
Abstract
One approach to understand how neural circuits contribute to behavior is to dissect the function of discrete neuronal components of the network. The transcriptional profile of a neuron is a starting point to infer morphological, biochemical, and physiological properties that determine its functionality. This chapter aims to provide an overview on the challenges and advances to gain genetic access to distinct neuronal cell types, and the transcriptional profiling methods used to query their gene expression. In addition, it also surveys the contribution of transcriptional profiling experiments to our knowledge on aspects of circuit structure and function, which include dendritic morphology, wiring specificity, synaptogenesis, remodeling, and physiological states and functional properties of neurons. Based on the limitations of the current transcriptional profiling approaches, this chapter also addresses the perspectives and new developments that are expected to push cell type-specific gene expression profiling to new frontiers.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1 Introduction
To understand how neural circuits generate behavior, it is necessary to identify the neuronal cell types within a circuit and determine their connectivity and function. One way to reveal the molecular basis of neural function is to characterize the gene expression blueprint that determines the highly specialized phenotype of different types of neurons. Neuronal phenotypes are determined by molecules that regulate the morphological, biochemical, and physiological properties of a cell. Thus, the unique phenotype of different types of neurons is expected to be the result of their differential gene expression. Hence, comparison of gene expression profiles between neurons should identify key molecular components that specify their distinct functions.
Neuroscientists have long desired to be able to measure cell type-specific gene expression. However, two main issues have slowed down progress in this direction: difficulties in genetically manipulating specific neuronal cell types and in obtaining their gene expression profiles.
A neuronal cell type can be defined as a group of neurons that carry out a distinct task. Most often the way to identify a neuronal cell type is through its shape, determined by the dendritic arborization and projection pattern of the axon. This approach is based on the fundamental premise that a neuron’s shape is a direct reflection of its connectivity, and hence of its unique function. It is reasonable to imagine that the distinct spatial position of neurons classified as belonging to the same cell type could result in further subdivision of that cell type into distinct subpopulations. This could be due to unidentified subtle morphological changes or physiological differences that would go undetected. Thus, one could argue that each neuron is unique. While the scientific community is aware of the drawbacks of morphological classification, at this point, in most cases, morphology is the easiest feature to score.
In the case of Caenorhabditis elegans and Drosophila, early studies identified distinct cell types through their morphology. A complete reconstruction of the C. elegans nervous system was undertaken using EM serial sections (White et al. 1986), and Golgi staining was used in Drosophila to characterize cell types in the optic lobe (Fischbach and Dittrich 1989). This information served as a mere, although very informative, catalog until experimental tools were developed that provided genetic access to specific neuronal cell types.
The nervous system is characterized by containing numerous highly intermixed cell types with irregular morphology. Many neuronal cell types are found in small numbers and are frequently difficult to access manually. For these reasons, cell type-specific gene expression analysis has also been dependent on the development of techniques that enable isolation of transcripts in a cell type-specific fashion. Technical advances in high-throughput gene expression analysis platforms have also been crucial to the success of these approaches.
This chapter aims to review the efforts to gain genetic access to specific neuronal cell types, an essential step to then apply profiling technologies since these depend on the expression of transgenes in a cell-specific fashion. In addition, it intends to provide an overview of the different types of profiling techniques that have been applied in C. elegans and Drosophila, with emphasis on the neuronal cell types to which these different techniques have been applied. To conclude, examples are given of biological questions related to the function of neural circuits that have been addressed through gene expression profiling.
2 Labeling Specific Neuronal Cell Types
Most methods used for cell type-specific profiling rely on the expression of some sort of transgene that distinguishes the cell type of interest from the rest of the neurons in the tissue. The nature of the transgene expressed will differ depending on the profiling approach taken, and this will be addressed in the corresponding section. Transgenesis techniques employed in C. elegans and Drosophila are well established and beyond the scope of this section, thus they will not be discussed. Here, we present the genetic approaches to molecularly mark specific neuronal populations.
2.1 Genetic Toolkit for Labeling Neurons
The preferred genetic methods to label any cell type of interest, in this case neurons, can be divided in two main types: regulatory sequence/reporter fusions and binary systems.
2.1.1 Regulatory Sequences/Reporter Fusions
In this strategy, the regulatory sequence of a known gene that is highly expressed in the neuronal cell type of interest is placed upstream of the coding sequence of a marker.
Identification of the regulatory sequence of the gene of interest is not necessarily a simple feat. Complementary approaches such as in situ hybridization and/or immunohistochemistry, if an antibody against the protein is available, can determine the correlation between the expression of the regulatory sequences/reporter fusion transgene and the endogenous expression of the gene.
The 7.4 kb 5′ regulatory sequences of the Drosophila choline acetyltransferase (ChAT) gene, which labels the cholinergic neuronal population, was determined through the generation of fusions of different lengths of 5′ flanking sequences of ChAT to the lacZ reporter gene, and comparison to the distribution of endogenous ChAT protein. Smaller fragments directed the lacZ expression in selected subsets of cholinergic neurons (Kitamoto et al. 1992). For cell type-specific genes of sensory neurons, such as opsins and odorant receptors, fusions of regulatory sequences to reporters have been quite successful (Couto et al. 2005; Fortini and Rubin 1990; Tahayato et al. 2003), probably due to the smaller size of their cis-regulatory regions.
The compacted nature of the C. elegans genome, and hence the fact that regulatory regions might be smaller, has facilitated the widespread use and success of regulatory sequences/reporter fusion transgenes in the worm. A significant number of cell type-specific fusions are available, among which there are many examples of mechano- and chemosensory neurons (Zaslaver et al. 2015).
To use direct fusion transgenes in profiling experiments it is necessary that the transgene used is expressed at levels compatible to the profiling approach that will be used. In this front, another reason for the success of regulatory sequences/reporter fusion transgenes in the worm is the presence of multiple transgene copies when the transgenesis approach involves extrachromosomal arrays.
The ease of genome editing using CRISPR technology, available both in the fly and the worm (Li and Ou 2016; Paix et al. 2015; Xu et al. 2015), could facilitate the generation of reporter lines where the marker expression is under endogenous regulation. This could be achieved by substituting one copy of the gene by the marker of choice or introducing the marker upstream of the translational start of the gene. This approach would be useful as long as the level of expression of the marker is sufficient for the profiling approach to follow.
2.1.2 Binary Transactivator/Responder Systems
Binary systems consist of a transactivator that binds to a specific DNA sequence to promote the transcription of a downstream responder. Spatial control of the expression of the responder is dictated by the choice of regulatory sequences that control the transactivator expression. The main virtue of this approach is the ability to control temporal and/or level of expression of the responder. This is achieved, thanks to the existence of transactivator repressors and compounds that positively or negatively modulate transactivator or repressor activity. Another advantage of this system is amplification of responder expression levels.
The main binary systems used in Drosophila are:
-
1.
GAL4-UAS: The yeast GAL4 transcription factor binds to the Upstream Activating Sequences (UAS) placed upstream of the responder (Brand and Perrimon 1993; Fischer et al. 1988). Additionally, the GAL4-UAS system is repressible by the GAL80 protein (Lee and Luo 1999; Ma and Ptashne 1987). The most widely used strategy to regulate temporal expression of GAL4 is to use the temperature sensitive GAL80 repressor (GAL80ts) (McGuire et al. 2001). This mutant version of the protein represses GAL4 transcriptional activity at 17 °C and releases repression at 29 °C or higher temperatures.
-
2.
LexA-lexAop: This system is based on the LexA bacterial repressor that binds to specific lexA operator (lexAop) sequences. LexA DNA binding domain (DBD) has been fused to several activation domains (AD). Fusions to GAL4 AD render the system sensitive to GAL80, conferring temporal control through the use of GAL80ts (Lai and Lee 2006; Szüts and Bienz 2000). Fusions to viral VP16 and human p65 strong activation domains result in chimeric proteins that transcribe high levels of responder expression and are insensitive to GAL80 (Lai and Lee 2006; Pfeiffer et al. 2010). The lexA system has been optimized to obtain better inducible expression and reduce leakiness and toxicity (Pfeiffer et al. 2010; Yagi et al. 2010).
-
3.
QF-QUAS: This recently developed system relies on components identified from the fungus Neurospora crasa (Potter et al. 2010). The QF transactivator binds to QF upstream activating sequences (QFUAS), triggering the transcription of downstream responders. The activity of the QF system can be temporally controlled through the presence of the suppressor QS in the genetic background and addition of quinic acid (QA) to the fly food. Interestingly, repressor activity can be titrated by varying the concentration of QA fed to the animal, adding an extra layer of regulation. Recent modifications of this system have generated less toxic versions of QF AD that have been proven to function in GAL4-QF AD and LexA-QF AD chimeras (Riabinina et al. 2015), enriching the tools available for responder expression regulation.
Binary expression systems are starting to become available in C. elegans but are not yet widely used. One of these approaches is a binary system employing heat shock induction (Bacaj and Shaham 2007). This is based on cell type-specific rescue of mutants defective in the heat shock response. The heat shock response factor (HSF) is expressed under a cell type-specific promoter in the cell of interest. The HSF transactivator activity is regulated by heat shock stress, which results in the formation of transcriptionally active trimers. The presence of an additional transgene containing HSF binding sites upstream of a marker gene triggers its expression in a cell type-specific manner. Transient or sustained heat shock pulses allow for temporal control of marker expression. In addition, a repressible Q binary system has been developed (Wei et al. 2012). Efforts to adapt the GAL4-UAS system for its use in the worm have entailed the systematic comparison of the transcriptional efficacy of three major components of this system—the DNA-binding domain, the activation domain, and UAS copy number. The Sternberg laboratory has found that performance of GAL4 is heavily dependent on temperature, acting poorly at 20 °C or below. Through evolutionary analysis they have identified Saccharomyces kudriavzevii GAL4, which functions robustly across the 15–25 °C range. Their optimized GAL4 system is capable of driving expression in a variety of tissues, including neurons (Wang et al. 2017). Long desired by the community, the GAL4/UAS system is expected to become widely used in the near future.
Alternatively, a two-part system for conditional FLP-out of FRT-flanked sequences in the worm has been developed to control gene expression in a spatially and/or temporally regulated manner (Davis et al. 2008; Voutev and Hubbard 2008). In this system, transcription is blocked by the presence of an “off cassette”, composed of a transcriptional terminator flanked by FLP recognition targets (FRT), between the promoter and the coding sequence of the desired product. FLP-mediated excision of the cassette brings together the promoter and coding sequence activating transcription. Temporal control of marker expression can be regulated through heat shock-mediated expression of FLP. In addition, this system could be used to spatially restrict expression in a subset of cells that can only be addressed as the intersection of two available promoters (Davis et al. 2008). In this context, FLP expression would be under a cell type specific promoter.
2.2 Endeavors to Gain Access to Neuronal Cell Types
2.2.1 Searching for Regulatory Sequences
A key factor in implementing the above approaches is to identify regulatory regions that label the cell type of interest. It is relatively easy to find regulatory regions that label a large population of neurons based on a molecular characteristic (e.g., neurotransmitter used). A gene expression analysis of such a population may reveal broad characteristics, but this knowledge will be obtained at the expense of understanding the diversity of cell types comprising the population. In consequence, concerted efforts have been made to gain access to smaller populations of neurons.
Over the years, the Drosophila community has made enormous progress in gaining genetic access to specific cell types. A first approach was based on the random insertion of transposable elements and their capacity to act as enhancer traps, enabling identification of genomic enhancers. Initial studies used P elements containing lacZ (O’Kane and Gehring 1987). The generation of P elements containing sequences coding for GAL4 (Brand and Perrimon 1993) paved the way for binary systems, and many GAL4 lines have been generated by this means (Brand and Perrimon 1993; Hayashi et al. 2002). Though not as extensive, similar collections have been made for GAL80 (Suster et al. 2004), and more recently for LexA (Miyazaki and Ito 2010). More recently, transposable element vectors have been designed that make it possible to swap DNA content through various methods. Thus, these new collections permit researchers to customize a pre-existing line according to their needs. MiMIC (minos-mediated integration cassette) lines (Venken et al. 2011) contain two inverted attP sites that allow DNA replacement using RMCE (recombinase-mediated cassette exchange) (Bateman et al. 2006). MiMIC lines inserted in the first noncoding intron can be replaced with the transactivator or suppressor of choice. G-MARET (GAL4-based mosaic-inducible and reporter-exchangeable enhancer trap) (Yagi et al. 2010) and InSITE (integrase swappable in vivo targeting element) (Gohl et al. 2011) insertion collections allow replacement of GAL4 with other transactivators. A recurrent finding with all these transposable element collections is that the expression patterns obtained often tend to be broad because the same gene can be expressed in more than one cell type. Since these lines often include different neural cell types, their usefulness for profiling is limited.
