
Abstract
Sphingolipids (SLs) are sphingoid base-containing lipids, which are enriched in plasma membrane microdomains. Some SLs behave as bioactive molecules, modulating cell signalling in various pathophysiological contexts. Whereas ceramide triggers apoptosis and impairs cell migration, sphingosine-1-phosphate (S1P) induces the opposite effects. CD95/Fas, TRAIL-R1 (DR4), and TRAIL-R2 (DR5) are death receptors (DRs) and members of the TNF-R1 superfamily. DRs trigger apoptosis of various cell types, including cancer cells. Over the last two decades, a growing body of evidence indicates that SLs modulate DR signalling. DR stimulation triggers the generation of SLs, including ceramide, sphingosine, and gangliosides. Ceramide has been reported to facilitate DR clustering into lipid rafts upon pro-apoptotic DR agonists. Moreover, ceramide and its metabolites likely contribute to the mitochondrial route of apoptosis. More recently, SLs have been shown to modulate CD95-mediated cell migration of triple negative breast cancer cell lines and Th17 lymphoid cells in response to a nonapoptotic form of CD95L. Herein, we review the role of SLs in DR signalling, including apoptotic and migration pathways.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
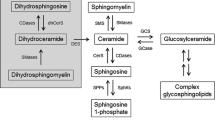
10.1 Sphingolipid Metabolism at a Glance
Sphingolipids (SLs) are one of the most complex and structurally diverse classes of lipids. They are defined by an aliphatic amino alcohol sphingoid backbone called sphingosine . Modification of this core results in the formation of a multitude of SLs that play important roles in membrane biology and signal transduction.
SL metabolism begins with the “de novo” biosynthesis pathway of ceramide. This reaction occurs on the cytosolic surface of the endoplasmic reticulum (ER) membrane, via the condensation of l-serine and palmitoyl coenzyme A (palmitoyl-CoA ) into 3-ketodihydrosphingosine (3-KDS ), catalyzed by the enzyme serine palmitoyltransferase (SPT) [1]. 3-KDS is subsequently reduced to form dihydrosphingosine (sphinganine) , which is then N-acylated to generate dihydroceramide (dhCer ) by the action of six mammalian dhCer synthases [also known as LASS or ceramide synthases (CerS )], each of which synthesizes dhCer with a specific chain length fatty acyl CoA. dhCer desaturase next desaturates dhCer to give ceramide, the central molecule in SL metabolism [2] (Fig. 10.1).

Subcellular compartmentalization of sphingolipid metabolism. Whereas most of the sphingolipid synthesis occurs in the endoplasmic reticulum and Golgi apparatus , most of the sphingolipid catabolism takes place in the lysosomes. Sphingolipid metabolism also occurs at the plasma membrane
Following transport to the Golgi apparatus , ceramide can then serve as a precursor for the biosynthesis of sphingomyelin (SM ) and glycosphingolipids (GSLs ), by the addition of various polar head groups. SM synthases (SMS) 1 and 2 transfer a phosphocholine headgroup from phosphatidylcholine to ceramide, thereby generating SM and diacylglycerol (DAG ) [3]. On the other hand glucosylceramide synthase (GCS ) attaches glucose to ceramide, thus generating glucosylceramide (GlcCer). Ceramide can also be glycosylated by galactosylceramide synthase (GalCerS ) to galactosylceramide (GalCer ) at the ER. GlcCer and GalCer are the precursors of the hundreds of the most complex GSLs including gangliosides, which are with SM the major plasma membrane lipid raft components. There, SM can be hydrolyzed by acid sphingomyelinase (aSMase ) (on the outer leaflet) and neutral sphingomyelinases (nSMase) (on the inner leaflet) to produce ceramide. SM can be also catabolized to ceramide in the lysosomal compartment by aSMase . Another important source of ceramide is provided by the hydrolysis of GSLs by specific β-glucosidases and galactosidases [4] (Fig. 10.1).
By the action of ceramide kinase, ceramide can be phosphorylated to form ceramide-1-phosphate (C1P ), which in turn can be recycled by a C1P phosphatase [5]. Ceramide can be also deacylated by ceramidases to sphingosine, which in turn can be phosphorylated to sphingosine-1-phosphate (S1P) by sphingosine kinases 1 and 2 [6]. Ceramidases are also distinguished according to their optimum pH and subcellular localization. Neutral ceramidase is located at the plasma membrane, acid ceramidase is lysosomal, and the alkaline ceramidase is located at the ER and Golgi apparatus . Ceramidase has been shown to be also present in the mitochondria [7].
Various alterations of SL catabolism due to gene mutations lead to lysosomal storage diseases. For instance, aSMase deficiency is responsible for type A and B Niemann-Pick diseases (NPD), the type A being the most severe form associated with the strongest loss of aSMase enzyme activity and lysosomal accumulation of SM [8]. Moreover, human genetic disorders of SL biosynthesis have been recently described in patients [9].
10.2 Role of Sphingolipids in CD95-Mediated Cell Apoptosis
10.2.1 Ceramide-Mediated CD95 Clustering in Lipid Rafts
The cell membrane of eukaryotic cells consists of a lipid bilayer containing predominantly SLs, cholesterol and glycerophospholipids with embedded proteins. Hydrophilic interactions between the polar head groups of SLs on one hand, and interactions between SLs and cholesterol on the other hand, result in the separation of cholesterol and SLs from the other lipids within the bilayer of the cell membrane and the formation of distinct microdomains named lipid rafts [10]. These small domains can float freely within the cellular membrane bilayer or assemble together to form large ordered platforms that play a central role in many cellular processes, including CD95-mediated apoptosis [11]. An important early event at the plasma membrane following the initiation of CD95-mediated apoptosis involves the clustering of CD95 in these lipid rafts for efficient and rapid transmembrane signalling [12] (Fig. 10.2). One should note, however, that CD95 constitutively resides or not in lipid rafts depending on the cell type. Indeed, CD95 was located in lipid rafts in type I cells but not in type II cells [13]. Whereas disruption of lipid rafts impaired CD95-induced apoptosis in type I cells [13], forced distribution of CD95 in rafts enhances type II cell death [14], arguing that CD95 location in lipid rafts is important for potent apoptosis signalling initiation.

