
Abstract
An applicability of 13C and 1H relaxation rates (1/T 1) to direct calculations of the characteristic reorientation time (τc) in imidazolium-based ionic liquids has been analyzed. It has shown that 13C NMR relaxation technique can be applied to ionic liquids as successfully as it took place for other liquid systems and allows one to get τc values for each carbon directly. The corresponding 1H data are affected by dipole-dipole interaction only at “higher temperature” range while at lower temperatures spin-diffusion process controls the proton relaxation. Both carbon and hydrogen 1/T 1 dependences are suitable for calculation of τc and reveal equal numerical values for a number of functional groups of [emim]Ac at the proper temperature range. On the other hand, 1H relaxation curves of some functional groups allow one to detect motions, unobservable in the carbon relaxation and, thereby, to extract more information concerning details of the dynamics of the [amim]+ cations.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


1 Introduction
For many decades, NMR T1-relaxation has been used successfully to study a variety of fluid systems, including both pure liquids and various solutions, see e.g. [1,2,3] and references within. In the most cases one needs to obtain a temperature dependence of the 1/T 1 with one or more maxima, and this dependence can be transformed to the temperature dependence of the characteristic reorientation time (τ c ) for each chemical/functional group of the system under investigation.
During 2 or 3 last decades a new class of liquid systems drew much attention, and the systems are known now as “Ionic Liquids” (ILs, RTILs). A lot of reviews were dedicated both to the unique properties of ionic liquids [4,5,6] and to results of an application of the NMR technique to ILs [7,8,9,10,11,12,13]. Keeping in mind these reviews the main purpose of this work was not a comprehensive survey of all the publications but a discussion of some trends in the application of NMR, especially of NMR relaxation, to understanding of structure and dynamics of ILs of various kinds with focusing on some differences/problems appearing during an evaluation of the NMR relaxation data in ILs. An accent has been made on critical analysis of common approach to the interpretation of 13C T1 relaxation data for imidazolium-based ILs because for these systems an erroneous interpretation is replicated for ca. last 15 years.
In view of the above only articles directly related to the designated task i.e., those connected to carbon or proton relaxation in ionic liquids of [amim]X kind, were cited in full (within the limits of our knowledge). Of the rest of the literature, only articles over the last 2–3 years were cited as well as some articles from previous years which were not included in the reviews listed above.
2 Basics of the Theory of NMR T1 Relaxation
First, we would like to recall briefly the basics of NMR relaxation for fluid systems taking into consideration that similar information was already included in a number of recent reviews and textbooks. In particular for the case of 1H and 13C nuclei the dipole-dipole interaction is the main one which affects the relaxation rate [1,2,3].
For the theoretical description of T1 frequency and/or temperature dependences the Bloembergen-Purcell-Pound theory (further BPP) is usually used. The BPP as well as the other NMR relaxation theories is based on the concept of spectral density function, J(ω, T). This function generally can be written using a set of relaxation times, τ i , and their contributions, C i ; note that the sum of all C i equals 1.
Here ω is a cyclic resonant frequency (2πυ 0 ) for the nucleus investigated.
In the case of dipole-dipole mechanism of relaxation with one correlation time τ c this theory reduced to expressions (2) for 13C and (3) for 1H [1,2,3, 14,15,16].
where ω H and ω C —cyclic resonant frequency (2πυ 0 ) for 1H and 13C, respectively; sc 2 and sH 2 (≤1) are so called order parameters which will be discussed later; A 0 is a constant which does not depend on temperature and frequency. For hydrogen and carbon nuclei this constant is given by expressions (3) and (4):
where ħ is the reduced Plank constant (h/2π), r HH is the distance between the interacting H-atoms, r CH is a length of the C–H bond, γ H and γ C are magnetogyric ratios for 1H and 13C nuclei.
Formulas (2) and (3) describe a relaxation under interaction of two isolated nuclei. The theory considering interaction of several nuclei leads to quite complicated formulas however, it appears that rather good approximation turns out multiplication of the right part of the (2) and (3) by number of pair interactions of the nucleus with an environment (interacting couples), and this approach will be used in further calculations. Only the nearest neighbor spins were thus considered due to a strong dependence of the dipole-dipole interaction intensity on the distance (r −6).
