
Abstract
The multifunctional protein Chromogranin A (CGA) is a major marker of the sympatho-adrenal neuroendocrine (SAN) activity. Stored in neuroendocrine chromaffin secretory granules with several prohormones and their proteolytic enzymes, with noradrenaline and adrenaline, it is released with catecholamines upon stimulation. It is also present in other cell types, including myocardiocytes of various vertebrates, and humans, particularly in the presence of cardiomyopathy and heart failure. Due to the processing into a number of biologically active peptides, it represents a prohormone with an important modulatory role on endocrine, cardiovascular, metabolic, and immune systems. Circulating CGA increases in the presence of stress-induced excessive SAN activation and of pathologies such as neuroendocrine tumors, and cardiovascular diseases including hypertension, coronary syndrome, and heart failure. Thus, the protein is considered a promising biomarker for a number of severe diseases. Recently, it was found that in the heart of normotensive and hypertensive rats (SHRs), CGA is processed under hemodynamic and excitatory stimuli, and the exogenous full length protein directly affects myocardial and coronary performance by Akt/NOS/NO/cGMP/PKG pathway. We here illustrate the emerging role elicited by CGA in the control of circulatory homeostasis with particular focus on its cardiovascular action under physiological and physio-pathological conditions. These actions contribute to extend our knowledge on the sympatho-chromaffin control of the cardiovascular system and its integrated “whip-brake” circuits.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Chromogranin A
- Vasostatin 1
- Catestatin
- Serpinin
- Adreno-sympathetic control
- Cardioprotection
- Vasoactive peptides
- Endothelial signaling
1 Introduction
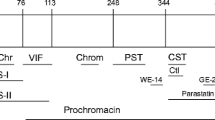
The spectrum of biological functions attributed to CGA includes the full-length 49-kDa protein implication as a major marker of the sympatho-adrenal neuroendocrine activity (SAN) and, at the same time, its prohormone ability to generate several biologically active peptides through partial processing (Fig. 1). In view of this capability of acting both as sensor of the organism stress- induced perturbations, and homeostatic counter-regulatory effector, CGA appears to posses intriguing cytokine and endocrine properties. Here we aim to illustrate these two facets of full- length CGA with particular reference to the cardio-vascular system under normal and physio-pathological conditions. Since CGA generates at least three peptides with relevant cardiotropic sympatho-adrenergic influence, i.e. Vasostatin 1 (VS-1), Catestatin (CST) and Serpinin (SERP), we will very briefly refer also to their cardio-vascular effects to provide an integrated information on how the CGA system, i.e. the full-length protein and its fragments, may monitor and influence circulatory homeostasis, especially under SAN overactivation. Detailed knowledge on these peptides is reported in other chapters of this Volume.

Human Chromogranin-A (CGA) sequence with post-translational modifications and the derived biologically active peptides. In Italic, the synthetically generated fragment Cateslytin
A physiological hallmark of the CGA system is represented by its involvement in the stress response in which it appears to closely interact with SAN activation. Such interaction is topologically reflected by its subcellular localization. In fact, together with other granins, CGA is stored in the secretory (chromaffin) granules of the diffuse neuroendocrine system and is released with catecholamines (CAs). Within the granules, CGA is also co-stored with neuropeptide Y, cardiac natriuretic peptide hormones, several prohormones and their proteolytic enzymes. The evidence that CGA is also present in the secretory granules of the heart (Tota et al. 2010 and references therein), including the human myocardium (Pieroni et al. 2007), is consistent with an emerging modulatory role of the prohormone and its derived fragments at the cardiac level.
To highlight for the non-expert reader the cardio-vascular interactions between CGA and the sympatho-chromaffin system, we will briefly summarize SAN involvement to maintain circulatory homeostasis under normal and physio-pathological conditions.
2 Physio-Pathological Aspects of SAN Overactivation
The cardiovascular system is intimately linked to the brain through two pathways, the hypothalamus-pituitary-adrenal (HPA) axis and the autonomic nervous system consisting of two limbs, i.e., the sympathetic and parasympathetic pathways. SAN and its end-products, the CAs, play a central role in the stress response (“fight or flight” reaction), characterized by Selye (1936) as the “general adaptation syndrome” (Samuels 2007 and references therein). The peripheral limbs of the stress system, the SAN and the HPA axis, maintain stress-related homeostasis through increased peripheral levels of CAs and glucocorticoids which act synergistically. However, their consequent and prolonged overstimulation can lead to visceral organ dysfunction, experimentally exemplified in the heart by the electrolyte-steroid-cardiopathy with necrosis (Selye and Bajusz 1958; Raab 1969). Therefore, through the SAN-induced overactivation of positive reverberatory networks, e.g., the Renin-Angiotensin System (RAS), in which the activation of one pathway tends to activate another excitatory one, the stress response itself could threaten the homeostasis of target organs and tissues.
