
Abstract
The current standard of care for initial treatment of adults with newly diagnosed glioblastoma (GBM) constitutes maximal safe resection followed by concurrent chemoradiation therapy and adjuvant chemotherapy with temozolomide; recent studies have shown improvement in survival benefit with the addition of tumor treatment fields (TTF) to this regimen. For recurrent disease, lomustine and bevacizumab yield benefit in progression-free survival but not in overall survival. Recent advances in the understanding of the biology of GBM have provided the basis for new therapeutic approaches against these tumors. However, the initial promise of agents targeted against specific pathways active in GBM failed to yield the expected improvement in outcome likely due to intra- and inter-tumoral heterogeneity. Current efforts are focused on immunotherapy, biological agents and combination targeted therapies to overcome these challenges. Additionally, ongoing research to better understand the basis of tumor heterogeneity is expected to provide new insights that can help broadly target GBMs that can translate into improved survival and quality of life for these patients. This review provides an outline of current treatments and examines the newer approaches that bear promise to provide a meaningful improvement in outcome of patients with GBM.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
Glioblastoma (GBM) is the most common primary brain tumor in adults and is associated with a dismal outcome. Several new therapeutic strategies have been developed over the past 4 decades that have provided new standards of care which have resulted in improvement in overall survival (OS). However, these improvements have been only incremental in nature and no significant shifts in paradigms of care have yet emerged to provide the expected dramatic improvements in survival. There is a growing realization that new approaches to treatment are needed to address the complexity and heterogeneity of GBM; this in turn has led to exploring exciting new avenues of treatment including biological therapies, immunotherapies, nanotherapies, and technology-based treatment modalities. Ongoing clinical trials are expected to better characterize the relative efficacies of such approaches and identify the ones that may potentially provide the anticipated paradigm-shifting therapy that significant improves survival with good quality of life. This review provides a comprehensive outline of the rationale for current standards of care for newly diagnosed and recurrent GBM and examines the development of promising novel approaches to therapy against these tumors.
3.2 Newly Diagnosed Glioblastoma
Current treatment for patients with newly diagnosed GBM includes maximal safe resection followed by concurrent chemoradiation therapy and adjuvant chemotherapy (chemoRT) with temozolomide (TMZ) , a monofunctional alkylating agent. This multimodal approach leads to a median OS of 15–17 months and notably improved 2- and 5-year survival compared to radiation therapy (RT) alone. New standards have also been established in elderly patients with GBM who have a good functional status. While these standards have provided a modest yet significant improvement in outcome for GBM patients, much remains to be achieved in further meaningfully improving survival and quality of life for these patients. Novel insights into genetic and epigenetic characteristics of GBM gained in recent years aim to provide new therapeutic strategies to bring a paradigm shift in treatment in these patients (Hegi et al. 2005) (Table 3.1).
3.2.1 Surgical Resection
Surgical resection in patients with newly diagnosed GBM is aimed initially at providing sufficient tissue for histologic and molecular diagnosis, reducing mass effect, and relieving symptoms. Whether extent of resection can improve overall survival has been less clear due to the absence of level 1 evidence for the same. However, cumulative evidence from several studies has strongly supported the potential for survival benefit with a greater extent of surgical resection. In the first major study assessing the effect of extent of resection in patients with newly diagnosed GBM, Lacroix et al. reported a single center retrospective study of 416 patients which showed that increasing extent of resection of the enhancing portion of the tumor ≥89% provided improved survival for every unit increase in volumetric resection (Lacroix et al. 2001). A subsequent retrospective study of a series of 500 patients by Sanai et al. again supported this finding with benefit being seen even in patients who underwent a partial resection (≥78%) and improving in a stepwise manner with greater extent of resection (Sanai et al. 2011). In a more recent large retrospective study expanding on the analysis by Lacroix et al. from the same center, Li et al. not only confirmed improvement in survival resulting from maximal resection of the enhancing part of the tumor in 1229 patients with newly diagnosed GBM, but also reported that increasing the extent of resection to safely remove the non-enhancing hyperintense portion on fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) yielded additional improvement in survival (Li et al. 2016). These results suggest that maximal safe resection of the enhancing and non-enhancing components of newly diagnosed GBM should be attempted in all patients who are surgical candidates. While these studies provide a proof-of-principle for maximal safe resection, it should be noted that these results may also be strongly influenced by the neurosurgeons’ experience with brain tumor surgeries and the availability of high quality MRI techniques and of advanced neurosurgical instrumentation, which are not always readily available in the community when the patient initially presents with symptoms. The development of intraoperative tools such as fluorescence-guided surgery with 5-aminolevulinic acid (5-ALA), which allows visualization of residual tumor cells, and intraoperative MRI that allows monitoring of residual tumor during surgery have helped improve the extent of resection and thus contribute to survival outcomes (Stummer et al. 2006; Kubben et al. 2011). Further, to preserve neurologic function during aggressive surgeries, awake craniotomies can be performed with real-time intra-operative neurologic and language assessments with an option for local stimulation to detect and avoid functional areas when operating on tumors near eloquent areas. However, when gross total resection is not possible due to the location or extent of the tumor or the patient’s clinical condition, subtotal resection or stereotactic biopsy may serve to relieve mass effect and to establish definitive histologic diagnosis.
3.2.2 First Line Therapy for Newly Diagnosed GBM
Given the infiltrative nature of GBM, maximal surgical resection has to be followed by adjuvant therapy to maximize tumor control and improve survival. The initial use of adjuvant whole brain radiation therapy (WBRT) in the late 1970s (Andersen 1978; Walker et al. 1978, 1980) was replaced by involved field radiation therapy (IFRT) to offer maximal treatment to the tumor while minimizing radiation to normal brain tissue, given that tumor recurrence following WBRT usually occurs within 2–3 cm of the original lesion (Wallner et al. 1989) and that less than 10% of patients develop multifocal recurrence (Choucair et al. 1986). Dose escalation studies failed to show any survival benefit for total doses above 60 Gy (Chan et al. 2002; Nelson et al. 1988). The current standards for radiotherapy consist of IFRT to the gross tumor volume with a 2–3 cm margin for the clinical target volume delivered with linear accelerators.
Chemoradiation Therapy
In a landmark study that established the current standard of care, Stupp et al. reported a significant improvement in OS and progression free survival (PFS) in adults between the ages of 18 and 70 years of age with newly diagnosed GBM who received concurrent chemoRT compared with those who received radiation therapy (RT) alone (Stupp et al. 2005). The treatment constituted fractionated involved-field radiation (2 Gy per day, 5 days a week, for a total dose of 60 Gy) with concurrent TMZ (75 mg/m2 of body surface area daily for 6 weeks) followed by six cycles of TMZ at 150–200 mg/m2 for 5 days during each 28 day cycle. Median OS was 14.6 months with RT plus TMZ versus 12.1 months with RT alone (HR 0.63 [95% CI, 0.52 to 0.75; P < 0.001]). More strikingly, there was improvement in 2-year survival from 10.4% on RT alone to 26.5% with chemoRT.
A companion retrospective study by Hegi et al. demonstrated the relevance of promoter methylation of O-6 methyl-guanine DNA methyl-transferase (MGMT) , the protein product of which is involved in repair of DNA lesions induced by methylating agents such as temozolomide (Hegi et al. 2005). Of 206 evaluable patients, those with methylated MGMT promoter (44.7%) had a significantly better OS compared to those with unmethylated promoters, regardless of treatment (18.2 vs. 12.2 months, respectively; HR 0.45; 95% CI 0.32–0.61), supporting a prognostic role for this marker. Patients with MGMT promoter methylated tumors who received RT plus TMZ had an improved median survival (21.7 months [95% CI, 17.4–30.4]) compared to those who received RT alone (15.3 months [95% CI, 13.0–20.9]) (p = 0.007). In the promoter methylated cohort, the 2-year survival rate was 46% for the group treated with RT plus TMZ compared with 22.7% for those receiving RT only. A five-year analysis of the patient cohort in this study showed an OS of 27.2% at 2 years and 9.8% at 5 years for the RT plus TMZ arm compared with 10.9% and 1.9% with RT alone (hazard ratio 0.6, 95% CI 0.5–0.7; p < 0.0001) (Stupp et al. 2009). Patients with MGMT promoter methylated GBM had the best survival outcomes.
Dose escalation of standard IFRT beyond 60 Gy has not yielded improved outcome and indeed has resulted in increased radiation toxicity (Chan et al. 2002; Nelson et al. 1988). Alternative strategies such as brachytherapy, involving interstitial delivery of radioactive isotopes such as 125Iodine (I-125), has been used as a local boost in conjunction with IFRT in the setting of newly diagnosed GBM and as treatment for recurrent GBM. However, the paucity of prospective randomized studies of brachytherapy and the confounding study designs and mixed results of nonrandomized prospective studies have resulted in this modality not being actively used against GBM (Barbarite et al. 2016). Similarly, stereotactic radiosurgery (SRS) , which involves the precise delivery of high dose radiation to a specified lesion by either in a single fraction or as fractionated stereotactic radiotherapy (FSRT) used in the setting of deep-seated lesions or those abutting eloquent brain have not yielded definite survival benefit in GBM patients (Nwokedi et al. 2002; Souhami et al. 2004). Radioimmunotherapy , the use of radiolabeled antibodies to target cancer cells, has been most notably used to target tenascin, an extracellular glycoprotein highly expressed in GBM but not in normal brain tissue. In a phase II trial of a I-125-conjugated murine anti-tenascin monoclonal antibody delivered by direct injection into the resection cavity during surgery, Reardon et al. reported a median survival of 79.4 weeks, which exceeded that of historical controls. Given the promising outcomes and tolerable toxicity, a phase III study is currently being planned (Reardon et al. 2006; Zalutsky et al. 2008).
BCNU Wafers
Interstitial biodegradable bis-chloroethyl-nitrosourea (BCNU, carmustine) wafers were developed as a way to initiate chemotherapy immediately after tumor resection and avoid the side-effects of systemic administration of BCNU. Using a biodegradable polymer containing 3.85% BCNU (Gliadel®, Arbor Pharmaceuticals, Atlanta, GA, USA) in a randomized trial of 240 patients with newly diagnosed high grade gliomas, Westphal et al. demonstrated the survival advantage of BCNU wafers compared to control (median OS 13.9 months vs. 11.6 months), which led to the FDA approval of Gliadel wafers for these patients (Westphal et al. 2003). Adverse events were similar to placebo although BCNU treated patients were more likely to experience cerebrospinal fluid leaks (5.0% vs. 0.8%) and intracranial hypertension (9.1% vs. 1.7%). The survival advantage remained consistent at long-term follow up 3 years following the initial analysis (Westphal et al. 2006). Despite these results, the use of BCNU wafers has declined in routine surgical practice since the advent of the current chemoRT regimen.