In an attempt to generate lines with more restricted expression patterns, Rubin and colleagues at Janelia Research Campus took the following approach. They selected a group of 925 genes for which available expression data or predicted function indicated expression in neurons in the adult brain. These genes included transcription factors, neuropeptides, receptors, and ion channels, among others. The approach consisted of cloning relatively small fragments of genomic DNA upstream of these genes to a promoter and the GAL4 coding sequence (Pfeiffer et al. 2008). These plasmids were integrated at a specific docking site in the genome using phiC31 integrase, yielding thousands of GAL4 lines (Jenett et al. 2012) and LexA lines (Bloomington FBrf0222940). These lines were then curated for expression in the embryonic, adult and larval CNS, providing an excellent resource for the community (http://flweb.janelia.org/cgi-bin/flew.cgi). The entire collection of lines covers most Drosophila neurons, and over half of the fragments drive unique expression in 10–200 cells in the brain (Pfeiffer et al. 2008). Plasmids are available to clone the identified enhancers for fusion to various transactivators or fluorescent proteins. A complementary collection of GAL4 and LexA lines has been generated by the Dickson and Stark research groups (Kvon et al. 2014), and their expression pattern in the nervous system has been cataloged (VDRC Vienna tiles http://brainbase.imp.ac.at/bbweb/#6?).
The C. elegans community has undertaken several genome-wide gene expression projects. Hope and colleagues pioneered these studies using lacZ reporters and later developed the “promoterome”: a genome-wide resource of C. elegans promoters to generate transgenic animals expressing GFP (Dupuy et al. 2004; Hope 1991; Lynch et al. 1995). Together with other groups, a collection of over 2000 transgenic lines carrying promoter: GFP fusions have been created and their spatiotemporal expression patterns curated (350 TF and almost 1900 genes) (Dupuy et al. 2007; Hunt-Newbury et al. 2007; Reece-Hoyes et al. 2007). Transgenic C. elegans strains for studying miRNA expression have also been generated (Isik et al. 2010; Martinez et al. 2008). Expression patterns are compiled in several databases: the Hope Lab Expression Patten Database: http://bgypc059.leeds.ac.uk/~web/; C. elegans Promoter/Marker Database: http://www.grs.nig.ac.jp/c.elegans/promoter/index.jsp?lang=english; the Promoterome Database: http://worfdb.dfci.harvard.edu/promoteromedb/; the BC C. elegans Gene Expression Consortium: http://gfpweb.aecom.yu.edu; and the Localizome Project: http://localizome.dfci.harvard.edu/index.php?page=home. Together with lines generated by researchers for their specific studies, these collections have expanded the catalog of regulatory regions with characterized expression patterns.
A cautionary note on expression patterns derived from transgenic enhancer/promoter constructs in C. elegans and Drosophila: besides the difficulty of defining regulatory regions that recapitulate the endogenous expression pattern of the gene of choice, factors such as integration site and surrounding chromatin structure can affect transgene expression. Thus, it is advisable to verify that expression of the reporter matches the endogenous gene expression. It is worth noting that the approach taken by the Rubin group was aimed at identifying small fragments in the putative upstream regulatory sequences of neuronal genes that would label subsets of neurons. It is possible that some of these fragments label subsets of neurons where the gene is actually not expressed. This situation could occur if the identified fragment lacked repressor sequences that under normal conditions repressed expression of the gene in those cells. Provided that the identified fragment labels neurons of interest for the researcher, the reporter is a valid reagent to genetically manipulate those neurons.
In addition, the modENCODE project aims to identify all of the sequence-based functional elements in the C. elegans and Drosophila melanogaster genomes. The work of this consortium (Gerstein et al. 2010; modENCODE Consortium et al. 2010; Nègre et al. 2011) and other laboratories (Kvon et al. 2012, 2014; Shi et al. 2009) could, in principle, aid researchers in the search of regulatory regions functioning as enhancers for neurons or specific neuronal populations in their gene of interest.
2.2.2 Applying Intersectional Strategies
All these efforts have yielded an exceptional collection of transgenic lines and a catalog of expression patterns in the nervous system in C. elegans, and especially in Drosophila. However, while some cell type-specific lines exist, many still label several neuronal populations. To overcome this issue, intersectional strategies have been developed. These are aimed at defining an expression domain that is cell type-specific. When reporter expression cannot be restricted to the cell type of interest using one particular regulatory sequence, the combined use of two or more unrelated regulatory sequences is employed to define a cell type-specific expression domain.
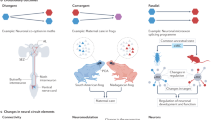
In Drosophila, the ample collection of enhancer and binary factor lines available, together with the fact that binary systems are specific and do not cross talk, and can be combined either together or with other genetic techniques, renders intersectional strategies a useful approach to label specific neuronal cell types (del Valle Rodríguez et al. 2012). Through intersectional strategies, cell type-specific expression domains can be obtained as a result of addition, intersection, or subtraction of the expression domains of the combined binary systems and/or other elements used. Below, we describe some of the possible combinations used in intersectional strategies (Fig. 19.1).

Examples of intersectional strategies used to restrict expression to the cell type-specific neuronal population of interest in Drosophila. (A) Addition strategy combining both Gal4 and LexA binary systems and the use of the same reporter for both of them. (B) Intersection strategy based on the split GAL4 approach (GDBD GAL4 DNA binding domain; AD activation domain; zip zipper). (B′) Intersection strategy based on the combination of the GAL4 and LexA binary systems and system-specific fluorescent reporters. Cells in the common domain are identified by the coexpression of Gal4 and LexA reporters. (B″) Flip-based example of an intersection strategy where an FRT flanked ORF is eliminated. (C) Example of subtraction strategy using the GAL4 system and the GAL80 repressor. (C′) Flip-based example of a subtraction strategy where the FLP-out of an interruption cassette results in the expression of a downstream ORF
When independent lines each label a different subset of cells of the same type, addition is the simplest strategy to label the entire cell-type population. This can be achieved by the combination of transactivators of the same or different type, for instance GAL4 + GAL4 or LexA + GAL4. In the latter case, the given responder transgenes for the two types of transactivators should be present in the background (Fig. 19.1A).
Regulatory sequences driving transactivators usually label more than one cell type; however, different intersectional strategies can restrict expression to the cell type of interest. When the cell type of interest falls within the expression domain common to the two regulatory sequences used, split binary systems are a useful option (Fig. 19.1B). This variation was pioneered by the split-GAL4 system (Luan et al. 2006), and has recently been developed for LexA (Ting et al. 2011). The transactivator is separated into two hemi-proteins, each of which is expressed from a different regulatory sequence. One hemi-protein contains the GAL4 DNA-binding domain (DBD) or LexA, while the other hemi-protein contains the activation domain (AD). The use of distinct ADs renders these split systems GAL80-sensitive or insensitive. A functional transactivator will only reconstitute and activate the transcription of the responder when expressed together in the same cell. One drawback of this approach is that it often requires the generation of new hemi-lines. A considerable improvement offered by the split-LexA system is that it can leverage the wealth of pre-existing GAL4 lines by placing the expression of one of the hemi-lines under UAS control (UAS-split-LexA or UAS-split-AD), while the other hemi-driver can be expressed from a direct fusion (Ting et al. 2011). Alternatively, based on the fact that binary systems do not cross talk, they can be combined. The use of binary system-specific responder transgenes encoding for different fluorescent proteins enables identification of cells in the common domain as the double-labeled cell type (Fig. 19.1B′).
When the cell type of interest falls within a specific expression subdomain driven by a regulatory sequence, it is possible to restrict expression by subtraction. The simplest method is by expression of a transactivator repressor, such as GAL80 when using the GAL4 or LexA GAL80 sensitive binary systems (Fig. 19.1C).
A combination of binary systems and FLP recombinase can be used to define intersecting domains and in subtraction strategies (Fig. 19.1B″, C′). In these scenarios, expression of the transactivator, repressor, or responder is regulated by recombinase activity removing an intervening FRT stop cassette. Many creative genetic designs have emerged from the combined use of the FLP recombinase and binary systems (for a review, see del Valle Rodríguez et al. 2012). The recent development of new recombinases and recognition sites has increased the numerous combinatorial options already available to researchers (Hadjieconomou et al. 2011; Nern et al. 2011).
In C. elegans, the most commonly used method to express transgenes is based on regulatory sequences/reporter fusions. By combining regulatory sequences yielding overlapping expression patterns, researchers can engineer worm strains that label specific subsets of neurons. Addition strategies can be pursued with transgenes expressing the same reporter under different regulatory sequences (Fig. 19.2A).

Examples of intersectional strategies used to restrict expression to the cell type-specific neuronal population of interest in C. elegans. (A) Addition strategy. (B) Intersection strategy based on multicolor labeling. Cells in the common domain are identified by the coexpression of fluorescent proteins. (B′) Split GFP intersection strategy. Cells in the common domain are identified by reconstitution of GFP fluorescence. (B″) Split Q system intersection strategy. (B″′) FLP-based refinement of expression patterns. Excision of an FRT flanked cassette containing a fluorescent protein and stop sequence results in the expression of a downstream ORF, normally not expressed, that codes for a different fluorescent protein. (C) Example of subtraction strategy using the Q system and the QS repressor. This approach is useful to label a subset of neurons in the X expression domain for which there is no available regulatory sequence that labels them. This can be achieved if there is a promoter that labels the complementary subset of neurons and is used to express the QS repressor. The cells of interest for which there is no specific regulatory sequence are identified as double labeled
One intersectional strategy that can be also applied using regulatory sequences/reporter fusion transgenes is multicolor labeling. Triple color combinations (CFP, YFP, DSRed) have been successfully employed to label separate classes of neurons using cell type-specific regulatory regions (Hutter 2003). Similarly, one could use this strategy to identify the neuronal type of interest when distinct regulatory sequences/GFP variant fusions are combined in the same organism. In this scenario, specific cell types can be detected by their distinct fluorescent marker combination (Fig. 19.2B). Similarly, the recent addition of the GAL4/UAS system to the worm toolkit promises to expand the possibilities with regard to intersectional strategies. For example, doing combinations of the GAL4 and Q systems, or either of these systems with a regulatory sequence/reporter fusion transgene, where the two regulatory sequences label a common set of neurons.
Another intersectional strategy is based on the split approach. This approach has been applied to obtain cell type-specific GFP reconstituted expression (Fig. 19.2B′). Identified N-GFP and C-GFP peptides fused to leucine zippers can reconstitute GFP fluorescence in vivo when expressed in the same cell type (Zhang et al. 2004). Expression vectors have been constructed that are suitable for cloning regulatory sequences. Alternatively, the split Q system has been generated and used in worms to label neurons common to two distinct promoters (Fig. 19.2B″) (Wei et al. 2012).
The FLP-out system also offers the possibility of using intersectional strategies in the worm (Davis et al. 2008). This could be achieved when FLP expression and the FLP-out cassette are under regulatory sequences that label a set of common neurons (Fig. 19.2B″′).
Finally, the subtraction approach has been achieved by combining QF, QS, and two distinct fluorescent reporters, for example, mCherry and GFP. By means of this strategy, neurons can be distinguished based on their single or double reporter expression pattern (Fig. 19.2C).
3 Methods to Profile Transcriptional Activity
A wide variety of techniques are currently available to profile the transcriptomes of specific cell types. Recent excellent reviews have discussed the key issues that influence their choice (McClure and Southall 2015; Otsuki et al. 2014). Yield, accuracy, technical difficulty, and cost are among the factors to consider. Since each method has its own strengths and limitations (Table 1), researchers must reach a decision based on the physical limitations of the biological material (ability to access the cell type of interest, abundance of the cell type), the biological question to address, and the type of information that can be obtained from the selected methodology. In this section, we will present these methods, discuss the nature of the transgene required to label neurons, and describe their use to profile specific types of neurons in C. elegans and Drosophila.
