
Abstract
Myeloid-derived suppressor cells (MDSCs) are a major immunosuppressive population in the tumor microenvironment. After being generated in the bone marrow, in tumor-bearing hosts, MDSCs migrate toward secondary lymphoid organs and the tumor where they contribute to the establishment of an immunosuppressive state. MDSCs directly support tumor growth and metastasis. The elimination of this cell population has been the focus for several years. We here review the recent findings exploring how various pharmaceutical compounds can target MDSCs and reduce their resultant immunosuppression.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
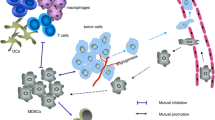


1 Introduction
Myeloid-derived suppressor cells (MDSCs) are a population of myeloid origin that exert immunosuppressive functions. MDSCs are distinct from terminally differentiated myeloid cells such as macrophages, dendritic cells (DCs), or neutrophils. MDSCs are hematopoietic cells generated in the bone marrow that can be divided into two subtypes. Monocytic MDSCs come from the macrophages/DC progenitor, while polymorphonuclear (PMN) MDSCs arise from the granulocytic arm of myeloid differentiation. Both subtypes can be found in humans and mice. In humans, M-MDSCs are characterized by their expression of CD14, CD11b, and CD33 and their lack of lineage marker as well as a low expression for HLA-DR. PMN-MDSCs express CD15, CD11b, and CD33 and are negative for lineage markers and HLA-DR. PMN-MDSC could be distinguished from PMN thanks to their difference in density. Although additional markers have been studied to further identify MDSCs, none of them is yet considered as a specific MDSC marker. However, recently, Dmitry Gabrilovich’s group demonstrated that the LOX1 marker could differentiate PMN-MDSC from PMN in human [1]. In mice, different molecules are used to delineate MDSCs populations. CD11b and Gr-1 expression identify both subsets of MDSC. Gr-1 is a combination of two markers, Ly6C and Ly6G. Using these, MDSC populations can be separated more accurately, M-MDSCs being Ly6C+ and Ly6G, while PMN-MDSCs are found out to be Ly6G+ and Ly6C.
In healthy individuals, MDSCs are almost undetectable. However, under certain circumstances such as acute infection (sepsis choc), chronic infection (tuberculosis), or cancer, MDSCs accumulate. MDSC expansion involves multiple factors like GM-CSF, G-CSF, SCF, or S100A8 and S100A9 that can be secreted among others by tumor cells [2]. The uprising of MDSCs in cancer patients can be seen in the tumor bed and the secondary lymphoid organs but also in the bone marrow where accumulation of MDSCs has been observed in several studies [3, 4]. In both humans and mouse, PMN-MDSCs represent the majority of MDSCs in most type of cancer. MDSCs support tumor growth and metastasis in various ways. MDSCs help establish a microenvironment favorable to tumor growth, thanks to their production of proangiogenic mediators like VEGF, bFGF, Mmp9, or PDGF, critical molecules for the development of new vessels that are essential to maintain tumor growth. MDSCs also play an important role in the initiation of metastasis process. A study showed an increase in MDSCs in the lung of mice bearing mammary adenocarcinoma up to 2 weeks before the arrival of tumor cells [5], an increase in MDSCs that was dependent on the production of Mmp9. The pro-inflammatory proteins S100A8 and S100A9 whose organ expression is induced by the primary tumors can attract MDSCs to a pre-metastasized niche. As MDSCs accumulate in a new organ, their production of S100A8 and S100A9 can then amplify the mechanism and favor tumor cell migration and metastasis [6, 7]. Recent studies also revealed MDSC potential to enhance stemness of cancer cells, thus facilitating their epithelial-to-mesenchymal transition (EMT), supporting metastasis [8].
Aside from promoting tumor cell growth and metastasis, MDSCs also support the tumor thanks to their immunosuppressive properties. MDSCs exert immunosuppression through multiple mechanisms. MDSCs can produce IL-10 and TGF-beta [9] and induce regulatory T cells [10]; they also produce reactive oxygen species like O2 − and H2O2 [11] and NO [12]. MDSCs lower TCR formation and induce T cell cycle arrest by depleting the milieu of l-arginine, thanks to their expression of Arg1 [13] or by reducing the levels of available tryptophan because of their indoleamine 2,3 dioxygenase (IDO) activity [14]. M-MDSCs and PMN-MDSCs do not possess exactly the same immunosuppressive mechanisms. M-MDSCs, which have a higher immunosuppressive activity than PMN-MDSCs, express Arg1 and produce IL-10, TGF-beta, and NO when PMN-MDSCs tend to produce more ROS that are short-lived molecules, explaining the lesser suppressive potential of PMN-MDSCs compared to M-MDSCs [15]. The ratio between PMN-MDSCs and M-MDSCs, in favor of PMN-MDSCs at the periphery in most cancers, has been shown to notably vary inside tumor bed depending on the type of cancer, especially in humans.
MDSCs are one of the major immunosuppressive components in tumor-bearing animals and patients. Consequently, their elimination or their differentiation into effective dendritic cells and macrophages is a major issue in immuno-oncology. Many data show that such a strategy can enhance antitumor immunity, allowing T cells to attack tumor cells and reduce the tumor burden. We will here focus on the various ways existing to reduce MDSCs, using chemotherapies and upcoming immunotherapies.