Expressions (2) and (3) describe a dependence of the relaxation rates on the correlation time (τ c ) and resonance frequency (ω H or ω C ). There are several procedures in order to transform 1/T 1 values to τ c magnitudes. Let us discuss them in more detail.
The simplest way is to use the ratio 1/T 1 = constant * τ c which is valid at so-called “extreme narrowing” case, i.e. at τ c ω « 1. This situation is common for systems with low viscosity and/or at not high working frequency, and it was used for a lot of pure liquids, liquid mixtures, and electrolyte solutions. However in the case of ionic liquids, due to much higher viscosity, this procedure is often insufficient and can even lead to absolutely wrong results.
Another known procedure is based on the use of the total temperature dependence. In this case an achievement of the 1/T 1 maximum point is extremely essential, since at that point the τ c magnitude can be directly calculated using (6) and (7):
for hydrogen and carbon, respectively [1,2,3, 14]. At the same time the y value at the point of maximum allows an independent determination of the constant (s2A) in expressions (2) or (3) hence, a further calculation of τ c at any temperature.
Experimentally specified dependences can be obtained by “scanning” of 1/T 1 relaxation rates on temperature, and for their approximation one may use expressions (2) or (3) in common with standard Arrhenius dependence of the correlation time
where E a —is an activation energy for this type of movement, R—the gas constant, τ 0 (so-called “τ c at zero T”)—the parameter which isn’t making clear physical sense. At approximations of experimental dependences A 0 , E a и τ 0 are commonly used as adjustable parameters.
The approach described above is rather effective and is widely used for the analysis of orientation mobility of various liquid systems including ionic liquids. A shortcoming of the approach is a postulation of the τ c Arrhenius dependence with one energy of activation (5) for the whole temperature range studied while it is well known that for many liquid systems (some pure liquids, solutions and so forth) such approach is unfair.
At the same time it is obvious from (2) and (3) that 1/T 1 experimental dependences can be directly counted in values of the correlation times, and, thereby, give valid τ c dependences on temperature. In practice, however, there are a number of difficulties. First, the numerical values of A 0 are known with insufficient accuracy because of the strong dependence on the exact r HH and r CH distances values in a liquid or solution. Second, an introduction of the s2 ≤ 1 coefficient which takes into account possible contributions to the relaxation rates from other molecular movements in complex molecules is needed for some cases. In particular, according to (1), the J(ω, T ) in (2) is a combination of various contributions with different τ i values reflecting the nucleus reorientation. For two different correlation times e.g. τ c1 and τ c2 , (1) transforms to Woessner “anisotropic rotation” model [15, 16] or to Lipari-Szabo “model-free” approach [17]. Both models predict a function with two maxima but the experimental curves are often able to reflect only a part of the whole function because of temperature and/or frequency limits i.e., only one of two possible maxima is observed. Therefore the dependence might look like a curve with only maximum. However factors c1 and/or c2 in (1) and corresponding factors in (2) and (3) are unknown, may be sufficiently less than 1, and therefore a direct calculation of τ c may lead to wrong τ c values. In this regard rather wide circulation was received by a method of measuring of relaxation times in so-called “dispersive” area, i.e. in the area which is including 1/T 1 maximum. It allows one to determine a τ c value in the maximum point directly using ratios (6) and (7) and not to demand a preliminary knowledge of the A 0 (or s 2 A 0 ). On the contrary, the last value can be independently determined from 1/T 1 magnitude at the point of maximum.
Thus, the foregoing analysis revealed, that each of the existing procedures used for a conversion of relaxation rates to the correlation times (the characteristic times of the rotational reorientation) contains approximations that require the verification in each individual case. Nevertheless, the τ c magnitudes obtained are well suited for characterization of the average ion mobility as a whole and of each functional group, for which a separate line exists in the NMR spectrum.
Experimental
Three pure ionic liquids: [bmim]PF6, [bmim]BF4, and [emim]Ac were received from Sigma-Aldrich and used without further purification. All NMR measurements were carried out using NMR AVANCE-400 spectrometer at resonance frequencies (f 0) 100 MHz for 13C nuclei and 400 MHz for 1H ones.