Profiling techniques can be divided into two main classes. One set of methods involves physical cellular/nuclear isolation prior to transcriptional profiling. The other techniques rely on capturing the transcriptional activity of the cell type of interest while in its tissue context. In both cases, it is necessary to drive the expression of different types of transgenes in a cell type-specific fashion.
3.1 Profiling Using Physical Cellular/Nuclear Isolation
In these techniques, physical isolation is used to minimize contamination from other cell types in the tissue sample.
3.1.1 Manual Isolation
Conceptually, manual isolation and identification of cells is the most straightforward technique. This procedure usually consists of dissecting the tissue containing the cells of interest, dissociating the cells and diluting the suspension to a concentration where cells can be individually viewed, and extracting them by aspiration with a micropipette. In principle, provided that the researcher can differentiate cells based on shape and/or size, this approach achieves very high purity. In general though, manual isolation is aided by the expression of fluorescent proteins in the cells of interest. In particular, when cells cannot be distinguished in any other way, it is essential that fluorescent protein expression is strong enough to allow for in vivo sorting under the microscope and that there is no leaky expression outside the neurons of interest.
This approach has been successfully used in Drosophila to address transcriptional changes in distinct types of larval and adult neurons in the circadian circuit (Abruzzi et al. 2017; Abruzzi et al. 2015; Kula-Eversole et al. 2010; Nagoshi et al. 2010). This methodology is highly suitable for such studies since collecting cells from entrained brains at different circadian times requires rapid isolation protocols. In addition, given that some of these types of neurons are present in very reduced numbers, this approach reduces the signal-to-noise ratio and allows detection of mRNAs that would be masked by mRNA in the rest of the brain. Using this approach, the Rosbash group has profiled the transcriptomes of small and large PDF-expressing ventral lateral neurons (s-LNvs and l-LNvs) known to drive the morning activity period at different circadian times (Abruzzi et al. 2015; Kula-Eversole et al. 2010). These cells, 8 s-LNvs and 10 l-LNvs per brain, were labeled with GFP using a Pdf-GAL4 line and isolated by the size of their cell bodies. 100 cells obtained from around 100 brains provided sufficient material to perform microarray analysis (Kula-Eversole et al. 2010). The same researchers have recently developed an RNA amplification protocol that has enabled them to obtain enough mRNA to generate libraries for RNA deep sequencing (Abruzzi et al. 2015) and profile additional clock neurons as well as dopaminergic neurons (Abruzzi et al. 2017).
A recent study reports the harvesting of different types mushroom body neurons (a/b and g Kenyon cells (KC)) and mushroom body extrinsic neurons (V2, DAL, MBONa3, MBONg5b′2a, MBONb2b′2a) using GAL4 cell type-specific lines. In this case, GFP-labeled cell bodies were manually extracted in vivo, from intact brains, via patch clamp electrodes on an electrophysiology rig. RNA-seq was performed with material obtained from pooling approximately 100 cells from a single fly for each KC sample, and 4–14 neurons from one or two flies for each mushroom body extrinsic neuron sample (Crocker et al. 2016).
In theory, manual sorting could be performed for identifiable cells for which there are no cell type-specific lines available by filling them with fluorescent dyes. This strategy has been used to identify gene expression profiles through microarray analysis on single cells isolated from living embryos (Bossing et al. 2012).
3.1.2 Automated Isolation
Several alternative methods exist for automated isolation:
-
Fluorescence-Activated Cell Sorting (FACS)
This flow cytometry isolation technique is based on sorting dissociated cells according to their fluorescent properties. In the case of C. elegans and Drosophila profiling, fluorescence is provided by genetic means in the cell type of interest. This fluorescent label must be sufficiently strong to be detected by the sorter in live cells.
FACS has been extensively used in C. elegans, especially in studies involving profiling of embryonic neurons since embryonic dissociated tissues can be cultured. An extensive collection of different types of neurons, including olfactory, thermosensory, and motor neurons, has been profiled with microarray experiments (Blacque et al. 2005; Cinar et al. 2005; Colosimo et al. 2004; Etchberger et al. 2007; Fox et al. 2005; Hallem et al. 2011; Von Stetina et al. 2007a; Zhang et al. 2002). Recently, the development of culture protocols for larval tissues has facilitated the use of FACS to isolate larval neurons and perform RNA-seq analysis (Spencer et al. 2014). Starting material is not a limiting factor since large numbers of larvae are easily generated using standard culture conditions. Indeed, even neuronal cell types consisting of 2 neurons per worm have been profiled from worm cultures containing approximately 3 million larvae. The NSM serotonergic neurosecretory neurons (2 neurons/worm, 6 million neurons in 3 million larvae) have been purified with a yield of 0.85%; in other words, as few as 30,000–50,000 neurons have been isolated through FACS and used to generate sequencing libraries (Spencer et al. 2014).
In Drosophila, the use of FACS to isolate neuronal cell types has been more limited. One of the earliest instances was a study by Jasper and colleagues where they used SAGE to profile a subset of photoreceptor neurons in larval stages (Jasper et al. 2002). FACS has also been used to profile multidendritic neurons, wild type and mutant motor neurons (Parrish et al. 2014), and wild type and mutant LNvs pacemaker neurons (Mizrak et al. 2012; Ruben et al. 2012). In the latter case, microarray analysis was performed with as few as 150–300 cells obtained from 50 brains. Recent publications have reported profiling cell type-specific neurons using RNA deep sequencing, including seven different neuronal cell types from the fly visual system, which have been used to create libraries from as few as 8000 sorted cells (Tan et al. 2015), and ultralow input RNA-seq data from 100 larval multidendritic neurons (Williams et al. 2016).
-
Magnetic Activated Cell Sorting (MACS)
MACS is an affinity-based purification strategy. Magnetic particles coupled to antibodies are used to capture the cell of interest from a suspension of dissociated tissue. Sorting specificity is based on the use of antibodies against membrane-targeted antigens specific to the cell type of interest. After the incubation period, beads are recovered with a magnet and cells eluted for further processing. Given that cell type-specific membrane-targeted antigens are often not known or antibodies are not available, the use of this method in Drosophila has been facilitated by the GAL4-UAS system and the exogenous expression of UAS-mCD8-fluorescent protein transgenes in the cell of interest.
MACS has been adapted to isolate dendritic arborization (da) neurons from the larval peripheral nervous system (Hattori et al. 2013; Iyer and Cox 2010; Iyer et al. 2013a). Peripheral neurons are difficult to isolate due to their low numbers and difficult-to-reach location below the chitinous larval cuticle. This approach has enabled Iyer and colleagues to isolate 1500–2000 da neurons (classes I–IV), and 300–500 Class IV da neurons, from 30 to 40 larvae (Iyer and Cox 2010). Using an intersectional approach, Class I da neurons have also been isolated. Since the Class I driver faintly labeled Class IV neurons, Class I driver expression was restricted to Class I neurons using the regulatory sequence of the Class IV driver fused to GAL80 (Iyer et al. 2013b). With this reported amount of material, they performed transcriptional profiling on microarrays.
MACS has also been used to isolate the dopaminergic neuronal population in the adult brain (Iyer et al. 2013a).
-
Laser microdissection of cells
Laser-based dissection enables isolation of single cells or single-cell clusters from complex tissue without the need for cell dissociation. Where the cell type of interest presents a recognizable shape, there is no need to use antibodies or genetic labels, either. Laser capture microscopy is the most common procedure and involves positioning a thermoplastic film over the frozen and/or fixed tissue sections. While cells are visualized under the microscope, a low-power infrared laser is used to locally melt the membrane around the cells of interest, binding them to the film. Lifting the membrane separates the cells from the rest of the sample.
This technique has been applied to profile the transcriptomes of Drosophila larval and pupal mushroom body neurons (Hoopfer et al. 2008), and larval insulin producing cells (IPCs) (Cao et al. 2014). Both these neuronal populations are characterized by the fact that their cell bodies form clusters, rendering LCM a useful isolation approach. Both studies relied on the use of cell type-specific lines and fluorescent reporters to visualize the cell bodies in the tissue sections. Mushroom body studies were performed with material pulled from 40 captures at a rate of 100 cells/capture per replicate (i.e., 4000 cells/replicate), and these were used to perform microarray analysis (Hoopfer et al. 2008). The number of IPCs per brain is just 14, distributed into two 7-cell clusters. Using both membrane (GFP) and nuclear (RFP) reporters to label these cells significantly accelerated the process and increased the reliability of their identification. The spatial resolution provided by LCM made it possible to use just 23 IPC cells/replicate, and improved amplification protocols enabled the construction of sequencing libraries for RNA-seq analysis (Cao et al. 2014).
A protocol for LCM isolation of da neurons has also been established (Iyer and Cox 2010). In this case, given the difficulty of capturing sparse da cell bodies in transversal sections of the whole larva, these researchers opted to isolate the cuticle from internal larval tissues and section the cuticle pellet. This modification enabled them to increase the number of cells accessible in the sample and to isolate single da cell bodies.
-
Isolation of nuclei
When cells are hard to dissociate, nuclei isolation presents itself as an alternative. Nuclei may be gently released from tissue homogenates without the need for hard dissociation, and are relatively unaffected by changes in the cytoplasmic RNA and protein pool. Most importantly, microarray-based mRNA expression analysis using nuclear RNA samples yields results comparable to those obtained using total RNA (Barthelson et al. 2007; Zhang et al. 2008). Another advantage of nuclei isolation is that it can be used for other high-throughput genomic characterization protocols besides transcriptional profiling.
The isolation of nuclei tagged in specific cell types (INTACT) method involves the coexpression of a nuclear envelope protein modified to sustain biotinylation, and a biotin ligase in the cell of interest. Incubation of the nuclear suspension with streptavidin-coated magnetic beads provides a rigorous method to isolate the nuclei of interest. Although this approach was developed in Arabidopsis (Deal and Henikoff 2010), it has been adapted to isolate nuclei from muscle of adult C. elegans and mesoderm from Drosophila embryos (Steiner et al. 2012). Shortly after the publication of the above studies, similar conceptual approaches and adaptations of INTACT were developed to isolate the nuclei of C. elegans and Drosophila neurons (Haenni et al. 2012; Henry et al. 2012; Ma and Weake 2014).
The difficulty in accessing post-embryonic tissues in C. elegans, mainly due to its tough cuticle, small size, and extremely complex tissue dissection, prompted the development of fluorescent activated nuclei sorting (FANS) (Haenni et al. 2012). Similar to INTACT, this procedure is based on cell type-specific nuclear labeling. However, it uses fluorescent labeling that does not need to be targeted to the nuclear envelope per se, since isolation is based on fluorescent sorting and not on antibody recognition. To gauge the scope of the technique, the method was tested on distinct cell types, including neurons. Although these studies focused on intestinal gene expression, it is expected that sequencing of cell type-specific neurons will also be feasible. This nuclear isolation protocol has been set up for large-scale worm cultures, and thus starting material should not present a problem in the case of small neural populations.
In Drosophila, various groups have developed procedures to isolate nuclei from cells in the adult brain and larval central nervous system following the INTACT rationale. Taking advantage of the GAL4-UAS system, these approaches are based on the cell type-specific expression of GFP-tagged nuclear envelope proteins and the use of anti-GFP antibody-coated magnetic beads for their isolation (Henry et al. 2012; Ma and Weake 2014). Nuclei of neuronal populations as small as 100–150 neurons per brain can be isolated from 600 tagged heads as starting material, without the need to dissect the brain, with high purity and around a 50% yield (Henry et al. 2012).
