
Abstract
Radical S-adenosylmethionine (SAM) enzymes comprise an important and a versatile superfamily of enzymes. For more than a decade, a significant effort has been directed towards understanding these enzymes. Electron paramagnetic resonance spectroscopy has played a crucial role in such studies, helping to decipher intricate details about the identity of the active metallocofactors, their relations to the substrate(s) utilized and an understanding of the mechanisms of the enzymatic reactions. In this chapter we overview research milestones in the field of radical SAM enzymes achieved with the aid of EPR spectroscopy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- ENDOR
- HYSCORE
- ESEEM
- CW-EPR
- Cfr
- RlmN
- Lipoyl synthase
- Lysine 2,3-aminomutase
- HydG
- Pyruvate formate-lyase
- Glycyl radical
- S-adenosylmethionine
- Nucleic acid radical
- [4Fe–4S] cluster
- Biotin synthase
- MoaA
- Rapid-freeze quench EPR
- Methylase
Introduction
In the past decade, a multitude of studies have been performed to interrogate the structures and mechanisms of the radical SAM (RS) superfamily of enzymes, and electron paramagnetic resonance (EPR) spectroscopic techniques have been vital in these endeavors. Members of the RS superfamily of enzymes catalyze an incredibly diverse set of chemical reactions . To date there are almost 114,000 individual sequences that belong to the family, and the products of these genes catalyze forty-eight distinct and known reactions [1, 2].
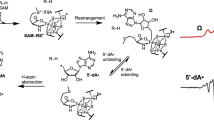
Most RS enzymes are easily identified by a CX3CX2C motif found in their primary structures [3]. The three cysteines in this motif coordinate three of the four iron ions of a [4Fe–4S] cluster. The fourth iron ion is not ligated by a protein residue, which allows S-adenosylmethionine (SAM) to bind to it in a bidentate fashion via its α-carboxylate and α-amino groups. Binding in this manner positions the sulfur atom of SAM to accept an electron from the reduced [4Fe–4S]+ cluster, resulting in homolytic cleavage of the C5′–S bond of SAM [4,5,6,7,8]. Homolytic cleavage of SAM results in the formation of methionine and a 5′-deoxyadensoyl radical (5′-dA•) , which is a potent oxidizing agent. The 5′-dA• is then used to abstract a specific hydrogen atom from a substrate molecule [9] (Scheme 1).

Reactions of S-adenosyl-L-methionine in radical SAM enzymes. (a) Reductive cleavage of SAM into L-methionine and 5'-deoxyadenosyl radical. (b) Methylgroup transfer in an SN2 reaction from SAM to a nucleophile (Nu) with a release of S-adenosyl-L-homocysteine (SAH)
The 5′-dA• is a common tool used by Nature to initiate radical chemistry. This same oxidizing agent is generated by the adenosylcobalamin (vitamin B12) cofactor, which undergoes a reversible homolytic cleavage of the C5′–Co bond to form the 5′-dA• [10]. SAM can also be reversibly cleaved to generate a 5′-dA•, allowing it to be used as a cofactor; however, it is most commonly consumed as a substrate in the reactions in which it participates. Upon abstraction of a hydrogen atom by the 5′-dA•, 5′-deoxyadenosine (5′-dA) and a substrate radical are formed. These substrates can be large biological polymers like proteins, RNA and DNA, or small molecules. In some cases the initial substrate radical formed is the final product of the RS reaction—as in the case of proteins that contain glycyl radical cofactors—but in most cases further bond-breaking and bond-making takes place. The complexity of the reactions catalyzed by RS enzymes varies dramatically from a single hydrogen atom abstraction, as in the case of pyruvate formate–lyase activating enzyme ) (PFL-AE), to highly complex rearrangements, as observed in MoaA [9].
RS enzymes are uniquely suited to study by EPR spectroscopy because of their redox active [4Fe–4S] clusters and the presence of radical intermediates in their reactions. Additionally, a number of RS enzymes contain multiple Fe/S clusters that can participate in redox chemistry, bind intermediates, or interact with other EPR active species [11, 12]. Because of these features, EPR spectroscopy can be used to probe the active sites and key intermediates of RS enzymes, which allows for detailed mechanistic analyses that are simply not possible for enzymes that do not contain paramagnetic intermediates.
EPR is an invaluable tool for resolving structural and electronic properties of paramagnetic centers in general and catalytic intermediates in particular. In some cases the resolution of EPR spectra can be increased by a procedure called resolution enhancement (RE ), which takes advantage of Fourier deconvolution of Lorentzian or Gaussian lineshapes [13, 14]. First, an inverse Fourier transformation is applied to the original spectrum and followed by a division of the time-domain trace with an exponential function (deconvolution of the Lorentzian lineshape). An apodization window function is then applied and the RE spectrum is obtained by subsequent forward Fourier transformation. The original article by Kauppinen et al. mentions [1-|x|/L]2 as an apodization function; however, other common functions, e.g. Hamming, Gaussian etc., can provide similar results. Two input parameters are required for this procedure: the width of the unitary lineshape and the apodization parameter (L). It is important to note that because neither the function nor the linewidth of the unitary line is generally known, the best values must be obtained by trial and error. This necessity makes the outcome dependent on the judgement of the user and thus may cause overinterpretation of the experimental results. A comprehensive overview of this methodology has been published by Reed and Poyner [13].
Advanced EPR spectroscopic techniques provide a much more convenient means to enhance spectroscopic resolution substantially and allow for a deeper view into the fine details of weaker magnetic interactions. Continuous wave Electron Nuclear Double Resonance (CW ENDOR) spectroscopy has been extensively used to obtain extraordinary details on paramagnetic species in RS enzymes and their reactions. The technique takes advantage of the fact that the amplitude of the EPR spectrum will vary if a radio frequency (RF) excitation is resonant with an NMR transition associated with an EPR transition. By scanning radio frequencies, nuclear transitions associated with magnetic nuclei in the vicinity of paramagnetic species can be resolved. The main disadvantage of this technique is that the RF perturbation occurs at the same time as the detection of the EPR signal, which makes this technique very sensitive to heating artifacts. As a result, the experimental setup needs to be considered very carefully. Pulse EPR provides an alternative way of performing ENDOR measurements . In this case, a RF is applied as a pulsed perturbation well before the detection. The two most common pulse methods are Mims and Davies ENDOR. The main disadvantage of pulse ENDOR methods as compared to CW ENDOR is the presence of blind spot patterns that alter the “true” lineshape of the ENDOR signal and, especially for weakly coupled nuclei, may strongly suppress the ENDOR signal. Therefore, application of this technique to weak, purely anisotropic hyperfine (HF) interactions is limited.
Another frequently used double-resonance pulse EPR technique to resolve hyperfine interaction is called ELDOR detected NMR ; however, given that, to our knowledge, no application of such methodology to the field of RS enzymes has been reported, this technique will not be covered here.
A second set of pulse EPR techniques, electron spin echo envelope modulation (ESEEM ), provides another way of resolving HF interactions without using a second distinct excitation frequency. In this class of techniques, the paramagnetic NMR frequencies are encoded in the dependence of the electron spin echo on a delay between microwave pulses. The limitation of the technique is that the amplitude of the effect depends on the degree of anisotropy of the HF interaction. The technique is most suitable to study weak, anisotropic, hyperfine interactions, which makes it complementary to ENDOR methods. Hyperfine sublevel correlation spectroscopy (HYSCORE ) is a variation of ESEEM that allows for correlation of nuclear frequencies on a two-dimensional spectrum. Most importantly, HYSCORE can separate nuclear frequencies for weak and strong HF coupling regimes in different parts of the spectrum, such as when a hyperfine coupling constant (A) is smaller or larger than twice the Larmor frequency (2νL).
Detailed explanations of the techniques described within have been provided by others [15, 16]. In what follows, we present several milestones in studying this versatile superfamily of enzymes that were achieved by EPR spectroscopy. We use the case studies of various radical SAM enzymes to highlight how the tools of EPR spectroscopy can be used to (1) identify the forms of Fe/S clusters present and monitor the binding of substrates to the clusters, (2) identify substrate and intermediate radicals that result from catalysis, and (3) use radical intermediates to probe the active sites to determine fine structure and electronic properties.
EPR Study of Metalloclusters in RS Enzymes
Radical SAM-Binding Cluster
The core feature of RS enzymes is the presence of a [4Fe–4S] cluster ([4Fe–4S]RS) that is coordinated by three cysteine ligands that are typically arranged in a signature CX3CX2C motif. The fourth iron ion is not coordinated by an amino acid ligand and is labile. Most notably, this cluster is sensitive to oxidative degradation, leading to [3Fe–4S] or even [2Fe–2S] constructs. The degraded [3Fe–4S] cluster form is also commonly found in as-isolated RS enzymes, especially if the enzymes are purified aerobically (see Fig. 1, A) [9]. CW EPR spectroscopy has proven to be useful in identifying the forms of Fe/S clusters in RS enzymes and in detecting the binding of SAM to the unique iron site.

EPR spectra of various states of the FeSRS cluster. (A) [3Fe4S]1+ (magenta) observed in as-isolated DesII [18]; (B) [4Fe–4S]3+ (black) and [3Fe–4S]1+ (magenta) observed in as-isolated and oxidized LAM [17]; (C) [4Fe–4S]1+ in the absence (blue) and presence (red) of SAM in reduced RNR-AE [19]; (D) [4Fe–4S]1+ in absence (blue) and presence (red) of SAM in reduced PFL-AE [7]. Adapted with permission from corresponding sources
The first direct evidence for the unique iron site in the [4Fe–4S]RS cluster was obtained in studies of Lysine 2,3-aminomutase (LAM) , wherein its [4Fe–4S]RS cluster could be oxidized to a [3Fe–4S] cluster and then converted back to the [4Fe–4S]RS cluster upon addition of iron and a reductant. Frey and coworkers [17] reported that as-isolated LAM contains a [4Fe–4S] cluster in the [4Fe–4S]3+ state that becomes EPR silent upon reduction using dithionite paired with methylviologen as a redox mediator. Subsequent addition of ferricyanide or oxygen resulted in the appearance of an EPR signal typical of a [3Fe–4S]+ cluster that is accompanied by an EPR signal at g = 4.3, characteristic of adventitiously bound Fe(III), indicating oxidative decomposition (Fig. 1, B). Preincubation of the oxidized enzyme with Fe(II) and addition of a reducing system resulted in recovery of approximately 90% of the activity of the pre-oxidized enzyme, indicating that the active [4Fe–4S]RS form could be restored.
A similar study was performed on pyruvate formate lyase-activating enzyme (PFL-AE) using Mössbauer spectroscopy [20]. The Mössbauer spectrum of anaerobically isolated PFL-AE was found to contain a mixture of [4Fe–4S]2+, cuboidal [3Fe–4S]+ (majority), linear [3Fe–4S]+ and [2Fe–2S]2+ signals, and upon reduction by dithionite, 66% of the associated iron was converted to [4Fe–4S]2+ clusters and 12% of the associated iron was converted to [4Fe–4S]1+ clusters , with the remaining 22% of the iron being attributed to adventitiously bound Fe(III).
In a later study, Frey and coworkers [21] showed that the [4Fe–4S] cluster in LAM can be reduced further to a [4Fe–4S]1+ state in the presence of SAM. Moreover, addition of SAH instead of SAM resulted in a similar EPR spectrum of the [4Fe-4S]1+ cluster even though SAH does not support LAM-dependent turnover. It is worth noting that [4Fe–4S] clusters in the majority of metalloproteins can only access one of the two redox transitions, [4Fe–4S]1+/2+ or [4Fe–4S]2+/3+, at physiological redox potentials . The fact that the presence of SAM or SAH allowed for a further reduction of a cluster provides one of the earliest indications of direct interaction between the [4Fe–4S] cluster and the SAM molecule.
The redox properties of the [4Fe–4S]RS cluster of LAM make it an outlier in the RS superfamily. The observation of a HiPIP-like [4Fe–4S]3+ cluster in the as-isolated state and the inability to reduce the [4Fe–4S]2+ state to the [4Fe–4S]+ state using typical reducing systems in absence of SAM or SAH is unusual among the characterized members of the RS superfamily. Most commonly, the [4Fe–4S]RS cluster is in an EPR-silent [4Fe–4S]2+ state in the as-isolated enzyme and can be reduced to a [4Fe–4S]1+ form in the absence of SAM with reductants such as sodium dithionite. To date, LAM is the only example among the members of RS enzyme superfamily that exhibits a [4Fe–4S]3+ cluster in a steady state. However, it is worth noting that in the later study of LAM it was indicated that an extensive Fe/S reconstitution procedure eliminated signatures of the HiPIP-like EPR signal [22].
Several studies indicate that the binding of SAM to the [4Fe–4S]RS cluster increase its redox potential. Hinckley and Frey used coulometric titrations to measure the [4Fe–4S]+/2+ redox potential in reconstituted LAM and found values of −430 mV in the presence of SAM, −460 mV in the presence of SAH, [22, 23] and −479 mV in an as-reconstituted state [24]. In PFL-AE, only a fraction of the sample can be reduced using sodium dithionite in absence of SAM, but in the presence of SAM the reduction is much closer to complete, [25] which also indicates an increase in the [4Fe–4S]+/2+ redox potential upon SAM binding. A protein film electrochemical study of redox properties of an RS enzyme, performed by Maiocco et al. on BtrN, indicated an upshift of the redox potential of the [4Fe–4S]RS cluster by about 50 mV when SAM is present [26].
In the majority of reported cases, the effect that SAM binding has on the electronic structure of the [4Fe–4S]RS cluster is also evident from the shift of the principal g-values in the CW EPR spectrum (Fig. 1, C and D). In the absence of SAM, the g-values of [4Fe–4S]RS + clusters are typically similar to the values observed for other iron-sulfur cluster-containing metalloproteins such as ferrodoxins, whereas the presence of SAM results in a much lower gav, where gav = (gx + gy + gz)/3. In a few cases, all three g-values were found to be below ge = 2.0023, although the exact nature of this phenomenon and its connection to other properties of the [4Fe–4S] clusters are yet to be understood. Figure 2 shows a distribution of g-values in different RS enzymes with the values presented in Table 1. Overall, these data show that binding of SAM to RS enzymes is one of the triggering events that not only initiates a reaction, but also primes the metallocofactor for the reactivity needed.