The heart and the vasculature work as an integrative interface between the noradrenergic nerve terminals, mainly releasing norepinephrine (NE), and the circulating CAs secreted by the adrenal medulla. In addition, similarly to other organs, in the heart, CAs are co- stored and co-released with other neuropeptides and humoral autacoids, in the afferent, efferent, interconnecting short neurons and intracardiac ganglia, as well as in the chromaffin cells and in the cardiomyocytes themselves. The convergence of these SAN excitatory stimuli may contribute to explain why the heart and the vasculature represent a typical paradigm of a stress-threatened organ.
CAs plasma levels induce myocardial excitability, contractility and relaxation (Fig. 2) and its increased concentrations can induce necrotic damage in the heart (Samuels 2007 and references therein). Moreover, the initial heart response to prolonged and excessive stress is represented by cardiac hypertrophy, i.e., a morphological enlargement which tends to compensate or prevent progressive deterioration of cardiac function challenged by the hemodynamic overload (Tota et al. 2008 and references therein). Under extreme or prolonged stress conditions, positive feed-back loops can lead the hypertrophied heart to failing processes. A concomitant sustained heightened activation of SNS and RAS tend to control cardiac output and systemic blood pressure.

Representative diagram showing the effects of adrenergic and anti-adrenergic drugs on ventricular contraction and relaxation (expressed as LVP variations)
The spectrum of SAN-elicited cardio-circulatory effects ranges from hyperlipidemia, platelet adhesiveness, blood coagulation, vascular smooth muscle hyperplasia, vasomotor tone, denervated myocardium etc. to immunologic responses induced by cardiovascular changes. Accordingly, SAN overstimulation may involve the actions of numerous different targets, directly impinging the heart and the vasculature (Samuels 2007). Compelling clinical evidence shows that the initial heart response to prolonged SAN over-activity leads to compensatory remodeling, cardiac hypertrophy and, if the stress will overwhelm the system, heart failure (HF) (Triposkiadis et al. 2009 and references therein). Uncontrolled heightened SAN activation may lead to changes more deleterious than those resulting from the actual stress placed on the heart (Samuels 2007). A negative prognostic value in the evolution of human HF has been associated with chronic SAN activation, mainly via CAs, enhancing the pathological processes (Cohn and Yellin 1984). This knowledge has provided the rationale for the anti-adrenergic drug therapy, e.g., beta-adrenergic-blockers.
The emerging cardiovascular importance of the CGA system may widen our knowledge regarding the SAN-orchestrated regulation of the cardio-circulatory system, stimulating, at the same time, potential additional or alternative therapeutic drugs designed for protecting stress-targeted organs.
3 Cardio-Circulatory Implications of Full-Length CGA
It is methodologically relevant to be aware that estimation of plasma levels of CGA and its derived peptides can be obtained by serological determinations that, in addition to processing-independent radioimmunoassay, include region-specific processing-dependent analysis. Noteworthy, only the latter is able to analyse the plasma levels of the various CGA-derived fragments, showing sometime opposite biological effects, functions and prognostic significance (Crippa et al. 2013; Goetze et al. 2014). The CGA in vivo long half-life (~18 min) and its relatively elevated circulating concentrations (including normal conditions), reduce eventual false measurements and facilitate blood collection, pre-analytic handling and final determinations (O’Connor et al. 1989). However, caution is required in CGA detection and quantification, since a variability exists in the methodologies used for determinations and, in many cases, different results may be obtained on the same sample if different methods are used (i.e. RIA vs ELISA). As commented by Angelone et al. (2012), a definitive standardization of the methods for CGA determination is essential to have comparable, uniform, and thus clinically relevant measurements in blood and tissues.
Normal concentrations of circulating CGA range between 0.5 and 2 nM (Helle et al. 2007; Crippa et al. 2013). They increase under stress-induced SAN over-stimulation and physio-pathological conditions, e.g., chronic inflammation, neuroendocrine tumors, acute coronary syndromes and chronic HF. Based on this clinical evidence, CGA plasma levels have been employed as prognostic indicators in a number of these diseases (Helle et al. 2007; Angelone et al. 2012; D’Amico et al. 2014).