Sphingolipids in CD95L-induced apoptosis . Binding of Membrane-bound CD95L (m-CD95L) to its cognate receptor CD95 leads to the activation of sphingomyelinase (SMase ). SMase catalyzes the hydrolysis of membrane sphingomyelin to ceramide, thus forming ceramide-enriched plasma membrane platforms that facilitate CD95 clustering in lipid rafts and DISC formation. m-CD95L/CD95 interaction rises also the intracellular ceramide level through different pathways. Whereas it enhances ceramide synthesis by the de novo pathway, it inhibits the metabolism of ceramide to sphingomyelin by inactivating sphingomyelin synthase 1 (SMS 1). Ceramide, sphingosine (So), and ganglioside 3 (GD3) activate the mitochondrial pathway leading to apoptosis
CD95 engagement triggers a rapid translocation of the aSMase from intracellular stores to the plasma membrane outer surface. ASMase is one of the enzymes expressed by the mammalian cells that are responsible for catalyzing the hydrolysis of SM to phosphocholine and ceramide. The SMases are characterized by their optimal pH: acid, neutral, or alkaline [15]. Of these enzymes, the aSMase is rapidly activated, within a few seconds to minutes, upon stimulation via CD95 [12]. ASMase showed strong activity with an optimum pH of 5.0, however, it can be active even at a neutral or slightly acidic pH. Indeed, an increase of the pH lowers the enzyme’s affinity for its substrate, while the activity of the enzyme remains unaffected. Since SM is essentially, but not exclusively, located in the anti-cytosolic leaflet of biological membranes, translocated aSMase , which localizes to SL-rich rafts, catalyzes the breakdown of SM to ceramide at the cell surface. Extracellularly orientated ceramides self-associate with each other and, therefore, induce the transformation of small existing rafts into large active signalling platforms called ceramide-enriched membrane domains that mediate selective clustering of CD95 and transmit the apoptotic stimulus from outside into the cell [12].
The formation of ceramide-enriched membrane platforms has been demonstrated in vitro by the treatment of SM -containing liposomes with immobilized SMase onto micro-beads [16] and in cellulo by the use of confocal microscopy employing anti-ceramide antibodies [17]. One should note, however, that the specificity of the anti-ceramide antibodies has not been firmly established, and the possibility that antibodies cross-react with complex SLs cannot be ruled out. The significance of these membrane platforms for CD95 signalling was indicated by the finding that the disruption of rafts using methyl-β-cyclodextrin , a compound that extracts cholesterol from the cell membrane, the neutralization of surface ceramides by treatment with anti-ceramide antibody or aSMase deficiency, prevented CD95 clustering and apoptosis. Moreover, providing cells with natural exogenous ceramide rescued aSMase-deficient cells and restored CD95 clustering and apoptosis [12]. Another interesting observation further argues that SLs in rafts play a critical role in CD95 pro-apoptotic signalling pathway activation. Indeed, SMS deficiency in murine leukemia cells, which altered plasma membrane composition and properties through SM depletion, impaired CD95 location into lipid rafts and conferred partial resistance to CD95-mediated apoptosis [18]. It is tempting to speculate that SM depletion impairs apoptosis because of the lack of substrate for aSMase -dependent ceramide generation in response to CD95 engagement.
The molecular mechanisms leading to aSMase activation and translocation to the cell surface upon CD95 stimulation have been investigated. FADD and caspase-8 are both required for CD95-induced aSMase activation [19] and overexpression of FADD (Fas-associated protein with death domain) or caspase-8 triggers ceramide production [20]. Upon CD95/CD95L interaction, the adaptor protein FADD is recruited to the CD95 death domain. Next, FADD activates a limited pool of caspase-8, which would be sufficient for the activation of aSMase and the mobilization of intracellular aSMase-containing vesicles to fuse with the cell membrane, resulting in the secretion or the exposure of the enzyme on the extracellular leaflet [21]. It was demonstrated that aSMase inactive mutants still translocate to the cell surface, indicating that aSMase cell surface exposure is independent on the enzyme activation [22]. This translocation is mediated by an exocytosis phenomenon implicating the t-SNARE protein Syntaxin 4 and requires an intact cytoskeleton [23, 24]. Following SM breakdown to ceramide, CD95 clustering, which results in a very high receptor density in the cell membrane, may stabilize the death-inducing signalling complex (DISC), leading to potent FADD -dependent caspase-8 activation and amplifying CD95-mediated cell death signalling [25].
Several studies have documented the significance of aSMase as a general requisite for the induction of CD95-mediated apoptosis. ASMase knockout mice were partially, yet significantly, resistant to fulminant hepatitis induced by the injection of anti-CD95, as compared to their wild-type counterparts [26]. Exogenous natural ceramide overcame resistance of aSMase-deficient hepatocytes towards CD95 agonist [27]. Furthermore, genetic studies employing lymphoblastoid cell lines (LCL) from NPD patients, which suffer from an in-born defect of aSMase, showed LCL resistance to apoptosis induced by an anti-CD95 agonist, while aSMase-positive control cells rapidly underwent programmed cell death. CD95-induced apoptosis was restored in aSMase-deficient cells by the addition of exogenous ceramide or aSMase [28]. However, others and we have questioned the role of aSMase in CD95-mediated cell death. Indeed, thymocytes, activated T and B cells derived from both aSMase -deficient and wild-type mice were equally sensitive to anti-CD95 or CD95L [26]. Moreover, LCL as well as SV40-transformed human fibroblasts deficient for aSMase died upon CD95 stimulation. Caspase-3 activation as well as ceramide production occurred to a similar extent in aSMase-deficient and -proficient cells in response to CD95 engagement [29, 30]. Thus, aSMase is not essential for the induction of CD95-induced apoptosis and ceramide increase. In this context, ceramide can be generated by different pathways including ceramide de novo synthesis [31], activation of nSMase [30], and inhibition of SM synthesis [32, 33].