Effect of SAM binding on g-values of the [4Fe–4S]RS 1+ cluster in RS enzymes. Values for the corresponding data points can be found in Table 1
The first evidence of SAM binding to the unique Fe site was obtained in studies of PFL-AE using 57Fe Mössbauer spectroscopy by taking advantage of the lability of this Fe site. Removal of the released fourth Fe ion by size exclusion chromatography in the oxidized sample and addition of 57Fe during the reductive reconstitution of the [4Fe–4S] cluster resulted in the installation of a single 57Fe ion at the unique Fe site of the [4Fe–4S]2+ cluster [6]. Subsequent 57Fe-Mössbauer measurements revealed that adding SAM dramatically changes the 57Fe isomer shift from 0.45 to 0.7 mm/s, indicating a direct binding of a strong ionic ligand. Using a reverse procedure of installing a natural-abundance Fe ion into uniformly 57Fe labeled PFL-AE resulted in Mössbauer parameters for the three other iron sites that are nearly unchanged upon SAM binding. The results indicate that SAM directly binds to the unique Fe site and that the unique Fe site is electronically decoupled or valence localized. Because the measurements were performed on the [4Fe–4S]2+ state, this study also indicated that SAM can bind to the cluster prior to the reduction event.
Hoffman and coworkers used pulse EPR techniques to further elucidate the binding mode of SAM to the unique iron site of the [4Fe–4S] cluster of PFL-AE [7, 8, 44]. Photoreduction of PFL-AE using 5-deazariboflavin afforded the [4Fe–4S]+ cluster in high yield. The EPR spectrum changed considerably in the presence of SAM with a decrease in the g-value and a change of the spectral envelope from a very rhombic signal to an axial signal. Using SAM labeled with 13C- and 2H-isotopes in the methyl group, the authors performed a series of ENDOR studies using Q-band Mims ENDOR to illustrate the proximity of the SAM molecule to the [4Fe–4S] cluster (Fig. 3b, c) [8].

(a) Schematic representation of SAM binding to the [4Fe–4S]RS cluster based on the X-ray crystal structure of PFL-AE (PDB: 3CB8). (b, c) Q-band ENDOR spectra of the [4Fe–4S]1+ RS cluster in the presence of 13C- and 2H-Methyl-SAM, respectively, adapted with permission from [8]; (d–f) ENDOR spectra of the [4Fe–4S]1+ RS cluster in the presence of 17O-, 13C-, and 15N-labeled SAM, adapted with permission from [7, 44]
Broad ENDOR signals found in the 2H region of the ENDOR spectrum were attributed to three 2H HF couplings, suggesting a close proximity of the methyl group to the [4Fe–4S] cluster. The field dependent ENDOR measurements revealed one stronger and two weaker 2H HF coupling constants with substantial anisotropy. More details about the relative positioning of SAM and the [4Fe–4S]1+ cluster were obtained from field-dependent 13C ENDOR measurements. The in-depth analysis of the obtained data accounted for the ESE signal intensity and the periodic τ-dependent blind-spot of the Mims ENDOR. The following principal values for the 13C HF coupling were extracted: A(13C) = [−0.6, 0.4, −0.5] MHz. The slight rhombicity and the presence of the isotropic component led the authors to conclude that a small amount of spin density must reside directly on the methyl group. It was also noted that the opposite sign of the local (due to spin density on the methyl group) and non-local (through space) contributions to A(13C) indicate that the spin density on the methyl group does not originate directly from the [4Fe–4S] cluster via spin delocalization but rather is a result of spin polarization through an intervening atom, e.g. the sulfonium atom of SAM. Because of the 1/R 3 dependence of the dipolar coupling constants on the distance (R), the authors concluded that only the distance to the nearest Fe site needs to be taken into account. Considering the uncertainty of assigning the dipolar interaction to a specific iron site of the [4Fe–4S] cluster and thus the uncertainty in the choice of the spin-projection factor, the authors provided a range of distances for R(Fe–2H) = 3.0–3.8 Å and R(Fe–13C) = 4–5 Å. To verify that SAM does not change its position relative to the [4Fe–4S] cluster upon reduction, the authors performed an experiment in which the [4Fe–4S]2+ was cryoreduced at 77 K by exposing the frozen sample to a 60Co source. This procedure allows for reduction of the Fe/S cluster in a frozen matrix and prevents any major structural rearrangements. The 2H and 13C ENDOR signals obtained for such samples were found to be almost identical to the ones obtained for the solution-reduced enzyme.
Later, Hoffman and coworkers extended the study of SAM binding to the [4Fe–4S]RS + cluster of PFL-AE using specific isotope labeling of the carboxyl group of the methionine moiety of SAM with 17O and 13C and the amino group with 15N [7, 44]. Q-band ENDOR measurements of the reduced PFL-AE sample prepared with 17O-labeled SAM exhibited strong and predominantly isotropic 17O HF coupling ([8.6, 14.4, 8.2] MHz) [44]. The overall magnitude of the 17O HF coupling constant was similar to that of the carboxylate-17O of citrate coordinated to the unique Fe in aconitase [45]. Orientation selective Q-band ENDOR measurements of carboxylate -13C –SAM revealed distinct 13C signals (A(13C) = [1.1,−0.95, −0.82] MHz). Once again, the authors decomposed the obtained 13C coupling constant into two components, non-local (Tnon-loc = [−1.28, 0.64, 0.64] MHz) and local interactions (Tloc = [−0.04, 0.09, −0.04]/aloc = 0.22 MHz) , i.e. A(13C) = Tnon-loc + (Tloc + aloc). In this case, the authors used a known spin-projection factor of 1.57 for the labile Fe site of the [4Fe–4S]1+ cluster of aconitase to provide an estimate of the distance between the unique Fe site and the 13C group R(Fe-13C) = 3.3 Å. Q-band ENDOR experiments using amino-15N-SAM showed the presence of a strongly coupled 14N/15N nucleus in the vicinity of the [4Fe–4S]RS cluster. Analysis of the orientation dependence of the 15N ENDOR signal yielded the following principal 15N HF coupling constants: A(15N) = [9.7, 6, 3.5] MHz. This coupling was found to be very similar to those typically found for histidines coordinated to the Fe(II) ion of the Rieske [2Fe–2S]+ cluster, thus indicating the direct binding of the amine to the unique Fe site in PFL-AE. From an analysis of all of their observations, the authors developed a SAM binding model, which was later verified by X-ray crystallographic data.
The metrics of SAM positioning with respect to the [4Fe–4S] cluster provided a hint about the mechanism of reductive homolytic cleavage of the S–C5′ bond . The fact that the sulfonium of SAM is in close proximity to the unique iron ion of the cluster suggests the possibility of a direct orbital overlap between the two. Consequently, this arrangement would allow for an efficient inner-sphere electron transfer from the [4Fe–4S]+ cluster to SAM to initiate the homolytic S–C5′ bond cleavage.
HYSCORE spectroscopy has proven to be another useful technique for observing the bidentate binding of SAM to the [4Fe–4S]RS cluster. This technique takes advantage of the fact that in the absence of SAM, the HYSCORE spectrum of the [4Fe–4S]RS + cluster contains only a minor signal (~4 MHz at X-band) resulting from “matrix” nitrogen atoms, likely from the peptide backbone of the cysteine ligands. A single HYSCORE measurement taken at the maximum absorption of the [4Fe–4S]RS + signal (at g = 1.92–1.93) was used in a variety of systems to identify such a binding mode. The first (to our knowledge) experiment of such a kind on a RS enzyme was performed on the activating enzyme of the class III ribonucleotide reductase (RNR) from Escherichia coli [46]. A broad and complex set of signals was found across the (++) and (+−) quadrants of the HYSCORE spectrum, which were absent in the sample without SAM. Using the known relation [47] between the frequencies of double quantum ridges in the (+−) quadrant of the HYSCORE spectrum and the 14N HF coupling (a) and quadrupole (K) coupling constant (Eq. 1), Gambarelli et al. [46] identified an isotropic component of the 14N HF coupling constant (aN) of 6.4 MHz. Similar studies of spore photoproduct lyase and TYW1 yielded aN values of 6.5 MHz [48] and 5.6 MHz, respectively [38].
A recent study of the cobalamin-dependent RS enzyme TsrM, a methyltransferase, provides the first example of an RS enzyme in which SAM does not bind to the [4Fe–4S] cluster, at least in the usual bidentate fashion [43]. The X-band HYSCORE spectrum of TsrM remained unchanged when SAM was added, although both cob(II)alamin and [4Fe–4S]+ EPR signals were found to be sensitive to the addition of SAM. Neither the 14N(amino) nor 13C(carboxylate) nuclei signals could be resolved. Additionally, the authors used the class A RS methylase RlmN as a positive control, for which both signals could be clearly observed in a single X-band HYSCORE measurement (see Fig. 4). Using the equation for extraction of the aN from above, we estimate the aN(RlmN) to be 5.4 MHz.

