
Abstract
At the crossroad of fuels utilization, the pyruvate dehydrogenase (PDH) complex plays a key role in substrate selection (primarily of glucose and fatty acids (FAs)) by mitochondria. In mammals, substrate selection takes place in all organs although to differing degrees depending upon nutritional conditions, physiological status (e.g., feed-fast, exercise), and the absence or presence of disease (e.g., metabolic disorder, cancer). Nutritional states such as those given by starvation, diabetes , caloric restriction, or aging can favor the oxidation of FAs over carbohydrates or vice versa, in which case modulation of the PDH complex is critically important for favoring or hindering the conservation of carbohydrate reserves. In this work, we review the literature in the context of the capacity of a cell or organ to adjust fuel selection as a function of nutrient availability and its influence on mitochondrial energetic-redox functions. We also present a computational model of PDH which includes its regulation by multiple effectors (AcCoA, CoA, NADH , NAD, ATP, ADP, Ca2+, pyruvate) targeting specific kinases and phosphatases that render the enzymatic complex phosphorylated (inactive) or dephosphorylated (active), respectively. Selection by mitochondria between glucose and FAs at different relative levels of both substrates, and its impact on respiration, ROS emission, effector levels, and the fluxes through PDH and β-oxidation, are also presented and analyzed.
This chapter was created within the capacity of an US governmental employment. US copyright protection does not apply.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Pyruvate dehydrogenase kinase and phosphatase
- Palmitoyl-CoA
- Acetyl-CoA
- NADH
- Reactive oxygen species
- Oxidative phosphorylation
- Redox environment
- Aging
- Diabetes
- Caloric restriction
1 Introduction
We increasingly recognize that complex gene-environment and gene-nutrient interactions underlie an organism’s response to physiological or pathophysiological stimuli. An individual genomic variant (Mendelian-type inborn error ) can affect subtly one primary metabolite flux, without evidence of clinical disease; however, in a complex disease, state variations perturbing a network of metabolite fluxes may attain the clinical threshold for disease, either alone or in combination with environmental factors (Lanpher et al. 2006). The capacity to comprehensively assess gene, protein, transcript, and metabolite profiles, including posttranslational modifications, through high-throughput “omics” studies, has opened new avenues of research possibilities, among them the characterization of metabolic remodeling associated with disease, e.g., diabetes , cancer, aging , or normal physiology such as observed in the fast-feed transition or caloric restriction (Kelley and Mandarino 2000; Mitchell et al. 2016).
Multiple environmental (e.g., nutrition) and genetic interactions produce different phenotypic patterns of which metabolites and metabolic fluxes are main functional readouts. Recently, the concept of metabolism-epigenome-genome axis was proposed to account for the dynamic and reciprocal feedback loops between the epigenome and the genome, in turn, driven by metabolic-elicited modifications , e.g., histones acetylation and deacetylation, DNA methylation, and the feedback from the genome to the epigenome, exemplified by the expression level of histone acetyltransferases, histone deacetylases, histone methyltransferases, and demethylating enzymes (Aon et al. 2016). Hypothetically, a metabolism-epigenome-genome axis provides a comprehensive framework for analyzing the modulation of genotype-phenotype interactions in response to different nutritional environments. According to this concept, the epigenome represents an interface between metabolism and the gene expression machinery of nuclear and mitochondrial DNAs (Donohoe and Bultman 2012; Keating and El-Osta 2015; Mcknight 2010; Wallace 2010; Wallace and Fan 2010), whereas its direct dynamic readout is embodied by the fluxome, defined as the ensemble of metabolic fluxes resulting from genes expressed and proteins translated including their posttranslational modifications (Aon 2013; Cascante and Marin 2008; Cortassa et al. 2015). The change in fluxome dynamics under different cellular functions or in response to nutritional status such as caloric restriction (Mitchell et al. 2016), starvation , or hypoxia has repercussions at both epigenetic and genetic levels thus retro-influencing metabolism. Mechanistically, the tricarboxylic acid cycle (TCA) metabolite citrate is important for acetyl-CoA (AcCoA) generation required for lipogenesis as well as for the acetylation of histones in nuclei (Salminen et al. 2014; Wallace and Fan 2010). Epigenetic modifications via histone acetylation and cellular energy metabolism are linked by ATP citrate lyase (ACLY) in a glucose-dependent manner (Wellen et al. 2009). In mitochondria, AcCoA is generated from pyruvate by PDH, and subsequently citrate synthase catalyzes the conversion of AcCoA and oxaloacetate into citrate in the TCA cycle . Citrate can be transported from mitochondria, via the citrate carrier, into cytoplasm, where ACLY generates AcCoA from citrate (Choudhary et al. 2014).