10.2.2 Relationship Between Ceramide and Caspases in CD95-Induced Apoptosis
The molecular connection between ceramide and caspases in CD95 signalling has been essentially documented in type II Jurkat leukemia T cells , in which CD95 stimulation triggers ceramide accumulation concomitantly to apoptosis induction [33,34,35,36]. Ceramide increase was prevented by the broad-spectrum caspase inhibitor zVAD-fmk [34, 36], arguing that CD95-mediated ceramide accumulation occurs in a caspase-dependent manner. One should note, however, that the effector caspase inhibitor DEVD-CHO, which completely prevented CD95-mediated apoptosis, failed to block ceramide increase. Thus, whereas CD95-induced ceramide increase is caspase-dependent, it occurs upstream of effector caspase activation and is unlikely a mere consequence of apoptosis induction [37]. Rather, ceramide may be instrumental in CD95 pro-apoptotic signalling pathway since exogenous short-chain ceramides potently activate caspase-3 and trigger apoptosis in Jurkat T cells [36,37,38].
The role of initiator caspases in ceramide generation upon CD95 stimulation has been investigated by diverse groups. Caspase-8 deficiency abolished anti-CD95-induced ceramide increase [35]. Moreover, caspase-8 and caspase-10 doubly deficiency totally abolished ceramide production in response to CD95L [38], further arguing that apical caspases are required for ceramide generation in CD95 signalling. In contrast, neither the caspase-9 inhibitor zLEHD-fmk [34] nor caspase-9 deficiency [38] impaired CD95-induced ceramide production. Accordingly, Bcl-xL overexpression, which impairs cytochrome c release from the mitochondria, did not impair CD95-induced ceramide production [36]. Thus, CD95-induced ceramide increase likely occurs in between the activation of apical caspases-8/-10 and mitochondrial events leading to cytochrome c release, which is required for caspase-9 activation.
10.2.3 Sphingolipids in CD95-Induced Intrinsic Pathway Activation
A growing body of evidence in the literature indicates that ceramide is instrumental in the activation of the intrinsic pathway, which involves cytochrome c release from the mitochondria and subsequent caspase-9 activation (Fig. 10.2). Cuvillier and coworkers have documented that treatment of Jurkat cells with exogenous ceramide and sphingosine led to cytochrome c release from the mitochondria and caspase-9 activation [36]. More recently, we have shown that exogenous ceramide-induced apoptosis of Jurkat cells occurred in a caspase-9-dependent manner [38]. Indeed, caspase-9-deficient Jurkat cells , which potently resisted CD95-mediated apoptosis [39], were also resistant towards C2- and C16-ceramides. Our data are in good agreement with the resistance of neuronal cells towards C2-ceramide conferred by caspase-9 pharmacological inhibition or overexpression of a dominant negative form of caspase-9 [40]. Of note, caspase-9 inhibition did not compromise exogenous ceramide-induced apoptosis in K562 leukemia cells [41], indicating that the role of caspase-9 might be dependent on the cell type or the experimental conditions. As a matter of fact, we observed that caspase-9 deficiency failed to confer resistance of Jurkat cells upon incubation with a high C2-ceramide concentration (i.e., 20 μM), which triggered necrosis rather than apoptosis [38]. We have also illustrated the role of endogenous ceramide in CD95L-induced caspase-9 activation in Jurkat cells. Indeed, SMS1 knockdown not only facilitated ceramide accumulation due to the reduced conversion of ceramide to SM but also enhanced cytochrome c release from the mitochondria and caspase-9 activation upon CD95L [38]. Moreover, the tricyclodecan-9-yl-xanthogenate D609, which inhibited SM synthesis and, albeit to a lesser extent, GlcCer synthesis in Jurkat T cells and in PHA-activated human T cells, increased endogenous ceramide levels and significantly enhanced caspase activation and apoptosis in response to CD95L. Interestingly, D609 not only overcame zVAD-fmk-conferred resistance to CD95L but also bypassed RIP deficiency. Mitochondrial events were likely involved, since Bcl-xL overexpression abolished D609 effects. Thus, the inhibition of ceramide conversion to SM enhanced CD95L-induced both caspase-dependent and -independent cell death in T cells [42].
The molecular mechanisms by which ceramide enhances CD95-induced mitochondrial pathway activation are not yet completely elucidated. Exogenous ceramides triggered mitochondrial outer-membrane permeabilization (MOMP) on purified mitochondria, most likely via ceramide channel formation. Moreover, SLs likely cooperated with canonical pathways, which involve pro-apoptotic Bcl-2 family members (Bid, Bax, Bad). Indeed, the cleavage of Bid by apical caspases-8 and -10 in response to CD95L [43] triggers cytochrome c release from the mitochondria in a Bax/Bak-dependent manner. Exogenous ceramides synergistically cooperated with Bax to induce the MOMP on purified mitochondria [44]. Moreover, the ceramide catabolites, S1P and hexadecenal, a product of S1P degradation by sphingosine-1-phosphate lyase, cooperated with Bak and Bax, respectively, to enhance tBid-induced MOMP on purified mitochondria [45]. Moreover, ceramide, which inhibits the PI3K/Akt pathway, likely facilitates the activation of Bad, another pro-apoptotic member of the Bcl-2 family involved in MOMP [46]. Finally, another interesting hypothesis is to consider that ceramide triggers an alternative splicing of the mRNA encoding caspase-9 and Bcl-xL, leading to pro-apoptotic caspase-9 and Bcl-x(s) expression [47].
Testi and coworkers have provided evidence that CD95 engagement led to GD3 ganglioside production, which disrupted mitochondrial transmembrane potential in hematopoietic cells. Pharmacological and epigenetic approaches inhibiting GD3 synthesis, impaired CD95-induced apoptosis [48]. The mechanism of GD3 production upon CD95 engagement remains a matter of debate. Whereas aSMase has been initially described as required for GD3 production in CD95 signalling [28], others did not confirm aSMase involvement [49]. Alternatively, GD3 production is due to an enhanced GD3 synthase gene expression as a consequence of NF-κB activation following CD95 engagement [50]. An intriguing point to consider is how a raft component (i.e., GD3) migrates to mitochondria upon CD95 stimulation. Interaction between GD3 and microtubules via CLIPR-59, a CLIP-170-related protein, has been shown to facilitate the redistribution of GD3 from rafts to mitochondria [51, 52]. GD3 is unlikely the only ganglioside produced in CD95 signalling. Moreover, the role of gangliosides in CD95-induced cell death has been challenged by the observation that inhibiting glucosylceramide synthase, which critically contributes to ganglioside production, did not abrogate the apoptotic process [49].
