
Abstract
Atherosclerosis leads to narrowing of vessels and acute coronary syndrome resulting in ischemic events due to either vasospasm or decreased blood flow. Atherosclerosis and acute coronary syndrome are more common in diabetes mellitus. Hyperglycemia and hypercholesterolemia in diabetes predispose the arteries to plaque development. Smoking, hypertension, male sex, and family history or genetic susceptibility are other predisposing factors for plaque development. Depending on the size, morphology, and symptoms of the patients, plaques can be classified as stable and unstable plaques. Unstable plaques are characterized by the presence of thin fibrous cap, necrotic core, and proliferation of vascular smooth muscle cells, angiogenesis and calcification. Plaque formation initiates with fatty streak and progresses through atheroma, atheromatous plaque to fibroatheromatous plaque. Fibroatheromatous plaques with thick fibrous cap are stable plaques. Thinning of the fibrous cap makes a plaque unstable, prone to rupture and thrombus formation. Mechanisms such as increased inflammation, foam cell deposition, impaired repair mechanism, endothelial cell dysfunction, vascular smooth muscle cell proliferation, angiogenesis, intra-plaque hemorrhage, and calcification which facilitate the plaque rupture are increased in diabetes mellitus. Thus, diabetes mellitus increases the prevalence of plaque formation and rupture. Diabetes mellitus affects various cellular and molecular effectors involved in plaque development and rupture. Understanding these cellular and molecular effectors and involved mechanisms in association with diabetes mellitus is essential for the development of potential therapeutic strategies. This review is a critical overview on the effect of hyperglycemia in diabetes mellitus on the pathogenesis of plaque formation and rupture.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Atherosclerosis
- Stable and unstable plaque
- Diabetes mellitus
- Hyperlipidemia
- Hyperglycemia
- Fibrous cap
- Plaque rupture
1 Introduction
The luminal narrowing due to plaque formation or precipitating thrombus in atherosclerosis results in adverse cardiac events (myocardial infarction , angina ), brain injury (ischemic stroke ) and peripheral vascular disease . Coronary artery disease (CAD) is the most common of all these, resulting in myocardial infarction and angina pectoris. An increased serum level of low-density lipoprotein (LDL) is sufficient to induce the atherosclerotic changes. The facilitating factors such as smoking , hypertension , diabetes mellitus, male sex, and family history or genetic susceptibility further add nuances to the disease presentation [1]. Coronary arteries, carotid bifurcations, abdominal aorta, iliofemoral arteries, the branch points of arteries, and the artery near the curvature are the common sites for atherosclerotic lesion due to the presence of low or oscillatory endothelial shear stress [2]. Depending on the clinical symptoms, the atherosclerotic plaques may be asymptomatic (subclinical disease), obstructive (stable angina , transient ischemic attack, amaurosis fugax), and symptomatic (acute thrombosis leading to acute coronary syndrome, stroke) [1, 3].
CAD is more common in diabetes mellitus type II (T2DM), or the individuals with persistent hyperglycaemia are more prone to CAD due to increased blood glucose and atheroma formation [4]. Further, it has also been documented that elevated glycosylated haemoglobin [5] and genetically driven hyperglycemia distinctly from T2DM also increases the risk of CAD [6], suggesting hyperglycemia as a major risk factor for CAD and can affect the pathogenesis as well as the stability of the plaque. Obesity is a chronic inflammatory disease and results in obesity-induced insulin resistance or impaired insulin secretion resulting in hyperglycemia, which further leads to functional and structural alterations of the vessel wall and culminates with diabetic vasculopathies [7]. Hyperlipidemia is another major risk factor for atherosclerosis . Deposition of LDL in the intima initiates the process of atherosclerosis . T2DM is associated with elevated triglycerides, decreased high density lipoprotein (HDL) and increased low density lipoprotein levels collectively characterizing the hyperlipidemia [8].