X-band HYSCORE spectra of the reduced RlmN in the absence (a) and the presence (b) of SAM [43]
In the most recent study of the reaction of PFL-AE by Hoffman, Broderick and coworkers, a new EPR species (denoted Ω) has been trapped by a rapid-freeze quench technique [49]. The signal has g-values of [2.035, 2.004, 2.004], that somewhat resemble the signals of a [4Fe–4S]3+ cluster with the intensity maximum at 500 ms of freeze-quench-delay time (Fig. 5). When annealed at temperatures up to 220 K, the EPR signal of Ω is replaced with a signal of a glycyl radical, indicating that this EPR active species precludes the hydrogen abstraction step. To verify the nature of intermediate Ω, the authors performed a series of ENDOR measurements using [adenosyl-13C10]- and 13CH3-SAM and 57Fe-labeled PFL-AE. When [adenosyl-13C10]-SAM was used to prepare this species, a strong 13C(adenosyl) coupling was observed (aiso(13C) = 9.4 MHz , T(13C) = 2.65 MHz). In contrast, when 13CH3-SAM was used, the corresponding 13C HF coupling constant was found to be about 20 times smaller than the one from the adenosyl moiety (~0.5 MHz). Moreover, 57Fe-isotope labeling of PFL-AE resulted in a 57Fe HF coupling of A(57Fe)~34 MHz that is comparable to the one observed for a [457Fe–4S]1+ cluster prepared without SAM. Overall, the results indicated that the observed intermediate signal results from both the [4Fe–4S] cluster and the adenosyl part of SAM. The magnitude of the 57Fe HF coupling indicates that the spin density of this EPR-active species resides on the Fe/S cluster, the adenosine is bound to the unique iron site by the C5′ of the adenosine (i.e. the strong 13C HF interaction observed is due to C5′), and SAM is cleaved such that the terminal methyl group of 13C-labeled methionine does not interact strongly with the EPR-active species. Overall, this conclusion points to a [4Fe–4S]3+-[5′-dA] configuration of the intermediate. Two alternative scenarios for the formation of this intermediate have been proposed by the authors: (1) the 5′-dA• formed by reductive cleavage of SAM adds to the Fe/S cluster; or (2) a nucleophilic attack of the Fea site of the [4Fe–4S]+ cluster on C5′ results in a formation of a Fe–C5′ bond and release of methionine. The authors propose that the Fe–C5′ bond is subsequently homolytically cleaved, resulting in a formation of the 5′-dA• and a [4Fe–4S]2+ cluster.

(a) X-band EPR spectra of PFL-AE indicating decay of the intermediate Ω and formation of the glycyl radical (G•) upon annealing at different temperatures. (b) Structural model of the intermediate Ω (c–e) Q-band ENDOR spectra of the intermediate Ω, prepared using [adenosyl-13C10] (c), 13CH3-SAM (d) and 57Fe-labeled PFL-AE (e). Adapted with permission from Horitani et al. [49]
Auxiliary Cofactors
In many cases, RS enzymes contain more than one Fe/S cluster or another metallocofactor. Most commonly, the auxiliary cofactor is another [4Fe–4S] cluster or a series of [4Fe–4S] clusters [50, 51]. Other Fe/S cluster forms, including a [2Fe–2S] cluster, can also be found in RS enzymes. Another subfamily of radical SAM enzymes requires a cobalamin cofactor for catalysis. In many reported cases, the auxiliary metallocenters act as active components of the reaction rather than as simple mediators of electron transfer; however, much still remains to be learned about their exact roles in many RS enzymes. Advanced EPR methods, however, have played an important role in deciphering the identity and the function of several auxiliary metallocenters in RS enzymes.
[4Fe–4S]aux as a Binding Site
MoaA is an RS enzyme that is involved in the biosynthesis of the biologically important molybdenum cofactor (Moco), and operates in concert with MoaC to convert guanosine triphosphate (GTP) to 1,1′-dihydroxy-2′,4′-cyclic pyranopterin monophosphate (cPMP).
MoaA contains two [4Fe–4S] clusters that are each ligated by three cysteines. The N-terminal cluster is coordinated by cysteines in the canonical CX3CX2C RS motif and facilitates the cleavage of SAM, while the auxiliary C-terminal cluster is ligated by cysteines in a CX2CX13C motif. The mode of GTP binding to the C-terminal [4Fe–4S]aux cluster was elucidated using Q-band CW ENDOR spectroscopy in concert with a C24S/C28S/C31S variant, which lacks the [4Fe–4S]RS cluster. This triple variant of MoaA allowed the authors to study the auxiliary cluster by pulse EPR in the absence of signals from the [4Fe–4S]RS cluster, which could complicate the analysis. ENDOR measurements on the S = 1/2 signal of the [4Fe–4S]aux + cluster of MoaA revealed the presence of a strongly coupled 14N nucleus, suggesting that a nitrogen atom of GTP binds to the cluster. The signal assignment was confirmed using uniformly 15N-labeled GTP, which shifted the signal by approximately 3 MHz (Fig. 6). Extensive orientation-dependent measurements were performed to extract the complete set of HF coupling constants. The resulting simulation yielded A(14N) = [2.4, 5.6, 4.1] MHz and A(15N) = [3.4, 7.8, 5.7]. To determine the exact binding orientation, the authors performed a similar study using a substrate analogue, inosine 5′-triphosphate (ITP), which differs from GTP by the absence of the exocyclic amino group. Similar 14N signals were observed in the orientation-selective ENDOR spectra of the ITP-treated MoaA with A(14N) = [0.0, 5.1, 4.0] MHz. Using pulse Q-band Mims ENDOR on the cysteine triple variant of MoaA in concert with 15N-labeled GTP, the authors were able to identify two other neighboring 15N nuclei with A(15N) ~0.5 and ~0.2 MHz that were attributed to the N2H2 (RFe-N ≈ 2.6–5.1 Å) and N3 (RFe-N > 3.6 Å) sites, respectively.

(a) Transformation catalyzed by MoaA and MoaC. (b) Q-band CW ENDOR spectra of the 1(14,15N)−MoaA triple variant at 2 K. (c) Proposed model for 5′-GTP binding (C, orange; N, blue; O, red; P, purple) to the Fe4 ion of [4Fe–4S]aux (S, yellow; Fe, green). The 5′-GTP model determined by X-ray crystallography (PDB entry 2FB3) is shown in white. Adapted with permission from Lees et al. [52]
Based on the data obtained, it was concluded that the auxiliary cluster in MoaA binds GTP via N1. The distances obtained allowed the authors to conclude that guanine binds as the enol tautomer rather than the typically favored keto form. Therefore, it was suggested that the binding-induced tautomerization is important for the mechanism of the catalytic transformation. Interestingly, the obtained metrics of GTP positioning (Fig. 6) differ from the ones observed in the X-ray crystallographic study of MoaA substrate binding, in which no direct binding was resolved. However, the authors argued that the procedures used to prepare MoaA samples for crystallography may have altered the binding mode.
A similar role for the [4Fe–4S]aux cluster in two methylthiotransferases, RimO and MiaB, has been hypothesized. MiaB installs a methylthiol group at C2 on an isopentenylated adenosine located at position 37 of certain tRNAs, while RimO installs a methylthiol group at C3 on Asp89 of bacterial ribosomal protein S12. Despite the difference in substrates, specifically the difference in hybridization of the target carbon atom, these two enzymes are postulated to catalyze their reactions by similar mechanisms. The mechanism by which these enzymes operate has been debated; however, the current hypothesis is that one SAM molecule donates a methyl group to an unknown thiol-containing group, which is then added to the substrate via a radical process that is initiated by reductive cleavage of another SAM molecule. It has been shown that exogenously added CH3S− or CH3Se− can be directly transferred to the corresponding products both in the MiaB and in the RimO reactions, indicating that a methylthiol molecule may be synthesized during catalysis rather than assembled directly on the substrate. Moreover, it was suggested that the auxiliary cluster does not act as a sacrificial donor but rather is a binding site for the synthesized methylthiol group. Consistent with this observation, Landgraf et al. showed that when RimO and MiaB are incubated with SAM in the absence of a low-potential reductant to initiate radical chemistry, time-dependent formation of methanethiol is observed upon addition of acid to the reaction mixtures.
Using 77Se-labeled CH3Se−, Forouhar et al. [53] demonstrated that this small molecule binds to the auxiliary cluster in both of these enzymes by observing the corresponding 77Se signals in X-band HYSCORE spectra (Fig. 7). The authors performed the same experiment on both wildtype MiaB as well as a variant in which the cysteine residues that coordinate the RS cluster were replaced with alanines. Unfortunately, they did not perform a complete characterization of the 77Se HF coupling, providing only a mean value of |A| = 3.8 ± 0.5 MHz. However, the magnitude of the HF interaction does suggest direct binding of CH3Se− to the [4Fe–4S] cluster. Rather interestingly, the X-ray crystallographic structure of chemically reconstituted RimO showed a polysulfide chain linking the unique iron sites of the two [4Fe–4S] clusters. Although it is most likely that such a construct is an artifact of the reconstitution process, it does indicate that the [4Fe–4S]aux cluster can bind exogenous sulfur atoms.

X-band HYSCORE spectra of the triple cysteine to alanine substitution of the RS motif MiaB variant without (a) and with (b) 77SeCH3Na. Adapted with permission from Forouhar et al. [53]
[4Fe–4S]aux as a Scaffold
HydG is one of the three maturation factors for [Fe-Fe] hydrogenases. It contains two Fe/S clusters: a typical N-terminal SAM-binding [4Fe–4S] cluster and an auxiliary Fe/S cluster coordinated by cysteines in a CX2CX22C motif. The role of HydG is two-fold: (1) to produce CO and CN− ligands through the breakdown of free tyrosine and (2) to form an [Fe(CO)x(CN)y] synthon using an exogenous source of iron [37, 54, 55]. The functional aspects of tyrosine cleavage will be covered later in this chapter; here we focus on the identity and the role of the auxiliary cluster. The first reports on HydG characterized the auxiliary cluster as a [4Fe–4S] cluster [56, 57]. However, a later study of the chemically reconstituted Shewanella oneidensis (So) HydG showed the presence of an unusual S = 5/2 EPR signal (geff: 9.5, 4.7, 4.1, 3.7) that persisted when the three cysteines of the canonical RS motif were substituted with serines (HydGXN), but was absent when the cysteines that coordinate the C-terminal cluster were altered (HydGXC). Based on the X-ray crystallographic structure of similarly prepared wild-type Thermoanaerobacter italicus (Ti) HydG, the S = 5/2 signal was attributed to a quite unprecedented construct of a cuboid [4Fe–4S] cluster attached to a dangling, histidine-coordinated, Fe ion via a μ-sulfide bridge [58]. In addition, a labile nonproteinaceous amino acid was found near the sulfide ion. The S = 5/2 state of the auxiliary cluster in SoHydG was explained using a simple exchange coupling model in which two spin systems S1([4Fe–4S]+1) = 1/2 and S2(Fe2+) = 2 are coupled ferromagnetically. The authors also noted that an additional observed signal at geff = 5.5 may originate from an S = 3/2 species due to an antiferromagnetic coupling mechanism. By simulating temperature dependent EPR signals, effective zero-field splitting parameters for the S = 5/2 species were extracted: Deff = +4.5 cm−1, Eeff/Deff = 0.255 and geff = 2.
The observation that tyrosine interacts with the auxiliary cluster of HydG suggests a scaffolding role for the auxiliary cluster. It was shown that addition of a reducing agent (dithionite), SAM, and tyrosine to SoHydG results in the decay of the S = 5/2 signal and the appearance of an S = 1/2 [4Fe–4S]aux + EPR signal (g = [2.09, 1.94, 1.93]), indicative of the loss of the dangling Fe (Fig. 8). HYSCORE measurements performed on the resulting [4Fe–4S]aux + cluster showed the presence of 15N and 13C signals when 13C9,15N-labeled tyrosine was used, indicating that the auxiliary cluster is coordinated by a fractured derivative of the backbone part of the tyrosine [37]. Adding K13CN to SoHydG resulted in a similar effect: a HYSCORE measurement of the [4Fe–4S]aux cluster in the presence of K13CN showed a clear signature of a strongly coupled 13C nucleus (A(13C) = [−5.0, −4.0, 0.9]), indicating direct binding of CN− to the auxiliary cluster [37, 59].

