
Abstract
Mixed phenotype acute leukemia (MPAL) is a rare disease and accounts for 2–5% of all acute leukemias. In 2008, the World Health Organization (WHO) classification of hematopoietic and lymphoid tumors proposed a simpler diagnostic algorithm, which relies on fewer and more lineage-specific markers to define MPAL. MPAL with t(9;22) and MLL rearrangement have been separated. The 2016 revision to the WHO classification has retained the same markers. Whole-exome sequencing in MPAL patients demonstrates frequent epigenetic regulatory genes and tumor suppressor genes, specifically DNMT3A, which is found in hematologic malignancies of both lymphoid and myeloid origin, as well as in age-related clonal hematopoiesis. The prognosis of patients with MPAL is considered poor, with long-term survival of <20%, and more uniform studies are needed to achieve better outcome in this disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Mixed phenotype acute leukemia is a rare disease and comprises 2–5% of all acute leukemias. These disorders have been historically labeled by a variety of names, such as mixed-lineage leukemia , bilineal leukemia , and biphenotypic leukemia [1]. Both the earlier 2008 and more recent 2016 World Health Organization (WHO) classifications have proposed a simpler diagnostic algorithm to define mixed phenotype acute leukemia (MPAL), which includes both biphenotypic and bilineal acute leukemias.
Morphology and Immunophenotype
Morphologically , MPAL blasts appear most often as undifferentiated medium-sized blasts with fine chromatin and indistinct-to-prominent nucleoli ; however, these blasts can show classical lymphoid features and appear smaller in size with variably condensed nuclear chromatin and very high nuclear-to-cytoplasmic ratios, or myeloid features with cytoplasmic granules, very fine nuclear chromatin, and large prominent nucleoli.
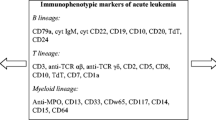
However, the diagnosis of MPAL rests on the immunophenotypic features of these blasts rather than morphology. Flow cytometry is the preferred method for recognizing MPAL. Even when there are not 2 distinctly separable populations, most cases of MPAL will show heterogeneity of expression of some antigens. For example, MPO expression will be expressed on the subset of blasts that show relatively brighter expression of myeloid markers and lower intensity of B-cell–associated markers . Figure 17.1 is an example of MPAL where the blasts are small with moderate cytoplasm and express myeloid markers (CD13, CD33, MPO) as well as strong CD19 and CD79a.


Case of B/myeloid mixed phenotype acute leukemia . Blasts are mostly small with moderate cytoplasm (part a) and expressed CD34, CD13, CD33, CD19, CD79a, TdT, and MPO (part b)
One of the first major attempts to define MPAL was the scoring criteria proposed by the European Group for the Immunological Characterization of Leukemias (EGIL) (Table 17.1) [3]. A numerical value, ranging from 0.5 to 2, was assigned for individual myeloid-associated or lymphoid-associated markers expressed by the blasts, and a biphenotypic acute leukemia was defined when a score over 2 points was achieved for each lineage [3]. In later years, the 2001 World Health Organization (WHO) classification of hematopoietic and lymphoid neoplasms incorporated the EGIL scoring system when defining acute leukemias of ambiguous lineage [4].
Then, in 2008, the WHO classification proposed a simpler diagnostic algorithm to define MPAL, which relies on fewer, more lineage-specific markers [5] (Table 17.2). Myeloid lineage requires the presence of myeloperoxidase as detected by flow cytometry , immunohistochemistry or cytochemistry, or evidence of monocytic differentiation (with at least 2 of the following markers being positive: non-specific esterase cytochemistry, CD11c, CD14, and CD64). T-lineage can be shown with cytoplasmic or surface CD3, at least as intense as background reactive T-cells, and multiple antigens are required for B-lineage including CD19, CD79a, CD22, and CD10. All possible combinations of MPAL can be observed including B/myeloid, T/myeloid, B/T, or even rarely B/T/Myeloid [5]. MPAL with t(9;22) and MLL rearrangement have been separated out as distinct subtypes. Acute leukemia of ambiguous lineage is reserved for cases of acute leukemia that show no clear evidence of differentiation along a single lineage.
In the 2016 revision to the WHO classification , no new entities were defined within this group of leukemias [6]. Although the list of lineage-specific markers is unchanged, it is now emphasized that in cases with 2 distinct blast populations, each population should meet criteria for B-lymphoblastic leukemia (B-ALL) , T-ALL, or acute myeloid leukemia but it is not necessary that specific markers are present [6]. It is also now more specifically stated that cases of otherwise typical B-ALL with only low-level expression of MPO (without other evidence of myeloid differentiation) should not be classified as MPAL. Furthermore, a specific statement is now included that cases of otherwise typical ALL or AML do not need to meet the strict lineage defining criteria listed for MPAL .