Hyperlipidemia and hyperglycemia are the risk factors for atherosclerosis , and increased deposition of lipids and inflammatory cells resulting in necrotic core within the atherosclerotic plaque renders the plaque vulnerable. Nearly 75 % of all acute coronary events and 90 % of all carotid plaques causing ischemic stroke results due to atherosclerotic plaque rupture [9]. Increased infiltration of inflammatory cells, thin fibrous cap , large necrotic core, and increased angiogenesis are the mechanisms involved in plaque rupture [10]. Since hyperglycemia and hyperlipidemia , both associated with T2DM causes the functional and structural alterations of the vessel wall, the core morphological alteration behind atherosclerosis , diabetes mellitus can affect the pathogenesis and process of atherosclerotic plaque formation and rupture. This chapter is focused on the effect of hyperglycemia on the various aspects of plaque pathogenesis and vulnerability.
2 Pathogenesis of Plaque Formation and Plaque Rupture
The process of plaque formation consists of adaptive intimal thickening with smooth muscle cell (SMC) proliferation, lipoprotein retention, intimal inflammation with inflammatory cell recruitment, foam cell formation, apoptosis and necrosis,matrix synthesis, calcification , angiogenesis , and arterial remodelling. Fibrous cap rupture results in thrombus formation and ischemic events [1]. LDL binding with the proteoglycans in the intima is important for initiation of the plaque formation [11]. The oxidation and aggregation of LDL lead to chronic innate and adaptive immune response resulting in induction of endothelial and smooth muscle cells. This results in expression of various adhesion molecules, chemo-attractants, and growth factors leading to enhanced homing, migration, and differentiation of the monocytes into macrophages and dendritic cells [12, 13]. Oxidized LDL also aids in pro-inflammatory macrophage (M1) predominance [14]. Further, macrophage and dendritic cells act as the deposits of the LDL. Deposition of LDL and foam cell formation leads to xanthoma formation, which further progresses to atherosclerotic lesion with the pathological intimal thickening involving deposition of acellular lipid-rich material in intima [15]. Deposition of collagen and extracellular lipid pools results in formation of fibroatheroma , characterized by the presence of a necrotic core, angiogenesis and fibrous cap . Calcification occurs in progressive atherosclerotic lesions which increase with age, and apoptotic cells, extracellular matrix, and necrotic core material in fibroatheroma act as nidus for calcification increasing the calcium deposits [16, 17] (Fig. 4.1).

Pathogenesis of plaque formation and plaque rupture. Deposition of the lipids in the intima leads to fatty streak formation and adaptive intimal thickening. Infiltration of inflammatory cells, lipid deposition, and vascular smooth muscle cells (VSMCs) proliferation results in progression of the fatty streak to atheroma and atheromatous plaque. Formation of the necrotic core due to increased apoptosis and necrosis in plaque; increased lipid deposition and angiogenesis and thinning of the fibrous cap results in the development of vulnerable fibroatheromatous plaque
The precipitating factor for acute coronary syndrome (ACS) is luminal thrombus or a sudden plaque hemorrhage within the atherosclerotic plaque. The ACS is not necessarily accompanied with concomitant vasospasm. Plaque rupture is the most frequent cause of thrombosis . Plaque rupture results in the exposure of highly thrombogenic, red cell–rich necrotic core material to the blood [18]. Plaque rupture mainly occurs in thin-cap fibroatheromas having an extremely thin fibrous cap . Infiltration of the foam cells or macrophages in the intima results in thinning of the fibrous cap, mainly in the cap margin or shoulder region. Thinning of the fibrous cap is mediated by gradual loss of SMCs from the fibrous cap and degradation of the collagen in fibrous cap via infiltrating macrophages /foam cells secreting proteolytic enzymes such as plasminogen activators, cathepsins, and matrix metalloproteinases (MMPs) . Thrombus formation can also occur in the areas of plaque erosion, most often in the areas of pathological intimal thickening. Coronary vasospasm is the frequent event responsible for plaque erosion and rupture [19]. Emotional stress or increased physical activity may be the precipitating event in plaque rupture [1, 18, 19]. Thin fibrous cap, thrombus formation, large necrotic core, neovascularisation, hemorrhage within the plaque, adventitial or perivascular inflammation , and spotty calcification characterize the vulnerable plaque (Fig. 4.1).
