
Abstract
We present a performance per watt analysis of CUDAlign 4.0, a parallel strategy to obtain the optimal alignment of huge DNA sequences in multi-GPU platforms using the exact Smith-Waterman method. Speed-up factors and energy consumption are monitored on different stages of the algorithm with the goal of identifying advantageous scenarios to maximize acceleration and minimize power consumption. Experimental results using CUDA on a set of GeForce GTX 980 GPUs illustrate their capabilities as high-performance and low-power devices, with a energy cost to be more attractive when increasing the number of GPUs. Overall, our results demonstrate a good correlation between the performance attained and the extra energy required, even in scenarios where multi-GPUs do not show great scalability.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The advent of the Human Genome Project has brought to the foreground of parallel computing a broad spectrum of data intensive biomedical applications where biology and computer science join as a happy alliance between demanding software and powerful hardware. Since then, the bioinformatics community generates computational solutions to support genomic research in many subfields such as gene structure prediction [5], phylogenetic trees [32], protein docking [23], and sequence alignment [12], just to mention a few of an extensive list.
Huge volumes of data produced by genotyping technology pose challenges in our capacity to process and understand data. Ultra high density microarrays now contain more than 5 million genetic markers, and next generation sequencing is enabling the search for causal relationship of variation close to the single nucleotide level. Furthermore, current clinical studies include hundreds of thousands of patients instead of thousands genetically fingerprinted few years ago, transforming bioinformatics into one of the flagships of the big data era.
In modern times of computing, when data volume pose a computational challenge, the GPU immediately comes to our minds. CUDA (Compute Unified Device Architecture) [21] and OpenCL [31] have established the mechanisms for data intensive general purpose applications to exploit GPUs extraordinary power in terms of TFLOPS (Tera Floating-Point Operations Per Second) and data bandwidth. Being GPUs the natural platform for large-scale bioinformatics, researchers have already analyzed raw performance and suggest optimizations for the most popular applications. This work extends the study to energy consumption, an issue of growing interest in the HPC community once GPUs have recently conquered the green500.org supercomputers list.
Our work focuses on biological sequences alignment in order to find the degree of similarity between them. Within this context, we may distinguish two basic approaches: Global alignment, in an attempt to align the entire length of the sequence when a pair of sequences are very similar in content and size, and local alignment, where regions of similarity between the two sequences are identified. Needleman-Wunsch (NW) [20] proposed a method for global comparison based on dynamic programming (DP), and Smith-Waterman (SW) [30] modified the NW algorithm to deal with local alignments. Computational requirements for SW are overwhelming, so researchers either relax them using heuristics as in the well-know BLAST tool [16], or rely on high performance computing to shorten the execution time. We have chosen commodity GPUs to explore the latter.
The rest of this paper is organized as follows. Section 2 completes this section with some related work. Section 3 describes the problem of comparing two DNA sequences. Section 4 summarizes our previous studies. Sections 5 and 6 introduce our infrastructure for measuring the experimental numbers, which are later analyzed in Sect. 7. Finally, Sect. 8 draws conclusions of this work.
2 Related Work
SW has become very popular over the last decade to compute (1) the exact pairwise comparison of DNA/RNA sequences or (2) a protein sequence (query) to a genomic database involving a bunch of them. Both scenarios have been parallelized in the literature [8], but fine-grained parallelism applies better to the first scenario, and therefore fits better into many-core platforms like Intel Xeon Phis [13], Nvidia GPUs using CUDA [26], and even multi-GPU using CUDAlign 4.0 [27], which is our departure point to analyze performance, power, energy and cost along this work.
On the other hand, energy consumption is gaining relevance within sequence alignment, which promotes methodologies to measure energy in genomic sequence comparison tools.
In [4], it is minimized the power consumption of a sequence alignment accelerator using application specific integrated circuit (ASIC) design flow. To decrease the energy budget, authors reduce clock cycle and scale frequency.
Hasan and Zafar [9] present performance versus power consumption for bioinformatics sequence alignment using different field programmable gate arrays (FPGAs) platforms implementing the SW algorithm as a linear systolic array.
Zou et al. [33] analyze performance and power for SW on FPGA, CPU and GPU, declaring the FPGA as the overall winner. However, they do not measure real-time power dynamically, but simplify with a static value for the whole run. Moreover, they use models from the first and second Nvidia GPU generations (GTX 280 and 470), which are, by far, the most inefficient CUDA families as far as energy consumption is concerned. Our analysis measures watts on physical wires and using Maxwell GPUs, the fourth generation where the energy budget has been optimized up to 40 GFLOPS/W, down from 15–17 GFLOPS/W in the third generation and just 4–6 GFLOPS/W in the previous ones.