MPAL with BCR-ABL Fusion Gene
Two genetic lesions are frequent enough in MPAL to now be considered as separate entities. The first is MPAL with t(9;22)(q34;q11.2) or BCR-ABL1 rearrangement. The t(9;22)(q34;q11.2) translocation results in a BCR-ABL1 fusion gene located on the Philadelphia chromosome (Ph), causing a constitutively active BCR-ABL1 tyrosine kinase. Acute leukemia with t(9;22) and blast phase of chronic myeloid leukemia (CML) have very similar clinical presentations and morphologic features. The 2008 WHO classification suggests caution when making the diagnosis of MPAL with t(9;22) [4]. Splenomegaly, peripheral leukocytosis due to maturing myeloid precursors and mature neutrophils, absolute basophilia, and a clinical history of CML may support the diagnosis of blast phase of CML with MPAL phenotype [4]. De novo MPAL with BCR-ABL rearrangement generally occurs more frequently in older patients. Although most studies found the frequency of MPAL with t(9;22) to be 28–35%, pediatric studies report it to be much lower at 3% [7]. Many of these cases show a dimorphic population of blasts, with most showing B and myeloid lineage [7]. Some studies suggest that this subtype of MPAL has a worse outcome [8].
MPAL with MLL Rearrangement
The second most frequent genetic lesion in MPAL is translocations involving MLL gene. MLL rearrangement juxtaposes the amino-terminus of the histone methyltransferase MLL to a variety of fusion partners, with the most common partner gene being AF4 on chromosome 4 band q21.35 in MPAL [9]. This tends to occur more commonly in children and is more frequent in infancy [9]. One study showed frequency of 10% in adults to 12–18% in pediatric MPAL [1]. These cases also tend to present with a dimorphic blast population, one resembling lymphoblasts and the other resembling monoblasts. By flow cytometry , the lymphoblasts usually have a CD19-positive, CD10-negative, B-precursor immunophenotype and are frequently positive for CD15. Usually, the flow cytometry identifies a separate population of myeloid blasts with monocytic differentiation. The prognosis of MPAL patients with MLL rearrangement is also poor [10].
Mixed Phenotype Acute Leukemia, Not Otherwise Specified
Cytogenetics and Molecular Findings
In a recent study, Yan et al., found that of 92 MAPL patients assessed, 64% presented with cytogenetic abnormalities [11]. The most prevalent aberration was the complex karyotype found in 24% of patients, followed by the t(9;22) chromosome in 15% (all B-myeloid phenotype) and translocations involving MLL gene at 11q23 in 4.3% of patients [11]. A specific reference was made in the 2008 WHO classification to exclude cases that can be classified in another category, either by genetic or clinical features. For instance, AML with t(8;21), t(15;17), and inv. [11] can express lymphoid-associated markers but should be classified as AML with recurrent genetic abnormalities. Cases of chronic myelogenous leukemia (CML) in blast crisis, AML with myelodysplasia-related changes, and therapy-related AML should be classified as their respective entities even if they happen to have a mixed phenotype.
In a study of 61 MPAL patients, Weinberg et al., found that 23 of 61 patients were under 21 years of age (38%), most showed a B/myeloid phenotype (67%), and had normal cytogenetics (44% of patients with cytogenetic information) [12]. Seven patients (or 22%) had t(9;22) or MLL rearrangement. This is a similar distribution to what Matutes et al., found in their study [13]. However, both Matutes et al., and Yan et al., included MPAL patients with complex karyotype (~24–32% of all their patients) in their series [11, 13]. In the 2008 WHO classification, the presence of a complex karyotype would be considered as AML with myelodysplasia-related changes if defined by cytogenetics alone, and such cases were excluded from the study by Weinberg et al.
Rubnitz et al., analyzed gene expression patterns in 13 pediatric patients with MPAL (as defined by EGIL) and found that 8 patients displayed gene expression patterns that were different from AML and ALL [14]. In contrast, using microRNA profiling studies, de Leeuw et al., demonstrated that 16 cases had microRNA expression profiles that clustered with AML or ALL [15]. Heesch et al., noted a higher expression of BAALC and ERG in 26 cases of MPAL as compared with other cases of AML [16]. Array-based comparative genomic hybridization analysis in 12 patients with MPAL demonstrated that all patients had at least 1 abnormality, including deletions of CDKN2A, IKZF1, MEF2C, BCOR, EBF1, KRAS, LEF1, MBNL1, PBX3, and RUNX1 [14].