Low-field (a) and high field (b) EPR signals of HydG, experimental spectra in black, where the red trace is the simulation of the S = 5/2 species and the blue trace is the simulations of [4Fe-4S]+-Cys species, (c) Q-band Mims ENDOR spectra of HydG in [4Fe–4S]−-3-13C-Cys state (black) with simulations (blue) (d) Evidence of the presence of CN-ligand to the [4Fe–4S]aux cluster by 15N- (left) and 13C- (right) HYSCORE, (e) Proposed scheme of formation of an [Fe(CO)x(CN)y] synthon on the auxiliary cluster of HydG, (f) Conversion of a HydG-generated synthon into the hydrogenase H-cluster by HydE and HydF. Adapted with permission from Dinis et al. [58] and Suess et al. [59]
Suess et al. [59] illustrated that the dangling Fe can be removed by adding a chelating agent such as EDTA, resulting in the appearance of the gmax = 2.09 signal from the S = 1/2 [4Fe–4S]aux + cluster. Subsequent addition of exogenous Fe2+ results in the restoration of the S = 5/2 [5Fe] species. The observations were also confirmed by a Mössbauer study [59]. Interestingly, Ni2+ could be installed as the fifth metal instead of Fe2+, giving rise to a high-spin signal that was attributed to an S = 3/2 state (geff = 4.8 and 3.6) due to ferromagnetic coupling between S1([4Fe–4S]) = 1/2 and S2(Ni2+) = 1.
Taking into account the previous work which illustrated that an addition of nonproteinaceous cysteine increases the maturation efficiency, [55] and that an unrecognized amino acid molecule was present near the auxiliary cluster in the X-ray crystal structure, [58] it was hypothesized that a cysteine molecule constitutes the Fe–S(Cys)–Fe bridge between the [4Fe–4S] cluster and the dangling Fe. Indeed, addition of cysteine and Fe2+ to EDTA-treated HydGXN results in a complete restoration of the S = 5/2 [5Fe]aux species. The use of other additives instead of l- cysteine, such as d-cysteine, l-homocysteine, l-alanine plus S2–, or l-serine, did not result in formation of the S = 5/2 species. To provide further evidence that cysteine binds to the [4Fe–4S]aux cluster, the authors performed a field-dependent ENDOR study of EDTA-treated HydG in the presence of 15N,13C3-Cys or 3-13C-Cys. The orientation selective Mims ENDOR measurements of the singly 13C-labeled samples revealed a pair of signals that were attributed to a 13C nucleus with a roughly isotropic HF coupling (A = [0.83, 0.83, 1.09] MHz). Similar measurements of HydG prepared with 15N,13C3-Cys revealed more complex signals, but the largest signals did not exceed the overall width of the signals obtained for the singly labeled sample, indicating that cysteine likely binds to the [4Fe–4S]aux cluster via the thiol group.
Finally, Suess et al. showed that cysteine can replace cyanide at the open coordination site by observing the change in the EPR signal for the auxiliary cluster from gCN = [2.09, 1.94, 1.93] to gCys = [2.06, 1.90, 1.87] when the samples were sequentially treated with KCN and Cys. Based on the obtained data, it was concluded that the auxiliary cluster is a scaffold for the formation of the [Fe(CN)2CO] synthon that is mediated by the presence of nonproteinaceous cysteine.
[2Fe–2S]aux as a Sacrificial Donor
Biotin synthase (BioB) is an RS enzyme that converts dethiobiotin (DTB) to biotin. Through extensive studies using a variety of biophysical methods, it was concluded that BioB acts as a dimer and contains one [4Fe–4S]RS and one [2Fe–2S]aux cluster per monomer [60, 61]. The stoichiometry and species of Fe/S clusters was later confirmed by X-ray crystallography [62].
The [2Fe–2S] cluster, coordinated by three cysteine and one arginine ligand, was shown to donate one of its μ-sulfide bridges to the substrate during turnover (Fig. 9) [63,64,65]. An important mechanistic insight was obtained by X-ray crystallography [62]. The structure of BioB in the presence of SAM and dethiobiotin revealed that dethiobiotin is located between the [4Fe–4S]RS and [2Fe–2S]aux clusters. C9 of dethiobiotin was located essentially in the middle between C5′ of SAM and one of the bridging thiols. Therefore, it was proposed that after the 5′-dA• abstracts a hydrogen atom from the C9 position, the substrate radical that is formed subsequently recombines with the thiol bridge of the [2Fe–2S] cluster and thus initiates the transfer of the sulfur atom to C9.

(a) Reaction of BioB with DTB . (b) Relative positioning of Fe/S clusters, SAM and DTB in BioB (PDB: 1R30), (c) Proposed structure of an intermediate. (d) X-band HYSCORE spectra of the [2Fe–2S]+ intermediate in BioB in natural abundance of N-isotopes (top) and with 15N-labeled Arg (bottom), insert shows HYSCORE spectra obtained using (9-methyl-13C)-DTB as a substrate. Adapted with permission from Fugate et al. [67]
Although no substrate radical intermediates have been identified, it was shown that the [2Fe–2S] cluster does get transiently reduced under turnover conditions with kinetics that correlate with the formation of a 9-mercaptodethiobiotin (MDBT) intermediate [65, 66]. The reduced [2Fe–2S]+ cluster was found to exhibit two distinct signals (g = [2.010, 1.955, 1.880] and g = [2.000, 1.940, 1.845]) that both have the same kinetic behavior, suggesting the possibility of structural heterogeneity. The identity of the paramagnetic species was verified by observation of nitrogen signals in ESEEM and HYSCORE that are characteristic of arginine ligation [65, 67]. Most notably, when BioB was expressed in the presence of guanidino-15N2–Arg, Fugate et al. [67] were able to resolve two 15N signals from the guanidino group in the X-band HYSCORE spectrum.
To clarify the structure of the intermediate further, Fugate et al. [67] prepared BioB with (9-methyl-13C)- dethiobiotin and performed a HYSCORE analysis of the [2Fe–2S]+ intermediate. The 13C HF coupling observed ([1.2, 1.2, 5.7] MHz ) provided direct evidence that dethiobiotin is bound to the reduced [2Fe–2S]+ cluster. Therefore, these measurements indicate that a dethiobiotin-radical is transiently stabilized as a bound adduct to the [2Fe–2S] cluster. Also, these results suggest that this Fe/S cluster is not just a sulfur donor but also is a catalytic cofactor that facilitates inner-sphere one-electron oxidation of the sulfide concurrent with formation of the new C–S bond. This mechanism provides a means for safe termination of the MDBT radical and, therefore, allows for a subsequent hydrogen abstraction at C6 using a second 5’-dA •.
Radical Intermediates
Because the mechanisms of RS enzymes imply the presence of organic radical intermediates, great effort has been devoted to trapping and characterizing such intermediates by EPR. In the following, we describe several examples of such work.
Pyruvate Formate-Lyase Activating Enzyme (PFL-AE)
PFL-AE was one of the earliest enzymes of the RS superfamily to be studied, and in fact preceded the bioinformatic identification of the superfamily. Soon after the identification of PFL-AE as an activator of PFL, it was found to cleave SAM into methionine and 5′-dA with concomitant formation of a protein-based radical on PFL [68,69,70].
The CW EPR spectrum of activated PFL was characteristic of a carbon-based radical that is dominated by a single 1H hyperfine splitting of 1.5 mT (Fig. 10) [71]. The doublet-shaped spectrum collapsed into a single line upon exchange into buffer prepared with D2O, indicating that the corresponding proton is solvent exchangeable [71, 72]. The spectrum also contained a fine structure that was not affected by H2O/D2O exchange but was diminished when PFL was expressed in deuteurated media indicating that this feature is due to non-exchangeable protons. The final identification was made by observing EPR line broadening upon selective labeling of glycine residues in PFL with either [2-13C]glycine or [1-13C]glycine [72]. Extracted spin-Hamiltonian parameters indicated that the spin density is localized on C2 and supported the notion that PFL-AE abstracts the Cα–H, pro-S H-atom of G734. These results, and the fact that only the reduced [4Fe–4S]+ state is the active form, illustrated that RS enzymes utilize reductive cleavage of SAM and use the 5′-dA• generated to abstract an H-atom from a (co)substrate; which, along with the early study of lysine 2,3-aminomutase , were the first indications of the key role SAM plays in the reaction of RS enzymes.

Generation of PFL-Gly• by PFL-AE (a). X-band EPR spectra of PFL-Gly• as generated by PFL-AE for (b) PFL in H2O (blue) and D2O (red); (c) 2H-labeled PFL in H2O (blue) and D2O (red); (d) [2-13C]-Gly enriched PFL; (e) [1-13C]-Gly enriched PFL in D2O with unlabeled serine (black) and [1-13C]-Ser (green). Adapted with permission from Unkrig et al. [71]
Lysine 2,3-Aminomutase (LAM)
LAM is perhaps the best spectroscopically characterized RS enzyme. LAM catalyzes the conversion of l-α-lysine to l-β-lysine [73]. In addition to SAM and a [4Fe–4S]RS cluster, its reaction also requires pyridoxal 5′-phosphate (PLP), which forms an external aldimine with the lysine substrate or β-lysine product. In the proposed reaction mechanism (Scheme 2), the SAM-derived 5′-dA• (1) initiates substrate-based catalysis by abstracting the 3-pro-R H-atom of lysine to yield a substrate radical intermediate (2), which undergoes formation of an azacylopropylcarbinyl radical (3). This quasi-symmetric radical intermediate can partition backwards to form the initial substrate radical or forward to form the product radical (4). The product radical is quenched upon abstraction of an H-atom from 5′-dA to regenerate the 5′-dA•, which, in concert with the [4Fe–4S]RS cluster (5), recombines with methionine to regenerate SAM (6). Catalysis is completed upon transimination to form an internal aldimine with a conserved lysyl residue. In the catalytic reaction of LAM, 5′-dA formation is not stoichiometric with product formation. SAM acts as a true cofactor rather than as a co-substrate, because it is regenerated after each turnover. The mechanism shown in Scheme 2 is supported by a number of EPR studies that provided evidence for various intermediates in the reaction.

Reaction of Lysine 2,3-aminomutase (LAM)
Identification of State “1”
Evidence for the involvement of the 5′-dA• in the LAM reaction was obtained in studies by Magnusson et al. using S-3′,4′-anhydroadenosyl-l-methionine (anSAM) , a SAM analogue that permits allylic stabilization of the radical generated upon reductive cleavage of anSAM [74, 75]. Upon reaction of anSAM with LAM, a slow formation of an organic radical was observed with a first order rate constant (kobs) of approximately 10 min−1. At 77 K, the radical exhibited a broad, roughly isotropic EPR signal with a fine structure that disappeared when [5′-2H2]-anSAM was used. Later, the authors reported that lowering the temperature to 4.2 K resulted in significant narrowing of the unitary lineshape, leading to a significant clarification of the fine structure to distinct lines. The unusual temperature effect was explained by a weak spin-spin interaction between a radical and low-lying excited states of the formally diamagnetic [4Fe–4S]2+ cluster that are partially populated at higher temperatures. Consequently, such an explanation implies a proximity of the radical to the [4Fe–4S]2+ cluster upon reductive cleavage.
The EPR spectrum broadened when the [1′,2′,3′,4′,5′-13C]-anSAM isotopolog was used to generate the radical, confirming that the radical is indeed located on the anhydroribosyl moiety (Fig. 11). Selective 2H isotope labeling of the molecule at C5′, C3′, and C2′ resulted in a significant perturbation of the EPR lineshape and permitted the authors to extract HF coupling constants for the corresponding protons, thus resolving the electronic structure of the radical species. Using McConnel’s relation for spin-polarized alpha carbons, aH = ρQ, in which Q is an empirical constant and ρ is the spin density population on the respective carbon, it was determined that the C3′ and C5′ carbons share near equally about 59% of the spin density, which is in a good agreement with the expected spin distribution for an allylic radical. It is worth noting that this study represents the most direct evidence for a 5′-dA• to date, as the direct observation of this radical species remains elusive due to its high reactivity.