10.3 Role of Sphingolipids in cl-CD95L-Mediated Cell Motility
Even though to date CD95/CD95L is one of the best characterized apoptosis-inducing receptor–ligand system, it can also trigger, in different pathophysiological contexts, nonapoptotic signalling [53]. Such signalling ranges from inflammation to carcinogenesis and even increased cell motility, which, in case of breast cancer, enables cancer cells to migrate and form metastatic tumors. In this context, it was shown that the level of the soluble form of CD95L is higher in the blood of patients with triple negative breast cancer (TNBC ) than that of patients affected with other breast cancer subtypes, and was associated with increased risk of developing distant metastases [54].
CD95L is a transmembrane member of the TNFα superfamily whose extracellular domain can be cleaved by metalloproteases or cathepsins, to produce a soluble ligand released into the connective tissue and the bloodstream [55]. This soluble form was initially described as an inert ligand competing with its membrane-bound and pro-apoptotic counterpart (m-CD95L) for binding to CD95, thus acting as an antagonist of the death signal [56, 57]. More recent studies have shown that, unlike m-CD95L, the metalloprotease-cleaved CD95L (cl-CD95L) fails to trigger apoptosis, but instead may exert nonapoptotic functions by promoting the survival and motility of cancer cells, and can also aggravate inflammation in chronic inflammatory disorders [54, 58].
From a molecular standpoint, binding of m-CD95L to CD95 leads to receptor clustering, which is crucial to promote the formation of DISC [59]. In contrast, cl-CD95L fails to induce DISC formation and instead promotes the formation of an atypical pro-migratory CD95-containing complex called motility-inducing signalling complex (MISC ), which is devoid of both FADD and caspase-8/-10 but contains the src kinase c-yes [60]. TNBC cells exposed to metalloprotease-cleaved CD95L showed enhanced cell migration demonstrated by using the Boyden chamber assay . In these cells, MISC formation induces Nox3 (nicotinamide adenine dinucleotide phosphate oxidase 3)-driven reactive oxygen species (ROS) generation. ROS activate Src kinase c-yes, which in turn implement Orai1-mediated increase of intracellular calcium levels and recruits and activates EGF receptor (EGFR ) in an EGF-independent manner, leading to PI3K signalling [61]. Moreover, in a very recent study it was shown that cl-CD95L activates NHE1, the Na+/H+ plasma membrane transporter whose main function is to regulate intracellular pH and cell volume, which thereby enhances cell spreading and migration [62].
Together with Legembre et al. and Micheau et al., we have shown, using electron paramagnetic resonance, that cl-CD95L treatment induced plasma membrane fluidity increase in TNBC cells and this phenomenon is impaired by C16-ceramide [63]. Our findings raise the possibility of an instrumental role played by some SLs in CD95-mediated pro-migratory signalling pathway owing to their role as essential plasma membrane components and/or bioactive molecules (Fig. 10.3).

Modulation of CD95L-induced cell motility by sphingolipids. Binding of cleaved CD95L (cl-CD95L) to CD95 promotes MISC (motility inducing signalling complex) formation, plasma membrane fluidity increase and calcium signalling, which triggers cell migration. Whereas sphingosine-1-phosphate receptor 3 (S1PR3) signalling is involved in cell migration triggered by cl-CD95L, the interconnection between plasma membrane fluidity increase, S1PR3 signalling and MISC remains to be evaluated. Cl-CD95L-triggered cell motility is blocked by C16:0 Ceramide (C16-Cer), which is produced by ceramide synthase 6 (CerS6) and increases plasma membrane rigidity
At the plasma membrane, in contrary to SM and GSLs , ceramide is not the major component. It can be generated via hydrolysis of SM by SMases or via ceramide synthase (CerS )-mediated de novo ceramide biosynthesis. The latter pathway involves acylation of sphinganine with fatty acyl-CoAs of chain lengths ranging from C14 to C26 to produce dihydroceramide. Alternatively, CerS can synthesize ceramide by the salvage pathway through direct acylation of sphingosine, which is derived from sphingolipid catabolism [64]. Ceramide synthesis is orchestrated by six mammalian CerS proteins, each of which produces ceramides with restricted acyl chain lengths [65]. Accumulation of ceramides, with N-acyl chain longer than C12, rigidifies the plasma membrane [66]. Accordingly, incubation of TNBC with exogenous C16-ceramide increases plasma membrane rigidity [63]. Furthermore, an increased membrane fluidity was observed in CerS2-null mice which was directly associated with changes in SL composition [67].
The epithelial–mesenchymal transition (EMT ) allows epithelial cells to acquire features of mesenchymal cells, increasing cell motility and invasiveness properties. EMT is associated with changes in SL metabolism, and SL metabolites, such as gangliosides, modulate EMT [68]. In this context, an integrative analysis of gene expression in National Cancer Institute (NCI) tumor cell lines revealed that the expression of CerS6 decreased during EMT. As a matter of fact, CerS6 expression was lower in cancer cell lines, which exhibit a mesenchymal gene signature such as TNBC cell lines, than in cancer cells having an epithelial gene signature. In epithelial cells, TGFβ-induced EMT was associated with a significant reduction of CerS6 transcript levels [63]. We next established that downregulation of CerS6 expression during EMT is instrumental in increasing plasma membrane fluidity and cell motility in response to cl-CD95L. Indeed, pharmacological and epigenetic approaches aiming at inhibiting CerS6 expression/activity in breast cancer cells with epithelial gene signature not only reduced the level of C16-ceramide but also increased plasma membrane fluidity and cell motility in response to cl-CD95L. Conversely, overexpression of CerS6 in TNBC cell lines, which exhibit mesenchymal-like gene signature, increased C16-ceramide levels, rigidified plasma membrane, and impaired cl-CD95L-induced cell motility [63]. Thus, we identified CERS6 as a novel EMT -regulated gene, which impairs cell migration in response to cl-CD95L.