3 Diabetes Mellitus and Atherosclerosis
Diabetes mellitus is one of the major risk factors for atherosclerosis and cardiovascular disease in the United States. Hyperglycemia increases the risk for atherosclerosis by a cumulative effect of various mechanisms (Fig. 4.2) discussed elaborately in the literature [7, 8]. Briefly, oxidized LDL enhances the oxidative stress in the intima which leads to activation of inflammatory cascade involving inflammatory receptors [receptor for advanced glycosylation end products (RAGEs), toll-like receptors (TRLs), and triggering receptor expressed on myeloid cells (TREMs) ], downstream signaling kinases [protein kinase C (PKCs), c-Jun NH2-terminal kinase (JNK), ERK, mitogen-activated protein kinase (MAPK ) etc.] and pro-inflammatory cytokines [interlekin (IL)-6, tumor necrosis factor (TNF)-α]. This leads to increased monocytic infiltration, M1 macrophage predominance and foam cell formation, which further enhances the inflammation and vascular smooth muscle cells (VSMCs) proliferation resulting in atherosclerosis (Figs. 4.2 and 4.3) [3]. Further, research studies have also demonstrated the involvement of inflammatory surface markers (TREMs and TLRs) [20,21,22] and pro-inflammatory cytokines (IL-6 and TNF-α) [22] in plaque vulnerability.The increased secretion of these pro-inflammatory cytokines [23], and increased expression of the inflammatory surface marker [24, 25] involved in the pathogenesis of atherosclerotic plaque formation and rupture suggest that persistent hyperglycemia in diabetes have a potential role in plaque formation and rupture.

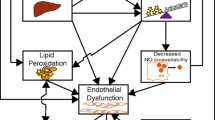
Pathogenesis of atherosclerosis and plaque vulnerability in diabetes mellitus. Persistent hyperglycemia in diabetes mellitus potentiate several mechanisms such as hyperlipidemia , increased angiogenesis , intra-plaque hemorrhage, proliferation of vascular smooth muscle cells (VSMCs), infiltration of inflammatory cells, foam cell formation, necrotic core formation due to oxidative stress , and hypercoagulability enhancing the thrombus formation along with thinning of the fibrous cap by altering the collagen content. These mechanisms make the plaque prone to rupture and thereby prevalence of acute coronary syndrome

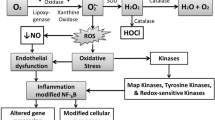
Schematic representation of the factors involved in the development of atherosclerosis in diabetes mellitus. Hyperglycemia in diabetes mellitus leads to increased free fatty acid (FFA) accumulation and oxidation. This results in increased oxidative stress due to the generation of reactive oxygen species (ROS) and the decreased availability of nitric oxide (NO ) . This leads to activation of various inflammatory pathways resulting in increased prevalence of atherosclerosis . Advanced glycosylation end products (AGEs) ; diabetes mellitus type 2 (T2DM); extracellular matrix (ECM); interleukin (IL)-6; Intercellular Adhesion Molecule (ICAM) 1;monocytes chemoattractant protein (MCP)-1; nuclear factor-kappa beta (NF-kB); protein kinase C (PKC); receptor for advanced glycosylation end products (RAGE);toll-like receptors (TLRs); triggering receptor expressed on myeloid cells (TREM ); transforming growth factor (TGF)-alpha (α)-beta (β); tumor necrosis factor (TNF)-α; vascular cell adhesion protein (VCAM)-1;vascular smooth muscle cells (VSMCs)
4 Diabetes and Plaque Vulnerability
4.1 Diabetes and Fibrous Cap
Fibrous cap of the plaque faces the lumen of the vessel and is responsible for the integrity of the plaque. Normally, fibrous cap is composed of VSMCs embedded in a collagen type I and III rich matrix. Thinning of the fibrous cap is associated with the plaque rupture and ACS. Studies have suggested the role of MMPs and macrophages in autolysis of the matrix content resulting in thinning of the fibrous cap, however, the exact mechanism underlying the MMP activity in under research. It has been found that symptomatic plaques have increased macrophage density, higher expression of MMPs, decreased VSMCs density, and decreased expression of collagens compared to asymptomatic plaques, and increased expression of MMPs with a decreased collagen expression is associated with plaque vulnerability [3, 20,21,22, 26]. Further, the association of diabetes with a higher prevalence of macrophage infiltration and thin-cap fibroatheroma suggest the proneness of plaque rupture in T2DM, as well as the role of persistent hyperglycemia in thinning of fibrous cap [27] (Fig. 4.2).