2 Mitochondrial Energy-Redox Functions
The interrelationship between mitochondrial energy and driving forces such as phosphorylation (ATP) , electrochemical (ΔΨm, ΔpH) , and redox (NAD(P)H, GSH, ROS ) is able to quickly change in response to substrate and ADP levels. These energetic changes determine intra- and extramitochondrial redox environments (RE) involving antioxidants level (e.g., glutathione peroxidase, peroxiredoxin), posttranslational modifications (glutathionylation, oxidation), and H2O2 emission , thus influencing the cytoplasmic redox status (Cortassa et al. 2014; Dey et al. 2016; Jones and Sies 2015; Kembro et al. 2013; Sies 2015; Swain et al. 2016). The energetic status responds to fluctuations in ADP availability modulated by nutrients (e.g., substrate type, abundance) and energy demand (e.g., sedentarism, physical activity) and may, in turn, generate rhythmically changing levels of mitochondrial H2O2 emission (Cortassa et al. 2014) thereby conveying the energetic status to ROS signaling, both converging to tune mitochondrial and cellular function.
The oxidative potential of the TCA cycle , via NADH , influences both respiration and the provision of NAD(P)H, to restoring the antioxidant systems. As a matter of fact, the mitochondrial redox potential of thioredoxin2 (Trx2) decreases (becomes more reducing) from −322 mV at baseline to −350 mV in state 4 and state 3 respiration, during the transition from non-energized to energized (Stanley et al. 2011). This corresponds to a tenfold decrease in the ratio of oxidized to reduced Trx2 and a 2.4-fold increase in the percentage of the Trx2 pool in the reduced form. Under these conditions, after glutamate/malate addition, Trx(SH)2 rises in parallel with ΔΨm, and NAD(P)H , as well as GSH in the mitochondrial matrix (Stanley et al. 2011). As another example, in isolated heart mitochondria, a lower respiratory flux is observed under oxidative stress than in its absence, at similar ΔΨm under both conditions (Cortassa et al. 2014). These results suggested that the NADH -electron donor capacity to respiration might diminish under stress likely due to redirection of electrons to the antioxidant systems. Indeed, the relationship between respiration and ROS is altered by oxidative stress, resulting in decreased mitochondrial energetic performance and higher levels of ROS emission (Cortassa et al. 2014). The intramitochondrial RE is highly influenced by the type of substrate that in the case of lipids is in the form of reducing equivalents, e.g., NADH and FADH2.
As a dynamic metric , the RE is a function of the different redox couples accounting for both their redox potential and the concentration of the reduced species (Jones 2002; Kembro et al. 2013; Schafer and Buettner 2001). Although specific for each subcellular compartment (Dey et al. 2016; Jones and Go 2010; Kaludercic et al. 2014; Swain et al. 2016), the RE dynamics between compartments (e.g., mitochondria, cytoplasm) is interdependent, mediated by exchange of ROS and redox-related components such as GSH and H2O2 (Dey et al. 2016; Jones and Go 2010; Kembro et al. 2014b). In the case of mitochondria, regeneration of glutathione (GSH) from its oxidized form (GSSG ) requires glutathione reductase harnessing the more negative reduction potential of NADPH , which, in turn, will be regenerated by the transhydrogenase coupling hydride transfer between NADH and NADP to the proton motive force (Aon et al. 2007; Hoek and Rydstrom 1988; Nickel et al. 2015; Rydstrom 2006). Since mitochondria cannot synthesize GSH, and the fact that GSSG cannot cross the membrane, the reduction of the latter strictly depends on compartmentalized mitochondrial NADPH generation, a crucial event in the ROS scavenging capacity by antioxidant systems (Aon et al. 2007; Dey et al. 2016; Swain et al. 2016).
