zerweck J, et al. Major histocompatibility complex class II molecule-human immunodeficiency virus peptide analysis using a microarray chip. Clin Vaccine Immunol. 2009;16(4):567–73.
Ferrara G, Valentini D, Rao M, Wahlstrom J, Grunewald J, Larsson LO, et al. Humoral immune profiling of mycobacterial antigen recognition in sarcoidosis and Lofgren’s syndrome using high-content peptide microarrays. Int J Infect Dis. 2017;56:167.
WHO. World cancer report 2014. Lyon: International Agency for Research on Cancer, World Health Organisation; 2014.
Al-Mansour Z, Nelson BP, Evens AM. Post-transplant lymphoproliferative disease (PTLD): risk factors, diagnosis, and current treatment strategies. Curr Hematol Malig Rep. 2013;8(3):173–83.
Karbach J, Gnjatic S, Pauligk C, Bender A, Maeurer M, Schultze JL, et al. Tumor-reactive CD8+ T-cell clones in patients after NY-ESO-1 peptide vaccination. Int J Cancer. 2007;121(9):2042–8.
Chen JL, Dawoodji A, Tarlton A, Gnjatic S, Tajar A, Karydis I, et al. NY-ESO-1 specific antibody and cellular responses in melanoma patients primed with NY-ESO-1 protein in ISCOMATRIX and boosted with recombinant NY-ESO-1 fowlpox virus. Int J Cancer. 2015;136(6):E590–601.
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–21.
Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21(5):1019–27.
Fonteneau JF, Brilot F, Munz C, Gannage M. The tumor antigen NY-ESO-1 mediates direct recognition of melanoma cells by CD4+ T cells after intercellular antigen transfer. J Immunol. 2016;196(1):64–71.
Jacquelot N, Enot DP, Flament C, Vimond N, Blattner C, Pitt JM, et al. Chemokine receptor patterns in lymphocytes mirror metastatic spreading in melanoma. J Clin Invest. 2016;126(3):921–37.
Munson DJ, Egelston CA, Chiotti KE, Parra ZE, Bruno TC, Moore BL, et al. Identification of shared TCR sequences from T cells in human breast cancer using emulsion RT-PCR. Proc Natl Acad Sci U S A. 2016;113(29):8272–7.
Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JL, et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci U S A. 2016;113(25):E3529–37.
Gilchuk P, Spencer CT, Conant SB, Hill T, Gray JJ, Niu X, et al. Discovering naturally processed antigenic determinants that confer protective T cell immunity. J Clin Invest. 2013;123(5):1976–87.
Tran E, Turcotte S, Gros A, Robbins PF, YC L, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Tran E, Ahmadzadeh M, YC L, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90.
Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K. Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–91.
Reshef R, Luskin MR, Kamoun M, Vardhanabhuti S, Tomaszewski JE, Stadtmauer EA, et al. Association of HLA polymorphisms with post-transplant lymphoproliferative disorder in solid-organ transplant recipients. Am J Transplant. 2011;11(4):817–25.
Subklewe M, Marquis R, Choquet S, Leblond V, Garnier JL, Hetzer R, et al. Association of human leukocyte antigen haplotypes with posttransplant lymphoproliferative disease after solid organ transplantation. Transplantation. 2006;82(8):1093–100.
Pourfarziani V, Einollahi B, Taheri S, Nemati E, Nafar M, Kalantar E. Associations of human leukocyte antigen (HLA) haplotypes with risk of developing lymphoproliferative disorders after renal transplantation. Ann Transplant. 2007;12(4):16–22.
Bertoletti A, Brunetto M, Maini MK, Bonino F, Qasim W, Stauss H. T cell receptor-therapy in HBV-related hepatocellularcarcinoma. Oncoimmunology. 2015;4(6):e1008354.
Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337–41.
Dvorak CC, Gilman AL, Horn B, Jaroscak J, Dunn EA, Baxter-Lowe LA, et al. Clinical and immunologic outcomes following haplocompatible donor lymphocyte infusions. Bone Marrow Transplant. 2009;44(12):805–12.
Neuenhahn M, Albrecht J, Odendahl M, Schlott F, Dossinger G, Schiemann M, et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia. 2017.
Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut. 2017;66(3):454–63.
Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21(1):15–25.
Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, YH X, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One. 2013;8(5):e62924.
Fan H, Lu X, Wang X, Liu Y, Guo B, Zhang Y, et al. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: a phase I/II report. J Immunol Res. 2014;2014:371087.
Siva S, MacManus MP, Martin RF, Martin OA. Abscopal effects of radiation therapy: a clinical review for the radiobiologist. Cancer Lett. 2015;356(1):82–90.
Silk AW, Bassetti MF, West BT, Tsien CI, Lao CD. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013;2(6):899–906.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–65.
D’Souza NM, Fang P, Logan J, Yang J, Jiang W, Li J. Combining radiation therapy with immune checkpoint blockade for central nervous system malignancies. Front Oncol. 2016;6:212.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174(12):7516–23.
Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–107.
Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer. 2016;4:51.
Cohen JV, Kluger HM. Systemic immunotherapy for the treatment of brain metastases. Front Oncol. 2016;6:49.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–31.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8.
Chen YY. Efficient gene editing in primary human T cells. Trends Immunol. 2015;36(11):667–9.
Arsenio J, Metz PJ, Chang JT. Asymmetric cell division in T lymphocyte fate diversification. Trends Immunol. 2015;36(11):670–83.
Gollwitzer ES, Marsland BJ. Impact of early-life exposures on immune maturation and susceptibility to disease. Trends Immunol. 2015;36(11):684–96.
Wertek F, Xu C. Digital response in T cells: to be or not to be. Cell Res. 2014;24(3):265–6.
Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity. 2013;39(5):846–57.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Rao, M., Zhenjiang, L., Meng, Q., Sinclair, G., Dodoo, E., Maeurer, M. (2018). Mutant Epitopes in Cancer. In: Zitvogel, L., Kroemer, G. (eds) Oncoimmunology. Springer, Cham. https://doi.org/10.1007/978-3-319-62431-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-62431-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62430-3
Online ISBN: 978-3-319-62431-0
eBook Packages: MedicineMedicine (R0)