Effect of temperature and 13C substitution on the X-band CW EPR spectrum of the anhydroadenosyl radical (5′-andA•): (a) using unlabeled anSAM at 77 K. (b) using unlabeled anSAM at 4.2 K. and (c) using [1′,2′,3′,4′,5′-13C5]anSAM at 4.2 K. Adapted with permission from Magnusson et al. [75]
More recently, Hoffman, Broderick, and coworkers analyzed the surrounding environment of the 5′-andA• in LAM in great detail using ENDOR and specific 13C and 2H isotope labeling [76]. The results indicated that C5′ is approximately 3 Å away from C2 of the lysine substrate, and although no signal from the C3 13C nuclei could be conclusively resolved, 2H measurements indicated that the 3-2H is only 2.4 Å away—a perfect distance for hydrogen atom abstraction. Interestingly, although in this state of the reaction no direct bonding should exist between C5′ and lysine, a substantial isotropic component to the corresponding 13C HF coupling of C2 was clearly evident from the corresponding ENDOR spectrum (Fig. 12). This effect was explained in terms of Pauli delocalization, which implies spin delocalization through space onto the neighboring atom when this atom is in tight van der Waals contact with the spin-bearing orbital of the allylic radical. Additionally, using 13C-methyl anSAM, the authors were able to resolve a distance between C5′ and the methyl group of l-methionine of about 3.6 Å. Overall, this study indicates that after reductive cleavage, SAM fragments are essentially fixed in the same position with the substrate located near the C5′ site as well. Such a conclusion coincides with the overall mechanism in which the fractured parts (l-methionine and 5′-dA) are reassembled to a complete SAM molecule in the final step of the reaction.

(a) Model of relative position of lysine , methionine and 5′-dA•. Position of SAM prior reductive cleavage is shown in gray (b, c) Q-band 13C ENDOR spectra of 5′-andA• in LAM prepared in the presence of 13C-lysine (B)or with 13C-methyl-anSAM (C) (d). Q-band 2H ENDOR spectra of 5′-andA• in the presence of 2H-labeled lysine. Adapted with permisson from Horitani et al. [76]
Identification of State “2”
As with the SAM analogue described above, reactions of LAM with substrate analogues resulted in a trapped substrate radical intermediate resulting from the abstraction of the 3-pro-R H-atom in the next step of the reaction. Upon reacting LAM with the lysine analogue, 4-thia-l–lysine (SLys), Miller et al. [77] observed formation of a new radical species by CW EPR. The spectral envelope of this radical (SLys•) was strongly perturbed by specific isotopic labeling; most notably, labeling of C3 with 13C resulted in significant broadening of the spectrum (Fig. 13, Traces A and E).

Left: X-band CW EPR spectra of LAM with SLys bound. (A), 4-thialysine; (B), 4-thia[5,5,6,6-2H4]lysine; (C), 4-thia[3,3-2H2]lysine; (D), 4-thia[3,3,5,5,6,6-2H6]lysine; (E), 4-thia[3-13C]lysine. Adapted with permission from Miller et al. [77]. Right: X-band CW EPR spectra of LAM with DHLys• bound using (A) unlabeled DHLys ; (B) [2-2H]-DHLys; (C) [4,5-2H2]-DHLys; (D) [3,3,4,5,6,6-2H6]-DHLys; (E) [2,3,3,4,5,6,6-2H7]-DHLys. Red spectra in panels (B) and (C) are the results of resolution enhancement. Adapter with permission from Wu et al. [78]
To improve spectral resolution, the authors applied a resolution enhancement (RE) data manipulation procedure [79]. Simulated resolution enhanced spectra obtained from various isotopically labeled substrate analogues (Fig. 13) revealed that 80% of the spin density of the radical was localized at the C3 position. However, as evident from the substantial 1H HF couplings of the C5 hydrogens (3.5 G and 4.0 G), moderate delocalization of the radical across S4 takes place. Rather surprisingly, the g-anisotropy reported by the authors is very small, thus indicating negligible contributions of the spin-orbit coupling of the sulfur atom to the overall electronic structure, which possibly indicates that sulfur carries no noticable spin density. In addition, interpretation of the EPR spectra required an isotropic HF coupling from an 14N nucleus of Aiso = 9.5 MHz. Resolving the 1H hyperfine coupling constants from the C2 proton allowed for the extraction of dihideral angles of 77° and 17° between the plane defining the spin-carrying p-orbital on C3 and the C2–H bond and the C2–N(PLP) bond, respectively. Due to hyperconjugation effects, this result implies that a strong 14N HF interaction with an α-amino group must take place, providing the assignment for the observed 14N HF coupling. Overall, this detailed study of the electronic structure confirmed that the observed radical species is analogous to state 2 in Scheme 2.
Further support for the presence of a C3-based substrate radical in LAM was provided with another substrate analogue, trans-4,5-dehydrolysine, that allowed for stabilization of an allylic 4,5-dehydrolysyl radical (DHLys•), formed upon abstraction of the 3-pro-R hydrogen atom [78]. The EPR spectrum of the observed radical contained fine structure that was affected by isotope labeling of trans-4,5-dehydrolysine with 2H. The HF coupling constants extracted from these experiments confirmed the delocalization of the spin density between C3 and C5 expected for an allylic radical. In addition, the authors labeled N6 with 15N, which resulted in no change in the spectrum, suggesting that the observed triplet splitting is due to the α-amino group (N2). Similarly to that observed in the SLys• radical, this splitting originates from a hyperconjugation with unpaired spin density on C3. The 14N2 and 1H6 HF coupling constants and the fact that the 1H2 signal is below the limit of detection suggests that the H6–C6 and C2–N2 bonds lie essentially in the plane perpendicular to the spin-bearing p-orbitals on C3 and C5.
The positioning of both substrate analogue radicals with respect to 5′-dA and PLP have been extensively analyzed by the Hoffman group [80]. Both the SLys• and DHLys• intermediates exhibited a relatively strong 13C HF coupling with 13C5′ when [5′-13C]-SAM was used in the reaction (SLys•: Aiso = 12.7 MHz and T = 2.5 MHz; DHLys•: Aiso = 2.0 MHz and T = 0.6 MHz, where T and Aiso are constants defining the principal components of the HF interaction Ax,y,z = [−1, −1, 2]T + Aiso). The strong isotropic character of both HF coupling constants suggests that the 5′ carbon is in tight van der Waals contact with the radical residing on the SLys and DHLys molecules. The magnitude of the HF coupling indicates that the interaction is stronger in the case of SLys than that of DHLys. The authors refrained from further analysis of the anisotropic HF coupling, because the almost-bonding positioning of the substrate and 5′-dA is too short to directly extract the distance between the radical and C5′ of 5′-dA. In addition, the authors observed a 2H signal in DHLys• originating from 2H at C5′, providing additional evidence for the proximity of the 5′ carbon to the DHLys•.
Analogously, the authors analyzed the position of methionine with respect to the substrate analogue radical species in LAM. Using a 13CH3-SAM isotopolog , a clear 13C Q-band ENDOR signal could be observed for SLys• and DHLys•. The interpretation yielded an anisotropic 13C HF coupling. Once again, SLys• showed a larger 13C HF coupling (T = 0.3 MHz and Aiso = 0.1 MHz) than DHLys• (T = 0.11 MHz and Aiso = 0.0 MHz). Using dipolar coupling constants (T), distances from the methyl group of SAM to C3 of SLys• and DHLys• were calculated to be ~4.1 Å and 5.5 Å, respectively, which is very similar to the distances observed in the X-ray crystallographic structure of LAM with all of its substrates and cofactors bound.
Another important structural detail was obtained by detecting 31P-PLP signals using the DHLys and SLys substrates. Both substrate radicals exhibited 31P signals in Q-band ENDOR. In this case, the DHLys• displayed a somewhat larger 31P HF tensor than the SCys• (DHLys•: T = 0.59 MHz and Aiso = 0.19 MHz; SCys• : T = 0.41 MHz and Aiso = 0.19 MHz). Using the dipolar coupling constant (T), the authors resolved R(C3–PPLP) distances of 4.3 Å and 3.6 Å for SCys• and for DHCys•, respectively. The authors pointed out that the distance obtained is considerably smaller than what was observed in the crystal structure (7 Å) regardless of the substrate analogue considered, indicating that the phosphate group of PLP moves closer to Cβ during the reaction and implying a possible isomerization of PLP (Fig. 14).

(a). Structure of the radical species generated from substrate analogues of LAM. (b) 31P ENDOR spectra of DHLys• and SLys•. (c). 13C ENDOR spectra of DHLys• and SLys• and native state “4” (4(Lys•)). (d) Schematic representation of the interaction of DHLys•, SLys•, and 4(Lys•) with 5′-dA , l-methionine, and PLP. Note that the numbering of states was changed from the original article to comply with the numbering on the Scheme 2. Adapted with permission from Lees et al. [80]
Identification of State “4”
During the reaction of LAM with its native substrate, a radical species formed in the steady state, which was absent when either lysine or SAM was omitted from the reaction. 2H isotope labeling of the substrate resulted in a perturbation of the EPR lineshape, indicating that the radical species is based on the substrate or a substrate derived molecule (Fig. 15) [81].

Conversion of the product radical in LAM from a deuterated to a non-deuterated form in the course of a freeze quench experiment schematically depicted in (b) with a time dependence shown in (c). (a) depicts EPR spectra obtained by quenching the reaction at 13.6 ms (A), 53 ms (B) and 5 s (C) Adapted from Chang et al. [82]
Since the activation of LAM by SAM is much slower than the enzymatic reaction (~5 s), the time dependence of the formation of the putative product radical is not represented by the catalytic kinetic rates but rather by the activation kinetics. Therefore, it is not possible to prove the kinetic competence of this radical species directly from the kinetics of rapid-freeze quench experiments . Instead, Chang et al. [82] performed a sequential mix rapid freeze-quench experiment to illustrate that the radical species turns over as fast as the overall enzymatic turnover. In this experiment, LAM was activated using [2-2H]-lysine and SAM for 5 s in the first mixing step. Then the reaction was mixed again with an excess of unlabeled lysine and freeze-quenched at various time points. If the radical is an intermediate state, the EPR spectrum of the deuterated radical species should be gradually replaced with the EPR spectrum of the unlabeled species with a rate of conversion comparable to the rate of product formation. The obtained rate of conversion of the EPR signal (24 s−1) was somewhat faster than the estimated LAM turnover rate determined from independent kinetic experiments , therefore proving the radical species observed is kinetically competent.
An extensive isotope labeling study in combination with CW EPR spectroscopy was performed to clarify the nature and the structure of the radical species. The most prominent perturbations to the EPR spectrum were achieved with 2H- and 13C-labeling at the C2 position of lysine, which resolved an isotropic 2H HF coupling constant and a significantly anisotropic 13C HF coupling constant. This result suggested that the radical resides on C2 with a single hydrogen atom. Further analysis of the radical using [2-15N1]- and [2-15N1, 2-2H2]-lysine substrates allowed the authors to assign the triplet hyperfine splitting observed in the EPR spectra to the nitrogen formally at the Cα position. However, the isotropic nature of the 14N HF coupling and the presence of a C2–H 1H HF coupling indicated that the substrate nitrogen atom is at the Cβ position in the radical species, meaning that the nitrogen atom shifted from the α to β position during the reaction. Therefore, the observed radical species is consistent with the product radical (species 4 in Scheme 2).
Because the nitrogen atom of the substrate migrates from C2 to C3 during the reaction, it was crucial to understand whether it was bound to PLP in the product radical species as an external aldimine. Pulse EPR measurements were performed to deduce the relative position of PLP and the product radical. Using [4′-2H] PLP, a 2H signal was observed by ESEEM spectroscopy . The field dependence of the 2H ESEEM signal was analyzed to provide an estimate for the principal components of the 2H HF tensor. Including the uncertainties in the determination of the HF coupling constants, the R(C2–H4′) distance was estimated to be 3–3.5 Å, meaning that Nβ and C4′ are within direct bonding distance [84]. Therefore, these results support the hypothesis that the PLP coenzyme facilitates radical-mediated rearrangements. More details about the overall positioning of the radical have been obtained through a series of ENDOR measurements analogous to the ones performed for the SLys and DHLys substrates (see above). Interestingly, the geometric metrics obtained were found to be very similar to the ones of SLys (Fig. 16) [80], indicating that despite the migration of the N-PLP moiety from Cα to Cβ, the substrate remains in the same place within the pocket. This once again illustrates the tight control that the protein environment imposes on the position of the (co)substrate molecules in this class of enzymes.