The mechanisms involving and promoting the thinning of the fibrous cap remains incompletely understood. The alteration of ECM matrix, impaired collagen content, and accumulation of lipids remains the cornerstone of the plaque rupture and thinning fibrous cap forming the rupture-prone plaque. A study on the diabetic and hypercholesterolemic swine reported that coronary regions exposed to low endothelial shear stress favour the collagen-poor, thin-capped fibrous plaque formation compared to high endothelial shear stress. This thinning of the fibrous cap was accompanied by reduced intimal SMC content; decreased procollagen-I gene expression; increased (MMP)-1, -8, -13, and -14 expression; and reduced collagen content [28]. Increased collagen loss in the fibrous cap suggests increased activity of MMPs in the symptomatic plaque whose prevalence is high in diabetics. Upregulated expression of IL-6, IL-8, and monocytes chemoattractant protein (MCP)-1 and the activities of MMP-2 and MMP-9 and downregulated expression of tissue inhibitor of metalloproteinase (TIMP)-2 with hyperglycemia via hyperglycemia-induced glycosaminoglycans alterations in the cell surface perlecan as well as in the extracellular matrix (ECM) have been reported [29], however, lower concentration of MMPs (MMP-2 and MMP-9) in T2DM has also been documented suggesting the complexity of the role MMPs [30]. Similarly, Fiaschi et al. [31] reported that hyperglycemia in association with angiotensin II enhances the collagen I production and deposition in ECM involving signal transducer and activator of transcription (STAT)3 in cardiac fibroblasts.
4.2 Diabetes and Lipid Deposition
T2DM and insulin resistance has been associated with reduced HDL cholesterol, a predominance of small dense LDL particles, and elevated triglyceride levels [8]. Deposition of the lipids in the intima initiates the process of atherosclerosis , and a large deposition of lipids (lipid core) within the fibroatheroma characterizes the vulnerable atherosclerotic plaque [26] (Fig. 4.2). Increased lipid deposition and larger lipid index have been reported in coronary plaques of diabetics compared to non-diabetics [27]. These results suggest that increased LDL levels are associated with increased atherosclerosis and plaque rupture . However, elevated plasma triglyceride levels but not the elevated plasma cholesterol levels have been reported with diabetes in hyperlipidemic pigs in association with increased atherosclerosis [32]. This suggests that diabetes is associated with hyperlipidemia (hypertriglyceridemia but not hypercholesterolemia) and increased prevalence of atherosclerosis , however, it has also been reported that isolated hypertriglyceridemia alone in not associated with increased CAD, but hypertriglyceridemia in association with hypercholesterolemia have a synergistic effect on CAD development [33]. Thus, increased LDL in diabetes is associated with increased prevalence of atherosclerosis , and increased lipid deposition in plaque is associated with plaque rupture correlates increased prevalence of plaque rupture with T2DM.