3 Carbon 1/T 1 Temperature Dependences
Regular publications on measuring the relaxation rates in ionic liquids started to appear from the beginning of the 90s, and most of them are already reflected in the reviews and books over the years. It is useful to note that many of pure ILs, especially the imidazolium-based ones (see above), are very suitable for investigation by NMR relaxation due to their much higher viscosity comparing to conventional pure liquids and electrolyte solutions. As the result a lot of investigations were made using 13C NMR, see reviews above. Some of the most recent works will be also cited at the end of the paper. The advantages of using the carbon resonance compared to hydrogen one look almost evident, since carbon is included in the cation skeleton and therefore directly reflects the cation mobility while the hydrogen relaxation can mask a relatively slow reorientation of the cation due to the additional rapid rotation of CHn-groups.
Probably the first regular investigations of 13C relaxation rates, 1/T 1C was made by Carper with coworkers in several imidazolium-based ILs [7, 18,19,20,21,22]. Indeed, these authors carried out measurements of temperature dependences of 1/T 1 for 13C nuclei in [amim]+ cations, showed that these dependences correspond to the expected ones (2), and calculated numerical magnitudes of the reorientation times (τ c ) for each group in the investigated ILs. Up to now these papers are cited in practically all reviews (including e.g. a review of 2016 [13]) as the basic works in the topic. However a number of questions and/or problems appeared if one decided to penetrate deeper into the interpretation procedure. So we reanalyzed ones more both the data and the interpretation procedure of the Carper’s group approach and would like to demonstrate below that some of the authors’ key assumptions used in the τ c calculation were wrong.
First, the authors reported about non-monotonic temperature dependence of the characteristic time (hereinafter correlation time, τ c ) for the imidazolium-ring carbons reorientation which appeared after direct calculations using (2), see Fig. 4.2 (Fig. 2 from [20]).
To try to explain and understand this non-physical dependence authors attributed the result to a strong influence of the chemical shift anisotropy (CSA) on ring carbons relaxation and suggested to use nuclear Overhauser effect (NOE) experiments in order to reach correct results, i.e. monotonic τ c increasing with the decreasing of temperature. To agree with the procedure one should declare at least three assumptions that require an additional verification:
-
(i)
Using (2) for direct transformation of 1/T 1C dependences to τ c ones reveals wrong results namely, non-monotonic temperature dependences of τ c ;
-
(ii)
A reason of (i) is a fact that 1/T 1C values are not of pure dipole-dipole origin but contain a sufficient contribution from CAS interaction, whereby the (2) ceases to be valid.
-
(iii)
NOE experiments require for right transformation of experimental 1/T 1C values to τ c ones.
Since the CSA contribution is proportional to the permanent magnetic field the assumption (ii) could be checked directly. Measuring the spin-lattice relaxation time (T 1) at two different magnetic fields [23] revealed undoubtedly an absence of the CSA contribution for any carbon of the [amim]+ cation (the CSA contribution was found less than 2%). As the result, this led to a situation where well-known and successfully used NMR relaxation approach could not be applied to ionic liquids due to unknown reasons. The situation was unclear up to 2014 when the assumption (i) was rejected [14]. And let us discuss this procedure in more detail.
The authors [14] recalculated τ c values by following the same procedure as in the works [7, 18,19,20,21,22] i.e. using the (2) for direct transformation a 1/T 1 magnitude to the τ c value. Initially non-monotonic τ c dependences were obtained similar to those obtained earlier, see Fig. 4.2. However, a deeper analysis revealed an error in the calculations. Namely (2) has two real roots at each temperature and both roots are positive. Temperature dependences of the roots, i.e. of the calculated τ c values, are shown in Fig. 3 from [14].
The root#1, marked by the dotted line, shows an increase of the molecular mobility under temperature increasing, i.e. corresponds to conventional models of molecular mobility. The second root, otherwise, shows an increase of the molecular mobility under temperature decreasing. It contradicts any existing theory and therefore this root doesn’t make physical meaning. It is worth emphasizing that such situation is not a feature of only ionic liquids as similar solutions of the (1) with two roots will turn out for any fluid system where maximum in the 1/T 1 temperature dependence is observed, e.g. in viscous liquids or concentrated electrolytes solutions.