2 Impact of Cytotoxic Chemotherapies on MDSCs
Some chemotherapies have been shown to directly kill MDSCs. Gemcitabine is a chemotherapy consisting in a nucleoside analog of the cytidine that acts as an antimetabolite agent. Gemcitabine is used to treat various cancers like ovarian, pancreatic, lung, and breast cancer and cholangiocarcinoma. In 2005, Suzuki made the first demonstration that gemcitabine-based chemotherapy could specifically target MDSC [16]. Using a dose equivalent to classical dose used to treat human patients, it was shown that gemcitabine could, in the spleen of tumor-bearing mice, decrease in a selective manner the number of MDSCs without impacting number and function of CD4 cells, CD8 cells, NK cell macrophages, or B cells. The antitumor activity of CD8+ T cells and NK cells after gemcitabine was increased, and less immunosuppression could be observed in the spleen of gemcitabine-treated mice [16]. However, no major impact of gemcitabine alone was observed on the tumor growth. These results were confirmed in 2009 by another team using a different tumor model. In addition to seeing a drop in the percentage of MDSCs in the spleen, they also observed a drop in MDSC both in the bone marrow and peripheral blood. The kinetic of treatment administration is of particular importance [17]. Only an early treatment with gemcitabine could delay tumor growth, suggesting that compensatory mechanisms limit the antitumor effect of MDSC depletion in established tumors.
Docetaxel and paclitaxel are the drugs of the taxane family used in clinic for cancer treatment. These two drugs both target tubulin, preventing the depolymerization of microtubules and thereby blocking mitosis. Docetaxel is a commonly used anticancer drug and was primarily developed for use against breast cancer in the 1990s. Now taxanes are used to treat various types of cancer including lung cancer, digestive cancer, and ovarian cancer. Docetaxel was demonstrated to have an effect on MDSCs. Mice bearing the mammary tumor model 4T1 and treated with docetaxel had significantly less MDSCs in their spleen and displayed an increased CTL response [18]. The decrease in MDSCs was partly due to the cytotoxic effect of docetaxel on PMN-MDSCs, while M-MDSCs differentiated toward an M1-like phenotype. M-MDSCs were later found to be resistant to docetaxel, thanks to their expression of secretory/cytoplasmic clusterin (sCLU) which expression prevented the induction of the apoptotic cascade by taxanes [19]. The analog of docetaxel, paclitaxel, has a cytotoxic activity weaker than its analog, but this drug is also largely used for the treatment of lung, breast, and ovarian cancers. Tumor-bearing mice, treated with paclitaxel at a low dose with non-cytotoxic effect, showed a decrease of MDSCs compared with non-treated mice. This decrease in MDSC was the consequence of their differentiation into DCs, and no MDSC cell death could be detected [20]. Using a model of spontaneous melanoma, the same team showed that low non-cytotoxic dose of paclitaxel could decrease the accumulation of MDSCs as well as their immunosuppressive activities (with less TNF-alpha and less S100A9 produced by the remaining MDSCs) [21].
Doxorubicin is a chemotherapy belonging to the anthracycline family of drugs. It interacts with DNA by intercalation and inhibits the progression of the topoisomerase II, thus blocking DNA replication. Doxorubicin is commonly used to treat sarcomas, breast cancers, leukemias, and non-Hodgkin’s lymphomas. Doxorubicin has been shown to selectively deplete MDSCs in the spleen, blood, and tumor bed of 4T1 mammary cancer-bearing mice. The residual MDSCs showed impaired suppressive functions, with a lesser production of ROS, arginase-1, and IDO, while a higher proportion of CD4 and CD8 lymphocytes and NK cells were observed [22]. However, it was recently shown that doxorubicin could also induce the secretion of prostaglandin E2 (PGE2) by cancer cells like 4T1 cells. PGE2 stimulates MDSC expansion and accumulation reestablishing a subsequent MDSC population and immunosuppression in the tumor-bearing host [23]. Such data underline that anthracyclines may have a contrasting effect on MDSCs.
Trabectedin is a cytotoxic agent that binds to the minor groove of DNA inducing a perturbation of the cell cycle. Trabectedin caused selective depletion of monocytes/macrophages in blood, spleens, and tumors, with an associated reduction of angiogenesis in different experimental models. Trabectedin activates caspase-8-dependent apoptosis selectivity in monocytic myeloid cells and not neutrophilic ones because of a differential expression of signaling and decoy TRAIL receptors. Such data underline the possibility to use trabectedin to target tumor-infiltrated M-MDSCs [24].
Local irradiation could change the tumor microenvironment and remove immunosuppressive cells. In the CT26 and MC38 mouse colon carcinoma models, high-dose radiation transformed the immunosuppressive tumor microenvironment resulting in an intense CD8(+) T cell tumor infiltrate and a loss of MDSC accumulation [25]. In a mechanistic point of view, CD8+ T cell production of IFN-γ, induced by radiotherapy, controlled the survival and infiltration of MDSCs in the tumor and reversed the immunosuppressive environment. Furthermore, antitumor immune CD8+ T cells can kill MDSCs via their production of TNF-α, IFN-γ, or expression of FasL and thereby reduce MDSC infiltration in tumor. In contrast, low dosage of radiotherapy did not positively affect MDSC number.