4.3 Diabetes and Inflammation
Infiltration of the inflammatory cells (monocytes , macrophages , dendritic cells etc.) is a key mechanism involved in fibroatheromatous plaque development (Figs. 4.2 and 4.3). Studies have suggested the association of increased infiltration and density of these cells in symptomatic plaque compared to asymptomatic plaque [21, 22, 26]. Further, it has also been suggested that inflammation not only enhances atherosclerosis and plaque formation but also the thrombus formation by affecting platelet function, coagulating factors and clotting mechanism, potentiated by diabetes being a chronic inflammatory disease [34]. Increased inflammation in the atheroma is also associated with hyperlipidemia in diabetic patients suggesting the synergism between hyperlipidemia and inflammation in diabetes resulting in enhanced vulnerability of plaque [1].
4.4 Diabetes and Calcification
Calcification is a histological feature of fibrous atherosclerotic plaque. Minimally oxidized LDL and T2DM are the risk factor for increased calcification in the pathogenesis of plaque development. Hyperglycemia influences the calcification in the vessel wall through ROS production. Increased expression of Cbfal (transcription factor for bone formation) and bone morphogenetic protein (BMP)-2 and enhanced calcification of VSMCs is associated with high serum glucose. Increased levels of BMP-2 exert pro-inflammatory and proatherogenic effects of BMP-2 induce the oxidative stress and endothelial dysfunction , leading to enhanced plaque calcification by inducing osteogenic phenotype in VSMCs [35, 36]. Avogaro et al. [37] have discussed various mechanism in association with diabetes such as upregulation of runt-related transcription factor 2 (Runx2), osterix, osteopontin (OPN), osteocalcin, and downregulation of smooth muscle-specific genes in VSMCs trans-differentiating it to more bone-forming cells. Increased calcification and higher prevalence of calcification have been reported in coronary plaques of diabetics compared to non-diabetics [27]. These studies suggest that diabetes is associated with increased calcification in the intima, which in turn is associated with increased prevalence of plaque rupture .
4.5 Diabetes and Thrombus Formation
Diabetes is a hypercoagulable disease due to an imbalance of pro- versus anticoagulation, and is associated with increased numbers of endogenous pro-coagulant triggers bearing circulating microparticles. Hypercoagulability in diabetes increases the risk of atherosclerosis and peripheral vascular disease [38]. Increased association of inflammation and oxidative stress with hypercoagulability state in diabetes has been established which leads to endothelial dysfunction , plaque formation, progression and rupture [39]. Further, increased thrombus formation in diabetes due to inflammation by affecting the platelet function, coagulating factors and clotting mechanism has been reported [34]. Higher prevalence of thrombus formation has been reported in coronary plaques of diabetics compared to non-diabetics [27]. These studies suggest that hypercoagulable state of diabetes promotes inflammation and thrombus formation (Fig. 4.2).
4.6 Diabetes and Intra-plaque Hemorrhage
The structural type I collagen, the predominant structural collagen, in the vessel wall is produced by smooth muscle cells and fibroblasts in the vascular media and intima. Type III collagen is present as a minor component in vessels. However, Purushothaman et al. [40] while comparing the diabetic and non-diabetic subjects reported that diabetes is associated with increased type III collagen instead of type I collagen, a feature of progressive atherosclerotic plaque, accompanied with inflammation , neovascularization , and intraplaque hemorrhage (IPH). Similar trends in collagen reversal in association with diabetes have been documented by various other studies [41,42,43]. Further mature plaques have a rich network of small vessels called as ‘ vasa vasorum ’ within the matrix of plaque, and rupture of these vessels within the plaque leads to IPH. Higher prevalence of IPH has been associated with symptomatic plaques [44], and plaque rupture [45] (Figs. 4.2 and 4.3).