Dose intensification of chemotherapy
Patients with MGMT promoter methylated GBM experience an improved outcome after treatment with temozolomide (Hegi et al. 2005; Esteller et al. 2000), which is believed to be due to their decreased ability to repair the O6-MG DNA lesion induced by TMZ therapy. These results suggested that MGMT-depletion could potentially sensitize GBMs to alkylating agents. Tolcher et al. found in two phase 1 trials that prolonged exposure to alkylating agents depleted intracellular MGMT in peripheral blood monocytes (Tolcher et al. 2003). Hypothesizing that a similar effect could be induced in tumor cells by dose intensification, Gilbert et al. conducted a multinational phase III trial (1173 patients registered, 833 randomized) comparing two schedules of adjuvant temozolomide: standard (5 days) or dose-dense (21 days) of a 28 day cycle, following concurrent chemoRT therapy in adults (≥18 years) with newly diagnosed GBM (Gilbert et al. 2013). However, the median survival in the dose-dense arm was not statistically different from that in the standard arm (16.6 months vs. 14.9 months, HR 1.03, p = 0.63); in addition, toxicity was also higher in the dose-dense schedule. The study did prospectively confirm the prognostic significance of MGMT promoter methylation in this patient population; MGMT promoter methylation was associated with improved median survival of 21.2 months versus 14 months in the unmethylated tumors (HR, 1.74; p < 0.001).
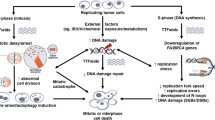
Alternating Electric Fields (Tumor Treatment Fields)
Kirson et al. developed technology that hampered cell division through the application of electric fields that alternated at a frequency in the range of 150–200 Hz and induced a cytotoxic effect on tumor cells in vitro in a dose-dependent manner in relation to the field intensity (Kirson et al. 2009a). Exposure to these tumor treatment fields (TTF) resulted in mitotic abnormalities, most notably membrane blebbing during entry into anaphase, which in turn resulted in aberrant mitotic exit and cell death (Gera et al. 2015). TTF were found to be most effective against protein targets that have high dipole moments, and its chief targets were the mitotic septin complex and the α/β-tubulin monomeric subunit of tubulin (Wong et al. 2015). The effects observed in vitro were additionally characterized in animal tumor models and human cancers, including colon adenocarcinoma, melanoma, and Lewis lung carcinoma (Kirson et al. 2007, 2009b). This was commercialized in the form of NovoTTF-100A (Novocure Ltd., Jersey Isle), a device with electrodes that could be applied directly to the scalp and generates a 50-V field (>0.7 V/cm at the center of the brain) that alternates at a frequency of 200 Hz and was intended to be worn continuously for at least 18 hours a day. In a multinational phase III trial by Stupp et al., adults with newly diagnosed GBM were randomized after chemoRT to receive adjuvant temozolomide with (n = 210) or without (n = 105) TTF therapy (Stupp et al. 2015). The trial was halted after a planned interim analysis demonstrated a significant improvement of PFS (7.1 vs. 4.0 months, HR 0.62, p = 0.001), which was the primary end-point of the study. Based on the results of this study, the US FDA approved the NovoTTF-100A for use with adjuvant temozolomide in patients with newly diagnosed GBM. Use of the NovoTTF-100A device was not associated with systemic toxic effects or increase in seizures, but it was associated with higher incidence of scalp irritation, anxiety, confusion, insomnia, and headaches compared to the control arm. The widespread use of this modality has been limited due to the slow acceptance of the potential utility of TTF, the cosmetic issues associated with the application of electrodes for over 18 hours a day, and the weight of the device (>3 kg). A newer version of the device has been released incorporating several improvements in design and weighing ~1 kg. In light of a separate randomized trial that found that TTF performed equally as well as physician’s choice chemotherapy in recurrent GBM (Stupp et al. 2012), the United States Food and Drug Administration (FDA) approved the device for use in patients with recurrent GBM.
Antiangiogenic strategies in initial therapy of GBM
Based on encouraging results with the use of bevacizumab in the setting of recurrent disease, two large randomized phase III studies examined the benefit of adding bevacizumab to standard treatment for newly diagnosed GBM both in the chemoRT and adjuvant settings. One study by Gilbert et al. reported the results of a multicenter Radiation Therapy Oncology Group (RTOG) trial in which 637 patients were randomized to either bevacizumab (10 mg/kg) or placebo beginning week 4 of chemoRT therapy with temozolomide and treatment was continued for up to 12 cycles along with adjuvant chemotherapy (Gilbert et al. 2014). The study also assessed the net clinical benefit of the treatments (including neurocognitive assessments, patient reported outcomes (PRO) , and health-related quality of life [HRQOL] measures). No significant difference in the median OS was seen between the bevacizumab group and the placebo groups (15.7 months vs 16.1 months, HR, 1.13). However, an improvement in PFS was seen in the bevacizumab group (10.7 months vs. 7.3 months; HR, 0.79) but did not reach the protocol specified threshold for significance. Despite this improvement in PFS, patients on the bevacizumab arm showed a decline in quality of life with increase in symptom burden and neurocognitive worsening compared with the placebo arm.
The other study (AVAglio) was a multinational trial which randomized 921 patients to either bevacizumab (n = 458) or to placebo (n = 463) (Chinot et al. 2014). In results that were strikingly similar to the RTOG study, no difference was seen in OS in the bevacizumab arm compared with the placebo arm (median OS 16.8 months vs 16.7 months, HR, 0.88; 95% CI, 0.76–1.02; p = 0.10), whereas an improvement in PFS was associated with bevacizumab treatment, which in this trial reached a pre-specified threshold for statistical significance (10.6 months vs 6.2 months; HR 0.64; 95% CI 0.55–0.74; p < 0.001). Both studies confirmed the value of MGMT promoter methylation as a prognostic marker associated with therapy in both arms. However, the two studies reported significant differences in HRQOL and PRO: results that remain to be fully understood. The AVAglio study showed an improvement in the quality of life measures associated with bevacizumab whereas the RTOG study showed a significant decline in both neurocognitive and PRO measures used in the study. A better understanding of the results of the two studies through an independent analysis will be critical in defining the significance of these net clinical benefit data.
3.3 Recurrent Glioblastoma
Despite aggressive initial therapy for newly diagnosed GBM, tumor recurrence is inevitable (Stupp et al. 2009). Recurrent GBM tend to be less sensitive to subsequent therapies due to development of emergent and adaptive resistance partly related to tumor heterogeneity. Several retrospective studies have shown that age and performance status at the time of recurrence were important independent prognostic factors for survival (Michaelsen et al. 2013). A small percentage of patients remain eligible for additional surgical resection (Weller et al. 2013). But for most patients, systemic therapies with agents such as bevacizumab, dose dense temozolomide, nitrosoureas or occasionally local therapies such as re-irradiation are required. This section provides a review of modalities used in patients with recurrent GBM.
Resection of Recurrent Glioma
Surgical resection of recurrent tumor is often required for individual patients to relieve symptomatic mass effect, recover function, or prevent neurological deterioration. However, there is a paucity of evidence to support a consistent role for re-resection in patients with recurrent GBM. While some prospective studies reported better OS with gross total resection (Suchorska et al. 2016; Yong et al. 2014), others have reported improvement in neither PFS nor post-recurrence OS with reoperation. A major role for repeat surgery is to provide histologic confirmation of recurrence, to differentiate the true progression from radiation necrosis and to identify biomarkers that may be useful in tailoring chemotherapeutic regimens. However, stereotactic biopsy can be a viable option to realize these goals even for the majority of patients who are not candidates for resection (Weller et al. 2013). Additional prospective and adequately powered trials are needed to evaluate whether re-resection provides a survival benefit in patients with recurrent GBM; however, such trials may be challenging due to the influence of other treatments being given to the patient subsequent to surgery as well as the issues related to patient selection for such resections.
Bevacizumab
Vascular proliferation and neoangiogenesis are hallmarks of GBM that are driven by its production of several angiogenesis promoting factors, especially vascular endothelial growth factor (VEGF) . Bevacizumab, a monoclonal antibody that binds VEGF to inhibit angiogenesis, was first reported to show unexpected activity in terms of radiological responses in a small series of patients with high grade gliomas (Stark-Vance 2005). This led to a more systematic evaluation of bevacizumab both as a single agent and in combination with irinotecan in a phase II open-label trial focused on patients with recurrent GBM. Of the 167 patients randomized to receive bevacizumab alone (10 mg/kg, n = 85) or bevacizumab plus irinotecan (n = 82) every 2 weeks, the PFS-6 rates (primary end point) were 42.6% and 50.3%, respectively with ORR of 28.2% and 37.8%, respectively. Median OS was 9.2 months with bevacizumab alone and 8.7 months with the combination. Treatment on these two arms were associated with significant toxicity with ≥grade 3 toxicities of 46.4% for the bevacizumab alone arm and 65.8% in the combination arm. Intracranial hemorrhage was seen in 2.4% of the patient in the bevacizumab-alone arm and in 3.8% in the combination arm, addressing the concerns about intracranial bleeding from such antiangiogenic therapies.
A second study examined the use of single agent bevacizumab in 48 patients with recurrent GBM and added irinotecan to bevacizumab when patients progressed on single agent bevacizumab therapy (Kreisl et al. 2009). The study showed a PFS6 of 29%, ORR of 35% (by Macdonald criteria), median PFS of 16 weeks, median OS of 31 weeks and OS6 rate of 57%. In this study, a higher rate of treatment related complication was reported, with 6 of the patients being removed from the trial due to thromboembolic events or intestinal perforation. Other adverse events included hypertension (12.5%), hypophosphatemia (6%), and thrombocytopenia (6%). Patients who progressed on bevacizumab had no objective responses when irinotecan was added at progression; median PFS was 30 days. The results of these two studies provided reasonable evidence that bevacizumab had activity in patients with recurrent GBM without significant toxicities and led to accelerated approval of bevacizumab by the US FDA as a single agent for treatment of this patient population. Of note, the approval was based on objective response rates that were also found to be durable; partial responses were observed in 25.9% and 19.6% respectively in these two studies with median response duration of 4.2 months and 3.9 months respectively. Bevacizumab is now considered the standard of care for patients with recurrent GBM without contraindications for this agent.