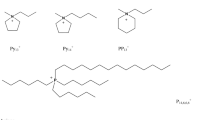
Demonstrates a chemical structure of the [emim]+ cation of the [emim]CH3COO ([emim]Ac) ionic liquid (IL) from the most common family of the imidazolium-based ILs. Numbering of lines in the Figure will be used further to describe the 1H relaxation in [emim]Ac

Reproduced with permission
From [20]. Corrected (upper curves) and initial (lower curves) correlation times (ns) for [BMIM][PF6] ionic liquid versus temperature. (bulls) Imidazolium ring C2 carbon; (open triangles) average of imidazolium ring C4 and C5 carbons.
Coming back to calculations in the [20], the authors used the smaller root magnitude at all temperatures as marked in Fig. 4.3 by the red line and thus calculated wrong values of the τ c at lower temperatures. On the contrary, if one uses the root#1 in the whole temperature range, then calculations lead to conventional temperature dependence of τ c for all spectral lines. Hence, 1/T 1 data are enough for calculation of right τ c magnitudes and no additional experimental measurements, such as NOE, required.

Hence, the results of [14] have shown that 13C NMR relaxation technique can be applied to ionic liquids as successfully as it took place for other liquid systems.
On the other hand, it is reasonable to expect that NOE data in the same systems can provide additional information on the systems properties under the condition of accurate analysis. But this task is not still carried out.
3.1 Comparison of the Behavior of Analogous Groups in the Studied ILs
At the next step one may try to compare 13C relaxation curves for the analogous groups in a few ILs namely, [bmim]PF6, [bmim]BF4, and [emim]Ac. For [bmim]PF6 the curves were firstly obtained by Carper with coauthors [18,19,20]. Later other group [14] repeated the measurements and found that Carper’s group experimental data had no errors and could be used without any correction. We carried out also additional measurements for [bmim]BF4 and [emim]Ac ILs. To provide an overall picture of differences in T1 relaxation, the comparison of 1/T 1 temperature dependencies, is presented in Figs. 4.4 and 4.5 [24].

Comparison of behavior of 1/T 1 temperature dependencies for C2 (a) and C4 (b) ring carbons in three ILs: [bmim]PF6, [bmim]BF4, and [emim]Ac

Comparison of 1/T 1 temperature dependencies for CH3-N carbon in three ILs, [bmim]PF6, [bmim]BF4, and [emim]Ac (a) and for two carbons of the CH3COO¯ anion in [emim]Ac ionic liquid
As follows from the Figures, each group reveals similar dependences in all three ILs, and the dependence corresponds to the expression (2), i.e. looks as a curve with one minimum. That is, the comparison of the carbon relaxation curves does not show a difference in the behavior of studied liquids. For [emim]Ac IL the carbon data allowed an observation of the anion as well. The 1/T 1 curves of acetate carbons (Fig. 4.4) reveal similar shape, close Tmax values but significantly different magnitudes of 1/T 1. This set of the data allows one to assume that COO-carbon relaxation is also affected by dipole-dipole interaction between a carbon atom and hydrogen ones while smaller 1/T 1 values of the COO-carbon correspond to higher distances between this carbon atom and hydrogen ones in the anion CH3-group.
Both carbons of the acetate anion have the maximum of relaxation rate at close temperatures (277.8 ± 3.5 K), relatively less than ring carbons. On one hand this is an evidence of the anion rotation (reorientation) as a whole so that the intramolecular (anisotropic) rotation of the methyl group does not make any significant contribution into the average correlation time of the methyl carbon. On the other hand a relatively high mobility of the anion may be a consequence of the incomplete association of the counter-ions. As a result, the correlation time which is determined from the position of the 1/T 1 relaxation rate maximum represents the weighted average value between the reorientation time of ion pair and of the characteristic time of the faster process, rotation of the dissociated anion.