The significant role of compartmentation in controlling ROS levels , the RE , and dynamic behavior also depends on the concerted and continuous function of the ROS scavenging systems, e.g., glutathione/thioredoxin, to keep low rates of H2O2 emission from mitochondria (Aon et al. 2012; Stanley et al. 2011). Therefore, the duplication of antioxidant defense systems in multiple compartments appears as a natural and efficient salvage mechanism to avoid or to reduce oxidative bursts (Kembro et al. 2013, 2014b). Failure to maintain NAD(P)H supply during oxidative stress or increased work is a key contributor to ROS overload which may lead to reperfusion-related arrhythmias after ischemic injury (Akar et al. 2005; Aon et al. 2009; Brown et al. 2010; Swain et al. 2016; Xie et al. 2013) or heart failure, the latter due, in part, to impaired mitochondrial Ca2+ signaling to the TCA cycle (Liu et al. 2014).
Current wisdom suggests that under high energy demand , e.g., exercise, in the absence of additional oxidative stress, mitochondria will function at relatively more reduced RE (Aon et al. 2015; Hafstad et al. 2015). However, a shift will happen under pathological conditions, displacing mitochondrial function toward a more oxidized RE (Alleman et al. 2014, 2016; Aon et al. 2014; Tocchetti et al. 2012, 2015). From this perspective, the TCA cycle function in response to nutrient availability can be viewed as a signaling task via its effects on NAD(P)H provision to the antioxidant systems and direct modulation of enzyme function via ROS , in addition to its well-known role as a source of metabolite precursors and NADH . For example, alpha ketoglutarate dehydrogenase and PDH are both deactivated by H2O2 (Crane et al. 1983; Mailloux 2015; Nulton-Persson et al. 2003) or influenced by the triggering of increased expression of antioxidant enzymes via the Nrf2 antioxidant response element (Nguyen et al. 2009).
3 Fatty Acids, Mitochondrial Function, and Oxidative Stress
FAs are main metabolic fuels , and β-oxidation represents their main degradation pathway, for example, in heart and skeletal muscle (Eaton 2002). FA beta-oxidation is a major pathway of energy metabolism providing ~80% of the ATP required for the liver and the heart (Eaton et al. 1996). The rate of β-oxidation is led by demand since an increase in work rate and ATP utilization leads to faster oxidative phosphorylation (OxPhos) and TCA cycle activity. In turn, the decrease in NADH and AcCoA levels leads to an increase in the β-oxidation flux (Eaton 2002; Eaton et al. 1996; Lopaschuk et al. 2010; Neely et al. 1969; Oram et al. 1973).
The FAs released during triacylglyceride (TAG) catabolism are mainly used for β-oxidation and subsequent ATP synthesis via OxPhos in mitochondria. In oxidative tissues such as the heart, TAG-derived FAs are utilized as an energy source, but they also serve as signaling molecules as well as building blocks for membranes and complex lipids. Hepatocytes , heart and skeletal myocytes, adrenocortical cells, enterocytes, adipocytes, and macrophages may all contain large amounts of lipid droplets (LDs) . Excessive LD accumulation is a hallmark of T2DM, obesity, atherosclerosis, hepatic steatosis, and other metabolic diseases (Aon et al. 2014; Singh and Cuervo 2012; Singh et al. 2009; Walther and Farese 2009; Walther and Farese 2012).
As a major energy source, FAs may provide up to two thirds of ATP synthesized via reducing equivalents derived from β-oxidation in mitochondria. The saturated FA palmitate (16:0) supplies about three times higher energy equivalents than glucose in the form of reducing power [7 NADH , 7 FADH2 plus 8 AcCoA ] with a net yield of 106 moles of ATP accounting for the energetic cost of activating the FA (-2 ATP), whereas unsaturated FA oleate (18:1) supplies 8 NADH, 7 FADH2 plus 9 AcCoA with a net ATP yield of 109 moles, in both cases assuming that OxPhos generates 1.5 ATP per FADH2 and 2.5 ATP per NADH oxidized (Nelson and Cox 2013). The reducing equivalents are not only able to contribute electrons to the respiratory/energetic machinery but also to the antioxidant systems via mitochondrial transhydrogenase that converts NADH to NADPH, the latter being a major electron donor to the glutathione and thioredoxin systems from mitochondria (Hoek and Rydstrom 1988).