The effect of isotope labeling on the X-band CW EPR spectra of the product radical. (A) unlabeled lysine, (B) L-[3,3,4,4,5,5,6,6-2H8]lysine (C) DL-[2-2H]lysine, and (D) L-[2-13C]lysine. The red spectrum in B is the result of resolution enhancement. Adapted with permission from Ballinger et al. [83]
HemN
An anaerobic pathway for the biosynthesis of porphyrins requires oxidative decarboxylation of the propionate side chains on the A and B pyrrole rings of coproporphyrinogen III to form the corresponding vinyl groups (see Scheme 3). This reaction is carried out by coproporphyrinogen III oxidase (HemN) , a radical SAM enzyme that contains a single [4Fe–4S] cluster. It was postulated that the 5′-dA• formed from the reductive cleavage of SAM abstracts the pro-S-hydrogen atom at the β-carbon of the substrate propionate side chain, which initiates cleavage of the Cα–COO− bond.

(a) Decarboxylation of the propionate side chains of coproporphyrinogen III by HemN. (b) Proposed substrate radical intermediate of HemN [85]
Therefore, an intermediate substrate radical species was expected during the catalytic reaction, and was indeed observed [85] when the substrate, coproporphyrinogen III, was added to a sample of reduced HemN along with an excess of SAM. The ability to detect this radical signal down to a temperature of 5 K suggested that the radical is in close proximity to the [4Fe–4S]2+ cluster. To provide further information about the identity of the radical, the authors synthesized 2H- and 15N-labeled coproporphyrinogens and proved that the obtained molecules are efficient substrates for HemN. While 15N-labeling did not result in any significant modification of the EPR spectra of the radical intermediate, selective deuterium labeling at different positions resulted in substantial narrowing of the EPR signal. The analysis of the spectra obtained allowed the authors to conclude that the radical species observed is a substrate-based with allylic character. The radical was also localized on the β-carbon of the carboxylate appendage and on the ring, as is evident from the effect of specific deuterium labeling of the meso-positions of coproporphyrinogen III on the EPR spectra.
Cfr/RlmN
Cfr and RlmN belong to the class A family of RS methylases. Both enzymes work on the same base, adenosine 2503 (A2503), of 23S rRNA. RlmN specifically installs a methyl group on the C2 position of the adenine ring of A2503 of 23S rRNA as well as on the C2 position of adenosine 37 (A37) in several tRNAs in E. coli and other organisms. Cfr, however, acts only on A2503 of 23S rRNA, but methylates both the C8 and the C2 carbons, with C8 methylation taking place before C2 methylation. C2 methylation of A2503 of rRNA and A37 of tRNA by RlmN plays a key role in translational fidelity and efficiency. Cfr-mediated C8 methylation, however, confers resistance to several classes of antibiotics that inhibit the peptidyl transferase activity of the bacterial ribosome, such as phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A. Since 2007, cases of cfr-positive staphylococcal isolates from hospitalized patients in several countries, including the USA, have been reported, suggesting that this mechanism of antibiotic resistance is readily spreading, even across different staphylococcal species.
Early studies of the RlmN and Cfr reactions demonstrated that two equivalents of SAM are necessary, one to produce the 5′-dA• and a second to donate a methyl group [86]. However, the proposed mechanisms would require abstraction of hydrogen atoms from sp 2-hybridized carbon centers that exhibit C–H bond-dissociation energies above 100 kcal/mol. An alternative mechanism for the reaction was postulated upon finding that the appended methyl carbon is first appended to a conserved cysteinyl residue in the active site of each enzyme as well as the results of isotope tracer experiments that showed that the 5′-dA• abstracts a hydrogen atom from the appended methyl group [87, 88]. Scheme 4 shows the transformations proposed for Cfr. For RlmN, a similar hypothesis has been proposed.

Proposed radical SAM reaction mechanism of Cfr
The key element of the proposed mechanism is the addition of the resulting Cys-appended methylene radical onto C8 (Cfr) or C2 (RlmN) of A2503 to generate a protein–nucleic acid cross-linked species containing an unpaired electron. By performing this radical addition, RlmN and Cfr transiently change the hybridization of the carbon center undergoing methylation from sp 2- to sp 3-hybridized.
An EPR study performed by Grove et al. provided direct evidence for such an intermediate in the reaction of Cfr with a small, 155-mer rRNA substrate analogue [88]. When Cfr was combined with the 155-mer RNA and SAM and then rapidly mixed with dithionite to initiate turnover before loading into an EPR tube and freezing, a doublet-shaped was observed by CW EPR that had spectroscopic characteristics typical for an organic radical species. The EPR spectrum was consistent with the presence of one strong isotropic 1H HF splitting and one anisotropic 14N HF splitting, which coincided with the proposed electronic structure of the radical intermediate, i.e. an N7-based radical and an sp 3-hybridized C8 that retains its proton. Use of a 155-mer RNA substrate that was deuterated in the adenosine rings resulted in a collapse of the doublet into a single line, confirming that the observed species is an adenosine-based radical. To confirm a cross-linked RNA-Cfr configuration, the authors prepared Cfr such that the enzyme was unmethylated as-isolated and then supplied 13C-met-SAM in vitro in order to install a 13C label onto the mCys338 (see Fig. 17). The spectrum of the radical species obtained using such protein is dominated by clear a signature of a strong 13C HF splitting when deuterated 155-mer RNA is used or adds to the 1H8 splitting when unlabeled substrate is used, thus confirming the addition of the mCys to the adenosine ring.

X-band CW EPR (a) and ENDOR (b) spectroscopic characterization of the organic cross-linked radical intermediate in the reaction of Cfr with 155-mer RNA [88]
Using a combination of Mims ENDOR and Davies ENDOR to observe 1H and 2H signals, five total HF coupling constants were extracted and assigned to the four protons of the adenosine ring and the C1′ proton. The obtained HF coupling constants indicated that although the spin density is significantly delocalized over the adenosine ring, the majority of the spin density is located on N7 and that C8 is sp 3-hybridized. The close resemblance of the DFT-calculated HF coupling constants to the ones obtained experimentally confirmed the overall structure of the cross-linked species.
The kinetic and chemical competence of the obtained radical species was confirmed by comparing the time dependence of the EPR signal and the product formation. The rate of product formation predicted from the time dependence of the EPR signal matched the one observed for product formation using liquid chromatography-mass spectrometry, thus providing the final proof that the radical species observed is indeed the intermediate proposed in Scheme 4.
In the case of RlmN, no cross-linked radical species could be observed for the wild-type protein. However, in an effort to understand role of a strictly conserved C118, Silakov et al. found that the inactive C118S and C118A variants of RlmN accumulate a substrate-derived paramagnetic species with an EPR lineshape very similar to the one observed in Cfr. Using an isotope labeling strategy similar to the one presented above permitted the assignment of the radical to a cross-linked species in which the radical is significantly delocalized over the adenine ring and an sp 3-hybridized C2 carbon. The analogous C108A Cfr variant was prepared and was found to stabilize the radical species as well. Therefore, it was concluded that the conserved residues, C108Cfr and C118RlmN, are crucial for resolving the cross-linked radical species.
HydG
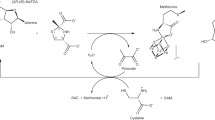
HydG and HydE are radical SAM enzymes that are involved in the maturation of the H-cluster active center of [Fe-Fe]-hydrogenases that catalyze the reversible heterolytic splitting of molecular hydrogen. The efficiency of the [Fe-Fe] hydrogenase as a hydrogen catalyst triggered large interest in this family of enzymes. The H-cluster is an unusual construct consisting of two subunits: a ferredoxin-like [4Fe–4S]H cluster and a unique 2FeH subcluster [89,90,91,92]. The 2FeH cluster is a dangling unit that is stabilized in the protein pocket by means of a H-bonding network, electrostatic interactions, and a single covalent link to the protein via one of the cysteines that coordinates the [4Fe–4S] “cubane”. Both irons in the bi-nuclear site are coordinated by CO and CN− ligands. Another unusual feature is the dithiolate bridge whose central atom was identified as a nitrogen by means of pulse EPR methods [93, 94] and proposed to serve as a base during heterolytic cleavage of molecular hydrogen. Using in vitro maturation, an exogenously added tyrosine was found to be crucial for the assembly of the cluster [95]. Isotopic labeling of tyrosine has confirmed that the backbone of this molecule is the source of the CO and CN− ligands [96]. It was also identified that free tyrosine is the substrate for HydG, which degrades tyrosine into p-cresol and dehydroglycine. Free CN− is released into the medium, where it was detected by upon being converted into a fluorescent derivative. Based on the biochemical and spectroscopic evidence, a reaction mechanism was proposed, in which abstraction of a hydrogen from the amino group by 5′-dA• triggers homolytic cleavage of the Cα–Cβ bond, resulting in the dehydroglycine intermediate that is used to generate the CO and CN− ligands. As described above, the auxiliary cluster acts as a scaffold that eventually forms a Fe(CN)x(CO)y unit, a building block of the [2Fe]H subcluster.
The radical species formed upon Cα–Cβ scission has been investigated by EPR. Reacting wildtype HydG with l-tyrosine, SAM and a reducing agent (DTH) resulted in the transient appearance of a complex signal in the g = 2 region that peaks at about 2s. The signal was susceptible to isotope labeling of tyrosine, illustrating that the observed signal corresponds to a molecule derived from the tyrosine. When the reaction was run with 3,5-2H2-labeled Tyr the EPR lineshape was unchanged, which is consistent with the predicted Cα–Cβ cleavage. Similarly, the authors noted no significant change when 17O-labeled Tyr was used. On the other hand, the majority of HF splitting vanishes when β,β-2H2 tyrosine was used. Therefore, the authors determined that the majority of the spin density is localized on Cβ. The fact that 13Cα labeling had no effect on the observed signal illustrated that the observed species is a 4-oxidobenzyl (4OB•) radical, therefore confirming that Cα–Cβ cleavage takes place. DFT calculations also confirmed this assignment. Overall, the study convincingly illustrated the presence of an intermediate 4-oxidobenzyl radical species that is a result of a cleavage of the tyrosine (see Fig. 18).