3.2 Correlation Times and Activation Energies of Different Groups
As noted above, a relative position of the maximum itself characterizes a mobility of the cation groups. Namely, a lower Tmax corresponds to the higher mobility. However, it is possible a direct calculation of τ c at each temperature using the corresponding 1/T 1 at current T and the s 2 A 0 value calculated at Tmax. A more detailed procedure was described in [14], and the results are presented on the Table 4.1. In order to demonstrate an adequacy of the calculation procedure the calculated τ c values for both carbons of the CH3COO¯ anion are shown in Fig. 4.6. As evident from the Figure the values are much closer despite a strong difference in the 1/T 1 magnitudes, Fig. 4.5b. Thus it supports a conclusion about the anion reorientation as a whole.

Comparison of behavior of τc temperature dependencies for CH3- and COO-carbons of the CH3COO¯ anion
The data from Table 4.1, i.e. 13C correlation times reveal a few trends which are valid for all studied ILs. First, the τ c values in the IL cations demonstrate similar dependences on a group location: (i) correlation time values of butyl chain groups increase moving towards the imidazolium ring of [bmim]BF4 and [bmim]PF6 ILs, (ii) the longest correlation time for every IL is observed for CH3-methyl group, (iii) CH-ring carbons, CH2-N, and CH3-N groups have close correlation times and thus characterize a mobility of the cation as a whole. Partly these trends were already observed and discussed in the literature.
4 Comparison of Information Obtained from 1H and 13C NMR Relaxation Data
As discussed above, the 13C relaxation looks more suitable to test the cation reorientation. On the other hand 1H measurements require much less spectrometer time and efforts. Therefore it was interesting to understand the limits up to which one can use 1H data in order to characterize both the cation reorientation as a whole and the reorientation of each functional group. To check the idea we have also analyzed 1H relaxation data for the same ILs.
4.1 General Comparison
First one can compare temperature dependences of the 13C and 1H relaxation rate. As it was already mentioned above the 1/T 1 curves for all carbon nuclei correspond to the expected dependence (2) over the entire temperature range. For 1H relaxation such kind of the dependence is observed only for the “high-temperature” part of the curves, and in the “low-temperature” area (below ca. 260 K) 1/T 1H values become practically independent of temperature and identical for all lines (see Fig. 4.7a and b).

Hydrogen 1/T 1 temperature dependences for some groups of [emim]CH3COO (a) and [bmim]PF6 (b) ionic liquids
Such behavior was attributed to the spin-diffusion interaction which becomes the main mechanism of 1H relaxation at low temperatures (under low molecular mobility), [25]; see also [26], where the same effect was observed for 1/T 1C but in much smaller measure.
Another apparent difference between carbon and proton relaxation manifests itself in a deviation of some proton curves from the (3). We will return to this effect later and now let us try to compare τ c values obtained from 1H and 13C data using the groups with similar 1/T 1C and 1/T 1H shapes. And we would like to start from a number of functional groups of the [emim]Ac IL following [25].
4.2 Carbon-Hydrogen Comparison for [Emim]Ac
The 1H NMR spectrum of the [emim]CH3COO IL is well known, see e.g. [25], and lines numbering coincides numbers in Fig. 4.1 (above) i.e. follows the chemical shift (δ) increasing. There are 5 groups for which the 1/T 1 maximum was observed both in 1H and 13C dependences namely: CH3(N), CH3(O), CH2(N) and two groups of the imidazolium ring: C(4)H and C(5)H; correspond spectrum lines: 2–6. These proton 1/T 1 dependences are similar to 13C ones at least for the higher temperature range where 1H relaxation affects by the dipole-dipole interaction, see also [25].
Calculations of τ c values for 1H were executed using the same procedure as for the carbon relaxation rates, see above. Two examples are shown in Fig. 4.8, pictures for other groups are similar [25].

Comparison of τ c curves calculated from 13C and 1H data for ring C(4)H/CH(5)H (a) and CH2-N (b) groups of the cation in the [emim]Ac ionic liquid
As evident from the Fig. 4.8, the τ c values calculated using 1H and 13C data coincide for ring groups as well as for aliphatic ones in the “high temperature” range. The same situation is valid for other functional groups mentioned above, see in more detail in [25], and this fact proves an adequacy of the used approach for the determination of characteristic times of the cation reorientation in the [emim]Ac ionic liquid. It means in turn that proton 1/T 1 data as well as 13C ones are suitable for calculation of τ c numerical values for a number of functional groups of [emim]Ac at the proper temperature range. This means also an identical nature of the orientation mobility process which controls the proton and carbon relaxation.