Information regarding the mutational landscape of MPAL is based on small patient numbers. Yan et al., analyzed 31 patients with MPAL and reported that 12 patients (39%) were found to harbor a known mutation [11]. These included IKZF1 deletion in 4 patients (all B-myeloid phenotype with evidence of BCR-ABL1 fusion gene), EZH2 in 3 (B- or T-myeloid; one case showing complex karyotype and another showing loss of chromosome 7), ASXL1 in 2 (both B-myeloid), TET2 in one (B-myeloid), and ETV6 and NOTCH1 in 1 patient each (both T-myeloid) [11]. A high rate of mostly biallelic mutations DNMT3A mutations were reported in 10 of 18 adults with T-myeloid MPAL [14]. No evidence of mutations in CBL, DNMT3A, FBXW7, FLT3, IDH1, IDH2, KIT, NPM1, PHF6, RUNX1, and WT1 were found in Yan’s study [11].
Whole-exome sequencing in 23 adult and pediatric patients with MPAL demonstrated that 35% patients had mutations in epigenetic regulatory genes ([17], Table 17.3). DNMT3A was the most common mutation (23%) followed by IDH2 (9%), TET3 (4%), and EZH2 (9%). All of the DNMT3A mutations involved the methyltransferase domain, three of which were missense mutations at Arg882, the hotspot common in AML . DNMT3A occurred in all immunophenotypic subtypes examined. Similar to reports in AML, MPAL patients with mutation in DNMT3A trended toward being older and having a normal cytogenetics [17]. Tumor suppressors were also frequently mutated and 5 patients (22%) had TP53 mutations ([17], Table 17.3). Mutations of DNMT3A and tumor suppressors showed high variant allele frequency (VAF), suggesting that these mutations arise early in the disease. Sixty-one percent of the patients also had mutually exclusive mutations of activating signaling genes including NRAS, KRAS, and NF1 [18]. NOTCH1 mutations were present in 5 of 16 (32%) with T-myeloid and B/T leukemia. Three samples (13%) also had WT1 mutations. In another series, clustering of FLT3 ITD and TKD mutations was reported in patients with T-myeloid MPAL. Seven of 15 patients (47%) were positive for FLT3 mutations (mostly ITD), all of which were CD117 + [19].
Prognosis and Therapy
There is no set therapy for MPAL patients, which is a result of the absence of prospective trials. In the few larger retrospective series of MPAL, the median overall survival is reported to range from 14.8 to 18 months and the rate of achieving long-term survival in patients with adult MPAL is poor (<20%) [17, 19, 20]. Most of the retrospective case series suggest that the complete remission rates are higher with ALL therapy or an ALL/AML combined regimen than with AML-type therapy [21, 22]. Children with MPAL are suggested to do better, although they do have inferior outcome compared with those diagnostic with typical ALL [22]. A few studies compared outcome of MPAL patients with that of matched control ALL or AML groups and most found that MPAL patients did worse than AML or ALL [1]. In a study of 61 patients, Weinberg et al., found that when compared with 177 patients with acute myeloid leukemia (AML) , MPAL patients had better overall survival (P = .0003) and progression-free survival (P = .0001). However, no difference in overall survival between MPAL and 387 patients with acute lymphoblastic leukemia was present (P = .599) [12]. For patients with t(9;22)-positive MPAL, a tyrosine kinase inhibitor (TKI) is usually added to treatment [23]. In his review, Wolach et al. suggested that the best approach for the non-t(9;22) MPAL patient is to treat with an ALL regimen and consolidate with an allogeneic stem cell transplant if a donor is available [23]. Shimizu H et al., have suggested that allogeneic hematopoietic stem cell transplantation may be an effective treatment for MPAL patients, especially early in the disease course [24].
Conclusion
Overall, acute leukemias with mixed phenotypes are uncommon and comprise 2–5% of all acute leukemias. Molecular studies showed frequent mutations in epigenetic regulatory genes and tumor suppressors in MPAL patients. The outcome of MPAL patients remains poor and mutations have been identified in this disease that are potentially targetable by agents that are currently available or are being tested in clinical trials, including epigenetically targeted agents, tyrosine kinase pathway inhibitors, and NOTCH1 inhibitors. Studies suggest that the best treatment of non-t(9;22) MPAL patient is to treat with an ALL regimen and consolidate with an allogeneic stem cell transplant if a donor is available. More studies are needed to address the biology and treatment of MPAL patients.
References
Weinberg OK, Arber DA. Mixed-phenotype acute leukemia: historical overview and a new definition. Leukemia. 2010;24(11):1844–51.
Weir EG, Borowitz MJ. Acute leukemias of ambiguous lineage. In: Jaffe ES, Harris NL, Vardiman JW, Campo E, Arber DA, editors. Hamtopathology. Philadelphia: Elsevier; 2010.
Bene MC, Castoldi G, Knapp W, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995;9:1783–6.
