
Abstract
Anticoagulation-related nephropathy (ARN) is an underdiagnosed complication of anticoagulation that is associated with increased renal morbidity and all-cause mortality. Originally defined by a distinct pattern of glomerular hemorrhage on kidney biopsy in the setting of anticoagulation use, ARN is clinically defined as acute kidney injury without obvious etiology in the setting of over anticoagulation. When caused by warfarin, it is often associated with supratheurapeutic International Normalized Ratio (i.e., greater than 3.0) however it can also be caused by direct oral anticoagulants (DOACs). The incidence of ARN due to warfarin has not been studied prospectively, but is estimated to be as high as 20% based on large-scale retrospective data. Higher rates are seen in patients with advances age, chronic kidney disease (CKD), hypertension, and diabetes. Prompt recognition of ARN is critical as it is associated with accelerated progression of chronic kidney disease (CKD) and decreased long- term survival. Given the significant impact of ARN morbidly and mortality, a thorough understanding of the pathophysiology, molecular mechanisms, clinical spectrum, and therapeutic interventions for ARN is crucial to balance the risks and benefits of anticoagulation and optimize treatment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Anticoagulation-related nephropathy
- Glomerular hemorrhage
- Obstructive tubular red blood cell casts
- Acute kidney injury
- Anticoagulation
- Warfarin
- Direct oral anticoagulants (DOACs)
Introduction and Importance
Anticoagulation-related nephropathy (ARN) is an under-recognized complication of anticoagulation that is associated with both irreversible kidney injury and increased mortality [1, 2]. The simplicity of ARN’s diagnostic criteria—acute kidney injury (AKI) in the setting of over anticoagulation without other identifiable etiology—camouflages a complex disease state with an unclear molecular mechanism, nuanced epidemiologic profile and multiple clinical manifestations. The only aspect of ARN that is clear is that as the total number of patients started on anticoagulation, both warfarin and direct oral anticoagulants (DOACs) continues to increase, the healthcare burden and costs associated with ARN will rise as well. In this chapter, we seek to review the epidemiology, pathogenesis, clinical feature, treatment and prevention of ARN.
Historical Perspective
While warfarin has been in use since 1954, its harmful effects on the kidneys have only recently been fully recognized [3, 4]. In the 1960s, Reilly and colleagues reported unexplained hematuria in 35 out of 200 patients on warfarin, however no association between hematuria, prothrombin time, and kidney function was observed [5]. Over the next 50 years, isolated case studies of patients with unexplained AKI associated with hematuria, usually in the presence of a supratherapeutic international normalized ratio (INR) were reported. However, the cases were attributed to underlying kidney disease (i.e., IgA nephropathy, lupus nephritis, etc.) and the role of anticoagulation was unknown [6,7,8,9]. In 2009, Brodsky and colleagues described nine patients without underlying kidney disease who developed unexplained AKI associated with elevated INRs [3]. Kidney biopsies from those nine patients showed glomerular hemorrhage and tubular injury associated with obstructive red blood cell casts. This clinic-pathologic constellation of AKI sans alternate etiology with these biopsy findings was termed “warfarin-related nephropathy” (WRN). This disease was thought to be exclusive to warfarin until 2013 when it was shown to occur in both rats and humans taking dabigatran [10, 11]. It has subsequently been renamed anticoagulant-related nephropathy (ARN) to reflect its wider association.
Epidemiology
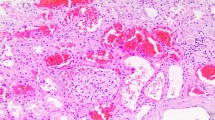
The incidence of ARN is difficult to determine due to its changing definition and diagnostic criteria. Originally, ARN was a pathologic diagnosis defined by (a) dysmorphic red blood cells (RBCs) implying injury to the glomerular filtration barrier, (b) uniform hemorrhage through all fields as to exclude biopsy artefact, (c) the presence of obstructive tubular RBC casts, and (d) absence of glomerulonephritis or other inflammatory changes that could account for glomerular hemorrhage (Fig. 28.1) [3]. However, given the risk of renal biopsies in the setting of anticoagulation, most cases of ARN are not biopsy-proven but presumptively diagnosed in patients who develop an unexplained AKI (increase in serum creatinine of more than 0.3 mg/dl or 1.5-fold greater than baseline) in the setting of warfarin use with an INR greater than 3.0, or use of a direct oral anticoagulant (DOAC) [2]. It is important to note that while the majority of the seminal work in ARN identified patients based on hematuria, the current definition does not require patients to have hematuria to be classified as ARN.