5-Fluorouracil (5-FU) is a thymidylate synthase inhibitor, preventing the synthesis of thymidine, a nucleoside essential for DNA replication. This antimetabolite drug is used to treat most digestive cancers and is a major drug for colon cancer. 5-FU can selectively deplete MDSCs, both PMN and monocytic, in several mouse cancer models (Fig. 11.1). MDSC depletion is due to the triggering of apoptosis after a 5-FU treatment. 5-FU selectively kills MDSCs because of their weak expression of the target enzyme of 5-FU, the thymidylate synthase. 5-FU is a competitive inhibitor of thymidylate synthase. Cells with a low expression of thymidylate synthase are very sensitive to cell death induced by 5-FU. In tumor-bearing mice, a 5-FU treatment significantly delayed tumor growth and induced a specific CD8 T cell activation in tumor bed [26]. A closer look at the impact of 5-FU on immune populations in tumor-bearing mice showed an increase in the number of Th17 cells 10 days after a 5-FU treatment accompanied by a return of the tumor growth. This increase in Th17 cells was due to the production of IL-1beta by dying MDSCs. Indeed, 5-FU induced BAX activation and lysosome permeabilization in MDSCs. The protein cathepsin B was released from the lysosomes into the cytoplasm where it interacted with NLRP3 and triggered the formation and activation of the NLRP3 inflammasome, leading to the production of cleaved and bioactive IL-1beta. The IL-1beta would then promote Th17 polarization of CD4 T cells. In vitro stimulation of CD4 T cells with 5-FU-treated MDSC promoted their capacity to produce IL-17. In vivo, 5-FU induced accumulation of Th17 cells in tumor-bearing mice in a NLRP3-dependent manner. Interestingly IL-17 promoted angiogenesis, and this neoangiogenesis seemed to be an essential effector of the deleterious effect of 5-FU [27]. The use of IL-1RA, a soluble receptor of IL-1, along with 5-FU, could block the action of IL-1beta. Such therapy reduced the generation of Th17 cells and neoangiogenesis and dramatically improved the efficacy of 5-FU on the tumor growth.

5-FU-dependent depletion of MDSCs. 5-FU specifically targets and depletes MDSCs, reducing the overall immunosuppression. However, 5-FU treatment also induces a permeabilization of the lysosome in MDSCs. Cathepsin B can then escape the lysosome and enter the cytoplasm where it interacts with NLRP3, inducing the formation and activation of the NLRP3 inflammasome. The activated NLRP3 inflammasome cleaves the pro-IL-1β into active IL-1β that activates CD4 T cells to produce more IL-17, enhancing neoangiogenesis and tumor growth
The impact of chemotherapies on MDSCs is often a double-edged sword, tumor cells and immunosuppressive immune cells being masters at finding a loophole allowing for the return of an immunosuppressive tumor environment as seen here. We should be careful not to make any precocious conclusion before the full picture is before our eyes.
3 Effect of Chemotherapies on MDSCs in Human
Studies in humans on the effect of chemotherapies on MSDCs have sometimes shown contradictory data. In patients with a pancreatic cancer, 5-FU and gemcitabine were first shown to reduce MDSC percentage in about 40% of patients and in most of patients when associated with the GV101 vaccine using GM-CSF as an adjuvant. MDSC number did however come up in some patients, and this was correlated with an increase of pro-inflammatory cytokines [28]. These results match with a more recent study where treatment with gemcitabine or 5-FU was associated with an upregulation of GM-CSF secreted by tumor cells inducing the differentiation of monocytes in MDSCs [29]. On the contrary, a positive effect of gemcitabine and 5-FU on MDSCs was observed when associated with an immunotherapy consisting in cytokine-induced killer cells [30]. In metastatic renal cell carcinoma and pancreatic cancer, the use of this chemotherapy in association with an immunotherapy successfully reduced the number of MDSCs in the peripheral blood of patients and increased the survival time. Interestingly, in colorectal cancer patients, the positive or negative outcome of a 5-FU treatment is dependent on the type of combination used in association with 5-FU. Indeed, 5-FU is not used alone in colorectal cancer. The use of 5-FU with folic acid and oxaliplatin (FOLFOX) was proven beneficial with a decrease of the overall immunosuppression and MDSC percentage, whereas the association of 5-FU with folic acid and CPT11 (FOLFIRI) was detrimental and even increased the number of MDSCs in patients [31]. In another study testing the effect of FOLFOX associated with bevacizumab, an anti-VEGF-A antibody, on MDSCs in patients treated in first line of metastatic colorectal cancer, authors observed a drop of PMN-MDSCs in 15 out of 25 patients [32]. As cancer chemotherapies always associate several agents, it is crucial to study the effects of these associations.
4 Tyrosine Kinase Inhibitors
Aside from cytotoxic chemotherapies, several other classes of anticancer agents have been studied for their capability to block MDSC proliferation or to enhance their differentiation. Tyrosine kinase inhibitors (TKIs) are a group of molecules aiming to target tyrosine kinases in various pathways. Targeted pathways, the RAS-RAF/MAPK pathway, the PI3K/AKT/mTOR pathway, and the EGFR pathway, are involved in the regulation of cell survival, proliferation, differentiation, migration, and angiogenesis [33, 34]. Mutations in these pathways are often found in cancer, explaining the rapid and ongoing development of TKI these past few years [35, 36]. The potential effects of TKIs on MDSCs have raised a growing interest.
Sunitinib is a TKI targeting multiple receptor tyrosine kinases including VEGF-R1 and VEGF-R2, PDGF-Rs, but also c-KIT. It was approved by the FDA for the treatment of advanced renal cell carcinoma (RCC) in 2007 and is currently used in the frontline treatment for RCC. In RCC, sunitinib reversed MDSC accumulation by affecting their viability and proliferation. The decrease in MDSCs was linked to an increase in IFN-gamma production by CD3 cells [37]. Sunitinib decreased the number of MDSCs and Tregs as well as the production of the immunosuppressive cytokines IL-10, TGF-beta. Interestingly, the expression of the negative costimulatory molecules CTLA-4 and PD-1 on CD4 and CD8 T cells was decreased after a sunitinib treatment [38]. Sunitinib may reduce the expansion of monocytic MDSC while inducing apoptosis in the granulocytic MDSC subset [39]. However, intratumoral MDSC number and function were not affected by sunitinib, as the high quantity of GM-CSF produced in the tumor bed was protecting MDSCs in a STAT5-dependent pathway [40, 41]. Sunitinib was reported to also affect other cell types than MDSCs as reduction in the percentage of neutrophils and monocytes and an increase in lymphoid cells can be observed [37] (Fig. 11.2).