Preservation of the intracellular RE is crucial for vital functions such as division, differentiation, contractile work, and survival, among others (Aggarwal and Makielski 2013; Aon et al. 2007, 2009; Brown et al. 2010; Fisher-Wellman and Neufer 2012; Jeong et al. 2012; Juhaszova et al. 2004; Lloyd et al. 2012; Muoio and Neufer 2012; Schafer and Buettner 2001). Mitochondria are main drivers of the intracellular RE (Alleman et al. 2014; Aon et al. 2015; Nickel et al. 2014) and together with peroxisomes constitute the main subcellular compartments where lipid degradation occurs. ROS imbalance can be transduced into redox-mediated posttranslational modifications and signaling via H2O2, a mild oxidant reacting with cysteine residues in proteins, affecting, e.g., protein traffic, enzyme, and receptor transcription factor activity, throughout compartmentalized cellular redox circuits (D’autreaux and Toledano 2007; Gauthier et al. 2013; Jones and Go 2010; Kaludercic et al. 2014; Kembro et al. 2013). The ability of H2O2 to freely diffuse throughout cellular compartments enables propagation of intracellular physiological and pathophysiological signals (Aon et al. 2004; Jeong et al. 2012; Juhaszova et al. 2004; Zhou et al. 2010).
A proper cellular/mitochondrial RE is also vital for optimal excitation-contraction (EC) coupling as well as energy supply in the heart (Burgoyne et al. 2012; Christians and Benjamin 2012). Mitochondrial lipid oxidation is a major determinant of the intracellular RE affecting, among other functions, Ca2+ handling by interfering with a wide range of proteins implicated in EC coupling (Fauconnier et al. 2007) including the SR Ca2+ release channels [the ryanodine receptors], the SR Ca2+ pumps, and the sarcolemmal Na+/Ca2+ exchanger (Dedkova and Blatter 2008; Zima and Blatter 2006). In this context, it becomes crucial to know about the impact of ROS on redox balance as a function of substrate oxidation.
The local balance between the ROS -generating and ROS-scavenging capacities in the dense and highly connected mitochondrial network of cardiac cells determines mitochondrion behavior. For instance, mitochondria oscillate when a threshold of ROS is attained, and their collective behavior is tuned via phase and frequency synchronization (Kurz et al. 2010). In turn, the synchronization process is influenced by the size of mitochondrial clusters: large clusters take longer to synchronize resulting in a lower common frequency compared to smaller clusters (Kurz et al. 2016). Importantly, mitochondrial cluster dynamics in cardiomyocytes can be altered by metabolic substrates (glucose, pyruvate, lactate, β-hydroxybutyrate) influencing the synchronization of mitochondrial dynamics, producing a larger frequency distribution and an inverse relation between cluster frequency and size implying a dynamic heterogeneity and functional fragmentation of the mitochondrial population into several localized, smaller clusters (Kurz et al. 2010, 2014, 2015, 2016).
In agreement with the prominent role of lipids on the intracellular redox status, it was shown that with palmitate as a fuel source, a transition from oxidized to reduced cellular redox status in cardiomyocytes from type 2 diabetic (db/db) hearts was determined, drastically abating ROS levels (Tocchetti et al. 2012). This effect was coupled to a marked GSH rise both in wild-type and db/db myocytes from mice. As a consequence of its favorable effect on cellular redox balance, Palm significantly improved ISO-induced contractile reserve in db/db, type 2 (Tocchetti et al. 2015) and type 1 (Tocchetti et al. 2015) diabetic cardiomyocytes, from mice and guinea pig, respectively, and heart trabeculae from Zucker diabetic fatty rat, type 2 diabetes animal model (Bhatt et al. 2015).
Beyond mitochondria, lipids exert a considerable impact on other cellular processes, influencing the functional status of several organs such as the liver, skeletal, and cardiac muscles (Lee et al. 2015; Muoio and Neufer 2012; Roul and Recchia 2015; Singh and Cuervo 2012; Sung et al. 2015). The impact of lipids on mitochondrial redox status and ROS emission, and their links to energetics, is not fully elucidated. At a most basic level, our knowledge remains quite incomplete about the action of lipids on mitochondrial energetic and redox functions. Lipids can act both as uncouplers and OxPhos inhibitors (Wojtczak and Schonfeld 1993), and the consequences of these counteracting effects on mitochondrial energetic, redox, and signaling functions are just starting to be unraveled (Aon et al. 2014; Kienesberger et al. 2013; Schonfeld and Wojtczak 2008). Recent data indicate that there is a concentration effect of lipids on the redox and energetic response of mitochondria; under the threshold concentration lipids can have beneficial actions as opposed to deleterious ones depending on the threshold levels achieved (Cortassa, Sollott, Aon, unpublished).