Progression after Bevacizumab Treatment
Patients developing progression after treatment with bevacizumab were clinically seen to have a possibly worse outcome and a lack of response to subsequent treatments, an issue which was addressed by several retrospective studies. Iwamoto et al. reported a median OS of 4.5 months for patients with GBM progressing after bevacizumab therapy; the subset of patients who received salvage chemotherapy after bevacizumab failure had a median PFS of 2 months, median OS of 5.2 months, and a PFS6 rate of 0% (Iwamoto et al. 2009). Similarly, in a similar population of patients, Lu-Emerson et al. reported a median PFS of 28 days, median OS of 78 days, and a PFS6 rate of 0%. These studies showed that outcome was dismal for patients who had progression after bevacizumab therapy and that salvage therapy in this setting was largely ineffective. In the absence of toxicity or progression, bevacizumab is often continued indefinitely, even after progression and often in combination with other agents due to concerns regarding tumor rebound seen in some patients with GBM and reported in patients with metastatic colorectal cancer (Bennouna et al. 2013). Quant et al. examined outcomes of patients who were continued on a bevacizumab containing regimen after progression on single agent bevacizumab therapy and reported a median PFS on the first regimen of 124 days and PFS6 rate of 33% and a median PFS on the second regimen of 37.5 days with a PFS6 of 2% (Quant et al. 2009). In another randomized phase II trial comparing bevacizumab with and without carboplatin, patient who progressed were randomized again to continue or cease bevacizumab; in this subset, there was no difference in median PFS (1.8 months vs 2.0 months) or median OS (3.4 months vs. 3.0 months) for those did or did not continue bevacizumab. There was also not definite evidence for a rebound effect (Hovey et al. 2015). Hence, the options for patients with bevacizumab failure remain limited prompting clinicians to postpone the use of this agent to the later stages of the disease.
Nitrosoureas
Nitrosoureas such as BCNU (carmustine), CCNU (lomustine), nimustine, and fotemustine, as well as another alkylating agent, procarbazine, were used frequently for first line therapy for newly diagnosed GBM precluding its use in the recurrent setting. However, following the approval of temozolomide for newly diagnosed glioblastoma, nitrosoureas were once again utilized more frequently to treat recurrence. Two phase II trials evaluating the efficacy of BCNU monotherapy in recurrent GBM showed PFS6 rates of 17.5% and 24%, though the second trial grouped patients who received temozolomide and BCNU together (Brandes et al. 2004; van den Bent et al. 2009). The response to BCNU in both studies was similar to historical reports, but patients were much more likely to experience hematologic, hepatic, and/or pulmonary toxicity. Serious toxicities such as irreversible pneumonitis or pulmonary fibrosis, prolonged myelosuppression, myelodysplasia and delayed secondary malignancies can occur in a minority of patients, which limit the cumulative dose of these agents. Locoregional therapy with placement of BCNU wafers at the time of reoperation for recurrent GBM was developed to reduce such systemic toxicity and was associated with a significantly longer OS in a randomized phase III trial compared to placebo (31 vs. 23 weeks, HR = 0.67, p = 0.006), without increased CNS toxicity (Brem et al. 1995), leading to its regulatory approval in the US.
Concurrent CCNU and bevacizumab showed improved PFS and OS in patients with recurrent GBM compared with those treated with CCNU (n = 46) or bevacizumab (n = 50) alone in initial results of the randomized phase II BELOB trial (Taal et al. 2014). However, results from a recently completed phase III trial comparing bevacizumab plus CCNU to CCNU alone showed no improvement in OS in the combination therapy arm (HR 0.95 CI 0.74, 1.21, p = 0.650), whereas PFS was longer with the addition of bevacizumab to CCNU (HR 0.49 (CI 0.39, 0.61). Median efficacy outcomes were: OS 9.1 (8.1, 10.1) versus 8.6 (7.6, 10.4) months and PFS 4.2 (3.7, 4.3) versus 1.5 (1.5, 2.5) months in the combination arm versus the CCNU arm, respectively. Toxicity was in the expected range with more events in the combination arm being also longer on treatment. These data suggest clinically relevant activity of CCNU as a single agent and in combination with bevacizumab. However, treatment with CCNU resulted in frequent hematologic toxicity (up to 50% of patients affected), particularly in combination with bevacizumab, leading to a dose reduction for CCNU with bevacizumab. Further phase III trials are needed to expand on this body of data.
Fotemustine is used mainly in Italy and France, and several studies found similar survival outcomes compared to CCNU (Brandes et al. 2009a; Fabrini et al. 2009). Hematologic toxicity was also not insignificant with fotemustine. Despite their toxicity profiles, nitrosoureas will likely continue to be utilized in the clinic and in trials.
Re-irradiation
Re-irradiation for recurrent GBM is most commonly considered in the setting of rapidly progressive symptomatic disease when there are few other treatment options available, but this decision has to be tempered by the potential risk of neurotoxicity from overlap of treatment fields with areas previously radiated. Since most recurrences occur within the target volume treated with IFRT, treatment planning needs to be adequately tailored to treat recurrence safely. Retrospective studies have suggested a potential benefit from re-irradiation with SRS or FSRT (typical doses 30–36 Gy) (Torok et al. 2011). Nevertheless, there is a lack of concordance in the literature regarding the time interval necessary from initial irradiation and a paucity of prospective and randomized trials to define properly the role of re-irradiation as monotherapy in the treatment of recurrent GBM (Seystahl et al. 2016). Combination therapy strategies have shown early promise; preliminary data regarding the use of SRS or FSRT with concurrent bevacizumab in recurrent GBM showed positive responses with minimal neurotoxicity (Gutin et al. 2009; Cabrera et al. 2013). The potential effects of this combination are hypothesized to be due to vascular normalization induced by bevacizumab that results in improved oxygenation of the tumor tissue and consequent increased radiation effect.
Laser-induced thermal therapy
If maximal resection is not feasible, newer modalities of tumor ablation have been developed as alternatives. Laser induced (or interstitial) thermal therapy (LITT ) is one such procedure which utilizes minimally invasive percutaneous insertion of an optical fiber into the tumor under intraoperative MR guidance and generates ablative heat, which induces targeted thermocoagulative necrosis of tumor cells, which may be particularly suitable for deep-seated brain lesions or lesions located in eloquent areas. A recent phase 1 trial of a commercial LITT device , NeuroBlate (Monteris Medical, Inc., Plymouth, MN, USA) in recurrent GBM demonstrated that LITT is a viable, safe option for treatment of GBM (Sloan et al. 2013). Additional trials are warranted to characterize fully the safety and efficacy profile of LITT systems in treatment of newly diagnosed GBM. Local thermal injury during intracranial use of LITT can result in serious neurologic morbidities or cerebrovascular complications. Further, the technique does not adequately address the extensive infiltrative disease usually seen in GBM.
3.4 Glioblastoma in Older Adults
The EORTC/NCIC trial established a new standard of care for adult patients between ages 18 and 70 years but did not provide information about patients >70 years with GBM (Stupp et al. 2005). Older age and poor performance status have been consistently shown to be associated with shorter survival (Buckner 2003). The median OS of patients age 65 and older with a new diagnosis of GBM is approximately six months (compared to 12–14 months in younger patients) (Paszat et al. 2001; Kita et al. 2009). However, this could be explained by differences in treatment administered, as older patients are less likely to be considered for more aggressive interventions (Paszat et al. 2001), and older adults were found to exhibit similar survival outcomes as younger adults when both groups receive the same treatments (Kita et al. 2009).
Current literature supports the role of maximal safe resection as an initial step in treatment of an older patient with newly diagnosed GBM, if the patient can tolerate the surgery. One prospective randomized trial reported significant benefits OS with maximal safe resection (n = 10) compared to biopsy alone (n = 13) in OS (171 vs. 85 days, respectively, p = 0.035), but no significant difference in PFS (105 vs. 72 days, respectively, p = 0.057) (Vuorinen et al. 2003). However if surgical resection is contraindicated, biopsy of the tumor is needed to establish histologic diagnosis and to assess molecular characteristics that can influence subsequent treatment (such as MGMT promoter methylation (Reifenberger et al. 2012)).
Two prospective trials demonstrated that postsurgical RT was safe and effective in older patients. A randomized phase III trial of 85 elderly patient with GBM by Keime-Guibert et al. reported that RT plus supportive care resulted in improved median survival compared with supportive care alone (29.1 weeks vs 16.9 weeks, HR 0.47, 95% CI 0.29–0.76, p = 0.002) with no significant differences in QOL and cognitive measures between the treatment groups (Keime-Guibert et al. 2007). To determine the optimal RT dose for elderly patients, Roa et al. conducted a phase III trial randomizing 98 patients ≥60 years of age with newly diagnosed GBM to either standard RT (60 Gy in 30 fractions over 6 weeks) or a shorter course of RT (40 Gy in 15 fractions over 3 weeks) and reported no significant difference in median OS between the two groups (5.1 months versus 5.6 months, log-rank test, p = 0.57), suggesting that the hypofractionated RT course may be reasonable for older patients with GBM (Roa et al. 2004).
The role of chemotherapy in this patient population was addressed in the Nordic study, a large multinational trial in which 291 patients aged ≥60 years were randomized to receive TMZ alone (200 mg/m2 days 1–5 of a 28 day cycle for up to six cycles, n = 93), hypofractionated RT (34 Gy over 2 weeks, n = 98), or standard RT (60 Gy over 6 weeks, n = 100), and an additional 51 were randomized to either TMZ alone (n = 26) or hypofractionated RT (n = 25) (Malmstrom et al. 2012). The study reported that treatment with TMZ alone yielded a longer median OS compared with standard RT (8.3 months vs 6 months, HR 0.70, p = 0.01), but not with hypofractionated RT (7.5 months vs 6 months, HR 0.85, p = 0.24). Overall survival was similar for patients who received TMZ (n = 119) or hypofractionated RT (n = 123), (8.4 months vs 7.4 months; HR 0.82, p = 0.12). However, in the subset of patients ≥70 years, better survival was noted with TMZ alone (HR 0.35, p < 0.0001) or with hypofractionated RT (HR 0.59, p = 0.02) compared with standard RT. In addition, in the subgroup receiving TMZ alone, patients with MGMT promoter methylation had significantly longer survival compared with those an unmethylated MGMT promoter (9.7 months vs 6.8 months; HR 0.56, p = 0.02), an improvement that was not seen in patients treated with RT (HR 0.97; p = 0.81). These results suggested that TMZ or hypofractionated RT may be considered as standard treatment options in elderly patients with GBM with MGMT promoter methylation status as a predictive marker for TMZ. This trial did not test the role of concurrent chemoRT in this population.
In an attempt to ascertain if dose intensification of TMZ may provide a greater benefit, the NOA-08 study compared the efficacy of RT versus dose dense TMZ in elderly patients (≥65 years) with good functional status with GBM or anaplastic astrocytoma. Patients were randomized to TMZ (100 mg/m2 daily on a week on week off schedule, n = 195) or standard RT (60 Gy in 30 fractions, n = 178) in a non-inferiority design. Median OS was 8.6 months in the TMZ group versus 9.6 months in the RT group (HR 1.09, pnon-inferiority = 0.033). The majority of patients had GBM (n = 331) and the rest anaplastic astrocytoma (n = 40) but there were no significant difference in survival based on histology (HR 0.69, p = 0.20). MGMT promoter methylation was associated with longer OS than was unmethylated status (11.9 months vs 8.2 months, HR 0.62, p = 0.014). Dose intensive temozolomide was found to be non-inferior to radiotherapy alone in elderly patients with malignant astrocytoma but was associated with increased toxicity.