Sunitinib immune effects. Sunitinib, a TKI targeting VEGF-Rs and PDGF-Rs, blocks MDSC accumulation by reducing their viability and proliferation. Sunitinib also targets Tregs and decreases IL-10 and TGF-β production while enhancing the proliferation of CD4 and CD8 T cells along with increasing the production of IFN-γ
A study using another VEGF pathway inhibitor, bevacizumab, an anti-VEGF-A mAb, showed that MDSCs were responsible for the refractoriness to clinical effect of anti-VEGF therapy [42], but no effect of bevacizumab on MDSC viability or differentiation was observed. This was later confirmed by another study showing that bevacizumab treatment did not decrease the percentage of MDSCs nor change their level of arginase-1 expression [43]. However, in patients with non-small cell lung carcinoma, three cycles of bevacizumab associated with chemotherapy regimens could reduce PMN-MDSC numbers in a bevacizumab-dependent way [44]. The impact of bevacizumab on MDSCs remains to be confirmed, and observed difference may be consequences of additional drugs used to treat cancer or due to the tumor types.
Sorafenib is an inhibitor directed against several kinases, among which are C-RAF, BRAF, and VEGF-R2 and VEGF-R3. Sorafenib was first demonstrated to have the capability to reduce Tregs and MDSCs in a murine liver cancer model, along with a slower tumor growth [45]. In addition, sorafenib was able to decrease the suppressive activity of MDSCs on CD8 T cells, while sunitinib, another inhibitor, could not [41]. Different protocols of administration with various doses were tested, and repetitive low doses of sorafenib appeared to enhance the efficacy of adoptive T cell therapy by decreasing MDSCs and Tregs but also by decreasing the expression of immunosuppressive molecules like IL-10 or TGF-beta [46]. Along with the selected dosage, the kinetic of treatment should also be considered as sorafenib could reduce the percentage of MDSC derived from monocytes but did not affect already differentiated MDSCs. Sorafenib effects might however not be restricted to MDSCs. Indeed, sorafenib decreased STAT1 and STAT5 phosphorylation in T cells, B cells, NK cells, Tregs, and MDSCs after stimulation with IL-2 or IFN-alpha [47]. Such data suggest that sorafenib could have deleterious effect on effector cells of the adaptive immune response. We probably should keep in mind these data when using sorafenib to deplete MDSCs.
Specific for c-kit and BCR-ABL, imatinib was the first TKI approved for chronic myeloid leukemia [48]. Imatinib efficiently reduced MDSC expansion and arginase-1 expression [49]. However, various reports also mention contradictory results in regard to its effects on other immune cell populations. Imatinib was shown to impair Tregs immunosuppressive functions [50], restore plasmacytoid dendritic cell function, and suppress tumor-induced CD4+ T cell tolerance [51, 52]. On the other hand, imatinib treatment was also shown to block the expansion of antigen-experienced CD8 T cells while leaving primary T and B cell responses unaffected [53]. Dasatinib is a second-generation compound used in patients with chronic myeloid leukemia who fail to respond to imatinib. Like imatinib, dasatinib blocked MDSC expansion [41, 49] and could trigger the development of a broad repertoire of tumor-associated CD8+ tumor-infiltrating lymphocytes when associated with a DC-based vaccine in a melanoma model [54]. However, in several studies dasatinib also inhibited CD4+ and CD8+ T cell activation and proliferation in a dose-dependent manner [55]. The beneficial use of imatinib and dasatinib against MDSCs is unambiguous, but the consequences of their use on the T cell compartment remain unclear.
Many other TKIs have been shown to deeply affect MDSCs, their proliferation, differentiation, as well as their suppressive functions. Vemurafenib was approved for the treatment of unresectable or metastatic melanoma with the BRAF V600-activating mutation by the FDA in 2011. Vemurafenib could decrease the proportion and absolute number of M- and PMN-MDSCs as well as Tregs in melanoma both in mice models and human. Following a vemurafenib treatment, an increase in tumor-infiltrating CD8 T cells was observed and was correlated with a reduction in tumor size [56,57,58]. Approved by the FDA in 2013 to use against some B cell lymphoma, the Bruton tyrosine kinase (BTK) inhibitor ibrutinib could reduce MDSC accumulation in the tumor bed and reduce the expression of IDO. These effects are likely to be a direct consequence of the inhibition of BTK in MDSCs [59, 60].
Recently a growing interest regarding the effects of the PI3K/AKT/mTOR pathway on MDSCs has arisen. The mTOR pathway activation in both tumor cells and MDSCs seems favorable to MDSCs. Indeed, rapamycin, an inhibitor of mTOR, has been shown to significantly decrease MDSC number as well as the immunosuppressive functions of M-MDSCs in tumor-bearing mice. mTOR appears to be an intrinsic factor involved in the differentiation and suppressive functions of MDSCs [61, 62]. Moreover, activation of the mTOR pathway in cancer cells could also favor the recruitment and accumulation of MDSCs in a G-CSF-dependent fashion in human breast cancer [63]. However, we have to keep in mind that mTOR activation is also essential for T cell activation and mTOR inhibitor could have some deleterious effect on CD8 antitumor immune response. So far only rapamycin derivatives are used in clinic to block the PI3K/AKT/mTOR pathway, but other inhibitors targeting this pathway are in development. Such drugs should also be tested to address their capacity to inhibit MDSCs or reduce their number.