4 Glucose Metabolism , Pyruvate Transport , and Pyruvate Dehydrogenase Complex Regulation
Cytoplasmic pyruvate is derived from multiple sources in the cytosol, namely, glycolysis, and precursors lactate and alanine. Pyruvate diffuses freely across the outer mitochondrial membrane through nonselective pores but, like other charged molecules, requires specialized transport across the inner membrane. The mitochondrial pyruvate carrier (MPC) conducts pyruvate across the inner mitochondrial membrane to the matrix and thereby occupies a critical link between cytosolic and mitochondrial metabolisms. The mammalian MPC protein complex comprises two obligate, paralogous subunits, designated MPC1 and MPC2, which are encoded by the MPC1 and MPC2 genes and highly conserved across eukaryotes (Bricker et al. 2012). In liver mitochondria, besides the TCA cycle , pyruvate can be channeled toward gluconeogenesis by carboxylation to oxaloacetate by the enzyme pyruvate carboxylase. This reaction regulates oxaloacetate supply to phosphoenolpyruvate kinase and, therefore, the overall gluconeogenic rate (Gray et al. 2015). In type 2 diabetes , elevated hepatic β-oxidation drives gluconeogenesis by raising mitochondrial levels of reducing equivalents and AcCoA which allosterically activates pyruvate carboxylase.
Metabolic flexibility denotes the capacity of a system to adjust fuel selection, primarily glucose and FAs , depending on nutrient availability (Kelley and Mandarino 2000; Zhang et al. 2014). Immediately downstream the MPC , sitting at the crossroad – utilization pathways of glucose-linked substrates (as sources of oxidative energy or as precursors of lipogenesis), or FAs (as preferred substrates for supplying AcCoA and NADH ) – the PDH complex plays a critical role in the use of either carbohydrate or fat as fuel. In animals, the PDH complex together with Mg2+, thiamin pyrophosphate, CoA, and NAD+ catalyzes the oxidative decarboxylation of pyruvate into AcCoA , CO2, and NADH , by an operationally nonreversible reaction (forward rate constant ~107 times higher than in the reverse direction, at pH 7) (Randle 1986). The PDH reaction has a role in ATP synthesis and in the biosynthesis of FAs and TCA cycle intermediates from glucose.
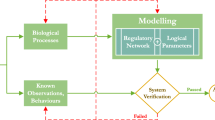
Regulation of the mammalian PDH complex proceeds via specific kinases (pyruvate dehydrogenase kinase, PDK ) and phosphatases (pyruvate dehydrogenase phosphatase, PDP ) that render the enzymatic complex phosphorylated (inactive) or dephosphorylated (active) (Fig. 13.1) (see Mailloux 2015; Roche et al. 2001 for reviews). The E1 component of the PDH complex is interconverted between active and inactive forms, resulting in activity proportional to the fraction of E1 tetramers that are not phosphorylated.

Scheme of the model used to test the influence of PDH regulation on mitochondrial substrate selection . The model encompasses (i) the uptake of pyruvate via the pyruvate carrier and its initial oxidation by pyruvate dehydrogenase (PDH) and the TCA cycle ; (ii) the uptake of palmitoyl-CoA (PCoA ) into mitochondria followed by its oxidation via β-oxidation, based on the model of van Eunen and collaborators (Van Eunen et al. 2013); Acetyl-CoA (AcCoA ) is at the branching point in which glucose and fatty acids degradation come together before entering the TCA cycle for complete oxidation. The model also includes (iii) oxidative phosphorylation , (iv) ionic transport for H+, Ca2+, and inorganic phosphate, (v) ATP/ADP exchange through the adenine nucleotide translocator, and (vi) the generation and scavenging of ROS as described in Kembro et al. (2013). Pyruvate dehydrogenase (PDH) complex regulation accounts for pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) modulation by all known effectors, as modeled by the factor αPDH described in the text (see Eqs. 13.1 and 13.2). The right box depicts the effectors (positive indicated with arrow heads, negative by blunt lines) and their targets PDK and PDP
The crucial role of PDH manifests differently depending upon the physiological condition or the body organ where tissue-selective control of this reaction is indicated by the distinct patterns of kinase isoform expression and the highly conserved primary structures of the different PDK isoforms (Holness et al. 2000; Roche et al. 2001; Roche and Hiromasa 2007). For example, among the four PDK isoforms, PDK2 is apparently the most widely distributed in various body tissues, namely, at high levels in heart, brain, liver, skeletal muscle, kidney, adipose tissue (brown and white), and lactating mammary gland, and at lower levels in lung and spleen (Bowker-Kinley et al. 1998; Wu et al. 1998, 2000).