Batch isolate tissue-specific chromatin for immunoprecipitation (BiTS-ChIP) is an alternative nuclei isolation procedure developed in Drosophila (Bonn et al. 2012a, b). It is particularly suitable for ChIP experiments since the tissue is fixed before nuclei isolation, and the method is based on cell type-specific expression of an epitope-tagged histone protein, immunostaining against the tag, and fluorescent sorting of the nuclei. Alternatively, given the advances in extraction and quantitation of RNA from fixed sorted cells, as well as its integrity (Nilsson et al. 2014; Russell et al. 2013), one can envisage that nuclear RNA could be obtained from these nuclei, which would expand the use of this procedure beyond ChIP analysis.
3.2 Profiling Without Cellular/Nuclear Isolation
These techniques are aimed at minimizing possible acute transcriptional changes due to the stress caused by physical isolation procedures. Techniques developed to capture the transcriptional activity of the cell of interest are based on tagging the RNA, proteins interacting with the RNA, or proteins interacting with the DNA in a cell type-specific fashion. This enables distinction of the transcriptional activity of the cell type of interest from the rest of the cells in the sample.
3.2.1 Tagging RNA
The most prominent technique for tagging RNA is TU tagging. This technique is based on the properties of the Toxoplasma gondii uracil phosphoribosyltransferase (UPRT) enzyme, which when provided with 4-thiouracil (4-TU) inserts this analog in place of uracil in nascent RNA (Cleary et al. 2005). Subsequent biotinylation of thio-RNA enables affinity purification using streptavidin-coated magnetic beads.
The TU-tagging method was developed in Drosophila and introduced spatial regulation of RNA tagging through the GAL4-UAS system (Miller et al. 2009). This was achieved by the cell type-specific expression of UPRT. Thus, even if RNA is isolated from the whole animal, tagged RNA from the cells expressing UPRT can be selectively recovered. In addition, this technique allows for temporal control by timing and duration of 4-TU administration. 4-TU has been provided to embryos by immersion and fed to larvae and adult flies. Though no reports are available of TU tagging during pupal stages, 4-TU could be provided to pupae by injection, as is done in mouse. Exposure to 4-TU for up to 8 h has enabled detection of 4-TU-tagged RNA from whole animal RNA extraction in neural populations as small as mushroom body neurons in larval and adult brains. For smaller populations (250 cells), dissection of the brain was necessary (Miller et al. 2009). As few as 50 larvae per sample have been used to perform TU tagging and RNA-seq analysis of wild-type and mutant larval neuroblasts (Lai et al. 2012). There are reports that TU feeding can lead to background incorporation into mRNA and is toxic to flies (Thomas et al. 2012). Oxonic acid can be added to prevent a salvage pathway, which can use 4-TU without the presence of UTPR (Lai et al. 2012).
3.2.2 Tagging Proteins Interacting with RNA
Once again, different techniques exist in this area as well:
-
Poly-A Binding Protein tagging
This approach uses the endogenous transcriptional machinery to isolate poly-adenylated mRNA. The cell type-specific expression of a FLAG-tagged poly(A) binding protein (PABP) enables isolation of poly-A mRNA from the cell of interest. Using FLAG antibodies, the cell type-specific mRNA can be immunoprecipitated from a total RNA lysate.
This technique was developed in C. elegans to overcome the difficulty of working with larval and adult worms where cell isolation was problematic. Initially developed for muscle cells (Roy et al. 2002), it was soon applied to the nervous system. In a first study of the nervous system, mRNA was isolated from ciliated sensory neurons, which comprise approximately 50 cells in the worm, confirming the applicability of this approach to small numbers of cells (Kunitomo et al. 2005). Subsequently, the procedure was successfully applied for profiling, using microarrays of different types of motor neurons (Petersen et al. 2011; Von Stetina et al. 2007a, b) and the two PVD multidendritic nociceptor neurons of C. elegans in wild type and mutant backgrounds (Chatzigeorgiou et al. 2010; Smith et al. 2010, 2013), and to identify differential gene expression between the gustatory neurons ASER and ASEL (Takayama et al. 2010).
Poly-A mRNA tagging has also been used in the fly to isolate mRNA from adult photoreceptors using Drosophila PABP (Yang et al. 2005). However, this study also reported toxicity effects upon expression of dPABP, depending on the spatiotemporal expression of the GAL4 lines used. This toxicity might be partially reduced by controlling temporal expression of GAL4. Additionally, toxicity caused by overexpression of dPABP could be due to deregulation of the translation initiation and mRNA stabilization/degradation roles that this protein might cause when interacting with other types of proteins.
The study showed that the use of hPABD, whose C-terminal interacting domain only shares 30% similarity to the fly, was an alternative to dPABP. In an attempt to use this technique in photoreceptor neurons with a different set of GAL4 lines, we detected developmental defects caused by dPABP overexpression (Morey and Zipursky, unpublished). Given the possible appearance of morphological defects and lethality when expressing dPABP, toxicity should be carefully assessed before opting for this approach.
-
Ribosome tagging
Translating ribosome affinity purification (TRAP) (Heiman et al. 2008) and RiboTag (Sanz et al. 2009) were developed in mouse and are based on tagging a ribosomal subunit with a tag antigen. Ribosomes and their attached RNA can then be isolated through immunoprecipitation with magnetic beads coated with antibodies against the tag antigen. While this approach does not recover noncoding RNAs, it offers a snapshot of the putative translatome: transcripts being actively translated. Thus, it provides a more relevant insight into the cellular environment as a proxy for the cellular proteome.
Integration into the GAL-UAS system has provided the means to isolate mRNA associated with ribosomes in a cell type-specific fashion in Drosophila. UAS transgenic lines expressing the mouse or Drosophila RpL10 ribosomal subunit tagged with EGFP have been generated and successfully used with cell type-specific GAL4 lines to analyze transcriptomes by RNA-seq. One of these studies successfully isolated ribosome-bound RNA from adult neurons, and a small population of around 200 neurons of the pars intercerebralis of the brain from whole head extracts using 500–1000 heads (Thomas et al. 2012). Another study documented the rhythmic translatome of clock neurons (150 cells/brain). Using a GAL80-based intersectional strategy to restrict GAL4 expression in clock neurons, ribosome-bound mRNAs were profiled at six different time points of the circadian cycle (Huang et al. 2013). In this case, 200 heads (30,000 clock neurons) were lysed for each affinity purification experiment.
One factor influencing the success of any affinity purification method is the signal-to-noise ratio. To this end, Zhang et al. (2016) have recently developed Tandem-TRAP (T-TRAP), which includes a second tag to facilitate an additional purification step. They generated a UAS line where the N-terminus of Drosophila RpL10 was modified with two tandemly arranged epitopes, 3X FLAG and GFP, separated by the tobacco etch virus (TEV) protease site. They expressed TRAP and T-TRAP transgenes in photoreceptor neurons and purified ribosomal mRNA from dissected retina–optic lobe complexes. They next assessed enrichment of photoreceptor specific versus optic lobe transcripts comparing TRAP and T-TRAP samples to reference RNA obtained from the retina–optic lobe complexes. Using two sequential purification steps resulted in higher cell type-specific enrichment for T-TRAP (TRAP 1–10 times, T-TRAP 25–500 times). Although this enrichment came at the cost of a 30% decrease in the mRNA yield compared with TRAP, the amount of material obtained with T-TRAP was sufficient to perform RNA deep sequencing. They used 40 retina–optic lobe complexes per sample to perform T-TRAP, which represents a total of 240,000 photoreceptor neurons (6000 photoreceptors/retina–optic lobe complex). Attempts to isolate cell type-specific mRNA from populations of 750 cells/retina–optic lobe complexes showed nonspecific mRNA presence. The transgene encoding T-TRAP has recently been modified to further reduce background noise by increasing expression via the inclusion of noncoding sequences enhancing translation (Pfeiffer et al. 2012), and by mitigating the effects of leaky expression of the UAS construct by inserting a transcriptional stop sequence flanked by FRT recombination sites (unpublished data). Intersectional strategies targeting FLP expression to the cell type of interest coupled with cell type-specific GAL4 expression will further increase the potential of this method.
-
RISC tagging
This strategy is based on tagging specific proteins of the RNA-induced silencing complex (RISC) where the miRNA and its mRNA target interact. Thus, pull down of RISC permits the identification of associated miRNAs and their targets. Among the first reports of this approach were studies performed on Drosophila and C. elegans (Easow et al. 2007; Zhang et al. 2007). Tissue-specific identification of miRNA has been reported in the worm using intestine and muscle-specific enhancers driving the expression of tagged RISC proteins (Kudlow et al. 2012). Cell type-based analysis of miRNA profiles has been successfully performed for glutamatergic and GABAergic neurons and subtypes in the mouse brain (He et al. 2012). Thus, in principle, this technique could be used to profile miRNAs and their targets in C. elegans and Drosophila neurons by targeting expression of the tagged RISC complex in the neuron of interest.
3.2.3 Tagging Proteins Interacting with DNA
Targeted DamID (TaDa) is an adaptation of the original DamID technique (van Steensel and Henikoff 2000; van Steensel et al. 2001). The DamID system is based on identifying methylation footprints generated by the DNA adenine methyltransferase (Dam) enzyme from Escherichia coli. When this enzyme is fused to a protein that binds DNA, it methylates GATC sites in the vicinity of the binding site. These methylated sites can be conveniently digested with the methyl-sensitive restriction enzyme DpnI, and the fragments amplified by PCR for profiling with microarrays or deep sequencing. DamID has been used to study chromatin-associated protein interactions with DNA to understand transcriptional regulation (through transcription factors-Dam fusions) and chromatin states and dynamics (for a review, see Aughey and Southall 2016).
In order to use DamID in a cell type-specific manner, Southall and colleagues developed targeted DamID in Drosophila using the GAL4-UAS system (Southall et al. 2013). To this end, it was necessary to limit the expression levels of the Dam fusion protein, since its inherent high activity causes cell toxicity. This was achieved by leveraging ribosome reinitiation constructing a UAS transgene that carried a fluorescent protein followed by the Dam fusion protein, which is expressed at very low levels. This approach has been used to profile RNA Pol-II occupancy, thus giving a readout of transcription (Southall et al. 2013), and was applied to study neuroepithelial and neuroblast populations in the fly brain using between 100 and 300 brains depending on the developmental stage analyzed. Combining use of the GAL4-UAS system with GAL80ts allowed temporal restriction of Dam expression.
Efforts are being made to maximize the potential of TaDa as a cell type-specific transcriptional profiling approach, when it uses a fusion of the Dam enzyme to Pol-II. From a technical standpoint, TaDa has many advantages over other methods for cell type-specific profiling. It does not require cell isolation, avoiding any possible transcriptional responses to tissue dissociation protocols, nor is crosslinking or antisera use necessary, eliminating the noise caused by these procedures. Furthermore, fixation artifacts are avoided, since TaDa profiles protein binding in vivo. This protein binding and methylation is achieved with very low levels of enzyme-fused protein, limiting the impact of protein overexpression. It also uses DNA as readout, avoiding the technical complications of working with RNA.
Two main aspects of the initial protocol have been modified and improved. The publication describing the transcriptional profiling application of TaDa used tiling arrays as a means of mapping expression. The new protocol includes preparing the material for next generation sequencing and can be accomplished in 5 days from collection of the tissue samples to generation of the sequencing libraries. In addition, the number of targeted cells required for TaDa is very low. This new protocol has achieved RNA-seq transcriptional profiling with approximately 10,000 cells in total from 100 Drosophila heads (100 neurons/head). At >200,000 cells per head, this represents a 1:2000 ratio of methylated DNA to total DNA (Marshall et al. 2016).
The main limitations of TaDa are: (1) it does not provide direction of transcription, which can be an issue for nearby genes transcribed in opposite directions, and (2) it does not provide quantitative levels of RNA produced. However, in addition to embryonic and larval neural stem cells, this method has been successfully used to profile larval and adult neurons (Southall et al. 2013; A. Estancio-Gomez and T.D. Southall, unpublished). Furthermore, the TaDa protocol has been used to compare the transcriptional states of distinct sets of neurons, enabling the identification of differentially expressed genes (A. Estacio-Gomez and T.D. Southall, unpublished). Importantly, other laboratories have used this protocol successfully. In a recent publication, the laboratory of Dr. Edgar used TaDa to identify target genes of the Capicua (Cic) transcriptional repressor (Jin et al. 2015). They generated a UAS-cic-Dam construct that was expressed specifically in Drosophila intestinal stem cells (ISC) for 24 h using an ISC GAL4 line and GAL80ts. Taken together, the preliminary data in neurons, and the easy implementation of the protocol, suggest that TaDa could become a widely used approach for cell type-specific transcriptional profiling, in addition to its many other applications (Aughey and Southall 2016).