Radical intermediate in the radical SAM reaction of HydG with tyrosine. (a) Proposed scheme of homolytic cleavage of tyrosine; (b) EPR spectra of the radical intermediate 4HOB• obtained using various isotopically-labeled Tyr substrates (indicated on the plot); (c) DFT-calculated spin distribution of the 4HOB•. Adapted with permission from Suess et al. [97]
LipA
Lipoyl synthase (LipA) catalyzes the final step in the de novo biosynthesis of the lipoyl cofactor—insertion of sulfur atoms at C6 and C8 of an n-octanoyl chain attached via an amide linkage to a target lysine residue of a lipoyl carrier protein (LCP). Recent studies have provided evidence that the auxiliary [4Fe–4S] cluster is the sacrificial donor of the requisite sulfur atoms [7]. Sequential addition of two equivalents of SAM resulted in the decay of the [4Fe–4S]aux first to a [3Fe–4S] cluster after the first equivalent and then to a [2Fe–2S] cluster and other mono-Fe species upon completion of the reaction. Two equivalents of the 5′-dA• are required to generate one equivalent of the lipoyl cofactor, and the two 5′-dA• have been shown to abstract H• sequentially from C6 and then C8 of octanoyl chain attached to LCPs or to short peptide substrate surrogates. These studies suggest that in the first half of the reaction, the octanoyl–LCP becomes cross-linked to LipA through the auxiliary [4Fe–4S] cluster.
To verify the proposed mechanism, Lanz et al. [98] used a two-carbon shorter substrate analogue, 2,4-hexadienoic acid, attached to the appropriate lysine residue of the EcH protein of the glycine cleavage system. The authors noted that this substrate does undergoes sulfur insertion by LipA , although at a much slower rate that the native substrate. Through the course of the reaction, a radical with a complex EPR lineshape was observed (Fig. 19). Using selective 2H-isotopic labeling, it was shown that the radical is located on the 2,4-hexadienoic acid. A total of six 1H HF coupling constants were extracted using a combination of EPR and pulse ENDOR, indicating a substantial delocalization of the radical, with the majority of spin population located on C3 and C5 positions.