Cardiac muscle preferentially uses FAs or ketone bodies but can rapidly upregulate glucose utilization by activating PDH during a rapid transition to exercise or postprandial elevation of blood glucose. Resting skeletal muscle has a lower proportion of PDH in the active form than the heart, but this enzymatic complex can be rapidly reactivated upon dephosphorylation during initiation of exercise, or its activity down-modulated as in sustained exercise, when the use of FAs increases (Egan and Zierath 2013, Roche et al. 2001). Within skeletal muscle, PDH regulation differs in slow- and fast-twitch muscles (Holness et al. 2000; Holness and Sugden 1990; Sugden et al. 1997, 2000), the latter exhibiting a greater reliance on glucose thus maintaining a greater portion of PDH in the active form. In slow-twitch muscle , the enhanced FA oxidation after feeding high-fat diets is mainly attributed to the upregulation of pyruvate dehydrogenase kinase 4, PDK4 (Zhang et al. 2014). During starvation , the need to protect glucose stores can be fulfilled by the overexpression of one or more PDK kinase isoforms as in liver (Denyer et al. 1986; Jones et al. 1992; Marchington et al. 1987; Sugden et al. 1996, 1998), kidney (Sugden et al. 1999), and lactating mammary gland (Baxter and Coore 1978) resulting in a shutdown or acute reduction of PDH activity in these tissues (Roche et al. 2001). In the liver, PDH activity is down-modulated immediately after feeding, even following a rise in insulin levels, until liver glycogen is replenished (Holness et al. 1988; Sugden et al. 1998). In fed animals , PDH activity in liver increases in response to the availability of mobile forms of glucose-linked substrates (e.g., glucose, alanine) rather than insulin-enhancing pyruvate dehydrogenase phosphatase (PDP) activity, leading to FA synthesis. In the brain, the PDH reaction plays a crucial role in the complete oxidation of glucose (Malloch et al. 1986).
4.1 General Effector Regulation of PDK and PDP Activities That Modulate Mammalian PDH
The PDH complex is negatively regulated allosterically by the enzymatic products AcCoA and NADH and activated by NAD+, ADP, and CoA (Fig. 13.1). These allosteric effectors modulate the kinase PDK and phosphatase PDP. The dedicated PDK/PDP system responds to metabolite and hormone signals to vary PDH activity in response to changes in nutritional state (Roche and Hiromasa 2007).
Among the PDK isoforms, only PDK2 exhibits strong sensitivity for all regulatory responses shown for PDK. PDK2 activity is greatly stimulated by NADH and AcCoA (Bao et al. 2004a, b), the products resulting from the PDH reaction and β-oxidation. Thus, elevation of PDK2 activity plays an important role in suppressing PDH activity to favor use of fat over carbohydrate as an oxidative fuel. ADP and pyruvate act synergistically to decrease PDK2 activity (Bao et al. 2004b).
PDP activity requires Mg2+ and effectors such as Ca2+, to decrease the Km of one or more PDP isoforms for Mg2+, and polyamines, an insulin second messenger (Roche et al. 2001). Ca2+ acts to directly upregulate the portion of active PDH by enhancing PDP1 activity up to tenfold (Denton et al. 1996; Thomas and Denton 1986; Yan et al. 1996). The possible regulatory role of Mg2+ is suggested by the sensitivity of the activation constant of PDP1 to Ca2+ depending on Mg2+ levels (~0.8 μM Ca2+ in the presence of saturating Mg2+, increasing to 2 μM at 1.0 mM Mg2+) (Roche et al. 2001). The PDP isoform activities are regulated by effector-altered sensitivities to Mg2+ level through very different mechanisms. With PDP1, its regulatory subunit plays a key role in modulating PDP1 catalytic site in response to Mg2+ level. PDP1 activity has been shown to occur in a wide range of tissues including the heart, skeletal muscle, kidney, brain, and liver. PDP2 was elevated in liver and adipose tissue suggesting its importance in fat-synthesizing tissues (Huang et al. 1998). PDP2 is probably the target for insulin activation of PDH in adipose tissue.