Plasma CGA concentrations increase up to 10–20 nM (500–1000 ng/ml) in patients with essential hypertension (Takiyyuddin et al. 1995), chronic HF (Ceconi et al. 2002), myocardial infarction (Omland et al. 2003), acute coronary syndromes (Jansson et al. 2009), acute destabilized HF (Dieplinger et al. 2009), and decompensated hypertrophic cardimyopathy (Pieroni et al. 2007). Ceconi et al. (2002) were the first to document that plasma CGA levels significantly parallel the severity of the dysfunction and represent an independent predictor for mortality. This clinical evidence highlights the role of CGA as a potentially new diagnostic and prognostic cardiovascular biomarker independent from conventional markers. Furthermore, various evidences strongly indicate the correlation between CGA and SAN activity. For example, studies by O’Connor and his group have shown in twins that basal plasma CGA level is heritable (Takiyyuddin et al. 1995). In addition, compared with age-matched normotensive counterparts, patients with essential hypertension exhibit augmented plasma CGA and enhanced release of stored CGA in response to adrenal medullary stimulation by insulin-elicited hypoglycemia (Takiyyuddin et al. 1995).
It is of cardiac relevance the observation that targeted ablation of the CGA gene makes CGA-KO mice hypertensive and hyper-adrenergic, with accompanied heart enlargement, increases reactive oxygen species production and consequent nitric oxide (NO) depletion (Gayen et al. 2010). A detailed information on this issue has been provided by Mahata in the present Volume.
Numerous studies, mainly by Corti and his group, revealed that the CGA system works at the interface of endothelial, angiopoietic and blood coagulation processes. It is implicated in the modulation of the endothelial barrier (Ferrero et al. 2002) and tumor-induced vascular remodelling (Veschini et al. 2011). Both CGA and its N-terminal fragment VS-1 are potent inhibitors of the thrombin-induced endothelial permeability and the pro-angiogenic Vascular Endothelial Growth Factor (VEGF) (Ferrero et al. 2002), also inhibiting the Tumor Necrosis Factor (TNF)-induced changes on endothelial cells, i.e. gap formation, disassembly of vascular endothelial-cadherin adherence junctions and vascular leakage (Ferrero et al. 2002; Dondossola et al. 2011). CGA can also affect host/tumor interactions (Colombo et al. 2002). For example, systemic administration of CGA (1 μg) to lymphoma-bearing mice potently reduces the TNF- induced penetration of the patent blue dye in tumor tissues (Dondossola et al. 2011).
In healthy subjects Crippa et al. (2013) detected the presence of biologically relevant plasma levels of full-length CGA, CGA 1-76 (antiangiogenic) and fragments lacking the C-terminal region (proangiogenic). Importantly, they demonstrated that blood coagulation activates a thrombin- dependent almost complete conversion of plasma CGA into fragments lacking the C-terminal region. Thus, the possibility exists that CGA functions as a homeostatic stabilizer of angiogenesis. Under conditions of perturbed angiogenesis (wound healing, cancer, etc.), it can contribute to circulatory and vascular protection via the opposite angiogenic actions of its fragments (possibly VS-1 and Catestatin: CST) produced by tightly spatio-temporally regulated proteolysis.
4 Cardiac CGA: Localization and Processing
Before illustrating the physio-pharmacological aspects of CGA cardio-circulatory activity, we will consider here the intracardiac localization of this granin and its significance.
Imunohistochemical evidence has shown in the myoendocrine granules of the rat heart the co-localization of CGA and Atrial Natriuretic Peptide (ANP) (Steiner et al. 1990). The immunoblotting data were consistent with a more extensive myocardial CGA processing compared to that of the adrenal medulla (Steiner et al. 1990). Possibly, additional source of cardiac CGA and/or CGA- derived peptides may result from the terminal innervations of the heart (Miserez et al. 1992). CGA has been also detected in rat Purkinje conduction fibers, in both rat atrium and ventricle, as well as in H9c2 rat cardiomyocytes (Weiergraber et al. 2000). CGA, Chromogranin B (CGB) and Secretogranin (SG) were found in the secretory granules of the mouse myocardium (Biswas et al. 2010). Of physio-pathological importance, Pieroni et al. (2007) provided histochemical evidence of CGA-positive intracellular staining in the human myocardium. Moreover, using confocal microscopy they showed that in ventricular cardiomyocytes of dilated and hypertrophic human hearts CGA is colocalized with Brain Natriuretic Peptide (BNP). This finding was confirmed by RT- PCR that documented the myocardial presence of CGA-mRNA; based on ELISA assays with four different monoclonal antibodies, more than 0.5 μg of CGA per gram of left ventricular myocardial tissue was measured. Therefore, it is conceivable that the myocardium under stimulated conditions may constantly release cardiac Natriuretic Peptide Hormones (NPs) together with its co-stored CGA, whose plasma half-life is 18.4 min. These observations emphasize the hypothesis of Corti et al. (1996) and Pieroni et al. (2007) who postulated a significant cardiac contribution to the increased circulating CGA levels reported in their patients. The possible cross-talk between CGA and the NPs system could be implicated in the cardiovascular control counteracting prolonged and reverberating excitatory stimuli, for example, exerting tonic vasodilation, hypotension and cardioprotection against SAN hyperactivation. Dieplinger et al. (2009, and references therein) considered the strong association between plasma CGA/NPs levels and the degree of hemodynamic dysfunction in HF and proposed both hormones as reliable prognostic indicators of the severity of HF. It has been suggested that the significant correlation between BNP levels and left ventricle end diastolic pressure can be indicative of an undefined stretch-elicited release and transcriptional up-regulation mechanisms; this could also be the case for myocardial CGA (Tota et al. 2010 and references therein).