3 DNA Sequence Comparison
A DNA sequence is represented by an ordered list of nucleotide bases. DNA sequences are treated as strings composed of characters of the alphabet \(\sigma = {A,T,G,C}\). To compare two sequences, we place one sequence above the other, possibly introducing spaces, making clear the correspondence between similar characters [14]. The result of this placement is an alignment.
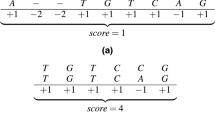
Given an alignment between sequences S0 and S1, a score is assigned to it as follows. For each pair of characters, we associate (a) a punctuation ma, if both characters are identical (match); or (b) a penalty mi, if the characters are different (mismatch); or (c) a penalty g, if one of the characters is a space (gap). The score is the addition of all these values. Figure 1 presents one possible alignment between two DNA sequences, where ma = +1, mi = 1 and g = 2.

Example of alignment and score.
3.1 Smith-Waterman
The SW algorithm [30] is based on dynamic programming (DP), obtaining the optimal pairwise local alignment in quadratic time and space. It is divided in two phases: calculate the DP matrix and obtain the alignment (traceback).
Phase 1. This phase receives as input sequences S0 and S1, with sizes \(\vert {S_0}\vert \) = m and \(\vert {S_1}\vert \) = n. The DP matrix is denoted \(H_{m+1,n+1}\), where \(H_{i,j}\) contains the score between prefixes S0[1..i] and S1[1..j]. At the beginning, the first row and column are filled with zeroes. The remaining elements of H are obtained from Eq. 1.
In addition, each cell \(H_{i,j}\) contains information about the cell that was used to produce the value. The highest value in \(H_{i,j}\) is the optimal score.
Phase 2 (Traceback). The second phase of SW obtains the optimal local alignment, using the outputs of the first phase. The computation starts from the cell that has the highest value in H, following the path that produced the optimal score until the value zero is reached.
Figure 2 presents a DP matrix with \(score=5\). The arrows indicate the alignment path when two DNA sequences with sizes \(m=12\) and \(n=8\) are compared, resulting in a 913 DP matrix. In order to compare Megabase sequences of, say, 60 Million Base Pairs (MBP), a matrix of size 60,000,001 \(\times \) 60,000,001 (3.6 Peta cells) is calculated.

DP matrix for sequences S0 and S1, with optimal \(score = 5\). The arrows represent the optimal alignment.
The original SW algorithm assigns a constant cost g to each gap. However, gaps tend to occur together rather than individually. For this reason, a higher penalty is usually associated to the first gap and a lower penalty is given to the remaining ones (affine-gap model). Gotoh [7] proposed an algorithm based on SW that implements the affine-gap model by calculating three values for each cell in the DP matrix: H, E and F, where values E and F keep track of gaps in each sequence. As in the original SW algorithm, time and space complexities of the Gotoh algorithm are quadratic.
3.2 Parallel Smith-Waterman
In SW, most of the time is spent calculating the DP matrices and, therefore, is a candidate process to be parallelized. From Eq. 1, we can see that cell \(H_{i,j}\) depends on three other cells: \(H_{i-1,j}\), \(H_{i-1,j-1}\) and \(H_{i,j-1}\). This kind of dependency is well suited to be parallelized using the wavefront method [22], where the DP matrix is calculated by diagonals and all cells on each diagonal can be computed in parallel.
Figure 3 illustrates the wavefront method. In step 1, only one cell is calculated in diagonal \(d_1\). In step 2, diagonal \(d_2\) has two cells, that can be calculated in parallel. In the further steps, the number of cells that can be calculated in parallel increases until it reaches the maximum parallelism in diagonals \(d_5\) to \(d_9\), where five cells are calculated in parallel. In diagonals \(d_{10}\) to \(d_{12}\), the parallelism decreases until only one cell is calculated in diagonal \(d_{13}\). The wavefront strategy limits the amount of parallelism during the beginning of the calculation (filling the wavefront) and the end of the computation (emptying the wavefront).

The wavefront method.
4 CUDAlign Implementation on GPUs
GPUs calculate a single SW matrix using all many-cores, but data dependencies force neighbour cores to communicate in order to exchange border elements. For Megabase DNA sequences, the SW matrix is several Petabytes long, and so, very few GPU strategies [11, 26] allow the comparison of Megabase sequences longer than 10 Million Base Pairs (MBP). SW# [11] is able to use 2 GPUs in a single Megabase comparison to calculate the Myers-Miller [15] linear space variant of SW. CUDAlign [26] obtains the alignment of Megabase sequences with a combined SW and Myers-Miller strategy. When compared to SW#, CUDAlign presents shorter execution times for huge sequences on a single GPU [11].