5 Other FDA-Approved Molecules with Impact on MDSCs
Molecules from different categories approved by the FDA are found to display activity against MDSCs. One of them is a phosphodiesterase 5 inhibitor named tadalafil. Tadalafil inhibited MDSC immunosuppressive functions via downregulation of iNOS and Arg-1, two key immunosuppressive enzymes of MDSCs [64]. In head and neck squamous cell carcinoma, tadalafil could reduce MDSCs and Tregs in both blood and tumor bed while increasing the concentration of CD8 T cells specific for tumor antigens in the blood [65, 66]. The dose at which tadalafil is used seems of importance as an important dose triggered off-target effects on PDE11 which may affect antitumor immunity by different ways [65]. A case report on a man with end-stage multiple myeloma showed that tadalafil reduced MDSC functions (Arg-1 and iNOS expressions downregulated) and established a durable anti-myeloma immune and clinical response although not complete [67].
CTL antigen-4 (CTLA-4) is a negative immune checkpoint expressed by T cells. Ipilimumab is a human antibody directed against CTLA-4, and it has been shown that in patients with metastatic melanoma, the frequency of PMN-MDSCs significantly decreased 3 weeks after a first treatment with ipilimumab [68]. In contrast, no impact on M-MDSCs was observed. Another study on melanoma patients showed a decrease in circulating MDSCs after an ipilimumab treatment and a positive association between decreased in MDSCs and a better PFS [69]. Complementary studies are required to understand the mechanisms by which ipilimumab affects MDSCs.
ATRA, for all-trans retinoic acid, is used to treat promyelocytic acute myeloid leukemia. This drug is also capable of inducing the maturation of MDSCs into DCs, macrophages, and granulocytes. As expected, the decrease in MDSCs by ATRA treatment improved CD4 and CD8 T lymphocyte tumor-specific response first in two mouse tumor models, DA3-HA adenocarcinoma and C3 fibrosarcoma [70], and then in patients with RCC [71] and small cell lung cancer [72].
6 Drugs in Developments
A broad spectrum of molecules from various origins have displayed an activity against MDSCs; they can either block the immunosuppressive functions of MDSCs, inducing their differentiation in dendritic cells or in M1-like macrophages, or deplete them.
In the MC38 colon carcinoma, the Lewis lung carcinoma and the EL-4 thymoma mouse models and in patients with RCC or soft tissue sarcoma, the triterpenoid CDDO-Me did not affect the size of the MDSC population but abrogated their immunosuppressive activities and improved immune responses [73]. Nitroaspirin also reduces MDSC functions by inhibiting NOS and Arg-1 activity, resulting in increased number and functions of tumor antigen-specific T lymphocytes [74]. The inhibitor of the ubiquitin receptor RPN13/ADRM1 RA190 was recently shown to lower the level of Arg-1, iNOS, and IL-10 in MDSCs. MDSCs treated by RA190 lost their capacity to suppress CD8+ T cells, thus enhancing antitumor immune response, in an ovarian mouse model [75]. TLR9 activation of MDSCs by CpG treatment was shown to block their suppressive activity on T cells in two mouse models of cancer and to induce MDSC differentiation [76]. The impact of CpG on MDSCs was confirmed in the Renca renal mouse tumor model [77]. On the opposite side, it is interesting to note that CpG emulsified in incomplete Freund’s adjuvant treatment expanded MDSCs and increased their expression of Arg-1 in aged mice free of tumor, suggesting contrasting effect of CPG on MDSCs [78].
Curcumin can differentiate MDSCs. Curcumin suppresses PMN-MDSC function and favors M-MDSC differentiation toward an M1-like phenotype [79] in a clusterin-dependent fashion [19]. Other molecules induce a depletion of MDSCs as the use of an antibody-targeting DR5, a death receptor present at the surface of MDSCs, can effectively eliminate them without affecting other myeloid populations and resulted in an enhanced antitumor immune response [80]. Beta-glucan-like curdlan can promote the differentiation of M-MDSCs into a mature CD11c+ F4/80+ population. That differentiation happens via a NF-κB-dependent dectin-1 pathway. A beta-glucan treatment diminished MDSCs in the tumor bed and increased infiltrated DCs and macrophages, leading to an enhanced CD8 and CD4 T cell responses and delayed tumor growth [81]. Two cationic polymers, cationic dextran (C-dextran) and polyethyleneimine (PEI), can differentiate MDSCs into an M1-like phenotype, decreasing IL-10 and TGF-beta production as well as suppressing Arg-1 expression while increasing M1-type cytokines production. This decrease in MDSC restored antitumor immunity and slowed tumor growth in the 4T1 mouse mammary carcinoma [82].
Depletion of MDSC can be achieved, thanks to a various set of molecule ever-expanding. However, the mechanisms behind that depletion are not always yet defined. A new therapeutic peptide, developed after identification and characterization of MDSC-binding peptides, depleted both monocytic and PMN-MDSCs in the blood and spleen of EL4 or EG7 thymoma-bearing mice and successfully delayed tumor growth [83]. The S100 protein family is a candidate target for this peptide, but a more thorough study may be needed in order to fully understand the mechanism of action of this peptide (Fig. 11.3).