Jaffe E, Harris N, Stein HVJ, et al. World Health Organization classification of tumours. Pathology and genetics of tumors of hematopoietic and lymphoid tissues 2nd printing. Lyon: IARC Press; 2001.
Borowitz MJ, Bene MC, Harris NL, et al. Acute leukemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2008. p. 150–5.
Arber DA, Orazi A, Hasserjian R, Thiele T, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Al-Seraihy AS, Owaidah TM, Ayas M, El-Solh H, Al-Mahr M, Al-Ahmari A, et al. Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica. 2009;94:1682–90.
Killick S, Matutes E, Powles RL, Hamblin M, Swansbury J, Treleaven JG, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84:699–706.
XQ X, Wang JM, Lü SQ, Chen L, Yang JM, Zhang WP, et al. Clinical and biological characteristics of adult biphenotypic acute leukemia in comparison with that of acute myeloid leukemia and acute lymphoblastic leukemia: a case series of a Chinese population. Haematologica. 2009;94:919–27.
Owaidah TM, Al Beihany A, Iqbal MA, Elkum N, Roberts GT. Cytogenetics, molecular and ultrastructural characteristics of biphenotypic acute leukemia identified by the EGIL scoring system. Leukemia. 2006;20:620–6.
Yan L, Ping N, Zhu M, et al. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 adult patients with mixed-phenotype acute leukemia defined by WHO-2008 classification. Haematologica. 2012;97(11):1708–12.
Weinberg OK, Seetharam M, Ren L, Alizadeh A, Arber DA. Mixed phenotype acute leukemia: a study of 61 cases using World Health Organization and European Group for the Immunological Classification of Leukaemias criteria. Am J Clin Pathol. 2014;142(6):803–8.
Matutes E, Pickl WF, Van’t Veer M, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood. 2011;117(11):3163–71.
Rubnitz JE, Onciu M, Pounds S, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children’s Research Hospital. Blood. 2009;113(21):5083–9.
de Leeuw DC, van den Ancker W, Denkers F, et al. MicroRNA profiling can classify acute leukemias of ambiguous lineage as either acute myeloid leukemia or acute lymphoid leukemia. Clin Cancer Res. 2013;19(8):2187–96.
Heesch S, Neumann M, Schwartz S, et al. Acute leukemias of ambiguous lineage in adults: molecular and clinical characterization. Ann Hematol. 2013;92(6):747–58.
Eckstein OS, Wang L, Punia JN, Kornblau SM, Andreeff M, Wheeler DA, Goodell MA, Rau RE, et al. Mixed-phenotype acute leukemia (MPAL) exhibits frequent mutations in DNMT3A and activated signaling genes. Exp Hematol. 2016;44(8):740–4.
Kern W, Grossmann V, Roller A, et al. Mixed Phenotype Acute Leukemia, T/Myeloid, NOS (MPAL-TM) has a high DNMT3A mutation frequency and carries further genetic features of both AML and T-ALL: results of a comprehensive next-generation sequencing study analyzing 32 genes. Blood. 2012;120:403.
Hoehn D, Medeiros LJ, Chen SS, et al. CD117 expression is a sensitive but nonspecific predictor of FLT3 mutation in T acute lymphoblastic leukemia and T/myeloid acute leukemia. Am J Clin Pathol. 2012;137(2):213–9.
Deffis-Court M, Alvarado-Ibarra M, Ruiz-Argüelles GJ, et al. Diagnosing and treating mixed phenotype acute leukemia: a multicenter 10-year experience in México. Ann Hematol. 2014;93(4):595–601.
Liu QF, Fan ZP, MQ W, et al. Allo-HSCT for acute leukemia of ambiguous lineage in adults: the comparison between standard conditioning and intensified conditioning regimens. Ann Hematol. 2013;92(5):679–87.
Gerr H, Zimmermann M, Schrappe M, et al. Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol. 2010;149(1):84–92.
Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125(16):2477–85.
Shimizu H, Saitoh T, Machida S, Kako S, Doki N, Mori T, Sakura T, Kanda Y, Kanamori H, Miyawaki S, Okamoto S, Kanto Study Group for Cell Therapy (KSGCT). Allogeneic hematopoietic stem cell transplantation for adult patients with mixed phenotype acute leukemia: results of a matched-pair analysis. Eur J Haematol. 2015;95(5):455–60.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Weinberg, O.K. (2018). Mixed Phenotype Acute Leukemia. In: Chang, CC., Ohgami, R. (eds) Precision Molecular Pathology of Myeloid Neoplasms. Molecular Pathology Library, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-62146-3_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-62146-3_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-62144-9
Online ISBN: 978-3-319-62146-3
eBook Packages: MedicineMedicine (R0)