Comparing Megabase DNA sequences in multiple GPUs is more challenging. GPUs are arranged logically in a linear way so that each GPU calculates a subset of columns of the SW matrix, sending the border column elements to the next GPU. Asynchronous CPU threads will send/receive data to/from neighbor GPUs while GPUs keep computing, that way overlapping the required communications with effective computations whenever feasible.
4.1 CUDAlign Versions
CUDAlign was implemented using CUDA, C++ and pthreads. Experimental results collected in a large GPU cluster using real DNA sequences demonstrate good scalability for up to 16 GPUs [27]. For example, using the input data set described in Sect. 5, execution time was reduced from 33 h and 20 min on a single GPU to 2 h and 13 min on 16 GPUs (14.8\(\,\times \,\)speedup).
Table 1 summarizes the set of improvements and optimizations performed on CUDAlign since its inception, and Table 2 describes all stages and phases for the 4.0 version, the one used along this work.
5 Experimental Setup
We have conducted an experimental survey on a computer endowed with an Intel Xeon server and an Nvidia GeForce GTX 980 GPU from Maxwell generation. See Table 3 for a summary of major features.
For the input data set, we have used real DNA sequences coming from the National Center for Biotechnology (NCBI) [19] database. Results shown in Table 4 use sequences from assorted chromosomes, whereas Tables 5 and 6 use as input a pair of sequences from the chromosome 22 comparison between the human (50.82 MBP - see [18], accession number NC_000022.11) and the chimpanzee (37.82 MBP - see [17], accession number NC_006489.4).

Wires, slots, cables and connectors for measuring energy on GPUs.
To execute the required stages on a multi-GPU environment, we performed two modifications in CUDAlign 4.0: (1) a subpartitioning strategy to fit each partition in texture memory, and (2) writing extra rows in the file system as marks to be used in later stages to find the crosses with the optimal alignment. Moreover, we focus our experimental analysis on the first three stages of the SW algorithm, which are the ones extensively executed on GPUs as Table 2 reflects.
6 Monitoring Energy
We have built a system to measure current, voltage and wattage based on a Beaglebone Black, an open-source hardware [3] combined with the Accelpower module [6], which has eight INA219 sensors [1]. Inspired by [10], wires taken into account are two power pins on the PCI-express slot (12 and 3.3 volts) plus six external 12 volts pins coming from the power supply unit (PSU) in the form of two supplementary 6-pin connectors (half of the pins used for grounding).
Accelpower uses a modified version of pmlib library [2], a software package specifically created for monitoring energy. It consists of a server daemon that collects power data from devices and sends them to the clients, together with a client library for communication and synchronization with the server (Fig. 4).
The methodology for measuring energy begins with a start-up of the server daemon. Then, the source code of the application where the energy wants to be measured has to be modified to (1) declare pmlib variables, (2) clear and set the wires which are connected to the server, (3) create a counter and (4) start it. Once the code is over, we (5) stop the counter, (6) get the data, (7) save them to a .csv file, and (8) finalize the counter.
7 Experimental Results
We start showing execution times and energy spent by four different sequences on a multi-GPU environment composed of four GeForce GTX 980 GPUs. Those sequences require around 5–6 h on a single GPU, and the time is reduced to less than a half using 4 GPUs. It is not a great scalability, but we already anticipated the existence of dependencies among GPUs, thus hurting parallelism.
Table 4 includes the numbers coming from this initial experiment. We can see that stage 1 predominates for the execution time, and that wattage keeps stable around 100 W for all sequences. Power goes down to less than 80 W in the third stage, but its weight is low (negligible for the case of the chrY sequence, where stage 2 also takes little time).
Once we have seen the behaviour of all these sequences, we have selected just chr22 as the more stable to characterize SW from now on.
Table 5 shows the results for chr22 when SW is executed on a multi-GPU environment. As expected, power consumed by each GPU remains stable regardless of the number of GPUs active during the parallelization process. Execution times keep showing the already announced scalability on stage 1. Those times are somehow unstable for stage 2, and finally reach good scalability on stage 3. Because GPUs keep computing on stage 1 most of the time, the overall energy cost is heavily influenced by this stage. Basically, entering multi-GPU from a single GPU execution doubles the energy cost, and then remains stable for 3 and 4 GPUs, where execution times are greatly reduces. That way, the performance per watt ratio is disappointing when moving from single to twin GPUs, but then evolves nicely for 3 and 4 GPUs.