Hormonal signaling cascades such as the insulin signaling pathway also play a part in modulating PDH activity in response to whole-body changes in nutrition and energy state. Insulin enhances PDH activity in fat-synthesizing tissues by producing a second messenger that enhances PDP activity by lowering the Km of a phosphatase for Mg2+ (Denton et al. 1986; Thomas et al. 1986). Hormonal signals can also alter the short-term control of PDH activity by altering kinase activity. Signals that increase the production of pyruvate from glucose (e.g., adrenalin in skeletal muscle) will enhance pyruvate inhibition of PDK activity (Randle 1998; Sugden and Holness 1994). Hormone signals that enhance triglyceride breakdown and therefore FA oxidation indirectly stimulate kinase activity due to the resulting elevation of the intramitochondrial levels of NADH and AcCoA (Randle 1998; Roche et al. 2001; Roche and Hiromasa 2007; Sugden and Holness 1994).
5 Computational Modeling of Mammalian PDH
5.1 Background
The critical role of PDH in redirecting catabolism toward the utilization of glucose or FAs is driven by the negative feedback of its own reaction products, AcCoA and NADH (also supplied by FAs, ketone bodies, and the degradation of several amino acids), and can enhance kinase activity up to fourfold (Cate and Roche 1978; Pettit et al. 1975). Figure 13.1 displays a scheme with the strategic location of PDH in the metabolic network, highlighting its dependence on multiple regulatory effectors from mitochondria.
Decreased PDH activity restricts carbohydrate consumption as a result of the increase in mitochondrial NADH /NAD+ and AcCoA/CoA ratios that stimulate PDH inactivation (Batenburg and Olson 1976; Hansford 1976). Stimulation of responsive PDK isoforms is produced by the use of these reactants to increase the proportion of reduced and acetylated lipoyl groups within the complex (Ravindran et al. 1996; Yang et al. 1998).
AcCoA inhibits PDH, potentially competing with CoA (Ki = 5–10 μM) (Quinlan et al. 2014). Reduced availability of AcCoA decreases malonyl-CoA, an inhibitor of lipid utilization, thus forcing β-oxidation which is facilitated by upregulation of PDK4 (Foster 2012; Sugden et al. 2000; Zhang et al. 2014). In isolated mitochondria, AcCoA can be removed by at least two mechanisms: the condensation of oxaloacetate with AcCoA to generate citrate through citrate synthase and, upon addition of carnitine, the conversion of AcCoA to acetylcarnitine, catalyzed by carnitine acetyltransferase. Removal of AcCoA by either pathway should promote flux through the PDH complex.
The effects of NADH/NAD+ and AcCoA/CoA are mediated by the oxidation, reduction, and acetylation state of the lipoyl group, an 80-amino acid, free-folding domain in the N-terminal region of E2 (Roche et al. 2001). Elevation of the NADH/NAD+ and AcCoA/CoA ratios facilitates the stimulation of the activity of certain kinase isoforms, including PDK2, through covalent changes in the E2 component. PDK2 binds to the lipoyl domain depending on the redox and acetylation status of the latter, determining the enzymatic activity (Roche and Hiromasa 2007; Steussy et al. 2001). The regulatory response of PDK2 is also positively dependent on K+ ions in the presence of physiologic levels of chloride and phosphate anions. It seems likely that, mechanistically, this product stimulation of PDK2 activity results from speeding up the rate of dissociation of ADP, which is reduced in the presence of elevated ions (Bao et al. 2004a, b). Potent synergistic inhibition of PDK2 activity by elevated ADP and pyruvate requires both K+ and Pi. The marked reduction in binding of PDK2 to the L2 domain of E2 due to binding of ADP and pyruvate (aided by K+ and phosphate) likely makes a major contribution (beyond slowing ADP dissociation) to the potent inhibition by these effectors (Roche and Hiromasa 2007).
5.2 Modular Analysis of PDH Activity and Its Regulation
Our model of PDH accounts for the regulatory influence of multiple effectors on its activity through αPDH, a comprehensive integrative factor that accounts for all well-known regulators targeting either PDK or PDP, including ATP, ADP, Ca2+, pyruvate in addition to AcCoA , CoA, NADH , and NAD (Fig. 13.1):
With
The symbols used in Eqs. (13.1 and 13.2) are defined in Table 13.1.