Microscopy pathology associated with ARN. RBCs and RBC occlusive casts in a patient with anticoagulation-related nephropathy. Image provided by Surya Seshan, Chief of the Division of Renal Pathology, Weill Cornell Medical Center, NY
Using this definition, between 17 and 20.5% of all patients on warfarin develop at least one episode of ARN during treatment [1, 12]. This incidence rate should be interpreted with caution for several reasons. First, ARN may be incorrectly diagnosed if the true underlying cause of the AKI is not identified (such as concomitant chronic kidney disease, heart disease, medications, etc.). Furthermore, the reported incidence is likely affected by sampling bias since the detection of AKI is dependent on measuring serum creatinine which is much more likely to occur in more ill patients who are at higher risk to develop ARN. Finally, since ARN can spontaneously resolve the incidence is likely much higher than the prevalence.
To date, only two cases of ARN have been reported in a patient using DOACs. In both cases, patients on dabigatran developed an unexplained AKI and were found to have diffuse interstitial hemorrhage and obstructing intratubular RBC casts consistent with ARN [13, 14]. While there are currently no epidemiologic studies investigating the rate of ARN in patients on DOACs, most studies record and publish rates of acute kidney injury, a prerequisite for ARN. A formal post hoc analysis of the RE-LY trial comparing two therapeutic doses of dabigatran (110 and 150 mg) and warfarin found that both doses of dabigatran were associated with smaller estimated glomerular filtration rate (eGFR) reductions at 12 and 24 months when compared with warfarin [15]. A recent meta-analysis of RE-LY (dabigatran vs. warfarin), ROCKET (rivaroxaban vs. warfarin) and ENGAGE AF (edoxaban vs. warfarin) trials and supplementary FDA data showed no significant difference in the rates of acute renal failure between the DOAC and warfarin arms [2]. Taken together this evidence suggest that the incidence of ARN associated with DOACs less than or equal to that of warfarin but further studies are needed to measure the true incidence.
The main risk factor for ARN associated with either warfarin or DOACs appears to be chronic kidney disease (CKD). Patients with documented CKD had twice the incidence of ARN compared to normal controls [1]. 5/6-nephrectomized rats, a well-established model for chronic kidney disease, treated with either warfarin or dabigatran, developed ARN like pathology at much greater frequency than control animals [10, 16]. Limited clinical evidence suggests that age, diabetes mellitus, and hypertension are also independent predictors of increased ARN risk [1, 17].
Pathophysiology and Mechanism
Based upon histologic analysis of kidney tissue obtained from patients and experimental animal models, the macroscopic pathophysiology of ARN is well understood. Disruption of the glomerular filtration barrier leads to hemorrhage into Bowman’s space and renal tubules. Underlying structural abnormalities in the glomerular basement membrane likely predispose kidneys to this anticoagulation-mediated hemorrhage consistent with the observation that patients with underlying chronic kidney disease are more likely to develop ARN [8, 9, 13]. As red blood cells from the glomerular hemorrhage reach the tubules, they coalesce into RBC casts, the hallmark feature of ARN [3] (Fig. 28.1). These RBC casts induce tubular injury and obliteration through multiple mechanisms including ischemia and oxidative stress due to free hemoglobin [18, 19].