Therapeutic approaches targeting MDSCs. There are three different ways to aim at MDSC. Molecules targeting MDSCs can directly kill them, like 5-FU or DR5 antibodies, or inhibit MDSC immunosuppressive functions as tadalafil does or also block MDSC accumulation or induce their differentiation like ATRA or curcumin does
CSF-1, also known as M-CSF, is overexpressed in many tumors and is a growth factor for M-MDSCs and macrophages. Several CSF-1 receptor inhibitors have been developed and, when tested in tumor-bearing mice, displayed the potency to deplete M-MDSC in tumor bed and spleen. Blockade of CSF1R increases antigen-specific T cell activity at the tumor site, delaying tumor growth in B16 melanoma [84, 85], RM-1, RM-9, and Myc-CaP prostate cancer-bearing mice [86].
Contradictory data can be found about histone deacetylase (HDAC) on MDSCs. It was first published that HDAC inhibition by TSA, a naturally occurring antifungal metabolite that potently inhibits HDAC, enhanced the expansion of MDSCs in a GM-CSF-dependent manner [87]. This was confirmed by another study showing that HDAC11 is a negative regulator of MDSC expansion and function and that EL4 tumor-bearing HDAC11 KO mice possessed a more suppressive MDSC population compared to wild-type mice [88]. However, in 2016, a team demonstrated that HDAC inhibitors depleted MDSCs induced by 4T1 mammary tumor both in vitro and in vivo in the spleen, blood, and tumor bed and increased the population of CD8 T lymphocytes. Interestingly, HDAC inhibitors also increased the apoptosis of MDSC precursor in the bone marrow, GR1+ cells [89]. Our understanding of the role of HDAC on MDSCs remains to be completed. Here the difference in models and inhibitors might be responsible for the difference in results, proving the complex interplay between MDSCs and the immune system state in tumor-bearing individuals.
7 Combination with Checkpoint Inhibitors
CTLA-4 and PD-1 (programmed death 1) are negative immune checkpoints regulating lymphocyte functions. CTLA-4 can be found in the cytoplasm of naïve T cells and is exported to the membrane after activation. Quantities of CTLA-4 found at the surface of activated T cells increase with their activation, allowing the creation of a negative feedback loop to avoid an overactivation of lymphocytes. CTLA-4 is a CD28 homologue binding CD80 and CD86 with a greater affinity than CD28. The ratio of CTLA-4/CD28 bound to costimulatory molecules will determine if the lymphocyte is activated or inhibited. Tregs express constitutively CTLA-4 which might play a part in the Tregs immunosuppressive functions. Blocking CTLA-4 results in an enhanced immune response explaining the interest it receives in oncology. PD-1 is found on highly activated T cells, NK cells, B cells, and monocytes. The binding of PD-1, with its ligands PD-L1 or PD-L2, found at the surface of tumor cells, and various tumor-infiltrated immune cells like MDSC and dendritic cells reduces T cell functions to avoid excessive immune responses [90]. PD-L1 plays a major role in the immunosuppression established in a tumor environment because of its expression on tumor cells and MDSCs; this is why the interaction PD-1/PD-L1 has led to the development of several antibodies aiming to block this interaction to restore potent antitumor immune responses [91]. Removing MDSC-dependent immunosuppression along with suppressing immune checkpoint blockade should induce a massive T cell response in tumor-bearing hosts. This is why several preclinical studies have tried to associate MDSC depletion or differentiation with antibody directed against negative checkpoint inhibitors (Fig. 11.4).

Combination with checkpoint inhibitors. Tumor-induced immunosuppression is dependent on three aspects. The direct immunosuppression mediated by tumor cells expressing PD-L1 on PD-1+ T cells, the induction of MDSCs by tumor cells and MDSC-dependent immunosuppression. By associating a molecule depleting MDSCs with a checkpoint inhibitor, it is possible to restore a potent antitumor immunity. (a) Ibrutinib depletes MDSCs, while the anti-PD-L1 blocks the PD-1/PD-L1 immunosuppression. This allows T cells to proliferate and establish an antitumor immunity, preventing tumor growth. (b) Trabectedin can directly suppress MDSCs and, when associated with an anti-PD-1, restores a proper T cell-mediated antitumor immunity. (c) The action of sorafenib against MDSCs in association with an anti-CTLA-4 allows T cells to develop an antitumor immune response leading to a reduced tumor growth
Ibrutinib, an inhibitor of BTK and ITK (interleukin-2-inducible T cell kinase) known to reduce MDSC accumulation in the tumor bed [59, 60], can enhance therapeutic antitumor immunity when associated with a PD-L1 treatment in the A20 lymphoma but also the CT26 colon carcinoma and 4T1 breast carcinoma models [59]. This was later confirmed by another study where the association of an anti-PD-L1 with ibrutinib almost completely abrogated tumor growth of 4T1 breast cancer [60]. In a model of ovarian cancer, trabectedin was associated with an anti-PD-1. This association cured about half of the mice and induced a strong tumor-specific immunity from CD4 and CD8 T cells. CD8 T cells exhibited tumor antigen-specific responses, and an increase in IFN-gamma was observed along with a reduction in immunosuppressive populations. Interestingly, in vivo trabectedin might be responsible for a rise in PD-L1 expression within tumor explaining the improved efficacy of the association over single therapies [92].