Glattard et al. (2006) provided in the rat heart biochemical characterization of intracardiac CGA and its processing. They submit the RPHPLC purified CGA-immunoreactive fractions from cardiac extracts to Western Blot and MS analysis (TOF/TOF technique) and identified four endogenous N terminal CGA-derived peptides, i.e. CGA4–113, CGA1–124, CGA1–135 and CGA1–199, containing the Vasostatin sequence. Importantly, among these and other C-terminal truncated fragments intact CGA was identified, highlighting the cell-specific proteolytic pattern of CGA, in contrast to the rat adrenal gland in which almost no intact CGA is detected. This and other comparative considerations suggest that in the heart the maturation process can be incomplete and specific. Noteworthy, Pasqua et al. (2013), showed in the rat heart that the cardioactive motif (the VS-1 sequence or a portion of it) is present among the identified low-molecular-mass fragments. These evidences support our view that, under normal or stressful conditions, the heart is able respond to a specific physical (e.g. stretch) or chemical (e.g. CAs, Endothelin-1: ET-1, Angiotensin) stimulus, triggering proteolytic CGA processing with subsequent increase in lower-molecular-mass cardioactive peptides (Pasqua et al. 2013, and references therein). Accordingly, CGA and its derived cardioactive peptides may work as a fine-tuned system, which, at both local and systemic levels, can exert endocrine, autocrine/paracrine cardiovascular modulations.
5 Physio-Pharmacological Profile of CGA Cardiovascular Activity
Evidence regarding the direct myocardial and coronary actions of CGA and its intracardiac stimulus-induced processing has only recently emerged. Pasqua et al. (2013) evaluated for the first time the influence exerted by the full-length human recombinant CGA on the cardiac performance of isolated and Langendorff perfused hearts of normotensive and spontaneously hypertensive rats (SHR), analyzing at the same time its proteolytic processing. The SHR heart is a well-known experimental model which mimics the hypertensive patho-physiological changes of the human heart (Doggrell and Brown 1998), representing an alternative and/or additional tool to the CGA/KO mice mentioned above. It was demonstrated that CGA at concentrations lower, or close to its physiological circulatory levels (1 nM), induces a mild but significant depression of mechanical performance and vasodilates the coronary arteries. These actions are mediated by an endothelium-dependent mechanism that involves PI3K-NO signalling. Moreover, intracardiac CGA appears subjected to proteolytic processing which generates smaller peptides including the cardioactive and vasoactive VS-1, the fragmentation being enhanced by chemical (Isoproterenol: ISO, or ET-1) stimulation. Here below we will detail these findings to highlight their putative physiological and physiopathological implications.
5.1 Myocardial and Coronary Actions
According to the analysis of the cardiac perfusates, exogenous CGA is not cleaved by the heart. Consequently, the myocardial and coronary actions induced by the granin can be attributed to the full-length protein, excluding the involvement of derived fragment, including VS-1.
Exogenous CGA starting from 1 to 4 nM concentrations directly depresses myocardial contractility (negative inotropism) and relaxation (negative lusitropy). The protein (from 100 pM to 4 nM) reduces major inotropic parameters, i.e., LVP and +(LVdP/dt)max, decreasing, at the same time, lusitropy, i.e., −(LVdP/dt)max and T/−t (at 100 pM, 1 and 4 nM). CGA is also able to elicit coronary vasodilation at 1 and 4 nM concentrations without influencing heart rate (HR). Both the inotropic and lusitropic effects disappear at higher concentrations (10–16 nM) (Fig. 3), pointing to a bell-shaped (or U-shaped) concentration/response curve. The underlying mechanism is unknown despite the phenomenon has been previously observed in several experiments with CGA and is common to a number of biological responses, e.g., endostatin-induced anti-tumor activity (Celik et al. 2005), interferon-alpha-mediated inhibition of angiogenesis (Slaton et al. 1999), including the biphasic influence of CGA on blood pressure levels and CAs secretion in mice. It is possible that the bell-shaped curve reflects a counter-regulatory mechanism activated at high concentrations. Another explanation may be related to changes in the quaternary structure of CGA that, at physiological pH, which can exist as a monomer or a dimer (Yoo and Lewis 1996).