Building on earlier studies of chemoRT using temozolomide , which suggested that this treatment was well tolerated in elderly patients with good functional status (Combs et al. 2008; Brandes et al. 2009b; Minniti et al. 2015), and to address whether the addition of TMZ to RT improves survival in elderly patients, a recent phase III trial enrolled 562 patients over 65 years old with newly diagnosed GBM and good functional status (ECOG 0-2) and randomized them to receive RT alone (40 Gy in 15 fractions, n=281) or RT (40 Gy in 15 fractions) with 3 weeks of concomitant TMZ plus monthly adjuvant TMZ (n=281) until progression or 12 cycles (Perry et al. 2016). RT combined with TMZ significantly improved OS over RT alone (median 9.3 months vs 7.6 months, HR 0.67, 95%CI 0.56-0.80, p<0.0001), a benefit that was seen in both MGMT methylated (OS 13.5 months and 7.7 months, respectively, HR: 0.53, p=0.0001) and MGMT unmethylated patients (OS 10.0 months vs 7.9 months, respectively, HR 0.75, p=0.055). The treatment was noted to be well tolerated, and patients with MGMT methylated tumors benefited the most from chemoRT with near doubling of median OS. This regimen is hence now considered the standard of care for elderly patients with a good functional status.
At the time of submission, no data exist specific to treatment of older patients with GBM recurrence. There are several ongoing trials investigating bevacizumab in this patient population.
In summary, although age remains a significant prognostic factor, age alone need not be the basis of exclusion for standard therapies in treatment of GBM in elderly patients with good functional status. However in some cases dose reduction may decrease toxicity while providing similar clinical benefits, particularly in combination chemoRT and in hypofractionated radiotherapy.
3.5 Gliomatosis Cerebrii
Gliomatosis cerebri (GC) is an uncommon, highly infiltrative presentation of gliomas first described in 1938 (Nevin 1938). Diagnosis requires histology demonstrating a glial-origin neoplasm and radiological evidence of involvement of more than two lobes of the brain on T2-weighted/FLAIR MRI. There is considerable variation in published literature regarding the prognostic factors and course of disease with varied reports of poor prognosis in some studies and promising outcomes compared to GBM. Given that GC is defined radiologically, the histology can vary from low grade infiltrative gliomas to GBM. These differences in reported interpretation and managements of patients with GC have made treatment decisions challenging in clinical practice. Given the diffuse nature of the disease, surgical resection is not indicated even in the presence of enhancing foci; instead, histological diagnosis is made by stereotactic or open biopsy often focusing on regions of enhancement if present to obtain the most accurate diagnosis. Radiation therapy is often the first line treatment with IFRT being used in cases where the cancer is more localized, and a focal boost given to contrast-enhancing lesion if present (e.g. total dose 50.4 Gy without a focal lesion, 45 Gy plus a 14.4 Gy boost in case of a focal lesion) although there is no standardized schedule. However, in many instances, the extent of the lesion may warrant the use of WBRT despite the risks of neurologic sequelae (Cozad et al. 1996). Although median survival ranges from 11 to 24 months following radiotherapy , a retrospective study found no difference in survival between patients receiving or not receiving radiotherapy (Herrlinger 2012). In contrast, the use of chemotherapy was a highly significant prognostic factor in an analysis of 296 patients with GC (Taillibert et al. 2006). The PCV regimen is most commonly used for these patients comprising of lomustine (110 mg/m2 on day 1), procarbazine (60 mg/m2 daily for days 8–21 of a 42-day schedule), and vincristine (1.4 mg/m2 on days 8 and 29). Since vincristine does not penetrate the blood-brain barrier, it is only used in patients with a focal contrast-enhancing lesion. Given the higher risks of toxicity associated with PCV, temozolomide has often been used as a substitute and has yielded similar survival. Overall, the scarcity of data regarding the natural progression of this disease and its response to multimodal therapy has resulted treatment being often directed to the histological nature of the tumor and has limited the ability to develop evidence-based treatment guidelines or prospective clinical trials for patients with GC.
3.6 Targeted Agents in Treatment of GBM
Insights into the specific signaling pathways in glioblastoma growth, invasion and angiogenesis, coupled with the development of technology platforms allowing for testing of the genetic mutations and proteins associated with these pathways has created enormous opportunity for the development of new therapeutics in this disease. The best studied of these targets are the Epidermal Growth Factor Receptor (EGFR) and its ligand (EGF), the angiogenic pathway best exemplified by the activity of the Vascular Endothelial Growth Factor (VEGF) and its receptor (VEGFR), and the phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathway.
3.6.1 EGFR Inhibitors
The majority of glioblastomas have a mutation, amplification, or deletion in at least one receptor tyrosine kinase, with mutations in epidermal growth factor receptor (EGFR) accounting for over half of these (Brennan et al. 2013). Roughly half of the patients who have EGFR amplification also have a specific deletion of the extracellular domain (exons 2–7): a genotype variant referred to as EGFRvIII (Pelloski et al. 2007; Del Vecchio et al. 2013). Numerous attempts have been made to capitalize on the deregulated expression and activity of EGFR in glioblastoma using small molecule inhibitors of EGFR (e.g. erlotinib and gefitinib) and monoclonal antibodies (e.g. cetuximab and nimotuzumab), though none have been particularly successful.
A phase II multi-center trial in newly diagnosed GBM by the North Central Cancer Treatment Group reported in 2008 that treatment of 97 patients with erlotinib 1 week prior to and concurrent with the standard Stupp protocol resulted in mean OS that was similar to TMZ era controls (median survival 15.3 months) (Brown et al. 2008). None of the tested genetic/molecular alterations were associated with survival, including EGFR amplification, combination EGFR and PTEN, and EGFRvIII (p > 0.05). In recurrent glioblastoma, a multi-center phase II trial randomized 110 patients to receive either erlotinib in the experimental arm or single agent temozolomide or BCNU if previously treated with TMZ as the control arm (van den Bent et al. 2009). The PFS at six months was 11.4% in the erlotinib arm and 24% in the control arm. EGFR expression, amplification, mutation in exons 18, 19, and 21, and EGFRvIII were not significant predictors of survival, though low levels of p-Akt was a borderline predictor of improved survival (p = 0.048).
Nimotuzumab , a monoclonal antibody against EGFR, was tested for newly diagnosed glioblastoma in a multi-center open label phase III trial that randomized 149 patients to receive either standard of care (Stupp regimen) or nimotuzumab in addition to the Stupp protocol (Westphal et al. 2015). The study found that 12 month PFS was 25.6% in the experimental arm, compared to 20.3% in the control arm (p = 0.53, Fisher’s exact test), with median OS at 22.3 and 19.6 months (p = 0.49, log-rank test), respectively. EGFR amplification was not associated with outcome (p = 0.88). Multiple phase II trials have failed to find significant benefit of cetuximab (monoclonal antibody to EGFR) in newly diagnosed and recurrent GBM.
3.6.2 PI3K and mTOR Inhibitors
The phosphoinositide 3-kinase (PI3K) /mammalian target of rapamycin (mTOR) signaling pathway plays a central role in cellular processes including cell growth, survival, and motility (Engelman 2009). Derangements in the PI3K/mTOR pathway are common in glioblastoma and can promote oncogenic activity (Choe et al. 2003). Such dysregulations can occur upstream (e.g. EGFR amplification or EGFRvIII leading to constitutive activation of PI3K), within PI3K (e.g. mutation or amplification of PI3K components), or in inhibitory regulatory processes (e.g. loss of phosphatase and tensin homolog [PTEN], a tumor suppressor) (Pitz et al. 2015; Wen et al. 2015). mTOR is a potential target for anti-cancer therapies, given its deregulation and role in cell growth in cancers.
In the only multi-center randomized controlled trial studying inhibition of the PI3K/mTOR pathway in glioblastoma, Wick and colleagues compared temozolomide to temsirolimus (an mTOR inhibitor) in newly diagnosed GBM (Wick et al. 2016). The investigators randomized 111 MGMT promoter unmethylated patients to receive either standard of care (Stupp regimen) or radiotherapy with weekly temsirolimus. Median PFS in the temsirolimus arm was 5.4 months versus 6.0 months in the control group. Median OS in patients treated with temsirolimus was 14.8 months, compared to 16.0 months in the control arm.
Two phase II single-arm trials investigating single-agent temsirolimus in recurrent glioblastoma reported did not find a survival benefit (Galanis et al. 2005; Chang et al. 2005). Galanis and colleagues found that in 65 patients treated with 250 mg of temsirolimus intravenously, PFS at 6 months was 7.8%. Chang and colleagues found that in 43 patients treated with 250 mg of temsirolimus (or 170 mg for those not on EIAED), PFS at 6 months was 2.4%.
Other phase II single-arm trials examining temsirolimus and everolimus (another mTOR inhibitor) in combination with chemotherapy and radiation sorafenib, and bevacizumab failed to find significant survival benefit of these mTOR inhibitors in both newly diagnosed and recurrent glioblastoma.
3.6.3 VEGF and VEGFR Inhibitors
Bevacizumab , a monoclonal antibody against VEGF, is FDA approved for single-agent treatment of recurrent glioblastoma patients.
To examine the strategy of pan-VEGFR inhibition to inhibit angiogenesis, the ‘Recentin in GBM alone and with lomustine’ (REGAL) trial studied the efficacy of cediranib, a potent orally bioavailable VEGFR inhibitor given singly or in combination with lomustine with a lomustine alone arm as control in a randomized phase III, placebo-controlled, trial in adults with recurrent glioblastoma who had failed radiation and temozolomide (Batchelor et al. 2013). The study randomized 325 patients to one of three arms: cediranib alone, cediranib plus lomustine or lomustine plus placebo in a 2:2:1 ratio. The final results of the study showed that there was no difference in PFS, the primary endpoint, for cediranib alone (median PFS 92 days, HR 1.05, p = 0.90) or cediranib plus lomustine (median PFS 125 days, HR 0.76, p = 0.16) compared with lomustine plus placebo (median PFS 82 days). Similarly, no improvement in median OS, the secondary endpoint, was seen for cediranib alone (8 months, HR 1.43, p = 0.10) or cediranib plus lomustine (9.4 months, HR 1.15 p = 0.50) compared with lomustine (9.8 months). The PFS6 was also not significantly different in the cediranib alone (16%) or cediranib plus lomustine (35%) arms compared with the lomustine arm (25%). Correlative studies showed that treatment on either of the cediranib arms showed sustained decreases in median serum soluble VEGFR2 (sVEGFR2) levels whereas treatment with lomustine alone was not associated with significant change in median sVEGFR2 levels; these results suggest that cediranib was able to hit its target at least in serum (although inhibition of tumor VEGFR remains unknown) but still lacked activity against recurrent GBM.
A multicenter open label phase III trial examined the efficacy of enzastaurin, an oral serine/threonine kinase inhibitor, which targets both protein kinase Cβ that mediates VEGF driven angiogenesis, and the PI3K/AKT pathways which drives proliferation in glioblastoma (Wick et al. 2010). The trial was designed to randomize 397 patients to enzastaurin or lomustine in a 2:1 ratio; however, the accrual was halted early after enrolling 266 patients (enzastaurin, n = 174; lomustine, n = 92) based on a planned interim futility analysis which showed no significant differences in median PFS (1.5 vs. 1.6 months; HR 1.28), median OS (6.6 vs. 7.1 months; HR 1.20), or PFS6 rate (11.1% vs. 19.0% p = 0.13) between the enzastaurin and lomustine arms.