More recently, Legembre and coworkers have demonstrated the involvement of the S1P receptor 3 (S1PR3) in cl-CD95L-induced T-cell helper 17 (Th17) endothelial transmigration [69] (Fig. 10.3). In this study, cl-CD95L likely facilitated the secretion of S1P, which stimulated S1PR3, thereby increasing migration properties of Th17 cells and contributing to autoimmune disorders in a CD95-driven lupus mouse model. Future studies are needed to decipher the molecular mechanisms by which cl-CD95L triggers S1P production. Considering that EGFR, which is part of the MISC complex, is known to activate both sphingosine kinase 1 and 2 [70], it is tempting to speculate that cl-CD95L-induced S1P production occurs in an EGFR-dependent manner. Moreover, it would be of interest to determine whether the S1P pathway is involved in the motility of cancer cells in response to cl-CD95L. The latter tenet gets further credence considering that sphingosine kinases 1 and 2 are both involved in breast cancer cell motility [70, 71].
10.4 Role of Sphingolipids in TRAIL Signalling
Tumor-necrosis-factor related apoptosis-inducing ligand (TRAIL) belongs to the tumor necrosis factor (TNF) superfamily. Also known as Apo2L, TRAIL plays an important role in multiple cellular processes including proliferation, differentiation, and apoptosis. A growing body of evidence in the literature indicates that TRAIL has a remarkable property: it is able to trigger apoptosis in a broad range of cancer cell types, while having no or little cytotoxicity to most of the normal cells and tissues [72]. Clinical trials indicate that TRAIL may serve as a potent anti-cancer molecule [72]. However, different molecular events impair TRAIL-induced apoptosis in cancer cells [73,74,75], thereby limiting the use of TRAIL or TRAIL analogs in oncology.
TRAIL is a type II transmembrane protein, which is expressed at the cell surface of different leukocyte populations, including T lymphocytes and natural killers [76, 77]. TRAIL can be secreted as a consequence of a proteolytic cleavage, leading to the generation of a soluble form [77]. Five TRAIL receptors have been identified so far [78]. Three receptors are type I transmembrane proteins, two of them being agonistic receptors [TRAIL-R1 (DR4) and TRAIL-R2 (DR5)] with a functional intracellular death domain, similar in structure to that of CD95. The third one, TRAIL-R4 (dcR2), contains a truncated and nonfunctional death domain, which is unable to trigger apoptotic signalling, and thus behaves as a decoy receptor [79, 80]. Another decoy receptor, TRAIL-R3 (dcR1), is associated to the plasma membrane via a glycosyl phosphatidyl inositol (GPI ) anchor. Finally, osteoprotegerin is a soluble TRAIL receptor known to neutralize TRAIL.
TRAIL triggers apoptosis through interacting with its receptors on the cell surface of target cells. Once bound to agonistic receptors (DR4 and DR5), TRAIL induces receptor oligomerization followed by the formation of the DISC, leading to intrinsic and extrinsic apoptotic signalling pathway activation [81].
SLs are also putative biologically active molecules in TRAIL signalling [8, 82, 83]. Cellular levels of ceramide and its metabolites are highly regulated by several SL-metabolizing enzymes (see Sect. 10.1) that may participate in regulating apoptotic responses to TRAIL [79]. For instance, aSMase , as for CD95L signalling pathway (see Sect. 10.2.1), plays a key role in TRAIL-induced apoptosis, through its ability to hydrolyze SM into ceramide. Upon TRAIL, aSMase translocates onto the cell surface via lysosome trafficking and fusion, enabling ceramide-enriched membrane platforms that contain DR4 and DR5 clusters . This process was impaired in aSMase-deficient splenocytes and coronary arterial endothelial cells (CAECs) isolated from aSMase knockout mice [79, 80].
Taking into account the various alterations of SL metabolism in cancer cells [84], it is tempting to speculate that some SL changes contribute to resistance of cancer cells to TRAIL-induced apoptosis. For instance, acid ceramidase, which is frequently overexpressed in cancer, catabolizes ceramide and prevents TRAIL-induced cytotoxicity in the murine fibrosarcoma L929 cell line [85]. One should note that TRAIL-induced L929 cell death is associated with necrosis features rather than apoptosis, indicating that ceramide is likely involved in TRAIL-induced necroptosis in this cell type. Furthermore, CerS6 is weakly expressed in SW620 colon cancer cells, which are resistant to TRAIL, and expressed at high levels in SW480 colon cancer cells, which are sensitive to TRAIL [86]. Whereas downregulation of CerS6 by siRNA in SW480 conferred resistance to TRAIL-induced apoptosis, the overexpression of this enzyme in SW620 had the opposite effect [86]. More recently, subtoxic doses of docosahexaenoic acid (DHA ), a fish oil fatty acid, which enhanced C16-ceramide intracellular contents, sensitized SW620 metastatic colon cancer cells to TRAIL. This effect was reduced by fumonisin B1, which prevented de novo ceramide synthesis [87]. One should note however that CerS6 knock-down or overexpression in breast cancer cell lines failed to modulate DR-mediated cell death [63], indicating that CerS6 function in DR apoptotic signalling cannot be generalized to all cancer subtypes.
Other studies argue that targeting sphingolipid metabolism sensitizes cancer cells to TRAIL. For instance, incubation of cells with exogenous ceramides enhanced TRAIL-induced apoptosis in different cell lines and restored sensitivity of resistant cancer cells to TRAIL [79, 88]. More recently, sphingosine kinase 2 was reported to be overexpressed in TRAIL resistant non-small cell lung cancer cell lines (NSCLC). Targeting sphingosine kinase 2 with the selective inhibitor ABC294640 or by siRNA strategy is associated with a significant sensitization of NSCLC cancer cell lines to TRAIL. This phenomenon is associated with an increase of DR4 and/or DR5 expression at the cell surface via unknown mechanisms [89]. Similar findings have been reported upon treatment of cancer cells with FTY720, a sphingosine analog used in the clinic to treat multiple sclerosis. FTY720 potently synergizes with TRAIL to trigger apoptosis in various cancer cell lines (renal, breast, and colon carcinoma) [90]. Mechanistically, FTY720 downregulates the anti-apoptotic Bcl-2 family member Mcl-1 and upregulates DR5. Moreover, renal carcinoma tumor growth in nude mice is abolished with FTY720 and TRAIL association [90]. Whereas the combination of FTY720 with TRAIL in cancer patients seems to be a promising therapeutic strategy, FTY720 is a potent immunosuppressor, which prevents lymphocyte egress from thymus and lymph nodes. Thus, FTY720-based therapy should be adapted with caution to avoid tumor progression due to immunosuppression.