4.3 Comparison of 13C and 1H Relaxation Data for [Bmim]PF6 and [Bmim]BF4
Some 1/T 1H temperature dependences for [bmim]PF6 IL are shown in Fig. 4.7b. For [bmim]BF4 the dependences are very close in their shape to [bmim]PF6 for each functional group differing only in numerical values and in the position of the maxima. However the 1H curves of these two ILs do not show a close similarity in shape to corresponding 13C curves though the main trends mentioned above remain.
Since the most of 1H dependencies have more or less pronounced maximum one can try to calculate the correlation times using the same procedure as above. Once more the obtained τ c are not equal to the corresponding 13C data. In more detail a description of 1H relaxation in the [bmim]PF6 and [bmim]BF4 ILs will be presented in [27].
4.4 1H and 13C Difference
Now let us return to the difference in the behavior of 13C and 1H curves for some functional groups of the studied ILs. There are two examples below, (see Fig. 4.9).

Comparison of 1H 1/T 1 curves for C(2)H groups in three studied ionic liquids (a) and for some groups in [emim]CH3COO (b)
One can observe a different temperature behavior of 1H relaxation of the C(2)H ring hydrogen in [emim]CH3COO from one side, and in [bmim]BF4 or [bmim]PF6 from another side (Fig. 4.9a). For the ILs with the BF4¯ and PF6¯ anions a behavior of the curves is similar and looks like an overlap of two or more broad unresolved lines, while in the case of the [emim]Ac a pronounced maximum is observed for C(4)H and C(5)H hydrogens (Fig. 4.9b). In addition, for the C(2) hydrogen one more maximum is clearly observable at higher temperature, and the maximum corresponds to the largest of the observed τ c i.e. reflects the slowest cation reorientation.
Thus we have found that 1H relaxation curves in some cases allowed one to detect motions, unobservable in the carbon relaxation and, thereby, to extract more information concerning details of a dynamics of the [amim]+ cations. In particular, a different 1/T 1 behavior for different anions leads to the hypothesis about different ways of the cation packaging in these systems. And it turned out that this assumption correlates well with the literature data on computer simulation of the same ionic liquids.
As the final point of our work a list of some most recent publications [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47] which were published after Damodaran’s review [13] is presented. We took into account the works where NMR—in any of its variants namely, spectra, relaxation, or diffusion—was used to study of ionic liquids. A very brief glance at the list as well as the recent reviews allows one to conclude that a sufficient part of the NMR publications during the recent years begins to be paid to the proton and inorganic ILs, to mixtures of an IL with other compounds, and to ILs in porous materials. Undoubtedly, it is due to the use of IL-based electrolytes in supercapacitors, ionic batteries etc. However the study of the basic physical and chemical properties of ILs of different kinds remains the essential aim of researchers.