High-dose ionizing irradiation (IR) results in direct tumor death and is used in many cancers. In the TUBO breast cancer and MC38 colon cancer models, IR also decreased the population of MDSCs but increased PD-L1 expression inside tumors. To overcome this issue, IR was used along with an anti-PD-L1. This association reduced MDSC population to close to zero percent in the tumor bed while enhancing cytotoxic CD8 T cells in a synergistic manner, delaying tumor growth [93]. In HPV-related oropharyngeal cancer, radiotherapy is often being associated with chemotherapy to treat patients. In a clinical trial, authors observed HPV-specific T cell responses in 13/18 patients prior to treatment. This immune response was lost in 10/13 patients within 3 months after chemoradiotherapy (CRT). CRT decreased circulating T cells and increased the MDSC population. PD-1 expression on CD4 T cells was also enhanced after CRT. The use of a PD-1 blocking antibody in ex vivo culture restored the HPV CD4 T cell-specific response, further encouraging the study of such association to help improve patient treatments [94].
As previously seen, sorafenib can effectively deplete MDSCs. It was associated with an anti-CTLA-4 in a RENCA mouse model and whereas the monotherapies did reduce tumor growth, the combination displayed a synergistic effect with the highest rate of tumor rejection and a strong increase in infiltrating CD4 and CD8 T lymphocytes in the tumor bed [95]. Unfortunately, the number of MDSCs was not assessed. There is an ongoing phase I study about the combination of sorafenib and ipilimumab (anti-CTLA-4) in patients with advanced hepatocellular cancer with a stable disease that should give us more information.
Ipilimumab is also often associated with an anti-PD-1 named nivolumab, and this combination is now used as standard treatment of metastatic melanoma in patients. While ipilimumab could decrease MDSC number, no study thoroughly examined the impact of this association on MDSCs. However, an increase of CD4 and CD8 T lymphocytes to MDSC ratio was observed in the mouse melanoma model B16 using a combination of anti-PD-1 and anti-CTLA-4 [96].
Sunitinib, a multi-target TKI that affects the viability and proliferation of MDSCs and Tregs, was used associated with IL-12, an activating cytokine and 4-1BB, a positive immune checkpoint expressed by T cells. In the MCA26 colon carcinoma mouse model, the combination of sunitinib, IL-12, and 4-1BB significantly improved long-term survival and had an efficacy superior to that of IL-12 and 4-1BB in association [38].
Conclusion
Targeting immunosuppression and particularly MDSCs in the setting of cancer is a major issue to improve the efficacy of immune therapy aimed at targeting CD8 T cell response. Many preclinical and clinical data underline that some cytotoxic chemotherapies and tyrosine kinase inhibitors could eliminate or decrease immunosuppressive functions of MDSCs leading to the rational that combination of such drugs with checkpoint inhibitor could improve their efficacy. However, a careful analysis of such data must be performed before moving to clinical trial because the type of tumor and the association of drugs frequently impact their effects on MDSCs. In addition, a large analysis of immune responses must be performed on many of these drugs as their positive effect on MDSCs could be accompanied by negative impact on other components of the immune system, resulting in a null effect.
References
Condamine T, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2):aaf8943.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–64.
Porembka MR, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61(9):1373–85.
Capietto AH, et al. Down-regulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. 2013;210(11):2257–71.
Yan HH, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70(15):6139–49.
Ichikawa M, et al. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011;9(2):133–48.
Hiratsuka S, et al. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8(12):1369–75.
Panni RZ, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother. 2014;63(5):513–28.
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–506.
Pan PY, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99–108.
Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Gehad AE, et al. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J Invest Dermatol. 2012;132(11):2642–51.
Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Investig. 2012;41(6–7):614–34.
Yu J, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–97.
Haverkamp JM, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity. 2014;41(6):947–59.
Suzuki E, et al. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–21.
Le HK, et al. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9(7–8):900–9.
Kodumudi KN, et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16(18):4583–94.
Zhou J, et al. Therapeutic targeting of myeloid-derived suppressor cells involves a novel mechanism mediated by clusterin. Sci Rep. 2016;6:29521.
Michels T, et al. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol. 2012;9(3):292–300.
Sevko A, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol. 2013;190(5):2464–71.
Alizadeh D, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–18.
Rong Y, et al. Doxorubicin resistant cancer cells activate myeloid-derived suppressor cells by releasing PGE2. Sci Rep. 2016;6:23824.
Germano G, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62.
Filatenkov A, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. 2015;21(16):3727–39.
Vincent J, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–61.
Bruchard M, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19(1):57–64.
Annels NE, et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother. 2014;63(2):175–83.
Takeuchi S, et al. Chemotherapy-derived inflammatory responses accelerate the formation of immunosuppressive myeloid cells in the tissue microenvironment of human pancreatic cancer. Cancer Res. 2015;75(13):2629–40.
Wang Z, et al. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget. 2016;7(4):4760–9.
Kanterman J, et al. Adverse immunoregulatory effects of 5-FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014;74(21):6022–35.
Limagne E, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016;76(18):5241–52.
Martinelli E, et al. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: the role of MEK. Cancer Treat Rev. 2017;53:61–9.
Mundi PS, et al. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82(4):943–56.
Kircher SM, Nimeiri HS, Benson AB 3rd. Targeting angiogenesis in colorectal cancer: tyrosine kinase inhibitors. Cancer J. 2016;22(3):182–9.
Bikas A, et al. Targeted therapies in thyroid cancer: an extensive review of the literature. Expert Rev Clin Pharmacol. 2016;15:1–15.
Ko JS, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–57.
Ozao-Choy J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69(6):2514–22.
Ko JS, et al. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res. 2010;70(9):3526–36.
van Cruijsen H, et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res. 2008;14(18):5884–92.
Heine A, et al. The induction of human myeloid derived suppressor cells through hepatic stellate cells is dose-dependently inhibited by the tyrosine kinase inhibitors nilotinib, dasatinib and Sorafenib, but not sunitinib. Cancer Immunol Immunother. 2016;65(3):273–82.