10.5 Concluding Remarks
The role of SLs in CD95L- and TRAIL-mediated apoptosis has been intensively investigated during the last two decades. CD95L and TRAIL trigger the activation of different metabolic pathways, including SMase activation , ceramide de novo synthesis increase, and SMS inhibition. Early ceramide production likely facilitates DR redistribution in lipid rafts and DISC formation. A growing body of evidence indicates that ceramide and possibly its metabolites contribute to the DR-mediated intrinsic signalling pathway. More recently, SLs have emerged as being involved in the modulation of CD95-induced cell migration. Deciphering the molecular mechanisms by which SLs modulate DR-driven cell motility and cell death may help better clarifying the role of SLs in cancer and autoimmune diseases. Interfering with SL metabolism may represent an interesting therapeutic strategy to limit cell motility and stimulate apoptosis triggered by DR.
References
Hanada K (2003) Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 1632:16–30
Levy M, Futerman AH (2010) Mammalian ceramide synthases. IUBMB Life 62:347–356. doi:10.1002/iub.319
Tafesse FG, Ternes P, Holthuis JCM (2006) The multigenic sphingomyelin synthase family. J Biol Chem 281:29421–29425. doi:10.1074/jbc.R600021200
Lahiri S, Futerman AH (2007) The metabolism and function of sphingolipids and glycosphingolipids. Cell Mol Life Sci CMLS 64:2270–2284. doi:10.1007/s00018-007-7076-0
Van Overloop H, Gijsbers S, Van Veldhoven PP (2006) Further characterization of mammalian ceramide kinase: substrate delivery and (stereo)specificity, tissue distribution, and subcellular localization studies. J Lipid Res 47:268–283. doi:10.1194/JLR.M500321-JLR200
Hait NC, Oskeritzian CA, Paugh SW et al (2006) Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta 1758:2016–2026. doi:10.1016/j.Bbamem.2006.08.007
Mao C, Obeid LM (2008) Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta 1781:424–434. doi:10.1016/j.Bbalip.2008.06.002
Sabourdy F, Kedjouar B, Sorli SC et al (2008) Functions of sphingolipid metabolism in mammals—lessons from genetic defects. Biochim Biophys Acta 1781:145–183. doi:10.1016/j.Bbalip.2008.01.004
Astudillo L, Sabourdy F, Therville N et al (2015) Human genetic disorders of sphingolipid biosynthesis. J Inherit Metab Dis 38:65–76. doi:10.1007/s10545-014-9736-1
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572. doi:10.1038/42408
Simons K, Ehehalt R (2002) Cholesterol, lipid rafts, and disease. J Clin Invest 110:597–603. doi:10.1172/JCI16390
Grassme H, Jekle A, Riehle A et al (2001) CD95 signaling via ceramide-rich membrane rafts. J Biol Chem 276:20589–20596. doi:10.1074/jbc.M101207200
Muppidi JR, Siegel RM (2004) Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat Immunol 5:182–189. doi:10.1038/ni1024
Legembre P, Daburon S, Moreau P et al (2005) Amplification of Fas-mediated apoptosis in type II cells via microdomain recruitment. Mol Cell Biol 25:6811–6820. doi:10.1128/MCB.25.15.6811-6820.2005
Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23
Nurminen TA, Holopainen JM, Zhao H, Kinnunen PKJ (2002) Observation of topical catalysis by sphingomyelinase coupled to microspheres. J Am Chem Soc 124:12129–12134
Grassmé H, Schwarz H, Gulbins E (2001) Molecular mechanisms of ceramide-mediated CD95 clustering. Biochem Biophys Res Commun 284:1016–1030. doi:10.1006/bbrc.2001.5045
Miyaji M, Jin Z-X, Yamaoka S et al (2005) Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J Exp Med 202:249–259. doi:10.1084/jem.20041685
Brenner B, Ferlinz K, Grassmé H et al (1998) Fas/CD95/Apo-I activates the acidic sphingomyelinase via caspases. Cell Death Differ 5:29–37. doi:10.1038/sj.Cdd.4400307
Grullich C, Sullards MC, Fuks Z et al (2000) CD95(Fas/APO-1) signals ceramide generation independent of the effector stage of apoptosis. J Biol Chem 275:8650–8656
Grassmé H, Cremesti A, Kolesnick R, Gulbins E (2003) Ceramide-mediated clustering is required for CD95-DISC formation. Oncogene 22:5457–5470. doi:10.1038/sj.Onc.1206540
Gulbins E, Kolesnick R (2000) Measurement of sphingomyelinase activity. Methods Enzymol 322:382–388
Perrotta C, Bizzozero L, Cazzato D et al (2010) Syntaxin 4 is required for acid sphingomyelinase activity and apoptotic function. J Biol Chem 285:40240–40251. doi:10.1074/jbc.M110.139287
Grassmé H, Bock J, Kun J, Gulbins E (2002) Clustering of CD40 ligand is required to form a functional contact with CD40. J Biol Chem 277:30289–30299. doi:10.1074/jbc.M200494200
Grassmé H, Riethmüller J, Gulbins E (2007) Biological aspects of ceramide-enriched membrane domains. Prog Lipid Res 46:161–170. doi:10.1016/j.Plipres.2007.03.002
Lin T, Genestier L, Pinkoski MJ et al (2000) Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J Biol Chem 275:8657–8663
Paris F, Grassmé H, Cremesti A et al (2001) Natural ceramide reverses Fas resistance of acid sphingomyelinase(−/−) hepatocytes. J Biol Chem 276:8297–8305. doi:10.1074/jbc.M008732200
De Maria R, Rippo MR, Schuchman EH, Testi R (1998) Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J Exp Med 187:897–902
Cock JG, Tepper AD, de Vries E et al (1998) CD95 (Fas/APO-1) induces ceramide formation and apoptosis in the absence of a functional acid sphingomyelinase. J Biol Chem 273:7560–7565
Bezombes C, Ségui B, Cuvillier O et al (2001) Lysosomal sphingomyelinase is not solicited for apoptosis signaling. FASEB J 15:297–299. doi:10.1096/fj.00-0466fje