3.7 Biological Agents in Clinical Trials Against Glioblastoma
3.7.1 Virus-Based Therapies
Viruses have been employed in clinical research for malignant glioma therapy for quite a few years with two main virus modifications made as follows: a) replication-deficient viral vectors which are used to deliver genes with therapeutic activity to the tumor environment and b) replication competent oncolytic viruses which function by infecting and replicating within a tumor cell, eventually causing tumor cell death and infecting other tumor cells. Broadly speaking, there are two main strategies underlying virus-based therapies in gliomas: The first is delivery of specific genes to modify the biology of the tumor and exert their antitumor effects; replication-deficient viruses such as the herpes simplex virus type I thymidine kinase (HSV-TK) construct are typically employed for therapy delivery. The second strategy is by replication of the virus within the glioma cell resulting in destruction of glioma cells after infection (oncolysis); this is achieved by use of oncolytic viruses which are replication (Table 3.2). Additionally, both virus categories of have the added benefit of immune activation , resulting in glioma cell destruction through secondary immune effects. Tables 3.2 and 3.3 highlight some of the completed and ongoing virus trials in high grade gliomas. Research in virus based therapies have yielded several valuable insights into this novel therapeutic approach
-
Virus therapies studied have so far been shown to be safe but not conclusively effective.
-
The most commonly studied gene therapy approach in gliomas has been that of suicide gene transfer; basically transfer of a gene encoding for enzymes with therapeutic activity. Thus, the herpes simplex type I – thymidine kinase construct (HSV-TK) allows for (systemically administered) ganciclovir to be converted to nucleotides that are incorporated into and are toxic to glioma cell DNA. Table 3.3 illustrates trials employing this strategy that have been completed.
-
Viruses must be engineered to be safe through genetic modifications that prevent normal cell infection. An example of such modification is the recombinant herpes simplex virus with deletions of both viral copies of the ICP34.5 gene. This modification removes virulence but allows the virus to destroy infected glioma cells.
-
Viruses can be genetically engineered and maintain replication competence. The ideal virus is one that is not virulent to normal cells, specifically infects gliomas cells and replicates within them, ultimately causing cell death (oncolysis), release of daughter virus particles and infection of neighboring glioma cells. The release of tumor and virus antigens stimulates an immune response causing further tumor cell destruction. DNX-2401, a replication competent oncolytic adenovirus engineered to exploit the interaction between the virus and the retinoblastoma (Rb) protein pathway has shown early promise in phase I and II trials in patients with recurrent GBM with long term control of disease in some patients; this virus replicates in cells with an impaired Rb pathway (seen in >80% of GBM cells) but not in those with an intact Rb pathway (normal brain cells). Oncolysis by this virus can trigger an immune response which is being now exploited by immune stimulants such as interferon-γ or pembrolizumab, a PD1 inhibitor.
-
Viruses can also be engineered to have a gene payload with therapeutic potential. An example of a virus construct that combines these qualities is Toca 511, a genetically engineered retrovirus that is replication competent and carries the gene for yeast cytosine deaminase (Ostertag et al. 2012). This enzyme converts 5-flucytosine (administered to patients orally) to 5-flurouracil, a chemotherapeutic agent that thus gets produced within the infected tumor cells where it exerts a chemotherapeutic effect and also diffuses to surrounding tumor cells. This is therefore another example of suicide gene therapy but has the potential added benefit of more efficient dissemination within the tumor due to virus replication and chemotherapy diffusion currently being tested in several trials (NCT01470794, NCT01156584).
-
Table 3.4 illustrates other examples of ongoing and completed clinical trials that employ replication competent viruses for the treatment of malignant gliomas.
3.7.2 Immunotherapies
Given the immune system’s role in human protection from infections and malignancy, the development of cancer could be viewed as a failure of this system. Additionally, once cancer develops it is associated with further immune suppression through effects of the tumor on immune system components. The strategy of immune therapy in central nervous system tumors was late in gaining acceptance compared to other cancers, mainly because of a perceived “immune privilege” of the central nervous system compared to other organ systems. However, clinical observations have shown that the immune system is activated specifically in response to central nervous system infections (meningitis and meningoencephalitis) and is in fact pivotal to the development and perpetuation of central nervous system pathologies (e.g. demyelinating disease). It is now known that intracranial malignancies do stimulate a T-cell response and that brain tumors, like systemic malignancies , do suppress the immune system in a variety of ways (Vauleon et al. 2010; Wilson et al. 2010). The following provides a brief overview of the most commonly employed strategies in currently ongoing clinical research in malignant gliomas.
Vaccine therapy
The most traditional way to stimulate an immune response is through the use of vaccines which may include cell-based or non-cell based approaches.
-
(a)
Cell-based vaccines :
-
Dendritic-cell vaccines : Dendritic cells are immune cells with very efficient antigen-presenting properties. In glioblastoma trials these cells are obtained from the patient and exposed to the tumor tissue obtained at surgery. DCVax-L® is an example of a dendritic-cell-based vaccine. The vaccine is a lysate consisting of peripheral blood mononuclear cells (PBMC) from the patient mixed with tumor tissue. The PBMC mature to dendritic cells a process that is encouraged by exposure to granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4. A Phase I trial showed this treatment to be safe and showed a promising overall survival of almost 32 months (Prins et al. 2011), leading to Phase III trial (NCT00045968). This study is ongoing although not currently recruiting patients.
-
Autologous vaccine : The strategy of autologous vaccination employs modification of the tumor cells or of immune cells (usually T-lymphocytes) and introduction of the altered cells into the patient to induce immune responses. An example of this strategy is the use of autologous formalin-fixed tumor vaccines in which T-cells are sensitized to the tumor. A recent study by Muragaki et al. employed this vaccine in newly diagnosed glioblastoma patients during radiation therapy (Muragaki et al. 2011). The median duration of overall survival was 19.8 months and the actuarial 2-year survival rate was 40%. The median duration of progression-free survival was 7.6 months leading the investigators to conclude that further clinical testing was warranted.
-
-
(b)
Non-cell based vaccines :
-
Peptide vaccines : EGFRvIII is a constitutively active mutant form of the epidermal growth factor receptor. It is present in about a third of glioblastoma specimens (Wong et al. 1992). A peptide-based vaccine was developed to induce a response to EGFR-VIII positive glioblastoma. A phase II trial examined the progression-free survival (PFS) , and overall survival (OS) in patients with newly diagnosed glioblastoma who received the vaccine after ascertaining EGFR-VIII expression in the tumor specimen (Sampson et al. 2010). There were a total of eighteen patients enrolled and the median PFS and OS were 14.2 and 26 months for those receiving the vaccine. This compared very favorably with a PFS of 6.3 months and an OS of 15 months for the unvaccinated controls. However, a phase III trial to confirm these results did not show any difference in survival between treated patients and the placebo group, leading the independent Data Safety and Monitoring Board (DSMB) to recommend study discontinuation (Celldex 2016).
-
Heat-shock protein vaccines : Heat-shock proteins are considered to be crucial to the survival of cancers such as glioblastoma due to their key roles in stabilizing proteins, facilitating protein conformational change, protein trafficking and breakdown as well as control of apoptosis (Powers et al. 2010). They are activated by the “stress” environment found in tumor beds and consisting mainly of hypoxia and inflammation (Young et al. 2004). It is therefore not surprising that they would be considered targets in an immune strategy for treatment of glioblastoma. A Phase I study of 12 patients with recurrent glioblastoma was designed to test the hypothesis that since heat shock protein peptide complexes (HSPPCs) carry tumor-specific antigenic proteins and facilitate immune responses, peptides bound to a 96 kD chaperone protein (HSP-96) from brain tissue containing glioblastoma can be used to immunize patients with recurrent disease (Crane et al. 2013) The study showed that this could be done safely; testing of peripheral blood leukocytes before and after vaccination showing a significant peripheral immune response specific for the peptides bound to HSP-96, in almost all (11 of the 12) patients treated. The study also included correlative brain biopsies of immune responders after vaccination showing focal CD4, CD8, and CD56 IFNγ positive cell infiltrates, consistent with tumor site specific immune responses. The immune responders had a median survival of 47 weeks after surgery and vaccination, compared with 16 weeks for one patient who did not show a response. The following Phase II trial was also promising with patients having a total resection of recurrent glioblastoma and then receiving vaccine with HSPPC-96 (Bloch et al. 2014). The median PFS of this cohort was 19.1 weeks with a median OS of 42.6 weeks. There is an ongoing randomized Phase II study examining this vaccine strategy with bevacizumab and comparing it bevacizumab alone [NCT01814813].
-
3.7.2.1 Immune Checkpoint Inhibitors
The immune system employs a system of checks and balances that include “checkpoints’, essentially proteins that down regulate the immune response to prevent damage to self. CTLA-4 controls T-cell activity and is a protein found on cytotoxic T-cells (CD8+) and subsets of helper (CD4+) T cells (Schwartz 1992; Rudd et al. 2009). Activation of the protein through ligand binding causes a reduction in IL-2 production, reduced IL-2 receptor expression, lymphocyte cell division (Alegre et al. 2001) and enhancement of T suppressor cell function (Wing et al. 2008; Peggs et al. 2009). The development of an antibody to CTLA-4, ipilimumab holds promise for inhibition of this checkpoint and has been approved for melanoma. Similarly, PD-1 is a protein expressed by T-cells, including regulatory T-cells (Tregs) (Francisco et al. 2009), B-cells and NK cells (Velu et al. 2009). Its expression serves as a “brake” on the immune response and it binds to PD-L1, a ligand that seems to be associated with derangements in the PI3K–Akt signaling pathway (Parsa et al. 2007) Nivolumab is a PD-1 antibody. There is an ongoing phase III trial (NCT02017717) comparing the efficacy of nivolumab with bevacizumab in patients with recurrent glioblastoma. There is also a phase I trial comparing ipilimumab, nivolumab, and the combination in patients with newly diagnosed glioblastoma (NCT02311920) and a randomized phase III open label study of nivolumab versus bevacizumab and multiple phase I safety cohorts of nivolumab or nivolumab in combination with ipilimumab (NCT02017717).
3.7.2.2 Genetically Engineered T-cells
The development of chimeric antigen receptor (CAR) technology has opened yet another door to the immune therapy possibilities in cancer. T-cells are engineered to recognize antigens on tumors by fusing an extracellular binding domain to the intra-cellular signaling domain of the T cell receptor (Eshhar et al. 1993). The extracellular domain is derived from an antibody to a tumor-associated antigen. CARs have been developed for HER2, IL-13Rα2, and EGFRvIII. The preclinical activity demonstrated by these cells led to clinical trial testing and these efforts have been comprehensively reviewed in recent publications (Thaci et al. 2014). CAR technology offers important advantages when compared to other immune therapies, including cytotoxicity that is independent of MHC class I expression. Given the variability of this expression in glioblastoma this may represent a significant therapeutic advantage. Additionally, CARs may have better penetration into blood vessel walls and tumor than other non-genetically engineered components of the immune system (Miao et al. 2014).