Power consumption in four GPUs for stages 1, 2 and 3 of the chr22 sequence comparison. The last chart shows the global results involving all of them.
Table 6 summarizes gains (in time reduction) and losses (as extra energy costs) on all scenarios of our multi-GPU execution for the chr22 sequence comparison. Stage 3 is the more rewarding one with the highest time savings and the lowest energy penalties, but unfortunately, SW keeps computing there just a marginal period of time. Stage 2 sets records in energy costs, and stage 1 keeps on an intermediate position, which is what finally characterizes the whole execution given its heavy workload. The sweetest scenario is stage 2 using 2 GPUs, where we are able to cut time in half and spend less energy overall. In the opposite side, the worst case goes to stage 1 using 2 GPUs, where time is reduced just one percent to almost double the energy spent. Finally, we have a solid conclusion on four GPUs, with time being reduced 50% at the expense of doubling the energy budget. Figure 5 provides details about the dynamic behaviour over time for each of the stages when running the chr22 sequence comparison on four GPUs.
8 Conclusions
Along this paper, we have studied GPU acceleration and power consumption on a multi-GPU environment for the Smith-Waterman method to compute, via CUDAlign 4.0, the biological sequence alignment for a set of real DNA sequences coming from human and chimpanzee homologous chromosomes retrieved from the National Center for Biotechnology Information (NCBI).
CUDAlign 4.0 comprises six stages, with the first three accelerated using GPUs. On a stage by stage analysis, the first one is more demanding and takes the bulk of the computational time, with data dependencies sometimes disabling parallelism and affecting performance. On the other hand, power consumption was kept more stable across executions of different alignment sequences, though it suffered deviations of up to 30% across different stages.
Within a multi-GPU platform, average power remained stable and execution times were more promising on a higher number of GPUs, with a total energy cost which was more attractive on those last executions. Overall, we find a good correlation between higher performance and additional energy required, even in those scenarios where multi-GPUs do not exhibit good scalability.
Finally, we expect GPUs to increase their role as high performance and low power devices for biomedical applications in future GPU generations, particularly after the introduction in late 2016 of the 3D memory within Pascal models.
References
Ada, L.: Adafruit INA219 Current Sensor Breakout. https://learn.adafruit.com/adafruit-ina219-current-sensor-breakout
Alonso, P., Badía, R., Labarta, J., Barreda, M., Dolz, M., Mayo, R., Quintana-Ortí, E., Reyes, R.: Tools for power-energy modelling and analysis of parallel scientific applications. In: Proceedings of 41st International Conference on Parallel Processing (ICPP 2012), pp. 420–429. IEEE Computer Society, September 2012
BeagleBone: Beaglebone black. http://beagleboard.org/BLACK
Cheah, R., Halim, A., Al-Junid, S., Khairudin, N.: Design and analysis of low powered DNA sequence alignment accelerator using ASIC. In: Proceedings of 9th WSEAS International Conference on Microelectronics, Nanoelectronics and Optoelectronics (MINO 2010), pp. 107–113, March 2010
Deng, X., Li, J., Cheng, J.: Predicting protein model quality from sequence alignments by support vector machines. J. Proteomics Bioinform. 9(2) (2013)
González-Rincón, J.: Sistema basado en open source hardware para la monitorización del consumo de un computador. Master thesis Project. Universidad Complutense de Madrid (2015)
Gotoh, O.: An improved algorithm for matching biological sequences. J. Mol. Biol. 162(3), 705–708 (1982)
Hamidouche, K., Machado, F., Falcou, J., Melo, A., Etiemble, D.: Parallel Smith-Waterman comparison on multicore and manycore computing platforms with BSP++. Int. J. Parallel Program. 41(1), 111–136 (2013)
Hasan, L., Zafar, H.: Performance versus power analysis for bioinformatics sequence alignment. J. Appl. Res. Technol. 10(6), 920–928 (2012)
Igual, F., Jara, L., Gómez, J., Piñuel, L., Prieto, M.: A power measurement environment for PCIe accelerators. Comput. Sci. - Res. Develop. 30(2), 115–124 (2015)