Chalfant CE, Ogretmen B, Galadari S et al (2001) FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 276:44848–44855. doi:10.1074/jbc.M106291200
Watanabe M, Kitano T, Kondo T et al (2004) Increase of nuclear ceramide through caspase-3-dependent regulation of the “sphingomyelin cycle” in Fas-induced apoptosis. Cancer Res 64:1000–1007
Lafont E, Milhas D, Carpentier S et al (2010) Caspase-mediated inhibition of sphingomyelin synthesis is involved in FasL-triggered cell death. Cell Death Differ 17:642–654. doi:10.1038/cdd.2009.130
Tepper AD, de Vries E, van Blitterswijk WJ, Borst J (1999) Ordering of ceramide formation, caspase activation, and mitochondrial changes during CD95- and DNA damage-induced apoptosis. J Clin Invest 103:971–978. doi:10.1172/JCI5457
Juo P, Woo MS, Kuo CJ et al (1999) FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ 10:797–804
Cuvillier O, Edsall L, Spiegel S (2000) Involvement of sphingosine in mitochondria-dependent Fas-induced apoptosis of type II Jurkat T cells. J Biol Chem 275:15691–15700. doi:10.1074/jbc.M000280200
Tepper AD, Cock JG, de Vries E et al (1997) CD95/Fas-induced ceramide formation proceeds with slow kinetics and is not blocked by caspase-3/CPP32 inhibition. J Biol Chem 272:24308–24312
Lafont E, Dupont R, Andrieu-Abadie N et al (2012) Ordering of ceramide formation and caspase-9 activation in CD95L-induced Jurkat leukemia T cell apoptosis. Biochim Biophys Acta 1821:684–693. doi:10.1016/j.Bbalip.2012.01.012
Samraj AK, Keil E, Ueffing N et al (2006) Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J Biol Chem 281:29652–29659. doi:10.1074/jbc.M603487200
Movsesyan VA, Yakovlev AG, Dabaghyan EA et al (2002) Ceramide induces neuronal apoptosis through the caspase-9/caspase-3 pathway. Biochem Biophys Res Commun 299:201–207
Nica AF, Tsao CC, Watt JC et al (2008) Ceramide promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving caspase-8 and JNK. Cell Cycle 7:3362–3370. doi:10.4161/cc.7.21.6894
Milhas D, Andrieu-Abadie N, Levade T et al (2012) The tricyclodecan-9-yl-xanthogenate D609 triggers ceramide increase and enhances FasL-induced caspase-dependent and -independent cell death in T lymphocytes. Int J Mol Sci 13:8834–8852. doi:10.3390/ijms13078834
Milhas D, Cuvillier O, Therville N et al (2005) Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem 280:19836–19842. doi:10.1074/jbc.M414358200
Ganesan V, Perera MN, Colombini D et al (2010) Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 15:553–562. doi:10.1007/s10495-009-0449-0
Chipuk JE, McStay GP, Bharti A et al (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148:988–1000. doi:10.1016/j.Cell.2012.01.038
Zundel W, Giaccia A (1998) Inhibition of the anti-apoptotic PI(3)K/Akt/Bad pathway by stress. Genes Dev 12:1941–1946
Chalfant CE, Rathman K, Pinkerman RL et al (2002) De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem 277:12587–12595. doi:10.1074/jbc.M112010200
De Maria R, Lenti L, Malisan F et al (1997) Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science 277:1652–1655
Popa I, Therville N, Carpentier S et al (2011) Production of multiple brain-like ganglioside species is dispensable for fas-induced apoptosis of lymphoid cells. PLoS One 6:e19974. doi:10.1371/journal.Pone.0019974
Kang N-Y, Kang Y, Kang S-K et al (2006) Transcriptional regulation of the human GD3 synthase gene expression in Fas-induced Jurkat T cells: a critical role of transcription factor NF-kappaB in regulated expression. Glycobiology 16:375–389. doi:10.1093/glycob/cwj087
Sorice M, Matarrese P, Tinari A et al (2009) Raft component GD3 associates with tubulin following CD95/Fas ligation. FASEB J 23:3298–3308. doi:10.1096/fj.08-128140
Sorice M, Matarrese P, Manganelli V et al (2010) Role of GD3-CLIPR-59 association in lymphoblastoid T cell apoptosis triggered by CD95/Fas. PLoS One 5:e8567. doi:10.1371/journal.Pone.0008567
Peter ME, Hadji A, Murmann AE et al (2015) The role of CD95 and CD95 ligand in cancer. Cell Death Differ 22:549–559. doi:10.1038/cdd.2015.3
Malleter M, Tauzin S, Bessede A et al (2013) CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res 73:6711–6721. doi:10.1158/0008-5472.CAN-13-1794
Fouqué A, Debure L, Legembre P (2014) The CD95/CD95L signaling pathway: a role in carcinogenesis. Biochim Biophys Acta 1846:130–141. doi:10.1016/j.Bbcan.2014.04.007
Suda T, Hashimoto H, Tanaka M et al (1997) Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J Exp Med 186:2045–2050
Schneider P, Holler N, Bodmer JL et al (1998) Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med 187:1205–1213
O’Reilly LA, Tai L, Lee L et al (2009) Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 461:659–663. doi:10.1038/nature08402
Kischkel FC, Hellbardt S, Behrmann I et al (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 14:5579–5588
Tauzin S, Chaigne-Delalande B, Selva E et al (2011) The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol 9:e1001090. doi:10.1371/journal.Pbio.1001090
Khadra N, Bresson-Bepoldin L, Penna A et al (2011) CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) β2, and prevents death-inducing signaling complex formation. Proc Natl Acad Sci U S A 108:19072–19077. doi:10.1073/pnas.1116946108
Monet M, Poët M, Tauzin S et al (2016) The cleaved FAS ligand activates the Na(+)/H(+) exchanger NHE1 through Akt/ROCK1 to stimulate cell motility. Sci Rep 6:28008. doi:10.1038/srep28008
Edmond V, Dufour F, Poiroux G et al (2015) Downregulation of ceramide synthase-6 during epithelial-to-mesenchymal transition reduces plasma membrane fluidity and cancer cell motility. Oncogene 34:996–1005. doi:10.1038/onc.2014.55