3.8 Conclusions
In contrast to the several decades of therapeutic strategies against GBMs during which only incremental advances in our knowledge of glioblastoma had been achieved, the past few years have seen an veritable explosion of knowledge of the basic biology of glioma and generated a high degree of enthusiasm and optimism in the field that we are closer to effective treatments that will dramatically improve outcomes of patients with GBM. In addition, the wealth of knowledge gained in modulation of the human immune system and the harnessing of biological therapies that are active regardless of tumor heterogeneity promise to transform therapeutic strategies against these aggressive tumors. These factors combined with the advances in technology and basic research are set to potentially shift the paradigm in therapeutic approaches for patients with GBM.
References
Alegre, M.L., K.A. Frauwirth, and C.B. Thompson. 2001. T-cell regulation by CD28 and CTLA-4. Nature Reviews. Immunology 1(3): 220–228. doi:10.1038/35105024.
Andersen, A.P. 1978. Postoperative irradiation of glioblastomas. Results in a randomized series. Acta Radiologica: Oncology, Radiation, Physics, Biology 17 (6): 475–484.
Barbarite, E., J.T. Sick, E. Berchmans, A. Bregy, A.H. Shah, N. Elsayyad, and R.J. Komotar. 2016. The role of brachytherapy in the treatment of glioblastoma multiforme. Neurosurgical Review 40 (2): 195–211. doi:10.1007/s10143-016-0727-6.
Batchelor, T.T., P. Mulholland, B. Neyns, L.B. Nabors, M. Campone, A. Wick, et al. 2013. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. Journal of Clinical Oncology 31 (26): 3212–3218. doi:10.1200/JCO.2012.47.2464.
Bennouna, J., J. Sastre, D. Arnold, P. Osterlund, R. Greil, E. Van Cutsem, et al. 2013. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. The Lancet Oncology 14 (1): 29–37. doi:10.1016/S1470-2045(12)70477-1.
van den Bent, M.J., A.A. Brandes, R. Rampling, M.C. Kouwenhoven, J.M. Kros, A.F. Carpentier, et al. 2009. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. Journal of Clinical Oncology 27 (8): 1268–1274. doi:10.1200/JCO.2008.17.5984.
Bloch, O., C.A. Crane, Y. Fuks, R. Kaur, M.K. Aghi, M.S. Berger, et al. 2014. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 16: 274–910.1093. doi:10.1093/neuonc/not203.
Brandes, A.A., A. Tosoni, P. Amista, L. Nicolardi, D. Grosso, F. Berti, et al. 2004. How effective is BCNU in recurrent glioblastoma in the modern era? A phase II trial. Neurology 63 (7): 1281–1284.
Brandes, A.A., A. Tosoni, E. Franceschi, V. Blatt, A. Santoro, M. Faedi, et al. 2009a. Fotemustine as second-line treatment for recurrent or progressive glioblastoma after concomitant and/or adjuvant temozolomide: A phase II trial of Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Cancer Chemotherapy and Pharmacology 64 (4): 769–775. doi:10.1007/s00280-009-0926-8.
Brandes, A.A., E. Franceschi, A. Tosoni, F. Benevento, L. Scopece, V. Mazzocchi, et al. 2009b. Temozolomide concomitant and adjuvant to radiotherapy in elderly patients with glioblastoma: Correlation with MGMT promoter methylation status. Cancer 115 (15): 3512–3518. doi:10.1002/cncr.24406.
Brem, H., S. Piantadosi, P.C. Burger, M. Walker, R. Selker, N.A. Vick, et al. 1995. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 345 (8956): 1008–1012.
Brennan, C.W., R.G. Verhaak, A. McKenna, B. Campos, H. Noushmehr, S.R. Salama, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155 (2): 462–477. doi:10.1016/j.cell.2013.09.034.
Brown, P.D., S. Krishnan, J.N. Sarkaria, W. Wu, K.A. Jaeckle, J.H. Uhm, et al. 2008. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. Journal of Clinical Oncology 26 (34): 5603–5609. doi:10.1200/JCO.2008.18.0612.
Buckner, J.C. 2003. Factors influencing survival in high-grade gliomas. Seminars in Oncology 30 (6 Suppl 19): 10–14.
Cabrera, A.R., K.C. Cuneo, A. Desjardins, J.H. Sampson, F. McSherry, J.E. Herndon 2nd, et al. 2013. Concurrent stereotactic radiosurgery and bevacizumab in recurrent malignant gliomas: A prospective trial. International Journal of Radiation Oncology, Biology, Physics 86 (5): 873–879. doi:10.1016/j.ijrobp.2013.04.029.
Celldex, Public Communication. 2016. http://ir.celldex.com/releasedetail.cfm?ReleaseID=959021.
Chan, J.L., S.W. Lee, B.A. Fraass, D.P. Normolle, H.S. Greenberg, L.R. Junck, et al. 2002. Survival and failure patterns of high-grade gliomas after three-dimensional conformal radiotherapy. Journal of Clinical Oncology 20 (6): 1635–1642.
Chang, S.M., P. Wen, T. Cloughesy, H. Greenberg, D. Schiff, C. Conrad, et al. 2005. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investigational New Drugs 23 (4): 357–361. doi:10.1007/s10637-005-1444-0.
Chinot, O.L., W. Wick, W. Mason, R. Henriksson, F. Saran, R. Nishikawa, et al. 2014. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. The New England Journal of Medicine 370 (8): 709–722. doi:10.1056/NEJMoa1308345.
Chiocca, E.A., K.M. Abbed, S. Tatter, et al. 2004. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Molecular Therapy 10: 958–966.
Chiocca, E.A., L.K. Aguilar, S.D. Bell, et al. 2011. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. Journal of Clinical Oncology 29: 3611–3619.
Choe, G., S. Horvath, T.F. Cloughesy, K. Crosby, D. Seligson, A. Palotie, et al. 2003. Analysis of the phosphatidylinositol 3′-kinase signaling pathway in glioblastoma patients in vivo. Cancer Research 63 (11): 2742–2746.
Choucair, A.K., V.A. Levin, P.H. Gutin, R.L. Davis, P. Silver, M.S. Edwards, et al. 1986. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. Journal of Neurosurgery 65 (5): 654–658. doi:10.3171/jns.1986.65.5.0654.
Combs, S.E., J. Wagner, M. Bischof, T. Welzel, F. Wagner, J. Debus, et al. 2008. Postoperative treatment of primary glioblastoma multiforme with radiation and concomitant temozolomide in elderly patients. International Journal of Radiation Oncology, Biology, Physics 70 (4): 987–992. doi:10.1016/j.ijrobp.2007.07.2368.
Cozad, S.C., P. Townsend, R.A. Morantz, A.B. Jenny, J.J. Kepes, and S.R. Smalley. 1996. Gliomatosis cerebri. Results with radiation therapy. Cancer 78 (8): 1789–1793.
Crane, C.A., S.J. Han, B. Ahn, J. Oehlke, V. Kivett, A. Fedoroff, et al. 2013. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clinical Cancer Research 19: 205–214. doi:10.1158/1078-0432.ccr-11-3358.
Del Vecchio, C.A., C.P. Giacomini, H. Vogel, K.C. Jensen, T. Florio, A. Merlo, et al. 2013. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 32 (21): 2670–2681. doi:10.1038/onc.2012.280.
Engelman, J.A. 2009. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nature Reviews. Cancer 9 (8): 550–562. doi:10.1038/nrc2664.
Eshhar, Z., T. Waks, G. Gross, and D.G. Schindler. 1993. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences of the United States of America 90: 720–410.
Esteller, M., J. Garcia-Foncillas, E. Andion, S.N. Goodman, Hidalgo OF, V. Vanaclocha, et al. 2000. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. The New England Journal of Medicine 343 (19): 1350–1354. doi:10.1056/NEJM200011093431901.
Fabrini, M.G., G. Silvano, I. Lolli, F. Perrone, A. Marsella, V. Scotti, et al. 2009. A multi-institutional phase II study on second-line Fotemustine chemotherapy in recurrent glioblastoma. Journal of Neuro-Oncology 92 (1): 79–86. doi:10.1007/s11060-008-9739-6.
Francisco, L.M., V.H. Salinas, K.E. Brown, V.K. Vanguri, G.J. Freeman, V.K. Kuchroo, et al. 2009. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. The Journal of Experimental Medicine 206: 3015–3029. doi:10.1084/jem.20090847.
Galanis, E., J.C. Buckner, M.J. Maurer, J.I. Kreisberg, K. Ballman, J. Boni, et al. 2005. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. Journal of Clinical Oncology 23 (23): 5294–5304. doi:10.1200/JCO.2005.23.622.
Gera, N., A. Yang, T.S. Holtzman, S.X. Lee, E.T. Wong, and K.D. Swanson. 2015. Tumor treating fields perturb the localization of septins and cause aberrant mitotic exit. PLoS One. 10 (5): e0125269. doi:10.1371/journal.pone.0125269.
Gilbert, M.R., M. Wang, K.D. Aldape, R. Stupp, M.E. Hegi, K.A. Jaeckle, et al. 2013. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. Journal of Clinical Oncology 31 (32): 4085–4091. doi:10.1200/JCO.2013.49.6968.
Gilbert, M.R., J.J. Dignam, T.S. Armstrong, J.S. Wefel, D.T. Blumenthal, M.A. Vogelbaum, et al. 2014. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England Journal of Medicine 370 (8): 699–708. doi:10.1056/NEJMoa1308573.
Gutin, P.H., F.M. Iwamoto, K. Beal, N.A. Mohile, S. Karimi, B.L. Hou, et al. 2009. Safety and efficacy of bevacizumab with hypofractionated stereotactic irradiation for recurrent malignant gliomas. International Journal of Radiation Oncology, Biology, Physics 75 (1): 156–163. doi:10.1016/j.ijrobp.2008.10.043.
Harrow, S., V. Papanastassiou, J. Harland, et al. 2004. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Therapy 11: 1648–1658. doi:10.1038/sj.gt.3302289.
Hegi, M.E., A.C. Diserens, T. Gorlia, M.F. Hamou, N. de Tribolet, M. Weller, et al. 2005. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England Journal of Medicine 352 (10): 997–1003. doi:10.1056/NEJMoa043331.
Herrlinger, U. 2012. Gliomatosis cerebri. Handbook of Clinical Neurology 105: 507–515. doi:10.1016/B978-0-444-53502-3.00005-7.