In contrast, the molecular mechanism of ARN is very poorly understood. Thrombin, the only vitamin K-dependent coagulation factor known to stimulate signaling cascades, binds and activates a family of proteinase-activated receptors (PARs) expressed on all endothelial cells including those within the glomeruli of the kidney [20]. It is hypothesized that reduction in thrombin levels by anticoagulation decreases PAR signaling which triggers the breakdown of endothelial cell-cell adhesions thus allowing glomerular hemorrhage. This hypothesis is supported by the observation that glomerular hemorrhage can be triggered in animal models of CKD by administration of PAR antagonists [10]. While appealing, this hypothesis fails to explain why in most epithelial model systems activation, not inhibition, of PAR results in endothelium retractions and increased paracellular flow [21, 22]. Furthermore, PAR knock-out mice do not have glomerular hemorrhage or other obvious renal phenotype [23]. Finally, the use of PAR antagonist voraxapar is not associated with higher rates of AKI or other renal effects when compared to placebo in initial clinical trials [24]. Taken together this data strongly suggest that anticoagulants are functioning through a non-PAR related mechanism. One potential mechanism could be the depletion of activated protein C, a potent but often less considered target of warfarin therapy, which is known to have trophic and anti-apoptotic effects in cultured podocytes [25, 26].
Diagnosis and Work-Up
ARN should be considered in the differential diagnosis of any patient on anticoagulation presenting with AKI especially in the setting of excessive anticoagulation (i.e., supratherapeutic INR or excessive DOAC dose). The initial work-up should consist of a clinical exam, careful medication history and urinalysis to assess for hematuria (Fig. 28.2). Subsequent diagnostic tests such as urine electrolytes, kidney ultrasound and renal biopsy may be need to exclude other causes of AKI or confirm the diagnosis in high risk individuals.

Clinical diagnostic algorithm for suspected ARN
The presence of hematuria (gross or microscopic), dysmorphic RBC cells or RBC casts in the urinalysis support a diagnosis of ARN but are not definitive. Glomerulonephritides, vacuities, urinary tract infections, and nephrolithiasis can all manifest with hematuria and must be excluded prior to the diagnosis of ARN being made. Since a sizable percentage of patients with ARN do not develop hematuria patients with an inactive urine sediment should still undergo further work-up to exclude ARN. Volume depletion or treatment with medications (e.g., angiotensin-converting enzyme/angiotensinogen receptor blocker) are often identified by a through clinical history, corroborated by pre-renal azotemia on urine electrolytes and proven to be the cause with a trial of volume repletion or medication abstinence. Urinary tract obstruction can be excluded via renal ultrasound. Other causes of AKI such as acute phosphate nephropathy, high-dose nonsteroidal anti-inflammatory drug (NSAID) use, crystal-induced nephropathy, myeloma cast nephropathy, acute tubular necrosis, and acute interstitial nephritis also should be considered and evaluated.
After excluding all other potential causes of renal dysfunction, a presumptive diagnosis of ARN can be made. A definitive diagnosis could be made using a renal biopsy, however this is often not performed due to the high risk of bleeding associated with renal biopsy in a patient on anticoagulation. However, there are two clinical scenarios where a biopsy would be indicated. First, if the patient’s creatinine remained elevated or continued to increase despite appropriate treatment a biopsy is indicated. The serum creatinine in patients with ARN improves within the first 1–2 weeks of reversing the coagulopathy, and persistent elevated or continually rising creatinine is highly suspicious for an alternative cause of kidney injury, which likely requires a biopsy for diagnosis. Second, persistent hematuria is uncommon in ARN and therefore patients with persistent hematuria after correction of their coagulopathy should undergo renal biopsy, after urologic causes have been excluded.
Treatment and Monitoring
The mainstay of ARN treatment is returning the anticoagulation to a therapeutic range. For patients who develop ARN while taking warfarin, this is accomplished by frequently monitoring and careful titration of their warfarin dose until the INR is below 3.0. While there is no evidence that rapid correction of a patient’s INR is beneficial, it is logical to assume that prolonged periods of elevated INR will result in continued glomerular hemorrhage and additional kidney tubular injury. For patients who develop ARN while taking DOACs, treatment involves confirming that the patient is taking the correct dose, adjusted for the patient’s renal function. Given that elevated blood pressure and concomitant antiplatelet use are both likely to exacerbate glomerular hemorrhage and production of RBC casts, it is reasonable to obtain tight blood pressure control and minimize antiplatelet therapy when feasible. Finally, anticoagulation levels and renal function should be closely monitored for the duration of the patient’s treatment.