Korpar, M., Sikic, M.: SW# GPU-enabled exact alignmens on genome scale. J. Bioinform. 29(19), 2494–2495 (2013)
Li, H., Homer, N.: A survey of sequence alignment algorithms for next-generation sequencing. Briefings Bioinform. 11(5), 473–483 (2010)
Liu, Y., Tam, T., Lauenroth, F., Schmidt, B.: SWAPHI-LS: Smith-Waterman algorithm on xeon phi coprocessors for long DNA sequences. In: IEEE Cluster, pp. 257–265 (2014)
Mount, D.: Bioinformatics: Sequence and Genome Analysis. CSHL Press, New York (2004)
Myers, E., Miller, W.: Optimal alignment in linear space. Comput. Appl. Biosci. (CABIOS) 4(1), 11–17 (1988)
NCBI: Blast: Basic local alignment search tool (2017). https://blast.ncbi.nlm.nih.gov/Blast.cgi
NCBI: NCBI Chimpanze Website (2017). https://www.ncbi.nlm.nih.gov/genome/gdv/?context=genome&acc=GCF_000001515.7&chr=22
NCBI: NCBI Human Website (2017). https://www.ncbi.nlm.nih.gov/genome/gdv/?context=genome&acc=GCF_000001405.36&chr=22
NCBI: NCBI Web Site (2017). http://www.ncbi.nlm.nih.gov
Needleman, S., Wunsch, C.: A general method applicable to the search for similarities in the aminoacid sequence of two proteins. J. Mol. Biol. 48(3), 443–453 (1970)
Nvidia: CUDA Home Page, October 2010. http://developer.nvidia.com/object/cuda.html
Pfister, G.: In Search of Clusters: The Coming Battle in Lowly Parallel Computing. Prentice Hall, Upper Saddle River (1995)
Pierce, B., Wiehe, K., Hwang, H., Kim, B., Vreven, T., Weng, Z.: ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. J. Bioinform. 30(12), 1771–1773 (2014)
Sandes, E., Melo, A.: CUDAlign: using GPU to accelerate the comparison of megabase genomic sequences. In: Proceedings of 15th ACM SIGPLAN Symposium on Principles and Practice of Parallel Programming (PPoPP 2010), pp. 137–146 (2010)
Sandes, E., Melo, A.: Smith-Waterman alignment of huge sequences with GPU in linear space. In: Proceedings of IEEE International Parallel and Distributed Processing Symposium (IPDPS 2011), pp. 1199–1211 (2011)
Sandes, E., Melo, A.: Retrieving Smith-Waterman alignments with optimizations for megabase biological sequences using GPU. IEEE Trans. Parallel Distrib. Syst. 24(5), 1009–1021 (2013)
Sandes, E., Miranda, G., Martorell, X., Ayguadé, E., Teodoro, G., Melo, A.: CUDAlign 4.0: incremental speculative traceback for exact chromosome-wide alignment in GPU clusters. IEEE Trans. Parallel Distrib. Syst. 27(10), 2838–2850 (2016)
Sandes, E., Miranda, G., Martorell, X., Ayguadé, E., Teodoro, G., Melo, A.: MASA: a multi-platform architecture for sequence aligners with block pruning. ACM Trans. Parallel Comput. 2(4), 28:1–28:31 (2016)
Sandes, E., Miranda, G., Melo, A., Martorell, X., Ayguadé, E.: CUDAlign 3.0: parallel biological sequence comparison in large GPU clusters. In: Proceedings of IEEE/ACM CCGrid 2014, pp. 160–169 (2014)
Smith, T., Waterman, M.: Identification of common molecular sequences. J. Mol. Biol. 127(1), 195–197 (1981)
The Khronos Group: The OpenCL Core API Specification, Headers and Documentation (2009). http://www.khronos.org/registry/cl
Wan, P., Che, D.: Constructing phylogenetic trees using interacting pathways. Bioinformation 9(7), 363–367 (2013)
Zou, D., Dou, Y., Xia, F.: Optimization schemes and performance evaluation of Smith-Waterman algorithm on CPU, GPU and FPGA. Concurrency Comput. Practice Experience 24, 1625–1644 (2012)
Acknowledgments
This work was supported by the Ministry of Education of Spain under ProjectTIN2013-42253-P and by the Junta de Andalucia under Project of Excellence P12-TIC-1741. We also thank Nvidia for hardware donations within GPU Education Center 2011–2016 and GPU Research Center 2012–2016 awards at the University of Malaga (Spain).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Pérez Serrano, J., De Oliveira Sandes, E.F., Magalhaes Alves de Melo, A.C., Ujaldón, M. (2017). Smith-Waterman Acceleration in Multi-GPUs: A Performance per Watt Analysis. In: Rojas, I., Ortuño, F. (eds) Bioinformatics and Biomedical Engineering. IWBBIO 2017. Lecture Notes in Computer Science(), vol 10209. Springer, Cham. https://doi.org/10.1007/978-3-319-56154-7_46
Download citation
DOI: https://doi.org/10.1007/978-3-319-56154-7_46
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56153-0
Online ISBN: 978-3-319-56154-7
eBook Packages: Computer ScienceComputer Science (R0)