4 Contributions of Profiling Experiments to Circuit Structure and Function
Profiling experiments have yielded an insight into molecular underpinnings regulating distinct developmental processes involved in the assembly of functional circuits. In addition, they have revealed distinct physiological states and properties of different types of neurons, which explain their unique functionality in the circuit.
4.1 Neural Circuit Architecture
4.1.1 Dendritic Morphology
Dendritic architecture is a neuronal feature with important functional implications in circuit assembly, signal processing, and neural function. Profiling approaches have contributed to the identification of molecular strategies regulating dendrite branching both in C. elegans and Drosophila.
Studies on C. elegans have identified a set of transcription factors regulating the morphology of PVD dendrites. Distinct transcription factors appear to control discrete steps in PVD dendritic morphogenesis and either promote or limit PVD branching at specific developmental stages (Smith et al. 2010, 2013).
In the fly, analyses of transcriptional differences between arborizations of two classes of dendritic neurons with uniquely distinct dendritic morphologies have been conducted (Hattori et al. 2013; Iyer et al. 2013b). Class I da neurons exhibit selective innervations of dendritic territories and occupy relatively small receptive fields, whereas Class IV da neurons exhibit an elaborate space-filling network of dendrites that completely and nonredundantly tile the larval body wall. Protein synthesis and proteolysis gene classes appear differentially expressed and directly correlate with the complexity of the dendritic arbors of the two classes. In addition, genes associated with oxidation and mitochondria appear enriched in Class IV, suggesting underlying differences in their metabolic demands. Similarly, more transcription factors appeared differentially expressed and showed phenotypes in Class IV than in Class I (Iyer et al. 2013b). Transcription factors had already been shown to regulate dendrite morphogenesis (Parrish et al. 2006). Profiling experiments have provided an insight into their cell type-specific diversity, and have revealed their context-dependent functions. For example, some differentially expressed transcription factors showed phenotypes in both classes of da neurons, and in some cases showed opposing effects (Iyer et al. 2013b). Additionally, distinct transcription factors can regulate the expression of the same target gene in different cell types, but do so at different levels resulting in distinct dendritic arborization patterns (Hattori et al. 2013).
4.1.2 Wiring Specificity
In order to assemble a functional neural circuit, neurites need to discriminate between one another and form connections with their specific synaptic partners. Langley and Sperry proposed that molecular differences between neurons would account for their specific connectivity. These molecular differences can be readily identified through cell type-specific profiling experiments.
In C. elegans, the expression profiles of wild type and mutant motor neurons have been compared to address the specificity of motor circuit synapses. In wild type animals, VA and VB motor neurons arise as sister cells that adopt distinctive morphologies and synapse with separate sets of interneurons. In UNC-4 mutants, morphological differences are preserved; VAs, however, are miswired with inputs from interneurons normally restricted to their VB sisters. Thus UNC-4, together with the corepressor UNC-37 (Groucho), explicitly controls synaptic choice and not axonal growth or process placement, which could indirectly alter wiring specificity. Comparison of wild type versus UNC-4 mutant VA transcriptional profiles identified VB genes to be negatively regulated in VA motor neurons (Von Stetina et al. 2007a). Of these, CEH-12, an HB9 family member, functions downstream of UNC-4 to regulate synaptic choice. This study revealed a developmental switch in which motor neuron input is defined by the differential expression of transcription factors that select alternative presynaptic partners.
A recent approach to investigate wiring specificity in Drosophila has been to obtain the transcriptional profiles of developmentally related neurons with distinct connectivity patterns (Tan et al. 2015). This study characterized the cell surface membrane and secreted molecule complement of neuronal types with distinct connectivity patterns, and proposed a molecular strategy underlying the selection of synaptic partners. How many cell surface and secreted molecules a neuron expresses has been a long-standing question in the field. The relevance of this question resides in the fact that these types of molecules are the final effectors of cell–cell interactions, since they mediate contact-dependent recognition (through attraction/adhesion or repulsion events) and synapse assembly. Lamina neurons (L1-L5) and photoreceptors R7 and R8 all have a unique morphology, including layer-specific arborizations and connectivity patterns in the medulla neuropil. Their expression profiles were obtained at a developmental time point just prior to (R7, R8) or in the early stages of synapse formation (L1-L5). Using stringent settings, these neurons express between one-quarter to one-third (247 for R7 and 322 for L3) of the 976 genes encoding cell surface membrane and secreted molecules (CSMs) in the fly genome. While these neurons express roughly the same amount of CSMs, marked differences in the type of CSMs are observed between neurons. Classification of CSMs into families led to the detection of particular families with unique paralog combinations expressed in a cell type-specific fashion. One of these is the Dpr family, comprising 21 members. Detailed immunohistochemistry analysis of this family and the Dpr interacting protein (DIP) family (9 members) revealed colocalization of interacting Dpr and DIP members in layers where Dpr expressing lamina neurons and photoreceptors R7 and R8 establish synapses with medulla neurons. This suggests that Dpr–DIP interactions could regulate synaptic connections within a layer. Indeed, this study identified cell type-specific Dpr–DIP interactions between lamina neurons and the R7 photoreceptor and a subset of their synaptic partners. Supporting this notion, a recent study (Carrillo et al. 2015) has shown defects in a subset of R7 photoreceptors that make connections with DM8 neurons. Defects observed in R7 cells, when analyzing either mutations for the Dpr expressed in them or mutations in the DIP expressed in Dm8 cells, are consistent with synaptic defects.
The simplest interpretation is that the matching of Dpr and DIPs between synaptic partners specifies connections between them. It is possible that these interactions regulate other aspects of wiring specificity in DM8, such as viability through trophic support, given that a reduction in the number of DM8 cells is observed when the DIP expressed in these cells is mutated. More detailed genetic analysis will be required to definitively establish the precise function of Dpr–DIP ligand receptor interactions in circuit assembly.
4.1.3 Synaptogenesis
Neural circuit assembly requires coordination of recognition events between synaptic partners and the establishment of synaptic connections. Presynaptic development is a complex process, the study of which can be hindered by the complex temporal dynamics of neural development, with different types of neuron being born and establishing synaptic connections at different time points.
A recent study adopted a profiling approach to analyze the conversion of growth cones to synaptic terminals (Zhang et al. 2016), taking advantage of the synchronicity of this process in the Drosophila photoreceptor population. An analysis was conducted of mRNAs bound to ribosomes over time, thus reflecting protein rather than gene expression during this process. Consistent with the coordination of recognition events and presynaptic development, substantial changes were observed in many mRNAs encoding CSM, including those implicated in recognition and synapse formation. The pattern of expression suggests a massive restructuring of the neuron cell surface in closely spaced time points, with a downregulation of CSM preceding the transformation of growth cones to presynaptic terminals (35–40hrs after puparium formation), and a strong upregulation of CSM correlating with the first morphological manifestation of presynaptic differentiation (40–45hrs after puparium formation). Interestingly, changes in the levels of transcripts of synaptic molecules were modest. However, a doubling in the length of the 3′ UTRs for these transcripts was correlated with an increase in the number of binding sites for RNA binding proteins implicated in the regulation of mRNA localization, stability, and translation, which were expressed at constant levels. These findings suggest strong post-transcriptional regulation of presynaptic differentiation.
4.1.4 Remodeling
Neural circuits are remodeled by developmental signals and experience. This plasticity is embodied in structural changes that include dendrite and axon pruning and synapse relocation. The study of developmentally regulated plasticity through profiling experiments can uncover molecular components of remodeling programs.
Pruning of neuronal connections is a widely used mechanism in metazoan nervous systems to achieve a mature connectivity pattern. In Drosophila, early born mushroom body gamma neurons undergo axon pruning at the onset of metamorphosis in a process regulated by ecdysone. Comparison of wild type and ecdysone receptor mutants identified the upregulation of genes in the UPS (ubiquitin proteasome system), providing a mechanistic link to pruning. Unexpectedly, an RNA-binding protein promoting translation was identified as a negative regulator or developmental axon pruning, which suggests that post-transcriptional regulation might be an important mechanism regulating axon remodeling (Hoopfer et al. 2008).
C. elegans Dorsal D (DD) GABAergic motor neurons undergo stereotypical synaptic changes during development. Initially formed ventral DD synapses are relocated to the dorsal side with no evident changes in DD process morphology. Ventral D (VD) GABAergic motor neurons, which are functionally and structurally related to DDs, do not remodel due to the action of UNC-55, the COUP transcription factor homolog, which has been shown to function as a negative regulator of transcription. UNC-55 mutant VDs relocate synapses to the dorsal side, similar to DD developmental synapse relocation, and thus UNC-55 target genes would be enriched in this scenario compared to wild-type VDs. Profiling experiments identified the Iroquois homeodomain protein IRX-1 as both necessary and sufficient for synaptic remodeling (Petersen et al. 2011).
4.2 Physiological States and Functional Properties of Neurons
The link between gene expression and behavior is best exemplified in circadian rhythms, which result in cycling physiological states of neurons in the circuit. Pacemaker neurons possess molecular clocks that control gene expression. The circadian function of clock molecules is regulated by negative feedback loops of transcription and post-transcriptional modifications that modulate their stability and activity in a rhythmical fashion. The core clock then regulates transcription of other output molecules, which also accumulate rhythmically or have rhythmic activity. These output molecules regulate electrical activity rhythms to more directly generate overt circadian behavior.
In Drosophila, the circadian circuit is comprised of about 75 clock neurons on each side of the adult brain. Of these, two key groups of neurons control adult locomotor activity, which peaks twice a day in anticipation of dawn and dusk transitions. Genetic screens and microarray analysis from whole fly heads collected at different circadian times have identified many cycling mRNAs (100–200). However, given the existence of seven classes of neurons in the circuit, it is possible that mRNA cycling in only a small number of clock neurons is masked by non-cycling mRNAs in other neurons and head tissues. Furthermore, mRNAs that are only expressed in the clock neurons or in a subset of these should only comprise a tiny fraction of head RNA, and may therefore escape detection in both cycling and non-cycling analyses of head RNAs. Cell type-specific profiling has provided a means to address the above issues and has indeed identified genes that are expressed in subsets of distinct clock neurons and that affect distinct aspects of rhythms (Abruzzi et al. 2017; Nagoshi et al. 2010). Moreover, potent oscillations of gene expression have been observed in clock neurons, as well as enrichment of certain transcripts important for the neural function of clock neurons themselves, suggesting that some physiological aspects such as firing rhythms and/or electrical excitability may be rhythmically regulated (Flourakis and Allada 2015; Kula-Eversole et al. 2010; Ruben et al. 2012). Interestingly, altered electrical activity of clock neurons results in overt transcriptional changes involving a large set of circadian genes. This suggests a positive feedback loop between transcription and electrical activity, which would add robustness and precision to circadian behaviors (Mizrak et al. 2012).
An analysis of the circadian translatome of clock neurons has revealed that translation of most rhythmic transcripts coincides with behavioral quiescence, prior to initiation of locomotor activity, and thus protein synthesis may occur predominantly at circadian phases associated with reduced metabolic expenditure. In addition, the synchronized translation of functionally related mRNAs suggests a clock-orchestrated activation of biological processes (Huang et al. 2013).
Taken together, the knowledge gained from these profiling studies has revealed distinct mechanisms that regulate the rhythmic physiological state of distinct neuronal populations in the circadian circuit.