Dose-dependent response curves of exogenous CGA (1 pM-16 nM) on +(LVdP/dT)max, −(LVdP/dT)max, T/−T, and CP, obtained on isolated and Langendorff perfused young normotensive rat heart preparations (Modified from Pasqua et al. 2013)
CGA-induced negative inotropism and lusitropism are more potent in SHR rats than in the normotensive young counterparts (Fig. 3). Likely, the higher responsiveness of the former to CGA may result from the enhanced sensitivity exhibited by the SHR heart toward inhibitory hormones such as Angiotensin II and NPs (Anand-Srivastava 1992). The suggested mechanisms include the activation of several inhibitory pathways, e.g., the overexpression of a Gi regulatory protein involved in the depressed vascular tone and impaired myocardial performance occurring in the hypertensive state (Anand-Srivastava 1992). In this context, it is relevant that exogenous CGA causes different coronary effects in young normal rats and SHR: while CGA (1 and 4 nM) elicits vasodilation in the former, the protein appears non-effective in SHR. Of note, CGA/KO mice are hypertensive (Mahapatra et al. 2005), and circulatory CGA protein levels are increased in human hypertension (Takiyyuddin et al. 1995). Therefore, the depressing influence induced by the full length granin might be interpreted as a compensatory response for increased blood pressure.
5.2 Obligatory Endothelium-NO Involvement
As shown by Triton-induced endothelial impairment, the cardiotropic modulatory actions of exogenous full-length CGA require the obligatory role of the endothelium. The fact that the endothelial involvement is also crucial for attaining the similar VS-1- and CST-dependent cardiotropic effects argues for a common signal transduction mechanism of CGA and its two cardio-inhibitory fragments. Results obtained with both whole organ and isolated cell (cultured endothelium, HUVEC) suggest that the intact granin, while perfusing the intracardiac circulatory bed, firstly encounters the endothelial barrier where a sill unidentified binding site is located, thereby triggering a downstream PI3K/Akt-dependent NO signaling that in turn modulates the responses of the myocardiocyte and coronary smooth muscle. Indeed, such endothelium- mediated mechanism appears compatible with the large dimensions of the protein that may prevent it to reach the cell targets subjacent to the endothelium. The involvement of the cardiac endothelium emphasizes the relevance of the interaction between this tissue and CGA mentioned previously and elsewhere in this Volume.
The vascular endothelium is a relevant source of eNOS-produced NO (Balligand et al. 2009). As shown by Pasqua et al. (2013) in ex vivo experiments, the specific chemical inhibition of Akt (an upstream activator of the NO pathway) and the eNOS isoform abolishes CGA cardioactivity. In agreement with this, in the perfused hearts of both normotensive and SHR, as well as in HUVEC, CGA exposure provokes eNOS phosphorylation and its induced actions require an NO-dependent obligatory mechanism. It is well recognized that NO modulates both the beat-to-beat and the long-term contractile performance of the heart (Balligand et al. 2009). Cardiac NOS-produced NO induces fine-tuned tonic depression of myocardial contractility through sGC-PKG signaling, thereby reducing L-type Ca2+ current and phosphorylating troponin I (Abi-Gerges et al. 2001). Furthermore, NO-cGMP-PKG activation can also modulate relaxation by inhibiting phospholamban (PLB) phosphorylation, hence attenuating sarcoplasmic reticulum Ca2+ reuptake (Stojanovic et al. 2001). In agreement with this knowledge, CGA depresses relaxation in both normotensive and SHR rats. Notably, in the latter, an impaired endothelium-dependent vasodilation has been related to structural changes at the level of myocardial arteries and/or a reduction in both capillary density and eNOS expression (Stojanovic et al. 2001). Intriguingly, Pasqua et al. (2013) have shown different coronary response to full-length CGA between young normotensive and SHR. In the absence of direct experimental explanation, we suggest that the lack of responsiveness observed in the hypertensive rats could result from reduced NO availability/capability in regulating SHR basal coronary flow due to a decreased shear stress-stimulated NO (Crabos et al. 1997; Kojda et al. 1998).