Shojaei F, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–20.
Rodriguez PC, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69(4):1553–60.
Koinis F, et al. Effect of first-line treatment on myeloid-derived suppressor Cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. 2016;11(8):1263–72.
Cao M, et al. Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Investig. 2011;91(4):598–608.
Chuang HY, et al. Serial low doses of Sorafenib enhance therapeutic efficacy of adoptive T cell therapy in a murine model by improving tumor microenvironment. PLoS One. 2014;9(10):e109992.
Martin del Campo SE, et al. The Raf kinase inhibitor Sorafenib inhibits JAK-STAT signal transduction in human immune cells. J Immunol. 2015;195(5):1995–2005.
Giallongo C, et al. Myeloid derived suppressor cells in chronic myeloid leukemia. Front Oncol. 2015;5:107.
Christiansson L, et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol Cancer Ther. 2015;14(5):1181–91.
Larmonier N, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008;181(10):6955–63.
Mohty M, et al. Imatinib and plasmacytoid dendritic cell function in patients with chronic myeloid leukemia. Blood. 2004;103(12):4666–8.
Wang H, et al. Imatinib mesylate (STI-571) enhances antigen-presenting cell function and overcomes tumor-induced CD4+ T-cell tolerance. Blood. 2005;105(3):1135–43.
Mumprecht S, et al. Imatinib mesylate selectively impairs expansion of memory cytotoxic T cells without affecting the control of primary viral infections. Blood. 2006;108(10):3406–13.
Lowe DB, et al. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology. 2014;3(1):e27589.
Fei F, et al. Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp Hematol. 2008;36(10):1297–308.
Steinberg SM, et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol Res. 2014;2(11):1044–50.
Ngiow SF, et al. Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAFV600E melanoma. Oncoimmunology. 2016;5(3):e1089381.
Schilling B, et al. Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. Int J Cancer. 2013;133(7):1653–63.
Sagiv-Barfi I, et al. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112(9):E966–72.
Stiff A, et al. Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by Ibrutinib treatment. Cancer Res. 2016;76(8):2125–36.
Wu T, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. 2016;6:20250.
Zhao T, et al. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(−/−) mice. Oncogene. 2015;34(15):1938–48.
Welte T, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18(6):632–44.
Serafini P, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203(12):2691–702.
Weed DT, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):39–48.
Califano JA, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(1):30–8.
Noonan KA, et al. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res. 2014;2(8):725–31.
Pico de Coana Y, et al. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their Arginase1 production. Cancer Immunol Res. 2013;1(3):158–62.
Tarhini AA, et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS One. 2014;9(2):e87705.
Kusmartsev S, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441–9.
Mirza N, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–307.
Iclozan C, et al. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother. 2013;62(5):909–18.
Nagaraj S, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16(6):1812–23.
De Santo C, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A. 2005;102(11):4185–90.
Soong RS, et al. RPN13/ADRM1 inhibitor reverses immunosuppression by myeloid-derived suppressor cells. Oncotarget. 2016;7(42):68489–502.
Zoglmeier C, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17(7):1765–75.
James BR, et al. CpG-mediated modulation of MDSC contributes to the efficacy of Ad5-TRAIL therapy against renal cell carcinoma. Cancer Immunol Immunother. 2014;63(11):1213–27.
Harman MF, et al. Expansion of myeloid-derived suppressor cells with arginase activity lasts longer in aged than in young mice after CpG-ODN plus IFA treatment. Oncotarget. 2015;6(15):13448–61.
Tu SP, et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res (Phila). 2012;5(2):205–15.
Condamine T, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626–39.
Rui K, et al. Curdlan blocks the immune suppression by myeloid-derived suppressor cells and reduces tumor burden. Immunol Res. 2016;64(4):931–9.
He W, et al. Re-polarizing myeloid-derived suppressor cells (MDSCs) with cationic polymers for cancer immunotherapy. Sci Rep. 2016;6:24506.
Qin H, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med. 2014;20(6):676–81.
Holmgaard RB, et al. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine. 2016;6:50–8.
Mok S, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–61.
Xu J, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–94.
Rosborough BR, et al. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol. 2012;91(5):701–9.
Sahakian E, et al. Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63(2):579–85.
Wang HF, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol Immunother. 2016;66(3):355–66.
Wang RF, Wang HY. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017;27(1):11–37.
Menon S, Shin S, Dy G. Advances in cancer immunotherapy in solid tumors. Cancers (Basel). 2016;8(12):E106.
Guo Z, et al. Combined trabectedin and anti-PD1 antibody produces a synergistic antitumor effect in a murine model of ovarian cancer. J Transl Med. 2015;13:247.
Deng L, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–95.
Parikh F, et al. Chemoradiotherapy-induced upregulation of PD-1 antagonizes immunity to HPV-related oropharyngeal cancer. Cancer Res. 2014;74(24):7205–16.
Motoshima T, et al. Sorafenib enhances the antitumor effects of anti-CTLA-4 antibody in a murine cancer model by inhibiting myeloid-derived suppressor cells. Oncol Rep. 2015;33(6):2947–53.
Curran MA, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–80.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Bruchard, M., Ghiringhelli, F. (2018). Effect of Pharmaceutical Compounds on Myeloid-Derived Suppressor Cells. In: Zitvogel, L., Kroemer, G. (eds) Oncoimmunology. Springer, Cham. https://doi.org/10.1007/978-3-319-62431-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-62431-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62430-3
Online ISBN: 978-3-319-62431-0
eBook Packages: MedicineMedicine (R0)