Early detection and prompt treatment are critical to minimize kidney damage. For patients on both warfarin and DOACs, kidney function should be monitored regularly throughout treatment and with increased frequency during the first 3 months when patients are at the highest risk (Table 28.1). Any patient with a supratheurapeutic INR should have their renal function assessed as soon as possible and renal function should be closely monitored until the INR returns to the therapeutic range. There is some evidence that having an episode of ARN increases the likelihood of subsequent episodes of ARN so more frequent kidney function monitoring (i.e., every 2–3 months) after an initial episode of ARN is prudent.
Clinical Consequences
ARN is independently associated with renal morbidity and overall mortality [1]. Even if the renal function returns to baseline following the ARN episode, some of the renal tubules will have been destroyed by the obstructive RBC casts thus permanently decreasing the nephron mass of the kidney. The tubules that do survive the ischemic and oxidative insult will likely manifest hyperfiltration injury that leads to accelerated CKD progression [27]. Brodsky and colleagues clearly demonstrated that patients who developed ARN had a serum creatinine level approximately 30% greater than matched patient 1 year following the ARN episode [17]. In the same study, 1-year mortality rates were 65% greater in patients with ARN compared to a match cohort (31.1% vs. 18.9% respectively). This association between increased mortality and ARN was confirmed in a subsequent study in Korean patients, which found that patients who developed ARN were at a twofold increased 1-year mortality (32.4% vs. 15.9%) [12]. It must be noted that all of the data linking ARN and mortality has been based on retrospective studies and there is currently no data establishing the causality. In fact, clinical factors that predispose patients to ARN such as age, diabetes, heart failure, and CKD are also associated with increased mortality, and it is plausible that the impact of ARN on all-cause mortality is reflective of a more chronically ill patient population.
Prevention
Despite the importance of prevention of ARN, there is currently no evidence to guide our interventions. Likely the most important measure to prevent ARN is correct dosing and titration of anticoagulants. Warfarin doses should be titrated judiciously to avoid rapid increases in INR and the dose of each DOACs should adjusted for renal function. Furthermore, it is logical to assume that optimization of a patient’s kidney function and co-morbid conditions, such as diabetes mellitus and hypertension, would decrease the risk of ARN.
Conclusions and Recommendations
The use of anticoagulation with warfarin and the new DOAC agents is increasing exponentially and thus the incidence and morbidity of ARN is only going to increase in the coming years. Given the significant impact of even a subtle decline in GFR on overall mortality, cardiovascular events and quality of life, it is imperative that all clinicians prescribing anticoagulants rigorously monitor and aggressively treat ARN to preserve kidney function. Joint collaborative efforts between cardiologists, nephrologists and hematologists will help elucidate the clinical and pathophysiological aspects of anticoagulation related nephropathy and help personalize anticoagulation to the individual patient for the future.
References
Brodsky SV, Nadasdy T, Rovin BH, Satoskar AA, Nadasdy GM, Wu HM, et al. Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int. 2011;80(2):181–9.
Wheeler DS, Giugliano RP, Rangaswami J. Anticoagulation-related nephropathy. J Thromb Haemost. 2016;14(3):461–7.
Brodsky SV, Satoskar A, Chen J, Nadasdy G, Eagen JW, Hamirani M, et al. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: a report of 9 cases. Am J Kidney Dis. 2009;54(6):1121–6.
Holbrook AM, Pereira JA, Labiris R, McDonald H, Douketis JD, Crowther M, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165(10):1095–106.
Reilly EB, Perry A, Fujita K, Nakamura RM. Haematuria and anticoagulants. Lancet. 1964;1(7332):554.
Praga M, Gutierrez-Millet V, Navas JJ, Ruilope LM, Morales JM, Alcazar JM, et al. Acute worsening of renal function during episodes of macroscopic hematuria in IgA nephropathy. Kidney Int. 1985;28(1):69–74.