4.7 Diabetes and Angiogenesis
Angiogenesis is a morphological feature of fibroatheroma . Lipid deposition and inflammation causes oxidative stress and increased ROS. Increased oxidative stress is a precursor for angiogenesis and arteriogenesis. Similarly, the toxic metabolites in metabolic syndrome and diabetes induce angiogenesis via oxidative stress, which further accelerates the progression of atherosclerosis [46]. Increased infiltration of inflammatory cells and angiogenesis increases the size of the necrotic core and IPH rendering the plaque prone to rupture [47]. A greater degree of plaque intimal neovascularization and inflammatory infiltrate leading to plaque vulnerability has been reported in diabetic subjects compared to non-diabetic subjects [48]. Although, the distribution, density and the role of vasa vasorum have been discussed in the context of plaque progression, atherosclerosis , and IPH, the causative or the only reactive role of vasa vasorum in atherogenesis needs to be elucidated (Figs. 4.2 and 4.3).
4.8 Diabetes and Impaired Endothelial Repair
Inflammation of the atheroma and plaque rupture is the cornerstone of the ACS. Plaque rupture occurs at the thinnest part of the fibrous cap , which is due to ECM and collagen loss. ECM and collagen loss leading to thin fibrous cap occurs due to impaired repair mechanism. Edsfeldt et al. [10] studied the carotid endarterectomy specimens in T2DM and non-diabetic patients analyzing the plaque structure, connective tissue proteins, inflammatory cells, and inflammatory markers, and reported the increased proneness of the atherosclerotic plaques to rupture in subjects with T2DM because of impaired repair responses rather than to increased vascular inflammation . The plaques in T2DM patients had lower collagen and elastin content, decreased levels of the VSMC growth factor , platelet-derived growth factor (PDGF) , decreased levels of inflammatory cells and decreased levels of MMP2. However, Ruiz et al. [49] reported the increased proliferation of VSMCs and decreased apoptosis leading to enhanced arterial remodeling in diabetic patients with upregulated expression of Bcl-2 gene with glucose (Figs. 4.2 and 4.3).
5 Conclusion
Unstable plaques and plaque rupture precede the acute coronary syndrome. Unstable plaque is characterized by the presence of necrotic core, lipid deposition, angiogenesis and thin fibrous cap . Inflammation in the atheroma, infiltration of inflammatory cells, angiogenesis and collagen loss renders a stable plaque to unstable plaque. Diabetes mellitus is a chronic inflammatory disease, associated with increased inflammation , oxidative stress , hyperlipidemia and increased angiogenesis , thereby increasing the prevalence of atherosclerosis as well as plaque vulnerability. Higher prevalence of atherosclerosis and plaque vulnerability has been reported in various studies along with the improved modalities of imaging to assess the plaque volume and its proneness to rupture. Although, there are reports about the most common arteries involved in atherosclerosis , diabetes induced structural changes in intima and intimal thickening, predominance of collagen III instead of collagen I, and presence of vasa vasorum and IPH, still further studies are needed to elucidate the molecular mechanism more clearly underlying these changes in diabetes to develop a potential therapy for plaque stability in diabetes mellitus.