Hovey, E., J., Field, K., M., Rosenthal, M., Nowak, A., K., Cher, L., Wheeler, H. et al. 2015. Continuing or ceasing bevacizumab at disease progression: Results from the CABARET study, a prospective randomized phase II trial in patients with recurrent glioblastoma. 2015 ASCO Annual Meeting: American Society of Clinical Oncology.
Immonen, A., M. Vapalahti, K. Tyynela, et al. 2004. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: A randomised, controlled study. Molecular Therapy 10: 967–972. doi:10.1016/j.ymthe.2004.08.002.
Iwamoto, F.M., L.E. Abrey, K. Beal, P.H. Gutin, M.K. Rosenblum, V.E. Reuter, L.M. DeAngelis, and A.B. Lassman. 2009. Patterns of relapse and prognosis after bevacizumab failure in recurrent glioblastoma. Neurology 73 (15): 1200–1206. doi:10.1212/WNL.0b013e3181bc0184.
Keime-Guibert, F., O. Chinot, L. Taillandier, S. Cartalat-Carel, M. Frenay, G. Kantor, et al. 2007. Radiotherapy for glioblastoma in the elderly. The New England Journal of Medicine 356 (15): 1527–1535. doi:10.1056/NEJMoa065901.
Kirson, E.D., V. Dbaly, F. Tovarys, J. Vymazal, J.F. Soustiel, A. Itzhaki, et al. 2007. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proceedings of the National Academy of Sciences of the United States of America 104 (24): 10152–10157. doi:10.1073/pnas.0702916104.
Kirson, E.D., R.S. Schneiderman, V. Dbaly, F. Tovarys, J. Vymazal, A. Itzhaki, et al. 2009a. Chemotherapeutic treatment efficacy and sensitivity are increased by adjuvant alternating electric fields (TTFields). BMC Medical Physics 9: 1. doi:10.1186/1756-6649-9-1.
Kirson, E.D., M. Giladi, Z. Gurvich, A. Itzhaki, D. Mordechovich, R.S. Schneiderman, et al. 2009b. Alternating electric fields (TTFields) inhibit metastatic spread of solid tumors to the lungs. Clinical & Experimental Metastasis 26 (7): 633–640. doi:10.1007/s10585-009-9262-y.
Kita, D., I.F. Ciernik, S. Vaccarella, S. Franceschi, P. Kleihues, U.M. Lutolf, et al. 2009. Age as a predictive factor in glioblastomas: Population-based study. Neuroepidemiology 33 (1): 17–22. doi:10.1159/000210017.
Kreisl, T.N., L. Kim, K. Moore, P. Duic, C. Royce, I. Stroud, et al. 2009. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. Journal of Clinical Oncology 27 (5): 740–745. doi:10.1200/JCO.2008.16.3055.
Kubben, P.L., K.J. ter Meulen, O.E. Schijns, M.P. ter Laak-Poort, J.J. van Overbeeke, and H. van Santbrink. 2011. Intraoperative MRI-guided resection of glioblastoma multiforme: A systematic review. The Lancet Oncology 12 (11): 1062–1070. doi:10.1016/S1470-2045(11)70130-9.
Lacroix, M., D. Abi-Said, D.R. Fourney, Z.L. Gokaslan, W. Shi, F. DeMonte, F.F. Lang, I.E. McCutcheon, S.J. Hassenbusch, E. Holland, K. Hess, C. Michael, D. Miller, and R. Sawaya. 2001. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. Journal of Neurosurgery 95 (2): 190–198. doi:10.3171/jns.2001.95.2.0190.
Li, Y.M., D. Suki, K. Hess, and R. Sawaya. 2016. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? Journal of Neurosurgery 124 (4): 977–988. doi:10.3171/2015.5.JNS142087.
Malmstrom, A., B.H. Gronberg, C. Marosi, R. Stupp, D. Frappaz, H. Schultz, et al. 2012. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. The Lancet Oncology 13 (9): 916–926. doi:10.1016/S1470-2045(12)70265-6.
Markert, J.M., M.D. Medlock, S.D. Rabkin, et al. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: Results of a phase I trial. Gene Therapy 7: 867–874. doi:10.1038/sj.gt.3301205.
McKie, E.A., A.R. MacLean, A.D. Lewis, et al. 1996. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours—evaluation of a potentially effective clinical therapy. British Journal of Cancer 74: 745–752.
Miao, H., B.D. Choi, C.M. Suryadevara, L. Sanchez-Perez, S. Yang, G. De Leon, et al. 2014. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One 9 (4): e94281. doi:10.1371/journal.pone.0094281.
Michaelsen, S.R., I.J. Christensen, K. Grunnet, M.T. Stockhausen, H. Broholm, M. Kosteljanetz, et al. 2013. Clinical variables serve as prognostic factors in a model for survival from glioblastoma multiforme: An observational study of a cohort of consecutive non-selected patients from a single institution. BMC Cancer 13: 402. doi:10.1186/1471-2407-13-402.
Minniti, G., C. Scaringi, G. Lanzetta, I. Terrenato, V. Esposito, A. Arcella, et al. 2015. Standard (60 Gy) or short-course (40 Gy) irradiation plus concomitant and adjuvant temozolomide for elderly patients with glioblastoma: A propensity-matched analysis. International Journal of Radiation Oncology, Biology, Physics 91 (1): 109–115. doi:10.1016/j.ijrobp.2014.09.013.
Muragaki, Y., T. Maruyama, H. Iseki, M. Tanaka, C. Shinohara, K. Takakura, et al. 2011. Phase I/IIa trial of autologous formalin-fixed tumor vaccine concomitant with fractionated radiotherapy for newly diagnosed glioblastoma. Journal of Neurosurgery 115: 248–255. doi:10.3171/2011.4.JNS10377.
NCT00805376. https://clinicaltrials.gov/ct2/show/NCT00805376?term=NCT00805376&rank=1.
NCT01582516. https://clinicaltrials.gov/ct2/show/NCT01582516?term=NCT01582516&rank=1.
Nelson, D.F., M. Diener-West, J. Horton, C.H. Chang, D. Schoenfeld, and J.S. Nelson. 1988. Combined modality approach to treatment of malignant gliomas--re-evaluation of RTOG 7401/ECOG 1374 with long-term follow-up: A joint study of the Radiation Therapy Oncology Group and the Eastern Cooperative Oncology Group. NCI Monographs 6: 279–284.
Nevin, S. 1938. Gliomatosis cerebri. Brain 61: 170–191.
Nwokedi, E.C., S.J. DiBiase, S. Jabbour, J. Herman, P. Amin, and L.S. Chin. 2002. Gamma knife stereotactic radiosurgery for patients with glioblastoma multiforme. Neurosurgery 50 (1): 41–46. discussion 46–7.
Ostertag, D., K.K. Amundson, F. Lopez Espinoza, B. Martin, T. Buckley, A.P. Galvao da Silva, A.H. Lin, D.T. Valenta, O.D. Perez, C.E. Ibanez, C.I. Chen, P.L. Pettersson, R. Burnett, V. Daublebsky, J. Hlavaty, W. Gunzburg, N. Kasahara, H.E. Gruber, D.J. Jolly, and J.M. Robbins. 2012. Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro-Oncology 14 (2): 145–159. doi:10.1093/neuonc/nor199.
Papanastassiou, V., R. Rampling, M. Fraser, et al. 2002. The potential for efficacy of the modified (ICP 34.5(−)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: A proof of principle study. Gene Therapy 9(6): 398–406. doi:10.1038/sj.gt.3301664.
Parsa, A.T., J.S. Waldron, A. Panner, C.A. Crane, I.F. Parney, J.J. Barry, et al. 2007. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature Medicine 13: 84–88. doi:10.1038/nm1517.
Paszat, L., N. Laperriere, P. Groome, K. Schulze, W. Mackillop, and E. Holowaty. 2001. A population-based study of glioblastoma multiforme. International Journal of Radiation Oncology, Biology, Physics 51 (1): 100–107.
Peggs, K.S., S.A. Quezada, C.A. Chambers, A.J. Korman, and J.P. Allison. 2009. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. The Journal of Experimental Medicine 206: 1717–1725. doi:10.1084/jem.20082492.
Pelloski, C.E., K.V. Ballman, A.F. Furth, L. Zhang, E. Lin, E.P. Sulman, et al. 2007. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. Journal of Clinical Oncology 25 (16): 2288–2294. doi:10.1200/JCO.2006.08.0705.
Perry, J.R., N. Laperriere, C.J. O’Callaghan, A.A. Brandes, J. Menten, C. Phillips, M.F. Fay, R. Nishikawa, J.G. Cairncross, W. Roa, D. Osoba, A. Sahgal, H.W. Hirte, W. Wick, F. Laigle-Donadey, E. Franceschi, O.L. Chinot, C. Winch, K. Ding, and W.P. Mason. 2016. A phase III randomized controlled trial of short-course radiotherapy with or without concomitant and adjuvant temozolomide in elderly patients with glioblastoma (CCTG CE.6, EORTC 26062-22061, TROG 08.02, NCT00482677). Journal of Clinical Oncology 34 (15, supplement (2016 ASCO Annual Meeting)):LBA2.
Pitz, M.W., E.A. Eisenhauer, M.V. MacNeil, B. Thiessen, J.C. Easaw, D.R. Macdonald, et al. 2015. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncology 17 (9): 1270–1274. doi:10.1093/neuonc/nou365.
Pol, J.G., M. Marguerie, R. Arulanandam, et al. 2013. Panorama from the oncolytic virotherapy summit. Molecular Therapy 21: 1814–1818. doi:10.1038/mt.2013.207.
Powers, M.V., K. Jones, C. Barillari, I. Westwood, R.L. van Montfort, and P. Workman. 2010. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 9: 1542–1550. doi:10.4161/cc.9.8.11204.
Prins, R.M., H. Soto, Odesa S.K. KonkankitV, A. Eskin, W.H. Yong, et al. 2011. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clinical Cancer Research 17(6): 1603–1615. doi:10.1158/1078-0432.ccr-10-2563.
Quant, E.C., A.D. Norden, J. Drappatz, A. Muzikansky, L. Doherty, D. Lafrankie, A. Ciampa, S. Kesari, and P.Y. Wen. 2009. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro-Oncology 11 (5): 550–555. doi:10.1215/15228517-2009-006.
Rainov, N.G. 2000. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Human Gene Therapy 11(17): 2389–2401. doi:10.1089/104303400750038499.
Rampling, R., G. Cruickshank, V. Papanastassiou, et al. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Therapy 7: 859–866.
Reardon, D.A., G. Akabani, R.E. Coleman, A.H. Friedman, H.S. Friedman, J.E. Herndon 2nd, et al.2006. Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: Phase II study results. Journal of Clinical Oncology 24 (1): 115–122. doi:10.1200/JCO.2005.03.4082.
Reifenberger, G., B. Hentschel, J. Felsberg, G. Schackert, M. Simon, O. Schnell, et al. 2012. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. International Journal of Cancer 131 (6): 1342–1350. doi:10.1002/ijc.27385.