5.3 Intracardiac CGA Processing
Pasqua et al. (2013) confirmed in the rat heart the presence of CGA and demonstrated its processing by detecting both in normal and SHR cardiac extracts smaller peptides including the cardioactive and vasoactive VS-1. It is physiologically relevant that the processing is enhanced by chemical (ISO or ET-1) stimulation.
As evidenced by Western Blotting analyses, the major immunoreactive bands of 80–50 kDa detected include the full-length CGA and the truncated fragments lacking the C-terminal region. These data confirm the granin identification and fragmentation reported by Glattard et al. (2006) in the rat heart, as well as the presence of CGA in the human ventricle (Pieroni et al. 2007).
In both normal and diseased SHR hearts, CGA processing appears responsive to physical (perfusion) and chemical (ISO and ET-1) stimuli able to provoke proteolytic fragmentation of cardiac CGA into shorter peptides. Adrenergic (ISO 100 nM) or ET-1 (110 nM) exposure enhances the processing and, compared with the untreated counterparts, the full-length/large fragments appear reduced, while short N-terminal fragments of a size corresponding to that of VS-1 are increased.
The chromaffin granules of the bovine adrenal medulla have provided information regarding the CGA maturation with its starting points at both the N terminus and the C terminus (Metz-Boutigue et al. 1993). The major proteolytic cleavage sites were identified, including the highly conserved 64–65 bond present in the N-terminal moiety of CGA and included in the VS sequence (Metz-Boutigue et al. 1993; Cerra et al. 2008). According to this information, it might be expected that pro-hormone convertase 1/3 (PC1/3), PC2, and carboxypeptidase H/E are implicated in CGA processing in the rat heart. The latter, similarly to the bovine adrenal medulla paradigm, can represent an intriguing experimental model for exploring the major enzymatic events underlining CGA proteolytic processing and how these are regulated to fulfill the requirements of the stimulus (SAN)-CGA proteolysis coupling eventually accomplished by the normal or stressed organ.
6 Conclusions and Perspectives
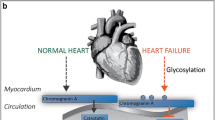
On the basis of the evidences here examined, it is conceivable that the full-length CGA and its derived cardioactive fragments work as a multilevel integrated system able to sense and, at the same time, counter-regulate overall circulatory homeostasis as well as local organ function, i.e. the beating heart. Therefore, the sensor and effector attitudes of the full-length CGA may indeed represent two faces of the same coin. In particular, the findings that both the full-length CGA and its derived peptides VS-1, CST and SERP exert direct myocardial and coronary effects provide a conceptual link between the granin-induced systemic and intracardiac modulatory influences. They appear to implicate paracrine/autocrine mechanisms, hence expanding the classical concept of the heart as an endocrine organ, especially in relation to elevated SAN outflow and cardiovascular stress (see also Tota et al. 2014). This scenario will be detailed by other chapters of this Volume and is schematically anticipated in Fig. 4.

Scheme of the possible sites of interventions for the CGA system. The protein and its derived fragments may interact at the systemic level with factors such as CAs, ANGII, NPs, etc., thus participating to the stress response, as in the case of the neuroendocrine scenario activated in HF. At the local (heart) level, the direct modulation of the myocardial and the coronary performance may contribute to cardiac homeostasis under basal conditions and in the presence of stress challenges
References
Abi-Gerges N, Fischmeister R, Mery PF (2001) G protein-mediated inhibitory effect of nitric oxide on L-type Ca2current in rat ventricular myocytes. J Physiol 531(Pt 1):117–130
Anand-Srivastava MB (1992) Enhanced expression of inhibitory guanine nucleotide regulatory protein in spontaneously hypertensive rats. Relationship to adenylate cyclase inhibition. Biochem J 288(Pt 1):79–85
Angelone T, Mazza R, Cerra MC (2012) Chromogranin-A: a multifaceted cardiovascular role in health and disease. Curr Med Chem 19(24):4042–4050