References
Bentzon JF, Otsuka F, Virmani R, Falk E (2014) Mechanisms of plaque formation and rupture. Circ Res 114:1852–1866
Wentzel JJ, Chatzizisis YS et al (2012) Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc Res 96:234–243
Rao VH, Rai V, Stoupa S, Agrawal DK (2015) Blockade of Ets-1 attenuates epidermal growth factor-dependent collagen loss in human carotid plaque smooth muscle cells. Am J Physiol Heart Circ Physiol 309:H1075–H1086
Sarwar N, Gao P, Seshasai SR et al (2010) Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 375:2215–2222
Cahill LE, Jensen MK, Chiuve SE et al (2015) The risk of coronary heart disease associated with glycosylated hemoglobin of 6.5% or greater is pronounced in the haptoglobin 2-2 genotype. J Am Coll Cardiol 66:1791–1799
Merino J, Leong A, Dupuis J, et al. (2016) OR27-5: genetically driven hyperglycemia increases risk of coronary artery disease distinctly from type 2 diabetes. Diabetes Care 40(5):687–693
Paneni F, Beckman JA, Creager MA, Cosentino F (2013) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J 34:2436–2443
Gleissner CA, Galkina E, Nadler JL, Ley K (2007) Mechanisms by which diabetes increases cardiovascular disease. Drug Discov Today Dis Mech 4:131–140
Kubo T, Imanishi T, Takarada S et al (2007) Assessment of culprit lesion morphology in acute myocardial infarction: ability of optical coherence tomography compared with intravascular ultrasound and coronary angioscopy. J Am Coll Cardiol 50:933–939
Edsfeldt A, Goncalves I, Grufman H et al (2014) Impaired fibrous repair: a possible contributor to atherosclerotic plaque vulnerability in patients with type II diabetes. Arterioscler Thromb Vasc Biol 34:2143–2150
Skalen K, Gustafsson M, Rydberg EK et al (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417:750–754
Libby P, Ridker PM, Hansson GK (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473:317–325
Subramanian M, Tabas I (2014) Dendritic cells in atherosclerosis. Semin Immunopathol 36:93–102
Leitinger N, Schulman IG (2013) Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol 33:1120–1126
Kolodgie FD, Burke AP, Nakazawa G, Virmani R (2007) Is pathologic intimal thickening the key to understanding early plaque progression in human atherosclerotic disease? Arterioscler Thromb Vasc Biol 27:986–989
Stary HC (2001) The development of calcium deposits in atherosclerotic lesions and their persistence after lipid regression. Am J Cardiol 88:16E–19E
Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R (2014) Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol 34:724–736
Davies MJ (2000) The pathophysiology of acute coronary syndromes. Heart 83:361–366
Niccoli G, Fracassi F, Crea F (2014) Pathophysiology and clinical significance of plaque rupture. Medicographia 36:37–44
Rao VH, Rai V, Stoupa S, Subramanian S, Agrawal DK (2016) Data on TREM-1 activation destabilizing carotid plaques. Data Brief 8:230–234
Rai V, Rao VH, Shao Z, Agrawal DK (2016) Dendritic cells expressing triggering receptor expressed on myeloid cells-1 correlate with plaque stability in symptomatic and asymptomatic patients with carotid stenosis. PLoS One 11:e0154802
Rao VH, Rai V, Stoupa S, Subramanian S, Agrawal DK (2016) Tumor necrosis factor-alpha regulates triggering receptor expressed on myeloid cells-1-dependent matrix metalloproteinases in the carotid plaques of symptomatic patients with carotid stenosis. Atherosclerosis 248:160–169
Jagannathan-Bogdan M, McDonnell ME, Shin H et al (2011) Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol 186:1162–1172
Dasu MR, Devaraj S, Park S, Jialal I (2010) Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 33:861–868
van Beijnum JR, Buurman WA, Griffioen AW (2008) Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 11:91–99
Mughal MM, Khan MK, DeMarco JK et al (2011) Symptomatic and asymptomatic carotid artery plaque. Expert Rev Cardiovasc Ther 9:1315–1330
Kato K, Yonetsu T, Kim SJ et al (2012) Comparison of nonculprit coronary plaque characteristics between patients with and without diabetes: a 3-vessel optical coherence tomography study. JACC Cardiovasc Interv 5:1150–1158