References
A. Abragam, Principles of Nuclear Magnetism (Oxford University Press, Oxford, U.K., 1961)
G.C. Levy, D.J. Kerwood, Encyclopedia of Nuclear Magnetic Resonance, vol. 2 (Willey, New York, 1996)
Chizhik, V.I. et al., Magnetic Resonance and Its Applications (Springer, 2014). ISBN 978-3-319-05299-1
B. Kirchner, Ionic Liquids. Topics in current chemistry, vol. 290 (Springer, Berlin Heidelberg, 2010)
S.T. Handy, Ionic Liquids—Classes and Properties (InTech, 2011), http://www.intechopen.com/books/ionic-liquids-classes-and-properties (ISBN 978-953-307-634-8)
K. Ghandi, A review of ionic liquids their limits and applications. Green Sustain. Chem. 4, 44–53 (2014)
J.H. Antony, D. Mertens et al., Pure Appl. Chem., 76, 255–261 (2004)
R. Giernoth, NMR Spectroscopy in Ionic Liquids In Ionic Liquids, ed. by B. Kirchner. Topics in current chemistry, vol. 290, p. 263 (Springer, Berlin, Heidelberg, 2009)
V.P. Ananikov, Chem. Rev. 111, 418–454 (2011)
K. Hayamizu, Ionic Liquids—Classes and Properties (Chapter 10), in Translational and Rotational Motions for TFSA-Based Ionic Liquids Studied by NMR Spectroscopy, ed. by S.T. Handy, vol. 344 (In Tech, 2011). ISBN 978-953-307-634-8
A.-L. Rollet, C. Bessada, Annu. Reports NMR Spectrosc. 78, 149–207 (2013)
R. Giernoth, A. Bröhl et al., Interactions in ionic liquids probed by in situ NMR spectroscopy. J. Mol. Liq. 192, 55–58 (2014)
K. Damodaran, Recent NMR studies of ionic liquids. Annu. Reports NMR Spectrosc. 88, 215–244 (2016)
V.V. Matveev, D.A. Markelov et al., 13C NMR relaxation and reorientation dynamics in imidazolium-based ionic liquids: revising interpretation. Phys. Chem. Chem. Phys. 16, 10480–10484 (2014)
D.E. Woessner, J. Chem. Phys. 36, 1–4 (1962)
D.E. Woessner, Brownian motion and correlation times. eMagRes (Wiley online library, 2007). https://doi.org/https://10.1002/9780470034590.emrstm0047
G. Lipari, A. Szabo, J. Am. Chem. Soc. 104, 4546 (1982)
J.H. Antony, D. Mertens et al., ChemPhysChem 4, 588–594 (2003)
W.R. Carper, P.G. Wahlbeck et al., Anal. Bioanal. Chem. 378, 1548–1554 (2004)
W.R. Carper, P.G. Wahlbeck et al., J. Phys. Chem. A 108, 6096–6099 (2004)
J.H. Antony, D. Mertens et al., J. Phys. Chem. A 109, 6676–6682 (2005)
N.E. Heimer, J.S. Wilkes et al., J. Phys. Chem. A 110, 868–874 (2006)
M. Imanari, H. Tsuchiya et al., Magn. Reson. Chem. 47, 67–70 (2009)
E.V. Brui, 13C NMR investigation of molecular mobility of ion groups in some ionic liquids, Master’s thesis. Lappeenranta University of Technology, 2012
V.V. Matveev, D.A. Markelov et al., Molecular mobility of counterion functional groups in ionic liquid 1-ethyl-3-methylimidazolium acetate according to 1H and 13C NMR relaxation data. Russ. Chem. Bull. 62(9), 1985–1990 (2013)
N.E. Heimer, R.E. Del Sesto et al., Magn. Reson. Chem. 42, 71–75 (2004)
V.V. Matveev, D.A. Markelov et al., Magn. Reson. Chem., aсcepted
M. Shadeck, Translational and Rotational Diffusion in Ionic Liquids Through NMR Spectroscopy (The Pennsylvania State University, Thesis, 2015)
H. Abe, T. Takekiyo et al., Anomalous freezing of nano-confined water in room-temperature ionic liquid 1-Butyl-3-methylimidazolium nitrate. ChemPhysChem 17, 1136–1142 (2016)
N. Gjineci, E. Boli et al., Separation of the ethanol/water azeotropic mixture using ionic liquids and deep eutectic solvents. Fluid Phase Equilib. 424, 1–7 (2016)
K.S. Han, X. Wang et al., Distribution of 1-Butyl-3-methylimidazolium Bistrifluoromethylsulfonimide in mesoporous silica as a function of pore filling. J. Phys. Chem. C 117, 15754–15762 (2013)
H. Abe, T. Takekiyo et al., Direct evidence of confined water in room-temperature ionic liquids by complementary use of small-angle x-ray and neutron scattering. J. Phys. Chem. Lett. 5, 1175–1180 (2014)