Kincaid-Smith P, Bennett WM, Dowling JP, Ryan GB. Acute renal failure and tubular necrosis associated with hematuria due to glomerulonephritis. Clin Nephrol. 1983;19(4):206–10.
Kabir A, Nadasdy T, Nadasdy G, Hebert LA. An unusual cause of gross hematuria and transient ARF in an SLE patient with warfarin coagulopathy. Am J Kidney Dis. 2004;43(4):757–60.
Abt AB, Carroll LE, Mohler JH. Thin basement membrane disease and acute renal failure secondary to gross hematuria and tubular necrosis. Am J Kidney Dis. 2000;35(3):533–6.
Ryan M, Ware K, Qamri Z, Satoskar A, Wu H, Nadasdy G, et al. Warfarin-related nephropathy is the tip of the iceberg: direct thrombin inhibitor dabigatran induces glomerular hemorrhage with acute kidney injury in rats. Nephrol Dial Transplant. 2014 Dec;29(12):2228–34.
Moeckel GW, Luciano RL, Brewster UC. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin Kidney J. 2013;6(5):507–9.
An JN, Ahn SY, Yoon CH, Youn TJ, Han MK, Kim S, et al. The occurrence of warfarin-related nephropathy and effects on renal and patient outcomes in korean patients. PLoS ONE. 2013;8(4):e57661.
Moeckel GW, Luciano RL, Brewster UC. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin Kidney J. 2013 Oct;6(5):507–9.
Escoli R, Santos P, Andrade S, Carvalho F. Dabigatran-related nephropathy in a patient with undiagnosed IgA nephropathy. Case Rep Nephrol. 2015;2015:298261.
Bohm M, Ezekowitz MD, Connolly SJ, Eikelboom JW, Hohnloser SH, Reilly PA, et al. Changes in renal function in patients with atrial fibrillation: an analysis from the RE-LY trial. J Am Coll Cardiol. 2015;65(23):2481–93.
Ware KM, Vance JC, Muni N, Hebert LA, Satoskar AA, Nadasdy G, et al. Oral warfarin and the thrombin inhibitor dabigatran increase blood pressure in rats: hidden danger of anticoagulants? Am J Hypertens. 2015 Feb;28(2):182–9.
Brodsky SV, Collins M, Park E, Rovin BH, Satoskar AA, Nadasdy G, et al. Warfarin therapy that results in an International Normalization Ratio above the therapeutic range is associated with accelerated progression of chronic kidney disease. Nephron Clin Pract. 2010;115(2):c142–6.
Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol. 2007;18(2):414–20.
Ware K, Qamri Z, Ozcan A, Satoskar AA, Nadasdy G, Rovin BH, et al. N-acetylcysteine ameliorates acute kidney injury but not glomerular hemorrhage in an animal model of warfarin-related nephropathy. Am J Physiol Renal Physiol. 2013;304(12):F1421–7.
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64.
Malik AB, Fenton JW 2nd. Thrombin-mediated increase in vascular endothelial permeability. Semin Thromb Hemost. 1992;18(2):193–9.
Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, Aird WC. Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol. 2004;24(1):41–53.
Darrow AL, Fung-Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, et al. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost. 1996;76(6):860–6.
Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404–13.
Christiaans SC, Wagener BM, Esmon CT, Pittet JF. Protein C and acute inflammation: a clinical and biological perspective. Am J Physiol Lung Cell Mol Physiol. 2013;305(7):L455–66.
Madhusudhan T, Wang H, Straub BK, Grone E, Zhou Q, Shahzad K, et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood. 2012;119(3):874–83.
Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol. 2010;298(5):F1078–94.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Wheeler, D.S. (2017). Anticoagulation-Related Nephropathy: Tip of the Iceberg. In: Rangaswami, J., Lerma, E., Ronco, C. (eds) Cardio-Nephrology. Springer, Cham. https://doi.org/10.1007/978-3-319-56042-7_28
Download citation
DOI: https://doi.org/10.1007/978-3-319-56042-7_28
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56040-3
Online ISBN: 978-3-319-56042-7
eBook Packages: MedicineMedicine (R0)