Koskinas KC, Sukhova GK, Baker AB et al (2013) Thin-capped atheromata with reduced collagen content in pigs develop in coronary arterial regions exposed to persistently low endothelial shear stress. Arterioscler Thromb Vasc Biol 33:1494–1504
Chang SC, Vivian Yang WC (2013) Hyperglycemia induces altered expressions of angiogenesis associated molecules in the trophoblast. Evid Based Complement Alternat Med 2013:457971
Lewandowski KC, Banach E, Bienkiewicz M, Lewinski A (2011) Matrix metalloproteinases in type 2 diabetes and non-diabetic controls: effects of short-term and chronic hyperglycaemia. Arch Med Sci 7:294–303
Shamhart PE, Luther DJ, Adapala RK et al (2014) Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can J Physiol Pharmacol 92:598–604
Chait A, Bornfeldt KE (2009) Diabetes and atherosclerosis: is there a role for hyperglycemia? J Lipid Res 50(Suppl):S335–S339
Rajmohan L, Deepa R, Mohan A, Mohan V (2000) Association between isolated hypercholesterolemia, isolated hypertriglyceridemia and coronary artery disease in south Indian type 2 diabetic patients. Indian Heart J 52:400–406
Hess K, Grant PJ (2011) Inflammation and thrombosis in diabetes. Thromb Haemost 105(Suppl 1):S43–S54
Chen NX, Duan D, O’Neill KD, Moe SM (2006) High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol Dial Transplant 21:3435–3442
Li X, Yang HY, Giachelli CM (2008) BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 199:271–277
Avogaro A, Fadini GP (2015) Mechanisms of ectopic calcification: implications for diabetic vasculopathy. Cardiovasc Diagn Ther 5:343–352
Tripodi A, Branchi A, Chantarangkul V et al (2011) Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis 31:165–172
Domingueti CP, Dusse LM, Carvalho M et al (2016) Diabetes mellitus: the linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J Diabetes Complicat 30:738–745
Purushothaman KR, Purushothaman M, Muntner P et al (2011) Inflammation, neovascularization and intra-plaque hemorrhage are associated with increased reparative collagen content: implication for plaque progression in diabetic atherosclerosis. Vasc Med 16:103–108
Vranes D, Cooper ME, Dilley RJ (1999) Cellular mechanisms of diabetic vascular hypertrophy. Microvasc Res 57:8–18
Tang M, Zhong M, Shang Y et al (2008) Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell Mol Life Sci 65:2924–2932
Brings S, Zhang S, Choong YS et al (2015) Diabetes-induced alterations in tissue collagen and carboxymethyllysine in rat kidneys: association with increased collagen-degrading proteinases and amelioration by Cu(II)-selective chelation. Biochim Biophys Acta 1852:1610–1618
Carr S, Farb A, Pearce WH, Virmani R, Yao JS (1996) Atherosclerotic plaque rupture in symptomatic carotid artery stenosis. J Vasc Surg 23:755–766
Michel JB, Virmani R, Arbustini E, Pasterkamp G (2011) Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J 32:1977–1985. 1985a, 1985b, 1985c
Hayden MR, Tyagi SC (2004) Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: a malignant transformation. Cardiovasc Diabetol 3:1
Virmani R, Kolodgie FD, Burke AP et al (2005) Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol 25:2054–2061
Carter A, Murphy MO, Turner NJ et al (2007) Intimal neovascularisation is a prominent feature of atherosclerotic plaques in diabetic patients with critical limb ischaemia. Eur J Vasc Endovasc Surg 33:319–324
Ruiz E, Gordillo-Moscoso A, Padilla E et al (2006) Human vascular smooth muscle cells from diabetic patients are resistant to induced apoptosis due to high Bcl-2 expression. Diabetes 55:1243–1251
Acknowledgement
This work was supported by research grants R01 HL112597, R01 HL116042, and R01 HL120659 to DK Agrawal from the National Heart, Lung and Blood Institute, National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Crown Copyright
About this chapter
Cite this chapter
Rai, V., Agrawal, D.K. (2017). Pathogenesis of the Plaque Vulnerability in Diabetes Mellitus. In: Kartha, C., Ramachandran, S., Pillai, R. (eds) Mechanisms of Vascular Defects in Diabetes Mellitus. Advances in Biochemistry in Health and Disease, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-60324-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-60324-7_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60323-0
Online ISBN: 978-3-319-60324-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)