X-band CW EPR spectra of the radical species of LipA obtained using unlabeled (b) and different 2H isotopically labeled 2,4-hexadienoic acid (c, d) as well as a pseudomodulated Q-band FID-detected EPR spectrum of the same species (a) (a). (b) Q-band Davies ENDOR spectrum of the radical species (B) and the scheme of spin density distribution (A). (c) Q-band 57Fe HYSCORE spectra of the radical species in LipA and corresponding simulation accounting for the presence of two 57Fe HF couplings. Adapted from Lanz et al. [98]
The Q-band HYSCORE spectrum of the radical obtained using 57Fe-labeled LipA showed presence of at least two 57Fe signals, illustrating the proximity of the auxiliary [4Fe–4S] cluster to the substrate. The extracted HF coupling constants contained both isotropic and anisotropic components. The authors reported an effective distance extracted from the anisotropy of about 2 Å. Although this distance clearly does not represent any physical distance between atoms, it does illustrate the close proximity of the radical to the [4Fe–4S] cluster. The presence of a small isotropic component in the 57Fe HF coupling was rationalized by a weak Heisenberg exchange interaction with individual iron ions.
Concluding Remarks
In this chapter we have presented several examples of applications of EPR spectroscopic techniques that have been used to investigate the functionality of radical SAM enzymes. This is by no means a comprehensive review of all the studies that were performed on these enzymes. In this chapter, we hoped to provide an overview of the methods that were applied so far to understand this incredibly versatile superfamily. EPR spectroscopy was used for two major functions in studying these enzymes: (1) obtain structural information about the relative arrangement of various components of the reactions and (2) resolve the identity of and obtain detailed information about the reactive intermediates.
The data accumulated to date has proven that CW EPR spectra of the [4Fe–4S]RS clusters are generally very sensitive to SAM binding, and one can utilize this effect as a sign of the interaction. However, we would like to note that our recent study of the B12-dependent RS enzyme TsrM showed that exceptions do exist, and it is apparently possible to significantly perturb the EPR spectrum of the [4Fe–4S]RS cluster without bidentate SAM binding. Therefore, use of either ENDOR or HYSCORE techniques to detect 14N HF interaction from the amino group is necessary to confirm the bidentate binding of SAM to the RS cluster. The data from both our studies and others suggest that the simplest way to clarify whether SAM binds is to perform a single HYSCORE experiment at the maximum absorption of the EPR signal of the [4Fe-4S]RS 1+ cluster and use the position of the double quantum ridges to estimate the 14N isotropic HF coupling constant. Rather interestingly, this parameter seems to vary from system to system, which is an effect that needs to be investigated systematically in conjunction with mechanistic studies.
CW EPR methods remain the most convenient technique to study organic radical intermediates, especially when combined with isotope labeling. However, as has been shown above, advanced EPR methods can significantly improve our understanding of the electronic and structural environment around the radical. Although pulse EPR methods are far more demanding in terms of technology and expertise, the details obtained are worth the effort. Moreover, the work of Hoffman and coworkers on LAM showed that a stable radical generated from a SAM analogue—anSAM—upon reductive cleavage can provide an important probe to observe the molecules in the vicinity of the radical, and thus provide a "t = 0" snapshot of the structural arrangement within the inner pocket. One can utilize such a method in combination with site-specific isotope labeling to decipher the site of attack for 5′-dA•, which in some cases can be as important as resolving reaction intermediates for understanding the reaction mechanism.
References
Pegg SC-H et al (2006) Leveraging enzyme structure−function relationships for functional inference and experimental design: the structure−function linkage database. Biochemistry 45:2545–2555
Akiva E et al (2014) The structure–function linkage database. Nucleic Acids Res 42:D521–D530
Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res 29:1097–1106
Cosper NJ, Booker SJ, Ruzicka F, Frey PA, Scott RA (2000) Direct FeS cluster involvement in generation of a radical in lysine 2,3-aminomutase. Biochemistry 39:15668–15673
Chen DW, Walsby C, Hoffman BM, Frey PA (2003) Coordination and mechanism of reversible cleavage of S-adenosylmethionine by the [4Fe-4S] center in lysine 2,3-aminomutase. J Am Chem Soc 125:11788–11789
Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH (2002) Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: A Mossbauer spectroscopic study. J Am Chem Soc 124:912–913
Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM (2002) An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc 124:11270–11271
Walsby CJ et al (2002) Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe-4S](+) cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc 124:3143–3151
Broderick JB, Duffus BR, Duschene KS, Shepard EM (2014) Radical S-adenosylmethionine enzymes. Chem Rev 114:4229–4317
Frey PA, Magnusson OT (2003) S-adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem Rev 103:2129–2148
Lanz ND, Booker SJ (2012) Identification and function of auxiliary iron–sulfur clusters in radical SAM enzymes. Biochim Biophys Acta 1824:1196–1212
Lanz ND, Booker SJ (2015) Auxiliary iron–sulfur cofactors in radical SAM enzymes. Biochim Biophys Acta 1853:1316–1334
Reed GH, Poyner RR (2015) Fourier deconvolution methods for resolution enhancement in continuous-wave EPR spectroscopy. Methods Enzymol 563:23–36
Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG (1981) Fourier self-deconvolution: a method for resolving intrinsically overlapped bands. Appl Spectrosc 35:271–276
Harmer J, Mitrikas G, Schweiger A (2009) In: Berliner L, Hanson G (eds) High resolution EPR, vol 28. Springer, New York, pp 13–61
Schweiger A, Jeschke G (2001) Principles of pulse electron paramagnetic resonance. Oxford University Press, Oxford
Petrovich R, Ruzicka F, Reed G, Frey P (1992) Characterization of iron-sulfur clusters in lysine 2,3-aminomutase by electron-paramagnetic resonance spectroscopy. Biochemistry 31:10774–10781
Ruszczycky MW, Choi S, Mansoorabadi SO, Liu H (2011) Mechanistic studies of the radical s-adenosyl-l-methionine enzyme desii: EPR characterization of a radical intermediate generated during its catalyzed dehydrogenation of TDP-D-quinovose. J Am Chem Soc 133:7292–7295
Padovani D, Thomas F, Trautwein AX, Mulliez E, Fontecave M (2001) Activation of class III ribonucleotide reductase from E-coli. The electron transfer from the iron-sulfur center to S-adenosylmethionine. Biochemistry 40:6713–6719
Krebs C, Henshaw TF, Cheek J, Huynh BH, Broderick JB (2000) Conversion of 3Fe-4S to 4Fe-4S clusters in native pyruvate formate-lyase activating enzyme: mossbauer characterization and implications for mechanism. J Am Chem Soc 122:12497–12506
Lieder KW et al (1998) S-adenosylmethionine-dependent reduction of lysine 2,3-aminomutase and observation of the catalytically functional iron-sulfur centers by electron paramagnetic resonance. Biochemistry 37:2578–2585
Hinckley GT, Frey PA (2006) Cofactor dependence of reduction potentials for [4Fe-4S](2+/1+) in lysine 2,3-aminomutase. Biochemistry 45:3219–3225
Hinckley GT, Ruzicka FJ, Thompson MJ, Blackburn GM, Frey PA (2003) Adenosyl coenzyme and pH dependence of the [4Fe-4S](2+/1+) transition in lysine 2,3-aminomutase. Arch Biochem Biophys 414:34–39
Hinckley GT, Frey PA (2006) An adaptable spectroelectrochemical titrator: The midpoint reduction potential of the iron–sulfur center in lysine 2,3-aminomutase. Anal Biochem 349:103–111
Broderick JB et al (1997) Pyruvate formate-lyase activating enzyme is an iron-sulfur protein. J Am Chem Soc 119:7396–7397
Maiocco SJ, Grove TL, Booker SJ, Elliott SJ (2015) Electrochemical resolution of the [4Fe-4S] centers of the AdoMet radical enzyme BtrN: evidence of proton coupling and an unusual, low-potential auxiliary cluster. J Am Chem Soc 137:8664–8667
Liu A, Graslund A (2000) Electron paramagnetic resonance evidence for a novel interconversion of [3Fe-4S](+) and [4Fe-4S](+) clusters with endogenous iron and sulfide in anaerobic ribonucleotide reductase activase in vitro. J Biol Chem 275:12367–12373
Silver SC et al (2010) Complete stereospecific repair of a synthetic dinucleotide spore photoproduct by spore photoproduct lyase. J Biol Inorg Chem 15:943–955
Rebeil R, Nicholson WL (2001) The subunit structure and catalytic mechanism of the Bacillus subtilis DNA repair enzyme spore photoproduct lyase. Proc Natl Acad Sci U S A 98:9038–9043
Yang L et al (2012) Mechanistic studies of the spore photoproduct lyase via a single cysteine mutation. Biochemistry 51:7173–7188
Kriek M et al (2007) Thiazole synthase from escherichia coli: an investigation of the substrates and purified proteins required for activity in vitro. J Biol Chem 282:17413–17423
Paraskevopoulou C, Fairhurst SA, Lowe DJ, Brick P, Onesti S (2006) The elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol Microbiol 59:795–806
McGlynn SE et al (2010) Identification and characterization of a novel member of the radical AdoMet enzyme superfamily and implications for the biosynthesis of the HMD hydrogenase active site cofactor. J Bacteriol 192:595–598
Brindley AA, Zajicek R, Warren MJ, Ferguson SJ, Rigby SEJ (2010) NirJ, a radical SAM family member of the d(1) heme biogenesis cluster. FEBS Lett 584:2461–2466
Zhang Q et al (2011) Characterization of NocL involved in thiopeptide nocathiacin i biosynthesis a [4Fe-4S] cluster and the catalysis of a radical s-adenosylmethionine enzyme. J Biol Chem 286:21287–21294
Yokoyama K, Ohmori D, Kudo F, Eguchi T (2008) Mechanistic study on the reaction of a radical SAM dehydrogenase BtrN by electron paramagnetic resonance spectroscopy. Biochemistry 47:8950–8960
Kuchenreuther JM et al (2013) A Radical intermediate in tyrosine scission to the CO and CN- ligands of FeFe hydrogenase. Science 342:472–475
Perche-Letuvee P et al (2012) 4-Demethylwyosine synthase from pyrococcus abyssi Is a radical-S-adenosyl-L-methionine enzyme with an additional [4Fe-4S](+2) cluster that interacts with the pyruvate co-substrate. J Biol Chem 287:41174–41185
McCarty RM, Krebs C, Bandarian V (2013) Spectroscopic, steady-state kinetic, and mechanistic characterization of the radical SAM enzyme QueE, which catalyzes a complex cyclization reaction in the biosynthesis of 7-deazapurines. Biochemistry 52:188–198
Layer G et al (2005) Radical S-adenosylmethionine enzyme coproporphyrinogen III oxidase HemN - functional features of the [4Fe-4S] cluster and the two bound S-adenosyl-L-methionines. J Biol Chem 280:29038–29046
Szu P-H, Ruszczycky MW, Choi S, Yan F, Liu H (2009) Characterization and mechanistic studies of desll: a radical S-Adenosyl-L-methionine enzyme involved in the biosynthesis of TDP-D-Desosamine. J Am Chem Soc 131:14030–14042
Kamat SS, Williams HJ, Dangott LJ, Chakrabarti M, Raushel FM (2013) The catalytic mechanism for aerobic formation of methane by bacteria. Nature 497:132–136
Blaszczyk AJ et al (2016) Spectroscopic and electrochemical characterization of the iron–sulfur and cobalamin cofactors of TsrM, an unusual radical S -adenosylmethionine methylase. J Am Chem Soc 138:3416–3426
Walsby CJ et al (2005) Spectroscopic approaches to elucidating novel iron-sulfur chemistry in the ‘Radical-SAM’ protein superfamily. Inorg Chem 44:727–741
Werst MM, Kennedy MC, Beinert H, Hoffman BM (1990) Oxygen-17, proton, and deuterium electron nuclear double resonance characterization of solvent, substrate, and inhibitor binding to the iron-sulfur [4Fe-4S]+ cluster of aconitase. Biochemistry 29:10526–10532
Gambarelli S, Luttringer F, Padovani D, Mulliez E, Fontecave M (2005) Activation of the anaerobic ribonucleotide reductase by S-adenosylmethionine. Chembiochem 6:1960–1962
Tyryshkin AM, Dikanov SA, Reijerse EJ, Burgard C, Hüttermann J (1999) Characterization of bimodal coordination structure in nitrosyl heme complexes through hyperfine couplings with pyrrole and protein nitrogens. J Am Chem Soc 121:3396–3406
Chandor A et al (2007) Characterization of the DNA repair spore photoproduct lyase enzyme from Clostridium acetobutylicum: a radical-SAM enzyme. C R Chim 10:756–765
Horitani M et al (2016) Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-C]-deoxyadenosyl bond. Science 352:822–825
Grove TL, Lee K-H, St. Clair J, Krebs C, Booker SJ (2008) In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry 47:7523–7538
Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ (2010) A consensus mechanism for radical SAM-dependent dehydrogenation? BtrN contains two [4Fe-4S] clusters. Biochemistry 49:3783–3785
Lees NS et al (2009) ENDOR spectroscopy shows that guanine N1 binds to [4Fe-4S] cluster II of the S-Adenosylmethionine-dependent enzyme MoaA: mechanistic implications. J Am Chem Soc 131:9184–9185
Forouhar F et al (2013) Two Fe-S clusters catalyze sulfur insertion by radical-SAM methylthiotransferases. Nat Chem Biol 9:333–338
Kuchenreuther JM et al (2013) Nuclear resonance vibrational spectroscopy and electron paramagnetic resonance spectroscopy of Fe-57-enriched [FeFe] hydrogenase indicate stepwise assembly of the H-cluster. Biochemistry 52:818–826
Kuchenreuther JM, Britt RD, Swartz JR (2012) New insights into [FeFe] hydrogenase activation and maturase function. PLoS One 7:e45850
Rubach JK, Brazzolotto X, Gaillard J, Fontecave M (2005) Biochemical characterization of the HydE and HydG iron-only hydrogenase maturation enzymes from Thermatoga maritima. FEBS Lett 579:5055–5060
Czech I, Silakov A, Lubitz W, Happe T (2010) The [FeFe]-hydrogenase maturase HydF from Clostridium acetobutylicum contains a CO and CN- ligated iron cofactor. FEBS Lett 584:638–642
Dinis P et al (2015) X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc Natl Acad Sci 112:1362–1367
Suess DLM et al (2015) Cysteine as a ligand platform in the biosynthesis of the FeFe hydrogenase H cluster. Proc Natl Acad Sci 112:11455–11460
Ugulava NB, Gibney BR, Jarrett JT (2001) Biotin synthase contains two distinct iron-sulfur cluster binding sites: chemical and spectroelectrochemical analysis of iron-sulfur cluster interconversions. Biochemistry 40:8343–8351
Ugulava NB, Surerus KK, Jarrett JT (2002) Evidence from Mosssbauer spectroscopy for distinct [2Fe-2S](2+) and [4Fe-4S](2+) cluster binding sites in biotin synthase from Escherichia coli. J Am Chem Soc 124:9050–9051
Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL (2004) Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 303:76–79
Bui BTS et al (1998) Biotin synthase mechanism: on the origin of sulphur. FEBS Lett 440:226–230
Bui BTS, Mattioli TA, Florentin D, Bolbach G, Marquet A (2006) Escherichia coli biotin synthase produces selenobiotin. Further evidence of the involvement of the [2Fe-2S](2+) cluster in the sulfur insertion step. Biochemistry 45:3824–3834
Taylor AM, Stoll S, Britt RD, Jarrett JT (2011) Reduction of the [2Fe-2S] cluster accompanies formation of the intermediate 9-mercaptodethiobiotin in escherichia coli biotin synthase. Biochemistry 50:7953–7963
Jameson GNL, Cosper MM, Hernandez HL, Johnson MK, Huynh BH (2004) Role of the [2Fe-2S] cluster in recombinant Escherichia coli biotin synthase. Biochemistry 43:2022–2031
Fugate CJ et al (2012) 9-Mercaptodethiobiotin is generated as a ligand to the [2Fe-2S](+) Cluster during the reaction catalyzed by biotin synthase from escherichia coli. J Am Chem Soc 134:9042–9045
Knappe J, Bohnert E, Brummer W (1965) S-adenosyl-L-methionine a component of clastic dissimilation of pyruvate. Biochim Biophys Acta 107:603–605
Knappe J, Schmitt T (1976) Novel reaction of S-adenosyl-L-methionine correlated with activation of pyruvate formate-lyase. Biochem Biophys Res Commun 71:1110–1117
Knappe J, Neugebauer F, Blaschkowski H, Ganzler M (1984) Post-translational activation introduces a free-radical into pyruvate formate-lyase. Proc Natl Acad Sci U S A 81:1332–1335
Unkrig V, Neugebauer FA, Knappe J (1989) The free radical of pyruvate formate-lyase. Characterization by EPR spectroscopy and involvement in catalysis as studied with the substrate-analogue hypopthosphite. Eur J Biochem 184:723–728
Wagner A, Frey M, Neugebauer F, Schafer W, Knappe J (1992) The free-radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci U S A 89:996–1000
Chirpich T, Zappia V, Costilow R, Barker H (1970) Lysine 2,3-aminomutase - purification and properties of a pyridoxal phosphate and s-adenosylmethionine-activated enzyme. J Biol Chem 245:1778–1789
Magnusson OT, Reed GH, Frey PA (1999) Spectroscopic evidence for the participation of an allylic analogue of the 5 ’-deoxyadenosyl radical in the reaction of lysine 2,3-aminomutase. J Am Chem Soc 121:9764–9765
Magnusson OT, Reed GH, Frey PA (2001) Characterization of an allylic analogue of the 5 ’-deoxyadenosyl radical: an intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 40:7773–7782
Horitani M et al (2015) Why nature uses radical SAM enzymes so widely: electron nuclear double resonance studies of lysine 2,3-aminomutase show the 5′-dAdo• ‘free radical’ is never free. J Am Chem Soc 137:7111–7121
Miller J, Bandarian V, Reed GH, Frey PA (2001) Inhibition of lysine 2,3-aminomutase by the alternative substrate 4-thialysine and characterization of the 4-thialysyl radical intermediate. Arch Biochem Biophys 387:281–288
Wu WM et al (2000) Lysine 2,3-aminomutase and trans-4,5-dehydrolysine: Characterization of an allylic analogue of a substrate-based radical in the catalytic mechanism? Biochemistry 39:9561–9570
Reed GH, Ballinger MD (1995) [24] Characterization of a radical intermediate in the lysine 2,3-aminomutase reaction. Methods Enzymol 258:362–379
Lees NS et al (2006) How an enzyme tames reactive intermediates: positioning of the active-site components of lysine 2,3-aminomutase during enzymatic turnover as determined by ENDOR spectroscopy. J Am Chem Soc 128:10145–10154
Ballinger M, Reed G, Frey P (1992) An organic radical in the lysine 2,3-aminomutase reaction. Biochemistry 31:949–953
Chang CH, Ballinger MD, Reed GH, Frey PA (1996) Lysine 2,3-aminomutase: rapid mix-freeze-quench electron paramagnetic resonance studies establishing the kinetic competence of a substrate-based radical intermediate. Biochemistry 35:11081–11084
Ballinger M, Frey P, Reed G (1992) Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 31:10782–10789
Ballinger M, Frey P, Reed G, Lobrutto R (1995) Pulsed electron-paramagnetic-resonance studies of the lysine 2,3-aminomutase substrate radical - evidence for participation of pyridoxal 5’-phosphate in a radical rearrangement. Biochemistry 34:10086–10093
Layer G et al (2006) The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase HemN. J Biol Chem 281:15727–15734
Yan F et al (2010) RImN and Cfr are Radical SAM Enzymes Involved in Methylation of Ribosomal RNA. J Am Chem Soc 132:3953–3964
Grove TL et al (2011) A radically different mechanism for s-adenosylmethionine-dependent methyltransferases. Science 332:604–607
Grove TL et al (2013) A substrate radical intermediate in catalysis by the antibiotic resistance protein Cfr. Nat Chem Biol 9:422–427
Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC (1998) X-ray crystal structure of the Fe-only hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 angstrom resolution. Science 282:1853–1858
Lemon BJ, Peters JW (1999) Binding of exogenously added carbon monoxide at the active site of the iron-only hydrogenase (CpI) from Clostridium pasteurianum. Biochemistry 38:12969–12973
Nicolet Y et al (2001) Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans. J Am Chem Soc 123:1596–1601
Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC (1999) Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Struct Fold Des 7:13–23
Silakov A, Wenk B, Reijerse E, Lubitz W (2009) N-14 HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge. Phys Chem Chem Phys 11:6592–6599
Adamska-Venkatesh A et al (2015) Spectroscopic characterization of the bridging amine in the active site of [FeFe] hydrogenase using isotopologues of the H-cluster. J Am Chem Soc 137:12744–12747
Kuchenreuther JM, Stapleton JA, Swartz JR (2009) Tyrosine, cysteine, and S-adenosyl methionine stimulate in vitro [FeFe] hydrogenase activation. PLoS One 4:e7565
Kuchenreuther JM, George SJ, Grady-Smith CS, Cramer SP, Swartz JR (2011) Cell-free H-cluster synthesis and [FeFe] hydrogenase activation: All Five CO and CN- ligands derive from tyrosine. PLoS One 6:e20346
Suess DLM, Kuchenreuther JM, De La Paz L, Swartz JR, Britt RD (2016) Biosynthesis of the [FeFe] hydrogenase H Cluster: a central role for the radical SAM enzyme HydG. Inorg Chem 55:478–487
Lanz ND et al (2015) Characterization of a radical intermediate in lipoyl cofactor biosynthesis. J Am Chem Soc 137:13216–13219
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Silakov, A., Lanz, N.D., Booker, S.J. (2017). Characterization of Radical S-adenosylmethionine Enzymes and Intermediates in their Reactions by Continuous Wave and Pulse Electron Paramagnetic Resonance Spectroscopies. In: Hanson, G., Berliner, L. (eds) Future Directions in Metalloprotein and Metalloenzyme Research. Biological Magnetic Resonance, vol 33. Springer, Cham. https://doi.org/10.1007/978-3-319-59100-1_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-59100-1_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-59098-1
Online ISBN: 978-3-319-59100-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)