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150. doi:10.1038/nrm2329
Mullen TD, Hannun YA, Obeid LM (2012) Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J 441:789–802. doi:10.1042/BJ20111626
Goñi FM, Alonso A (2009) Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim Biophys Acta 1788:169–177. doi:10.1016/j.Bbamem.2008.09.002
Pewzner-Jung Y, Park H, Laviad EL et al (2010) A critical role for ceramide synthase 2 in liver homeostasis: I. Alterations in lipid metabolic pathways. J Biol Chem 285:10902–10910. doi:10.1074/jbc.M109.077594
Levade T, Andrieu-Abadie N, Micheau O et al (2015) Sphingolipids modulate the epithelial–mesenchymal transition in cancer. Cell Death Discov 1:15001. doi:10.1038/cddiscovery.2015.1
Poissonnier A, Sanséau D, Le Gallo M et al (2016) CD95-mediated calcium signaling promotes T helper 17 trafficking to inflamed organs in lupus-prone mice. Immunity 45:209–223. doi:10.1016/j.Immuni.2016.06.028
Hait NC, Sarkar S, Le Stunff H et al (2005) Role of sphingosine kinase 2 in cell migration toward epidermal growth factor. J Biol Chem 280:29462–29469. doi:10.1074/jbc.M502922200
Sarkar S, Maceyka M, Hait NC et al (2005) Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett 579:5313–5317. doi:10.1016/j.Febslet.2005.08.055
Ashkenazi A, Holland P, Eckhardt SG (2008) Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 26:3621–3630. doi:10.1200/JCO.2007.15.7198
LeBlanc H, Lawrence D, Varfolomeev E et al (2002) Tumor-cell resistance to death receptor—induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med 8:274–281. doi:10.1038/nm0302-274
Ndozangue-Touriguine O, Sebbagh M, Mérino D et al (2008) A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 27:6012–6022. doi:10.1038/onc.2008.197
Micheau O, Shirley S, Dufour F (2013) Death receptors as targets in cancer. Br J Pharmacol 169:1723–1744. doi:10.1111/bph.12238
Wiley SR, Schooley K, Smolak PJ et al (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3:673–682
Almasan A, Ashkenazi A (2003) Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14:337–348
Mérino D, Lalaoui N, Morizot A et al (2007) TRAIL in cancer therapy: present and future challenges. Expert Opin Ther Targets 11:1299–1314. doi:10.1517/14728222.11.10.1299
Dumitru CA, Gulbins E (2006) TRAIL activates acid sphingomyelinase via a redox mechanism and releases ceramide to trigger apoptosis. Oncogene 25:5612–5625. doi:10.1038/sj.Onc.1209568
Xiang H, Reyes AE, Eppler S et al (2013) Death receptor 5 agonistic antibody PRO95780: preclinical pharmacokinetics and concentration-effect relationship support clinical dose and regimen selection. Cancer Chemother Pharmacol 72:405–415. doi:10.1007/s00280-013-2200-3
Walczak H, Krammer PH (2000) The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res 256:58–66. doi:10.1006/excr.2000.4840
Pettus BJ, Chalfant CE, Hannun YA (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta 1585:114–125
Ségui B, Andrieu-Abadie N, Jaffrézou J-P et al (2006) Sphingolipids as modulators of cancer cell death: potential therapeutic targets. Biochim Biophys Acta 1758:2104–2120. doi:10.1016/j.Bbamem.2006.05.024
Truman J-P, García-Barros M, Obeid LM, Hannun YA (2014) Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta 1841:1174–1188. doi:10.1016/j.Bbalip.2013.12.013
Thon L, Mathieu S, Kabelitz D, Adam D (2006) The murine TRAIL receptor signals caspase-independent cell death through ceramide. Exp Cell Res 312:3808–3821. doi:10.1016/j.Yexcr.2006.08.017
White-Gilbertson S, Mullen T, Senkal C et al (2009) Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28:1132–1141. doi:10.1038/onc.2008.468
Skender B, Hofmanová J, Slavík J et al (2014) DHA-mediated enhancement of TRAIL-induced apoptosis in colon cancer cells is associated with engagement of mitochondria and specific alterations in sphingolipid metabolism. Biochim Biophys Acta 1841:1308–1317. doi:10.1016/j.Bbalip.2014.06.005
Voelkel-Johnson C, Hannun YA, El-Zawahry A (2005) Resistance to TRAIL is associated with defects in ceramide signaling that can be overcome by exogenous C6-ceramide without requiring down-regulation of cellular FLICE inhibitory protein. Mol Cancer Ther 4:1320–1327. doi:10.1158/1535-7163.MCT-05-0086
Yang J, Yang C, Zhang S et al (2015) ABC294640, a sphingosine kinase 2 inhibitor, enhances the antitumor effects of TRAIL in non-small cell lung cancer. Cancer Biol Ther 16:1194–1204. doi:10.1080/15384047.2015.1056944
Woo SM, Seo BR, Min K, Kwon TK (2015) FTY720 enhances TRAIL-mediated apoptosis by up-regulating DR5 and down-regulating Mcl-1 in cancer cells. Oncotarget 6:11614–11626. doi:10.18632/oncotarget.3426
Acknowledgements
This work was supported by Ligue nationale contre le cancer, Institut National du Cancer (INCa), INSERM and Paul Sabatier University (Toulouse III). FB is a recipient of a grant from Association de Spécialisation et d’Orientation Scientifique.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Bilal, F. et al. (2017). Role of Sphingolipids in Death Receptor Signalling. In: Micheau, O. (eds) TRAIL, Fas Ligand, TNF and TLR3 in Cancer. Resistance to Targeted Anti-Cancer Therapeutics, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-56805-8_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-56805-8_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56804-1
Online ISBN: 978-3-319-56805-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)