Functional specialization is a hallmark of sensory neurons. The C. elegans nervous system is richly endowed with sensory neurons. This organism navigates its environment by chemo-, thermo-, and aerotaxis, and thus exhibits behavioral responses to these types of stimuli. This is accomplished through 24 sensillar organs and some isolated sensory neurons. Most sensory neurons are characterized by the presence of ciliated endings. Many of the early studies focused on identifying chemotaxis mutants through genetic screens (Dusenbery 1974; Dusenbery et al. 1975; Ward 1973); however, this approach does not favor the detection of genes with redundant function or genes that give subtle phenotypes when mutated. Profiling complements genetic methods by providing a direct examination of genetic networks in a cell type-specific fashion (Blacque et al. 2005; Kunitomo et al. 2005; Zhang et al. 2002). Indeed, while it was through a genetic screen that the transcription factor DAF-19 was shown to regulate ciliated sensory neuron formation (Swoboda et al. 2000), gene expression analysis was necessary to obtain a transcriptome of ciliated neurons (Blacque et al. 2005; Kunitomo et al. 2005) and identify new ciliary components under the regulation of DAF-19 (Blacque et al. 2005). The genetic networks regulating the differentiation of touch receptor neurons and the ASE gustatory neuron have also been characterized. Cell type-specific profiling experiments combined with transcription factor motif discovery have started to unveil the regulatory logic behind sensory neuron differentiation programs (Etchberger et al. 2007; Zhang et al. 2002).
In addition, recent profiling studies have identified genes regulating the functional properties of particular types of sensory neurons. Using in vivo calcium imaging, Hallem and colleagues showed that CO2 specifically activates BAG neurons, and using profiling unveiled that their CO2-sensing function requires a particular type of cyclic nucleotide-gated ion channel and receptor-type guanylate cyclase (Hallem et al. 2011). Similarly, Chatzigeorgiou and colleagues have identified a distinct set of channels involved in responses to thermal and mechanical stimuli in polymodal nociceptor PVD neurons (Chatzigeorgiou et al. 2010). Thus, cell type-specific transcriptional analysis has shed light on the genetic programs regulating differentiation of sensory neurons and the molecular mechanisms that explain their physiological properties.
5 Perspectives and New Developments
Two of the main goals in the field are to achieve progress on the issue of neuronal classification and to work toward improving current profiling techniques.
The best functional classification of neurons would be one combining morphological and physiological data. Profiling based on cell types defined by morphology has revealed unknown physiological properties of the studied neurons. However, it has not given an overview of the physiological differences across morphologically defined cell types. In addition, cell type population profiling does not detect differences or variances among cells from a morphologically defined cell type. Two distinct complementary approaches are emerging as possible ways to address these issues: single-cell profiling, fluorescent in situ sequencing (FISSEQ) and Patch-seq.
In recent years, low input RNA-seq methods have been adapted to work in single cells (Tang et al. 2009). Single-cell RNA-seq (sc-RNA-seq) methods are now robust and economically practical, and are becoming a powerful tool for high-throughput, high-resolution transcriptome analysis (Liu and Trapnell 2016). Data analysis is not easy, since the low input material for scRNA-seq creates high levels of technical noise (Brennecke et al. 2013; Ding et al. 2015; Grün et al. 2014; Marinov et al. 2014). In addition, only around 10% of each cell’s transcript complement is represented in the final sequencing libraries (Islam et al. 2014), and this technique is unable to reliably detect low-abundance transcripts (Deng et al. 2014; Islam et al. 2014; Saliba et al. 2014). Many of the genes detected are housekeeping genes such as ribosomal subunits, and thus uninformative; therefore, reads from multiple cells must be combined to detect biologically meaningful gene expression differences between groups of single cells (Grün et al. 2014). Nevertheless, scRNA-seq has revealed intrapopulation heterogeneity in various tissues, including the brain (see the many references in the following reviews Poulin et al. 2016; Johnson and Walsh 2017).
Recently developed fluorescent in situ hybridization (FISH) techniques such as single-molecule FISH (sm-FISH) (Raj et al. 2008), which allows visualization of bright fluorescent spots that can be counted to determine the copy number of the gene of interest and its cellular location in individual cells, are typically performed on one RNA species at a time. Efforts to massively multiplex the sm-FISH imaging method have culminated in the development of multiplexed error-robust fluorescence in situ hybridization (MERFISH) (Chen et al. 2015). This method achieves large-scale multiplexing by assigning error-robust barcodes to different RNA species and then reading out these barcodes through successive rounds of hybridization and imaging on the same sample, so far up to 1000 genes. This technique can extend the benefits of sm-FISH toward the transcriptome scale. However, in situ hybridization techniques rely on a defined set of probes. Church and colleagues developed an unbiased and transcriptome-wide sampling method for quantitative visualization of RNA in situ. This technique is called fluorescent in situ sequencing (FISSEQ) (Lee et al. 2014, 2015), and combines the benefits of in situ hybridization with RNA-seq. It is based on the generation of stably cross-linked complementary DNA (cDNA) amplicons, which are sequenced manually on a confocal microscope within the biological sample. FISSEQ enriches biologically active genes, enabling the discrimination of cell type-specific processes with a small number of reads. However, it is not clear how such enrichment occurs. It has been proposed that active RNA molecules are more accessible to FISSEQ than ribosomal transcripts trapped in ribonucleoproteins, spliceosomes, or stress granules. Further elimination of still remaining transcripts of this nature (i.e., using random priming with rRNA depletion) will increase the number of cell-specific reads and enable FISSEQ to generate single-cell gene expression profiling that is biologically meaningful (Lee et al. 2015). Alternatively, sc-RNAseq is starting to be combined with tissue reference maps (for examples see the review by Moor and Itzkovitz 2017).
Patch-seq is a method that combines whole cell electrophysiological recordings, sc-RNA-seq and morphological characterization. This technique has been used to characterize pyramidal cells and cortical interneurons (Fuzik et al. 2016; Cadwell et al. 2016). While the efficiency of mRNA capture in Patch-seq is lower than that of in sc-RNA-seq on dissociated tissues is still sufficient to sample genes with low expression. This allowed to make inferences on the specificity and heterogeneity of afferent inputs for different cell types (Fuzik et al. 2016) and the identification of genes associated to neurological disorders such as autism and schizophrenia in particular neuronal subtypes (Cadwell et al. 2016). These studies were also able to render associations between the expression of ion channels and synapse-related proteins and biophysical parameters of action potentials. Thus, Patch-seq has an enormous potential in the vertebrate brain to precisely map neuronal subtypes and predict their network contributions in the brain.
Both sc-RNAseq and FISSEQ could in principle be easily adapted to invertebrates. Patch-seq will depend on the development of electrophysiological probes suitable for the small size of Drosophila and C. elegans neurons. Importantly, especially in Drosophila, Patch-seq could be done in vivo for behaviors that can be assessed in tethered flies.
6 Concluding Remarks
Gene expression profiling approaches are making important contributions to the understanding of neural circuit structure and function. Gene expression profiling experiments can address various biological questions depending on their design. Initial experiments characterized broad neuronal populations by identifying enriched transcripts versus the whole animal or neural tissue reference sample. However, as a result of advances in technology and knowledge, researchers are shifting their focus to discrete neuronal cell types. Thus, they are now addressing questions such as what genes determine the unique morphology or physiology of related neuronal cell types, by comparing their gene expression patterns, or what are the genetic programs and downstream molecular determinants that drive these differences when comparing wild type versus genetically manipulated gene expression in a particular neuronal cell type. These types of profiling experiments have shown clear potential for discovery and are becoming increasingly popular.
Nevertheless, the qualitative and quantitative information obtained from gene expression analysis of a particular neuronal cell type will always be dependent on two factors: (1) the definition of cell type and (2) the specificity of the data obtained depending on the profiling method used.
Cell types are often arbitrarily defined by the expression of markers or their morphology. However, it is possible that definition by these criteria can include heterogeneous neurons, even in a small population. This is exemplified in a recent study analyzing R7 photoreceptors and their major postsynaptic partner DM8 neurons. A specific subset of DM8 neurons was identified, and based on genetic analysis of mutants, suggested to be selectively targeted by a subset of R7 cells (yR7) (Carrillo et al. 2015). In these scenarios, discerning between different types of neurons might require complementary knowledge such as the electrophysiological properties of discrete neurons in the population or detailed connectivity maps. Obtaining this type of data might not be feasible for certain neuronal cell types and/or in certain organisms.
All profiling techniques have their advantages and disadvantages. A major concern in profiling approaches based on cell/nuclei isolation is the potential transcriptional changes caused by the cellular stress associated with dissociation procedures. It is assumed that in experiments designed to pinpoint differential expression between cell types, these transcriptional responses will be equal in both cell types, and thus will not interfere in the bioinformatic identification of differentially expressed transcripts. However, it is possible that distinct neurons present different sensitivity to cellular stress. In addition, if the aim is simply to characterize the gene expression profile of a particular cell type, these techniques will not differentiate between naturally expressed genes versus gene expression caused by dissociation stress. The main issue with profiling techniques that do not involve cell/nuclei isolation is nonspecific contamination by RNA in the total sample. Considerable efforts are being made to improve protocols and strategies in order to minimize this type of contamination. However, the smaller the size of neuronal cell type population under study versus the tissue sample used, the lower the signal-to-noise ratio. Where there is sufficient knowledge about the cell type being studied, unspecific contamination can be identified. For example, if the neurotransmitter identity is known, the presence of other neurotransmitters can be a sign of the presence of contaminating transcripts. However, in cases where there is little knowledge about the cell type being studied, caution is required when interpreting the data obtained.
Single-cell transcriptional profiling using microarrays and specially RNA-seq is emerging as a technology that sheds light on cell type identification in the nervous system. While signal-to-noise ratio can be an issue (Brennecke et al. 2013; Grün et al. 2014; Wu et al. 2014), improvements to reduce noise and the development of microfluidic technology to perform parallel sequencing of large numbers of cells simultaneously will contribute greatly to unveiling neural heterogeneity. In addition, the pursuit of knowledge will lead to the development of techniques that minimize both dissociation stress and nonspecific RNA contamination. This has already commenced with the development of fluorescence in situ sequencing (FISSEQ) (Lee et al. 2014, 2015). This technique has achieved RNA sequencing in cells within their tissue by combining biochemical with fluorescence imaging processing steps. One can envisage that high-throughput parallel single-cell FISSEQ would address both neural heterogeneity and the technical issues associated with profiling techniques. Moreover, Patch-seq is arising as an approach to classify neurons based on their physiology as well as their morphology and gene expression pattern. The endless creativity of researchers and multidisciplinary collaborative efforts will certainly push technology toward such new frontiers.