T.Y. Wu, Y.-H. Wang et al., Influence of LiTFSI addition on conductivity, diffusion coefficient, spin—lattice relaxation times, and chemical shift of one-dimensional NMR spectroscopy in litfsi-doped dual-functionalized imidazolium-based ionic liquids. J. Chem. Eng. Data 60, 471–483 (2015)
K. Hayamizu, S. Tsuzuki et al., Transport and electrochemical properties of three quaternary ammonium ionic liquids and lithium salts doping effects studied by NMR spectroscopy. J. Chem. Eng. Data 59, 1944–1954 (2014)
C.I. Daniel, F. Vaca Chávez, C.A.M. Portugal, J.G. Crespo, P.J. Sebastião, 1H NMR relaxation study of a magnetic ionic liquid as a potential contrast agent. J. Phys. Chem. B 119, 11740–11747 (2015)
L.M. Varela, T. Méndez-Morales, Carrete et al., Solvation of molecular cosolvents and inorganic salts in ionic liquids: a review of molecular dynamics simulations. J. Mol. Liq. 210, 178–188 (2015)
Y. Shimizu, Y. Wachi et al., NMR study on ion dynamics and phase behavior of a piperidinium-based room-temperature ionic liquid: 1-Butyl-1-methylpiperidinium Bis(fluorosulfonyl)amide. J. Phys. Chem. B, 120(25), 5710–5719 (2016). https://doi.org/10.1021/acs.jpcb.6b04095
C.A. Rumble, A. Kaintz et al., Rotational dynamics in ionic liquids from NMR relaxation experiments and simulations: benzene and 1‑Ethyl-3-methylimidazolium. J. Phys. Chem. B 120, 9450–9467 (2016)
Bradley J. Butler, Donald S. Thomas et al., NMR spectroscopy to follow reaction progress in ionic liquids. Magn. Reson. Chem. (2014). https://doi.org/10.1002/mrc.4161
M.N. Garaga, M. Persson et al., Local coordination and dynamics of a protic ammonium based ionic liquid immobilized in nano-porous silica micro-particles probed by Raman and NMR spectroscopy. Soft Matter (2016). https://doi.org/10.1039/c5sm02736e
I. Seymour, D.S. Middlemiss et al., Characterizing oxygen local environments in paramagnetic battery materials via 17O NMR and DFT calculations. J. Am. Chem. Soc. 138, 9405–9408 (2016)
S.K. Davidowski, F. Thompson et al., NMR characterization of ionicity and transport properties for a series of diethylmethylamine based protic ionic liquids. J. Phys. Chem. B 120, 4279–4285 (2016)
Danuta Kruk, Milosz Wojciechowski et al., Dynamics of ionic liquids in bulk and in confinement by means of 1H NMR relaxometry—BMIM-OcSO4 in an SiO2 matrix as an example. Phys. Chem. Chem. Phys. 18, 23184 (2016)
Sachin Thawarkar, Nageshwar D. Khupse et al., Comparative investigation of the ionicity of aprotic and protic ionic liquids in molecular solvents by using conductometry and NMR spectroscopy. ChemPhysChem 17, 1006–1017 (2016)
R. Nanda, Thermal dynamics of lithium salt mixtures of ionic liquid in water by PGSE NMR spectroscopy. RSC Adv. 6, 36394 (2016)
T.-Y. Wua, S.-G. Sub et al., Diffusion coefficients, spin-lattice relaxation times, and chemical shift variations of NMR spectra in LiTFSI-doped ether- and allyl-functionalized dicationic ionic liquids. J. Taiwan Inst. Chem. Eng. 60, 138–150 (2016)
X. Zhu, H. Zhang et al., Structural heterogeneities in solutions of triethylamine nitrate ionic liquid: 1H NMR and LC model study. J. Solut. Chem. 45, 359–370 (2016)
Acknowledgements
The NMR measurements have been partially carried out in the Center for Magnetic Resonance of Research Park of St. Petersburg State University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this paper

Cite this paper
Matveev, V.V., Tyutyukin, K.V. (2018). Peculiarities of NMR Relaxation in Ionic Liquids: Difficulties in Interpretation and Novel Possibilities. In: Bulavin, L., Chalyi, A. (eds) Modern Problems of Molecular Physics. Springer Proceedings in Physics, vol 197. Springer, Cham. https://doi.org/10.1007/978-3-319-61109-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-61109-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-61108-2
Online ISBN: 978-3-319-61109-9
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)