Balligand JL, Feron O, Dessy C (2009) eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 89(2):481–534
Biswas N, Curello E, O’Connor DT, Mahata SK (2010) Chromogranin/secretogranin proteins in murine heart: myocardial production of Chromogranin A fragment catestatin (Chga(364-384)). Cell Tissue Res 3:353–361
Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, Parrinello G, Corti A (2002) Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur Heart J 12:967–974
Celik I, Surucu O, Dietz C, Heymach JV, Force J, Höschele I, Becker CM, Folkman J, Kisker O (2005) Therapeutic efficacy of the endostatin exhibits a biphasic dose-response curve. Cancer Res 65(23):11044–11050
Cerra MC, Gallo MP, Angelonte T et al (2008) The homologous rat chromogranin A1–64 (rCGA1–64) modulates myocardial and coronary function in rat heart to counteract adrenergic stimulation indirectly via endothelium-derived nitric oxide. FASEB J 22(11):3992–4004
Cohn JN, Yellin AM (1984) Learned precise cardiovascular control through graded central sympathetic stimulation. J Hypertens Suppl 2:S77–S79
Colombo B, Curnis F, Foglieni C, Monno A, Arrigoni G, Corti A (2002) Chromogranin A expression in neoplastic cells affects tumor growth and morphogenesis in mouse models. Cancer Res 3:941–946
Corti A, Gasparri A, Chen FX, Pelagi M, Brandazza A, Sidoli A, Siccardi AG (1996) Characterisation of circulating Chromogranin A in human cancer patients. Br J Cancer 8:924–932
Crabos M, Coste P, Paccalin M et al (1997) Reduced basal NO-mediated dilation and decreased endothelial NO-synthase expression in coronary vessels of spontaneously hypertensive rats. J Mol Cell Cardiol 29(1):55–65
Crippa L, Bianco M, Colombo B, Gasparri AM, Ferrero E, Loh YP, Curnis F, Corti A (2013) A new Chromogranin A- dependent angiogenic switch activated by thrombin. Blood 2:392–402
D’amico MA, Ghinassi B, Izzicupo P, Manzoli L, Baldassarre A (2014) Biological function and clinical relevance of Chromogranin A and derived peptides. Endocr Connect 2:45–54
Dieplinger B, Gegenhuber A, Haltmayer M, Mueller T (2009) Evaluation of novel biomarkers for the diagnosis of acute destabilized heart failure in patients with shortness of breath. Heart 18:1508–1513
Doggrell SA, Brown L (1998) Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc Res 39(1):89–105
Dondossola E, Gasparri AM, Colombo B, Sacchi A, Curnis F, Corti A (2011) Chromogranin A restricts drug penetration and limits the ability of NGR-TNF to enhance chemotherapeutic efficacy. Cancer Res 17:5881–5890
Ferrero E, Magni E, Curnis F, Villa A, Ferrero ME, Corti A (2002) Regulation of endothelial cell shape and barrier function by Chromogranin A. Ann N Y Acad Sci 971:355–358
Gayen JR, Zhang K, Ramachandra Rao SP, Mahata M, Chen Y, Kim HS, Naviaux RK, Sharma K, Mahata SK, O’Connor DT (2010) Role of reactive oxygen species in hyperadrenergic hypertension: biochemical, physiological, and pharmacological evidence from targeted ablation of the chromogranin A (Chga) gene. Circ Cardiovasc Genet 3(5):414–425
Glattard E, Angelone T, Strub JM, Corti A, Aunis D, Tota B, Metz-Boutigue MH, Goumon Y (2006) Characterization of natural vasostatin-containing peptides in rat heart. FEBS J 14:3311–3321
Goetze JP, Alehagen U, Flyvbjerg A, Rehfeld JF (2014) Chromogranin A as a biomarker in cardiovascular disease. Biomark Med 1:133–140
Helle KB, Corti A, Metz-Boutigue MH, Tota B (2007) The endocrine role for Chromogranin A: a prohormone for peptides with regulatory properties. Cell Mol Life Sci 22:2863–2886
Jansson AM, Røsjø H, Omland T, Karlsson T, Hartford M, Flyvbjerg A, Caidahl K (2009) Prognostic value of circulating Chromogranin A levels in acute coronary syndromes. Eur Heart J 1:25–32
Kojda G, Kottenberg K, Hacker A, Noack E (1998) Alterations of the vascular and the myocardial guanylate cyclase/cGMP-system induced by long-term hypertension in rats. Pharm Acta Helv 73(1):27–35
Mahapatra NR, O’Connor DT, Vaingankar SM, Hikim AP, Mahata M, Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, Ross J, Mahata SK (2005) Hypertension from targeted ablation of chromograninAcan be rescued by the human ortholog. J Clin Invest 115(7):1942–1952
Metz-Boutigue MH, Garcia-Sablone P, Hogue-Angeletti R, Aunis D (1993) Intracellular and extracellular processing of chromogranin A. Determination of cleavage sites. Eur J Biochem 217(1):247–257