References
Abruzzi K, Chen X, Nagoshi E, Zadina A, Rosbash M (2015) RNA-seq profiling of small numbers of Drosophila neurons. Methods Enzymol 551:369–386
Abruzzi KC, Zadina A, Luo W, Wiyanto E, Rahman R, Guo F, Shafer O, Rosbash M (2017) RNA-seq analysis of Drosophila clock and non-clock neurons reveals neuron-specific cycling and novel candidate neuropeptides. PLoS Genet 13(2):e1006613. doi: 10.1371/journal.pgen.1006613 (eCollection 2017 Feb)
Aughey GN, Southall TD (2016) Dam it’s good! DamID profiling of protein-DNA interactions. Wiley Interdiscip Rev Dev Biol 5:25–37
Bacaj T, Shaham S (2007) Temporal control of cell-specific transgene expression in Caenorhabditis elegans. Genetics 176:2651–2655
Barthelson RA, Lambert GM, Vanier C, Lynch RM, Galbraith DW (2007) Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells. BMC Genom 8:340
Bateman JR, Lee AM, Wu C (2006) Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173:769–777
Blacque OE, Perens EA, Boroevich KA, Inglis PN, Li C, Warner A, Khattra J, Holt RA, Ou G, Mah AK et al (2005) Functional genomics of the cilium, a sensory organelle. Curr Biol 15:935–941
Bonn S, Zinzen RP, Perez-Gonzalez A, Riddell A, Gavin A-C, Furlong EEM (2012a) Cell type-specific chromatin immunoprecipitation from multicellular complex samples using BiTS-ChIP. Nat Protoc 7:978–994
Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczyński B, Riddell A, Furlong EEM (2012b) Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44:148–156
Bossing T, Barros CS, Fischer B, Russell S, Shepherd D (2012) Disruption of microtubule integrity initiates mitosis during CNS repair. Dev Cell 23:433–440
Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401–415
Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC et al (2013) Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods 10:1093–1095
Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, Sandberg R, Tolias AS (2016) Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat Biotechnol 34:199–203. doi: 10.1038/nbt.3445
Cao J, Ni J, Ma W, Shiu V, Milla LA, Park S, Spletter ML, Tang S, Zhang J, Wei X et al (2014) Insight into insulin secretion from transcriptome and genetic analysis of insulin-producing cells of Drosophila. Genetics 197:175–192
Carrillo RA, Özkan E, Menon KP, Nagarkar-Jaiswal S, Lee P-T, Jeon M, Birnbaum ME, Bellen HJ, Garcia KC, Zinn K (2015) Control of synaptic connectivity by a network of Drosophila IgSF cell surface proteins. Cell 163:1770–1782
Chatzigeorgiou M, Yoo S, Watson JD, Lee W-H, Spencer WC, Kindt KS, Hwang SW, Miller DM, Treinin M, Driscoll M et al (2010) Specific roles for DEG/ENaC and TRP channels in touch and thermosensation in C. elegans nociceptors. Nat Neurosci 13:861–868
Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X (2015) RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348:aaa6090
Cinar H, Keles S, Jin Y (2005) Expression profiling of GABAergic motor neurons in Caenorhabditis elegans. Curr Biol 15:340–346
Cleary MD, Meiering CD, Jan E, Guymon R, Boothroyd JC (2005) Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat Biotechnol 23:232–237
Colosimo ME, Brown A, Mukhopadhyay S, Gabel C, Lanjuin AE, Samuel ADT, Sengupta P (2004) Identification of thermosensory and olfactory neuron-specific genes via expression profiling of single neuron types. Curr Biol 14:2245–2251
Couto A, Alenius M, Dickson BJ (2005) Molecular, anatomical, and functional organization of the Drosophila olfactory system. Curr Biol 15:1535–1547
Crocker A, Guan X-J, Murphy CT, Murthy M (2016) Cell-type-specific transcriptome analysis in the Drosophila mushroom body reveals memory-related changes in gene expression. Cell Rep 1–17
Davis MW, Morton JJ, Carroll D, Jorgensen EM (2008) Gene activation using FLP recombinase in C. elegans. PLoS Genet 4:e1000028
Deal RB, Henikoff S (2010) A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell 18:1030–1040
del Valle Rodríguez A, Didiano D, Desplan C (2012) Power tools for gene expression and clonal analysis in Drosophila. Nat Methods 9:47–55
Deng Q, Ramsköld D, Reinius B, Sandberg R (2014) Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343:193–196
Ding B, Zheng L, Zhu Y, Li N, Jia H, Ai R, Wildberg A, Wang W (2015) Normalization and noise reduction for single cell RNA-seq experiments. Bioinformatics 31:2225–2227
Dupuy D, Li Q-R, Deplancke B, Boxem M, Hao T, Lamesch P, Sequerra R, Bosak S, Doucette-Stamm L, Hope IA et al (2004) A first version of the Caenorhabditis elegans promoterome. Genome Res 14:2169–2175
Dupuy D, Bertin N, Hidalgo CA, Venkatesan K, Tu D, Lee D, Rosenberg J, Svrzikapa N, Blanc A, Carnec A et al (2007) Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat Biotechnol 25:663–668
Dusenbery DB (1974) Analysis of chemotaxis in the nematode Caenorhabditis elegans by countercurrent separation. J Exp Zool 188:41–47
Dusenbery DB, Sheridan RE, Russell RL (1975) Chemotaxis-defective mutants of the nematode Caenorhabditis elegans. Genetics 80:297–309
Easow G, Teleman AA, Cohen SM (2007) Isolation of microRNA targets by miRNP immunopurification. RNA 13:1198–1204
Etchberger JF, Lorch A, Sleumer MC, Zapf R, Jones SJ, Marra MA, Holt RA, Moerman DG, Hobert O (2007) The molecular signature and cis-regulatory architecture of a C. elegans gustatory neuron. Genes Dev 21:1653–1674
Fischbach K-F, Dittrich AP (1989) The optic lobe of Drosophila melanogaster. I: A. Golgi analysis of wild-type structure. Cell Tissue Res 258:441–475
Fischer JA, Giniger E, Maniatis T, Ptashne M (1988) GAL4 activates transcription in Drosophila. Nature 332:853–856
Flourakis M, Allada R (2015) Patch-clamp electrophysiology in Drosophila circadian pacemaker neurons. Methods Enzymol 552:23–44. doi: 10.1016/bs.mie.2014.10.005
Fortini ME, Rubin GM (1990) Analysis of cis-acting requirements of the Rh3 and Rh4 genes reveals a bipartite organization to rhodopsin promoters in Drosophila melanogaster. Genes Dev 4:444–463
Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM (2005) A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genom 6:42
Fuzik J, Zeisel A, Máté Z, Calvigioni D, Yanagawa Y, Szabó G, Linnarsson S, Harkany T (2016) Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat Biotechnol 34:175–183. doi: 10.1038/nbt.3443
Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K et al (2010) Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330:1775–1787
Gohl DM, Silies MA, Gao XJ, Bhalerao S, Luongo FJ, Lin C-C, Potter CJ, Clandinin TR (2011) A versatile in vivo system for directed dissection of gene expression patterns. Nat Methods 8:231–237
Grün D, Kester L, van Oudenaarden A (2014) Validation of noise models for single-cell transcriptomics. Nat Methods 11:637–640
Hadjieconomou D, Rotkopf S, Alexandre C, Bell DM, Dickson BJ, Salecker I (2011) Flybow: genetic multicolor cell labeling for neural circuit analysis in Drosophila melanogaster. Nat Methods 8:260–266
Haenni S, Ji Z, Hoque M, Rust N, Sharpe H, Eberhard R, Browne C, Hengartner MO, Mellor J, Tian B et al (2012) Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res 40:6304–6318
Hallem EA, Spencer WC, McWhirter RD, Zeller G, Henz SR, Rätsch G, Miller DM, Horvitz HR, Sternberg PW, Ringstad N (2011) Receptor-type guanylate cyclase is required for carbon dioxide sensation by Caenorhabditis elegans. Proc Natl Acad Sci U S A 108:254–259
Hattori Y, Usui T, Satoh D, Moriyama S, Shimono K, Itoh T, Shirahige K, Uemura T (2013) Sensory-neuron subtype-specific transcriptional programs controlling dendrite morphogenesis: genome-wide analysis of Abrupt and Knot/Collier. Dev Cell 27:530–544
Hayashi S, Ito K, Sado Y, Taniguchi M, Akimoto A, Takeuchi H, Aigaki T, Matsuzaki F, Nakagoshi H, Tanimura T et al (2002) GETDB, a database compiling expression patterns and molecular locations of a collection of Gal4 enhancer traps. Genesis 34:58–61
He M, Liu Y, Wang X, Zhang MQ, Hannon GJ, Huang ZJ (2012) Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron 73:35–48
Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ et al (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell 135:738–748
Henry GL, Davis FP, Picard S, Eddy SR (2012) Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res 40:9691–9704
Hoopfer ED, Penton A, Watts RJ, Luo L (2008) Genomic analysis of Drosophila neuronal remodeling: a role for the RNA-binding protein Boule as a negative regulator of axon pruning. J Neurosci 28:6092–6103
Hope IA (1991) “Promoter trapping” in Caenorhabditis elegans. Development 113:399–408
Huang Y, Ainsley JA, Reijmers LG, Jackson FR (2013) Translational profiling of clock cells reveals circadianly synchronized protein synthesis. PLoS Biol 11:e1001703
Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, Halfnight E, Lee D, Lin J, Lorch A et al (2007) High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol 5:e237
Hutter H (2003) Extracellular cues and pioneers act together to guide axons in the ventral cord of C. elegans. Development 130:5307–5318
Isik M, Korswagen HC, Berezikov E (2010) Expression patterns of intronic microRNAs in Caenorhabditis elegans. Silence 1:5
Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S (2014) Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 11:163–166
Iyer EPR, Cox DN (2010) Laser capture microdissection of Drosophila peripheral neurons. J Vis Exp
Iyer EPR, Iyer SC, Cox DN (2013a) Application of cell-specific isolation to the study of dopamine signaling in Drosophila. Methods Mol Biol 964:215–225
Iyer EPR, Iyer SC, Sullivan L, Wang D, Meduri R, Graybeal LL, Cox DN (2013b) Functional genomic analyses of two morphologically distinct classes of Drosophila sensory neurons: post-mitotic roles of transcription factors in dendritic patterning. PLoS ONE 8:e72434
Jasper H, Benes V, Atzberger A, Sauer S, Ansorge W, Bohmann D (2002) A genomic switch at the transition from cell proliferation to terminal differentiation in the Drosophila eye. Dev Cell 3:511–521
Jenett A, Rubin GM, Ngo TTB, Shepherd D, Murphy C, Dionne H, Pfeiffer BD, Cavallaro A, Hall D, Jeter J et al (2012) A GAL4-driver line resource for Drosophila neurobiology. Cell Rep 2:991–1001
Jin Y, Ha N, Forés M, Xiang J, Gläßer C, Maldera J, Jiménez G, Edgar BA (2015) EGFR/Ras signaling controls Drosophila intestinal stem cell proliferation via capicua-regulated genes. PLoS Genet 11:e1005634
Johnson MB, Walsh CA (2017) Cerebral cortical neuron diversity and development at single-cell resolution. Curr Opin Neurobiol 42:9–16. doi: 10.1016/j.conb.2016.11.001
Kitamoto T, Ikeda K, Salvaterra PM (1992) Analysis of cis-regulatory elements in the 5′ flanking region of the Drosophila melanogaster choline acetyltransferase gene. J Neurosci 12:1628–1639
Kudlow BA, Zhang L, Han M (2012) Systematic analysis of tissue-restricted miRISCs reveals a broad role for microRNAs in suppressing basal activity of the C. elegans pathogen response. Mol Cell 46:530–541
Kula-Eversole E, Nagoshi E, Shang Y, Rodriguez J, Allada R, Rosbash M (2010) Surprising gene expression patterns within and between PDF-containing circadian neurons in Drosophila. Proc Natl Acad Sci U S A 107:13497–13502
Kunitomo H, Uesugi H, Kohara Y, Iino Y (2005) Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol 6:R17
Kvon EZ, Stampfel G, Yáñez-Cuna JO, Dickson BJ, Stark A (2012) HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev 26:908–913
Kvon EZ, Kazmar T, Stampfel G, Yáñez-Cuna JO, Pagani M, Schernhuber K, Dickson BJ, Stark A (2014) Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature
Lai S-L, Lee T (2006) Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat Neurosci 9:703–709
Lai S-L, Miller MR, Robinson KJ, Doe CQ (2012) The Snail family member Worniu is continuously required in neuroblasts to prevent Elav-induced premature differentiation. Dev Cell 23:849–857
Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22:451–461
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SSF, Li C, Amamoto R et al (2014) Highly multiplexed subcellular RNA sequencing in situ. Science (80-) 343:1360–1363
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Terry R, Turczyk BM, Yang JL, Lee HS, Aach J et al (2015) Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc 10:442–458
Li W, Ou G (2016) The application of somatic CRISPR-Cas9 to conditional genome editing in Caenorhabditis elegans. Genesis 54:170–181
Liu S, Trapnell C (2016) Single-cell transcriptome sequencing: recent advances and remaining challenges. F1000Research 5
Luan H, Peabody NC, Vinson CR, White BH (2006) Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron 52:425–436