Roa, W., P.M. Brasher, G. Bauman, M. Anthes, E. Bruera, A. Chan, et al. 2004. Abbreviated course of radiation therapy in older patients with glioblastoma multiforme: a prospective randomized clinical trial. Journal of Clinical Oncology 22 (9): 1583–1588. doi:10.1200/JCO.2004.06.082.
Rudd, C.E., A. Taylor, and H. Schneider. 2009. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunological Reviews 229: 12–26. doi:10.1111/j.1600-065X.2009.00770.x.
Sampson, J.H., A.B. Heimberger, G.E. Archer, K.D. Aldape, A.H. Friedman, H.S. Friedman, et al. 2010. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology 28: 4722–4729. doi:10.1200/jco.2010.28.6963.
Sanai, N., M.Y. Polley, M.W. McDermott, A.T. Parsa, and M.S. Berger. 2011. An extent of resection threshold for newly diagnosed glioblastomas. Journal of Neurosurgery 115 (1): 3–8. doi:10.3171/2011.2.JNS10998.
Sandmair, A.M., S. Loimas, P. Puranen, et al. 2000. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Human Gene Therapy 11(16): 2197–2205. doi:10.1089/104303400750035726.
Schwartz, R.H. 1992. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 71: 1065–1068.
Seystahl, K., W. Wick, and M. Weller. 2016. Therapeutic options in recurrent glioblastoma–An update. Critical Reviews in Oncology/Hematology 99: 389–408. doi:10.1016/j.critrevonc.2016.01.018.
Sloan, A.E., M.S. Ahluwalia, J. Valerio-Pascua, S. Manjila, M.G. Torchia, S.E. Jones, et al. 2013. Results of the NeuroBlate System first-in-humans Phase I clinical trial for recurrent glioblastoma: Clinical article. Journal of Neurosurgery 118 (6): 1202–1219. doi:10.3171/2013.1.JNS1291.
Souhami, L., W. Seiferheld, D. Brachman, E.B. Podgorsak, M. Werner-Wasik, R. Lustig, et al. 2004. Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: Report of Radiation Therapy Oncology Group 93-05 protocol. International Journal of Radiation Oncology, Biology, Physics 60 (3): 853–860. doi:10.1016/j.ijrobp.2004.04.011.
Stark-Vance, V. 2005. Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. Neuro-Oncology 7: 369.
Stummer, W., U. Pichlmeier, T. Meinel, O.D. Wiestler, F. Zanella, H.J. Reulen, et al. 2006. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. The Lancet Oncology 7 (5): 392–401. doi:10.1016/S1470-2045(06)70665-9.
Stupp, R., W.P. Mason, M.J. van den Bent, M. Weller, B. Fisher, M.J. Taphoorn, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England Journal of Medicine 352 (10): 987–996. doi:10.1056/NEJMoa043330.
Stupp, R., M.E. Hegi, W.P. Mason, M.J. van den Bent, M.J. Taphoorn, R.C. Janzer, et al. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology 10 (5): 459–466. doi:10.1016/S1470-2045(09)70025-7.
Stupp, R., E.T. Wong, A.A. Kanner, D. Steinberg, H. Engelhard, V. Heidecke, et al. 2012. NovoTTF-100A versus physician's choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. European Journal of Cancer 48 (14): 2192–2202. doi:10.1016/j.ejca.2012.04.011.
Stupp, R., S. Taillibert, A.A. Kanner, S. Kesari, D.M. Steinberg, S.A. Toms, et al. 2015. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. Journal of the American Medical Association 314 (23): 2535–2543. doi:10.1001/jama.2015.16669.
Suchorska, B., M. Weller, G. Tabatabai, C. Senft, P. Hau, M.C. Sabel, et al. 2016. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma-results from the DIRECTOR trial. Neuro-Oncology 18 (4): 549–556. doi:10.1093/neuonc/nov326.
Taal, W., H.M. Oosterkamp, A.M. Walenkamp, H.J. Dubbink, L.V. Beerepoot, M.C. Hanse, et al. 2014. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. The Lancet Oncology 15 (9): 943–953. doi:10.1016/S1470-2045(14)70314-6.
Taillibert, S., C. Chodkiewicz, F. Laigle-Donadey, M. Napolitano, S. Cartalat-Carel, and M. Sanson. 2006. Gliomatosis cerebri: A review of 296 cases from the ANOCEF database and the literature. Journal of Neuro-Oncology 76 (2): 201–205. doi:10.1007/s11060-005-5263-0.
Thaci, B., C.E. Brown, E. Binello, K. Werbaneth, P. Sampath, and S. Sengupta. 2014. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro-Oncology 16 (10): 1304–1312.
Tolcher, A.W., S.L. Gerson, L. Denis, C. Geyer, L.A. Hammond, A. Patnaik, et al. 2003. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. British Journal of Cancer 88 (7): 1004–1011. doi:10.1038/sj.bjc.6600827.
Torok, J.A., R.E. Wegner, A.H. Mintz, D.E. Heron, and S.A. Burton. 2011. Re-irradiation with radiosurgery for recurrent glioblastoma multiforme. Technology in Cancer Research & Treatment 10 (3): 253–258.
Vauleon, E., T. Avril, B. Collet, J. Mosser, and V. Quillien. 2010. Overview of cellular immunotherapy for patients with glioblastoma. Clinical & Developmental Immunology 2010: 689171. doi:10.1155/2010/689171.
Velu, V., K. Titanji, B. Zhu, S. Husain, A. Pladevega, L. Lai, et al. 2009. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458(7235): 206–210. doi:10.1038/nature07662.
Vuorinen, V., S. Hinkka, M. Farkkila, and J. Jaaskelainen. 2003. Debulking or biopsy of malignant glioma in elderly people – a randomised study. Acta Neurochirurgica 145 (1): 5–10. doi:10.1007/s00701-002-1030-6.
Walker, M.D., E. Alexander Jr., W.E. Hunt, C.S. MacCarty, M.S. Mahaley Jr., J. Mealey Jr., et al. 1978. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. Journal of Neurosurgery 49 (3): 333–343. doi:10.3171/jns.1978.49.3.0333.
Walker, M.D., S.B. Green, D.P. Byar, E. Alexander Jr., U. Batzdorf, W.H. Brooks, et al. 1980. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. The New England Journal of Medicine 303 (23): 1323–1329. doi:10.1056/NEJM198012043032303.
Wallner, K.E., J.H. Galicich, G. Krol, E. Arbit, and M.G. Malkin. 1989. Patterns of failure following treatment for glioblastoma multiforme and anaplastic astrocytoma. International Journal of Radiation Oncology, Biology, Physics 16 (6): 1405–1409.
Weller, M., T. Cloughesy, J.R. Perry, and W. Wick. 2013. Standards of care for treatment of recurrent glioblastoma–are we there yet? Neuro-Oncology 15 (1): 4–27. doi:10.1093/neuonc/nos273.
Wen, P.Y., A. Omuro, M.S. Ahluwalia, H.M. Fathallah-Shaykh, N. Mohile, J.J. Lager, et al. 2015. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro-Oncology 17 (9): 1275–1283. doi:10.1093/neuonc/nov083.
Westphal, M., D.C. Hilt, E. Bortey, P. Delavault, R. Olivares, P.C. Warnke, et al. 2003. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology 5 (2): 79–88. doi:10.1215/S1522-8517-02-00023-6.
Westphal, M., Z. Ram, V. Riddle, D. Hilt, E. Bortey, and Executive Committee of the Gliadel Study G. 2006. Gliadel wafer in initial surgery for malignant glioma: Long-term follow-up of a multicenter controlled trial. Acta Neurochirurgica 148 (3): 269–275. doi:10.1007/s00701-005-0707-z.discussion 275
Westphal, M., O. Heese, J.P. Steinbach, O. Schnell, G. Schackert, M. Mehdorn, et al. 2015. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. European Journal of Cancer 51 (4): 522–532. doi:10.1016/j.ejca.2014.12.019.
Wick, W., V.K. Puduvalli, M.C. Chamberlain, M.J. van den Bent, A.F. Carpentier, L.M. Cher, et al. 2010. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. Journal of Clinical Oncology 28 (7): 1168–1174. doi:10.1200/JCO.2009.23.2595.
Wick, W., T. Gorlia, P. Bady, M. Platten, M.J. van den Bent, M.J. Taphoorn, et al. 2016. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clinical Cancer Research. doi:10.1158/1078-0432.CCR-15-3153.
Wilson, E.H., W. Weninger, and C.A. Hunter. 2010. Trafficking of immune cells in the central nervous system. The Journal of Clinical Investigation 120: 1368–1379. doi:10.1172/jci41911.
Wing, K., Y. Onishi, P. Prieto-Martin, T. Yamaguchi, M. Miyara, Z. Fehervari, et al. 2008. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322 (5899): 271–275. doi:10.1126/science.1160062.
Wong, A.J., J.M. Ruppert, S.H. Bigner, C.H. Grzeschik, P.A. Humphrey, D.S. Bigner, et al. 1992. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences of the United States of America 89: 2965–2969. doi:10.1073/pnas.89.7.2965.
Wong, E.T., E. Lok, and K.D. Swanson. 2015. An evidence-based review of alternating electric fields therapy for malignant gliomas. Current Treatment Options in Oncology 16 (8): 40. doi:10.1007/s11864-015-0353-5.
Yang, L., R. Hwang, Y. Chiang, et al. 1998. Mechanisms for ganciclovir resistance in gastrointestinal tumor cells transduced with a retroviral vector containing the herpes simplex virus thymidine kinase gene. Clinical Cancer Research 4: 731–741.
Yong, R.L., T. Wu, N. Mihatov, M.J. Shen, M.A. Brown, K.A. Zaghloul, et al. 2014. Residual tumor volume and patient survival following reoperation for recurrent glioblastoma. Journal of Neurosurgery 121 (4): 802–809. doi:10.3171/2014.6.JNS132038.
Young, J.C., V.R. Agashe, K. Siegers, and F.U. Hartl. 2004. Pathways of chaperone-mediated protein folding in the cytosol. Nature Reviews. Molecular Cell Biology 5 (10): 781–791. doi:10.1038/nrm1492.
Zalutsky, M.R., D.A. Reardon, G. Akabani, R.E. Coleman, A.H. Friedman, H.S. Friedman, et al. 2008. Clinical experience with alpha-particle emitting 211At: Treatment of recurrent brain tumor patients with 211At-labeled chimeric antitenascin monoclonal antibody 81C6. Journal of Nuclear Medicine 49 (1): 30–38. doi:10.2967/jnumed.107.046938.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Wang, J.L., Mugge, L., Giglio, P., Puduvalli, V.K. (2017). Current Therapies and Future Directions in Treatment of Glioblastoma. In: Somasundaram, K. (eds) Advances in Biology and Treatment of Glioblastoma. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-319-56820-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-56820-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56819-5
Online ISBN: 978-3-319-56820-1
eBook Packages: MedicineMedicine (R0)