Miserez B, Annaert W, Dillen L, Aunis D, De Potter W (1992) Chromogranin A processing in sympathetic neurons and release of Chromogranin A fragments from sheep spleen. FEBS Lett 2:122–124
O’Connor DT, Pandlan MR, Carlton E, Cervenka JH, Hslao RJ (1989) Rapid radioimmunoassay of circulating Chromogranin A: in vitro stability, exploration of the neuroendocrine character of neoplasia, and assessment of the effects of organ failure. Clin Chem 35:1631–1637
Omland T, Dickstein K, Syversen U (2003) Association between plasma Chromogranin A concentration and long-term mortality after myocardial infarction. Am J Med 1:25–30
Pasqua T, Corti A, Gentile S, Pochini L, Bianco M, Metz-Boutigue MH, Cerra MC, Tota B, Angelone T (2013) Full- length human chromogranin-A cardioactivity: myocardial, coronary, and stimulus-induced processing evidence in normotensive and hypertensive male rat hearts. Endocrinology 9:3353–3365
Pieroni M, Corti A, Tota B, Curnis F, Angelone T, Colombo B, Cerra MC, Bellocci F, Crea F, Maseri A (2007) Myocardial production of chromogranin A in human heart: a new regulatory peptide of cardiac function. Eur Heart J 28(9):1117–1127
Raab W (1969) Myocardial electrolyte derangement: crucial feature of pluricausal, so-called coronary disease. Ann N Y Acad Sci 147:627–686
Samuels MA (2007) The brain-heart connection. Circulation 1:77–84
Selye H (1936) A syndrome produced by diverse nocuous agents. Nature 138:32
Selye H, Bajusz E (1958) Notes on stress studies in cardiology: cardiac necrosis and its prevention. Schweiz Med Wochenschr 88(46):1147–1155
Slaton JW, Perrotte P, Inoue K, Dinney CP, Fidler IJ (1999) Interferon-mediated down-regulation of angiogenesis- related genes therapy of bladder cancer are dependent on optimization of biological dose and schedule. Clin Cancer Res 5(10):2726–2734
Steiner HJ, Weiler R, Ludescher C, Schmid KW, Winkler H (1990) Chromogranins A and B are colocalized with atrial natriuretic peptides in secretory granules of rat heart. J Histochem Cytochem 6:845–850
Stojanovic MO, Ziolo MT, Wahler GM, Wolska BM (2001) Anti-adrenergic effects of nitric oxide donor SIN-1 in rat cardiac myocytes. Am J Physiol Cell Physiol 281(1):C342–C349
Takiyyuddin MA, Parmer RJ, Kailasam MT, Cervenka JH, Kennedy B, Ziegler MG, Lin MC, Li J, Grim CE, Wright FA, O’Connor DT (1995) Chromogranin A in human hypertension. Influence of heredity. Hypertension 26(1):213–220
Tota B, Angelone T, Mazza R, Cerra MC (2008) The chromogranin A-derived vasostatins: new players in the endocrine heart. Curr Med Chem 15(14):1444–1451
Tota B, Cerra MC, Gattuso A (2010) Catecholamines, cardiac natriuretic peptides and chromogranin A: evolution and physiopathology of a ‘whip-brake’ system of the endocrine heart. J Exp Biol 213(Pt 18):3081–3103
Tota B, Angelone T, Cerra MC (2014) The surging role of Chromogranin A in cardiovascular homeostasis. Front Chem 2:64
Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J (2009) The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 54(19):1747–1762
Veschini L, Crippa L, Dondossola E, Doglioni C, Corti A, Ferrero E (2011) The vasostatin-1 fragment of Chromogranin A preserves a quiescent phenotype in hypoxia-driven endothelial cells and regulates tumor neovascularization. FASEB J 11:3906–3914
Weiergräber M, Pereverzev A, Vajna R, Henry M, Schramm M, Nastainczyk W, Grabsch H, Schneider T (2000) Immunodetection of alpha1 E voltage-gated Ca (2+) channel in chromogranin-positive muscle cells of rat heart, and in distal tubules of human kidney. J Histochem Cytochem 6:807–819
Yoo SH, Lewis MS (1996) Effects of pH and Ca2 on heterodimer and heterotetramer formation by chromogranin A and chromogranin B. J Biol Chem 271(29):17041–17046
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Tota, B., Cerra, M.C. (2017). Full Lenght CgA: A Multifaceted Protein in Cardiovascular Health and Disease. In: Angelone, T., Cerra, M., Tota, B. (eds) Chromogranins: from Cell Biology to Physiology and Biomedicine. UNIPA Springer Series. Springer, Cham. https://doi.org/10.1007/978-3-319-58338-9_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-58338-9_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58337-2
Online ISBN: 978-3-319-58338-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)