Lynch AS, Briggs D, Hope IA (1995) Developmental expression pattern screen for genes predicted in the C. elegans genome sequencing project. Nat Genet 11:309–313
Ma J, Ptashne M (1987) The carboxy-terminal 30 amino acids of GAL4 are recognized by GAL80. Cell 50:137–142
Ma J, Weake VM (2014) Affinity-based isolation of tagged nuclei from Drosophila tissues for gene expression analysis. J Vis Exp
Marinov GK, Williams BA, McCue K, Schroth GP, Gertz J, Myers RM, Wold BJ (2014) From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res 24:496–510
Marshall OJ, Southall TD, Cheetham SW, Brand AH (2016) Cell-type-specific profiling of protein-DNA interactions without cell isolation using targeted DamID with next-generation sequencing. Nat Protoc 11:1586–1598. doi: 10.1038/nprot.2016.084
Martinez NJ, Ow MC, Reece-Hoyes JS, Barrasa MI, Ambros VR, Walhout AJM (2008) Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res 18:2005–2015
McClure CD, Southall TD (2015) Getting down to specifics: profiling gene expression and protein-DNA interactions in a cell type-specific manner. Adv Genet 91:103–151
McGuire SE, Le PT, Davis RL (2001) The role of Drosophila mushroom body signaling in olfactory memory. Science 293:1330–1333
Miller MR, Robinson KJ, Cleary MD, Doe CQ (2009) TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat Methods 6:439–441
Miyazaki T, Ito K (2010) Neural architecture of the primary gustatory center of Drosophila melanogaster visualized with GAL4 and LexA enhancer-trap systems. J Comp Neurol 518:4147–4181
Mizrak D, Ruben M, Myers GN, Rhrissorrakrai K, Gunsalus KC, Blau J (2012) Electrical activity can impose time of day on the circadian transcriptome of pacemaker neurons. Curr Biol 22:1871–1880
modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L et al (2010). Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330:1787–1797
Moor AE, Itzkovitz S (2017) Spatial transcriptomics: paving the way for tissuelevel systems biology. Curr Opin Biotechnol 46:126–133. doi: 10.1016/j.copbio.2017.02.004
Nagoshi E, Sugino K, Kula E, Okazaki E, Tachibana T, Nelson S, Rosbash M (2010) Dissecting differential gene expression within the circadian neuronal circuit of Drosophila. Nat Neurosci 13:60–68
Nègre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R et al (2011) A cis-regulatory map of the Drosophila genome. Nature 471:527–531
Nern A, Pfeiffer BD, Svoboda K, Rubin GM (2011) Multiple new site-specific recombinases for use in manipulating animal genomes. Proc Natl Acad Sci U S A 108:14198–14203
Nilsson H, Krawczyk KM, Johansson ME (2014) High salt buffer improves integrity of RNA after fluorescence-activated cell sorting of intracellular labeled cells. J Biotechnol 192 Pt A:62–65
O’Kane CJ, Gehring WJ (1987) Detection in situ of genomic regulatory elements in Drosophila. Proc Natl Acad Sci U S A 84:9123–9127
Otsuki L, Cheetham SW, Brand AH (2014) Freedom of expression: cell-type-specific gene profiling. Wiley Interdiscip Rev Dev Biol 3:429–443
Paix A, Folkmann A, Rasoloson D, Seydoux G (2015) High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics 201:47–54
Parrish JZ, Kim MD, Jan LY, Jan YN (2006) Genome-wide analyses identify transcription factors required for proper morphogenesis of Drosophila sensory neuron dendrites. Genes Dev 20:820–835
Parrish JZ, Kim CC, Tang L, Bergquist S, Wang T, Derisi JL, Jan LY, Jan YN, Davis GW (2014) Krüppel mediates the selective rebalancing of ion channel expression. Neuron 82:537–544
Petersen SC, Watson JD, Richmond JE, Sarov M, Walthall WW, Miller DM (2011) A transcriptional program promotes remodeling of GABAergic synapses in Caenorhabditis elegans. J Neurosci 31:15362–15375
Pfeiffer BD, Jenett A, Hammonds AS, Ngo T-TB, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR et al (2008) Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci U S A 105:9715–9720
Pfeiffer BD, Ngo T-TB, Hibbard KL, Murphy C, Jenett A, Truman JW, Rubin GM (2010) Refinement of tools for targeted gene expression in Drosophila. Genetics 186:735–755
Pfeiffer BD, Truman JW, Rubin GM (2012) Using translational enhancers to increase transgene expression in Drosophila. Proc Natl Acad Sci U S A 109:6626–6631
Potter CJ, Tasic B, Russler EV, Liang L, Luo L (2010) The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell 141:536–548
Poulin J-F, Tasic B, Hjerling-Leffler J, Trimarchi JM, Awatramani R (2016) Disentangling neural cell diversity using single-cell transcriptomics. Nat Neurosci 19:1131–1141. doi: 10.1038/nn.4366
Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5:877–879
Reece-Hoyes JS, Shingles J, Dupuy D, Grove CA, Walhout AJM, Vidal M, Hope IA (2007) Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genom 8:27
Riabinina O, Luginbuhl D, Marr E, Liu S, Wu MN, Luo L, Potter CJ (2015) Improved and expanded Q-system reagents for genetic manipulations. Nat Methods 12:219–222, 5 p following 222
Roy PJ, Stuart JM, Lund J, Kim SK (2002) Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418:975–979
Ruben M, Drapeau MD, Mizrak D, Blau J (2012) A mechanism for circadian control of pacemaker neuron excitability. J Biol Rhythms 27:353–364
Russell JN, Clements JE, Gama L (2013) Quantitation of gene expression in formaldehyde-fixed and fluorescence-activated sorted cells. PLoS ONE 8:e73849
Saliba A-E, Westermann AJ, Gorski SA, Vogel J (2014) Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 42:8845–8860
Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS (2009) Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106:13939–13944
Shi B, Guo X, Wu T, Sheng S, Wang J, Skogerbø G, Zhu X, Chen R (2009) Genome-scale identification of Caenorhabditis elegans regulatory elements by tiling-array mapping of DNase I hypersensitive sites. BMC Genom 10:92
Smith CJ, Watson JD, Spencer WC, O’Brien T, Cha B, Albeg A, Treinin M, Miller DM (2010) Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Dev Biol 345:18–33
Smith CJ, O’Brien T, Chatzigeorgiou M, Spencer WC, Feingold-Link E, Husson SJ, Hori S, Mitani S, Gottschalk A, Schafer WR et al (2013) Sensory neuron fates are distinguished by a transcriptional switch that regulates dendrite branch stabilization. Neuron 79:266–280
Southall TD, Gold KS, Egger B, Davidson CM, Caygill EE, Marshall OJ, Brand AH (2013) Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell 26:101–112
Spencer WC, McWhirter R, Miller T, Strasbourger P, Thompson O, Hillier LW, Waterston RH, Miller DM (2014) Isolation of specific neurons from C. elegans larvae for gene expression profiling. PLoS ONE 9:e112102
Steiner FA, Talbert PB, Kasinathan S, Deal RB, Henikoff S (2012) Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res 22:766–777
Suster ML, Seugnet L, Bate M, Sokolowski MB (2004) Refining GAL4-driven transgene expression in Drosophila with a GAL80 enhancer-trap. Genesis 39:240–245
Swoboda P, Adler HT, Thomas JH (2000) The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. elegans. Mol Cell 5:411–421
Szüts D, Bienz M (2000) LexA chimeras reveal the function of Drosophila Fos as a context-dependent transcriptional activator. Proc Natl Acad Sci U S A 97:5351–5356
Tahayato A, Sonneville R, Pichaud F, Wernet MF, Papatsenko D, Beaufils P, Cook T, Desplan C (2003) Otd/Crx, a dual regulator for the specification of ommatidia subtypes in the Drosophila retina. Dev Cell 5:391–402
Takayama J, Faumont S, Kunitomo H, Lockery SR, Iino Y (2010) Single-cell transcriptional analysis of taste sensory neuron pair in Caenorhabditis elegans. Nucleic Acids Res 38:131–142
Tan L, Zhang KX, Pecot MY, Nagarkar-Jaiswal S, Lee P-T, Takemura S-Y, McEwen JM, Nern A, Xu S, Tadros W et al (2015) Ig superfamily ligand and receptor pairs expressed in synaptic partners in Drosophila. Cell 163:1756–1769
Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A et al (2009) mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6:377–382
Thomas A, Lee P-J, Dalton JE, Nomie KJ, Stoica L, Costa-Mattioli M, Chang P, Nuzhdin S, Arbeitman MN, Dierick HA (2012) A versatile method for cell-specific profiling of translated mRNAs in Drosophila. PLoS ONE 7:e40276
Ting C-Y, Gu S, Guttikonda S, Lin T-Y, White BH, Lee C-H (2011) Focusing transgene expression in Drosophila by coupling Gal4 with a novel split-LexA expression system. Genetics 188:229–233
van Steensel B, Henikoff S (2000) Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol 18:424–428
van Steensel B, Delrow J, Henikoff S (2001) Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet 27:304–308
Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA et al (2011) MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods 8:737–743
Von Stetina SE, Fox RM, Watkins KL, Starich TA, Shaw JE, Miller DM (2007a) UNC-4 represses CEH-12/HB9 to specify synaptic inputs to VA motor neurons in C. elegans. Genes Dev 21:332–346
Von Stetina SE, Watson JD, Fox RM, Olszewski KL, Spencer WC, Roy PJ, Miller DM (2007b) Cell-specific microarray profiling experiments reveal a comprehensive picture of gene expression in the C. elegans nervous system. Genome Biol 8:R135
Voutev R, Hubbard EJA (2008) A “FLP-Out” system for controlled gene expression in Caenorhabditis elegans. Genetics 180:103–119
Wang H, Liu J, Gharib S, Chai CM, Schwarz EM, Pokala N, Sternberg PW (2017) cGAL, a temperature-robust GAL4-UAS system for Caenorhabditis elegans. Nat Methods 14:145–148. doi:10.1038/nmeth.4109 (Epub 2016 Dec 19)
Ward S (1973) Chemotaxis by the nematode Caenorhabditis elegans: identification of attractants and analysis of the response by use of mutants. Proc Natl Acad Sci U S A 70:817–821
Wei X, Potter CJ, Luo L, Shen K (2012) Controlling gene expression with the Q repressible binary expression system in Caenorhabditis elegans. Nat Methods 9:391–395
White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314:1–340
Williams CR, Baccarella A, Parrish JZ, Kim CC (2016) Trimming of sequence reads alters RNA-Seq gene expression estimates. BMC Bioinformatics 17:103
Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF et al (2014) Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 11:41–46
Xu J, Ren X, Sun J, Wang X, Qiao HH, Xu BW, Liu LP, Ni JQ (2015) A toolkit of CRISPR-based genome editing systems in Drosophila. J Genet Genomics 42:141–149
Yagi R, Mayer F, Basler K (2010) Refined LexA transactivators and their use in combination with the Drosophila Gal4 system. Proc Natl Acad Sci U S A 107:16166–16171
Yang Z, Edenberg HJ, Davis RL (2005) Isolation of mRNA from specific tissues of Drosophila by mRNA tagging. Nucleic Acids Res 33:e148
Zaslaver A, Liani I, Shtangel O, Ginzburg S, Yee L, Sternberg PW (2015) Hierarchical sparse coding in the sensory system of Caenorhabditis elegans. Proc Natl Acad Sci U S A 112:1185–1189
Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M (2002) Identification of genes expressed in C. elegans touch receptor neurons. Nature 418:331–335
Zhang S, Ma C, Chalfie M (2004) Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell 119:137–144
Zhang L, Ding L, Cheung TH, Dong M-Q, Chen J, Sewell AK, Liu X, Yates JR, Han M (2007) Systematic identification of C. elegans miRISC proteins, miRNAs, and mRNA targets by their interactions with GW182 proteins AIN-1 and AIN-2. Mol Cell 28:598–613
Zhang C, Barthelson RA, Lambert GM, Galbraith DW (2008) Global characterization of cell-specific gene expression through fluorescence-activated sorting of nuclei. Plant Physiol 147:30–40
Zhang KX, Tan L, Pellegrini M, Zipursky SL, McEwen JM (2016) Rapid changes in the translatome during the conversion of growth cones to synaptic terminals. Cell Rep 14:1258–1271
Acknowledgements
The author apologizes to those whose work was not cited due to space constraints, and thanks A. Ferrús, T. Southall, L. Broday, and A. Sapir for critical reading, thoughtful comments, and insightful suggestions on this manuscript. M. Morey is supported by a Ramón y Cajal contract (RYC-2011-09479) and a Ministerio de Economía y Competitividad grant (BFU2015-69689-P).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Morey, M. (2017). Transcriptional Profiling of Identified Circuit Elements in Invertebrates. In: Çelik, A., Wernet, M. (eds) Decoding Neural Circuit Structure and Function. Springer, Cham. https://doi.org/10.1007/978-3-319-57363-2_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-57363-2_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57362-5
Online ISBN: 978-3-319-57363-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)