The main modulators of the interconversion between the non-phosphorylated and phosphorylated forms of PDH are AcCoA/CoA and NADH/NAD molar ratios. An increase in either ratio augments the proportion of inactive PDH since the activity of the kinase PDK is stimulated by both AcCoA and NADH while it is inhibited by CoA and NAD. NADH renders PDH more inactive via inhibition of the phosphatase PDP, which is reversed by NAD (Pettit et al. 1975).
The 3D plots in Figs. 13.2 and 13.3 show the dependence of PDH activity (Figs. 13.2a and 13.3a) and the factor α PDH (Figs. 13.2b and 13.3b) as a function of its substrate pyruvate or Ca2+ and both AcCoA/CoA and NADH/NAD ratios, respectively. PDH activity increases exponentially at low AcCoA/CoA ratio concomitantly with the sensitivity to pyruvate. From the behavior of α PDH (Fig. 13.2b), it can be seen that the modulatory role of AcCoA/CoA appears to dominate the enzyme activity at low rather than high ratios, since at high AcCoA/CoA the activity of PDH is negligible, whereas α PDH, although low, is not, suggesting that other effectors may prevail. Comparatively, the degree of enzyme activation attained by NADH/NAD and Ca2+ is about fivefold lower than AcCoA/CoA and pyruvate (compare Figs. 13.2a and 13.3a).

Modular analysis of PDH activity and its modulatory factor αPDH as a function of pyruvate and AcCoA/CoA ratio. The PDH rate expression (Eq. 13.1) was studied as a function of pyruvate concentration in the range 0.01–2.5 mM, while CoA was varied between 5 × 10−4 and 0.05 mM while keeping AcCoA constant at 0.2 mM. Other parameters were as follows: NADH = 0.1 mM; NAD+ = 0.9 mM; ATP = 1.0 mM; ADP = 0.5 mM; Ca2+ = 0.2 μM. Maximal rate and other parameters utilized are indicated in Table 13.1

Modular analysis of PDH activity and its modulatory factor αPDH as a function of Ca2+ and NADH/NAD ratio. The PDH rate expression (Eq. 13.1) was analyzed as a function of Ca2+ (range 1 × 10−5–1.5 × 10−3 mM) and NADH (range 0.01–0.8 mM) concentrations, while NAD+ was kept constant at 0.9 mM. Other parameters were as follows: AcCoA = 0.2 mM; CoA = 0.01 mM; ATP = 1.0 mM; ADP = 0.5 mM; Pyr = 0.5 mM. Maximal rate and other parameters utilized are indicated in Table 13.1
The PDH model was able to simulate the experimental data of Pettit and colleagues (Pettit et al. 1975) obtained in highly purified preparations of PDH complexes and their component enzymes from bovine kidney and heart. The steady-state activity of PDH was modulated throughout a wide range of AcCoA/CoA (Fig. 13.4a) and NADH/NAD (Fig. 13.4b) ratios, both experimentally and computationally. The model correctly predicts a decrease in activity as a function of an increase in the ratios, with is only a minor impact at substantially low ratios (<0.1) (Fig. 13.4a, b, insets).

Comparative study of PDH activity as a function of the main modulatory ratios NADH/NAD and AcCoA/CoA. The expression of PDH rate (Eq. 13.1) was evaluated at (mM): Ca2+ = 2 × 10−4, ADP = 0.5, ATP = 1.0, and Pyr = 2.5. In panel A NADH was kept constant at 0.1 mM, AcCoA = 0.05, while CoA varied in the range 5 × 10−3 – 0.76 mM. In Panel B CoA was constant at 0.1 mM, while NADH changed from 1 × 10−4 to 0.975 and NAD changed in parallel to keep the total pyridine nucleotide pool at 1.0 mM
5.3 Integrated Analysis of PDH Activity and Regulation
Next, we analyzed the PDH behavior in an integrated model of mitochondrial metabolism (Cortassa, Sollott, Aon, unpublished) including simultaneous degradation of glucose-derived substrates (pyruvate, Pyr) and FAs (palmitoyl-CoA, PCoA ) (Fig. 13.1). We sought to understand the impact of substrate selection (glucose-FA ) on the regulation of PDH activity via AcCoA/CoA and NADH/NAD ratios, when the enzyme is integrated to mitochondrial metabolism. Figure 13.5 depicts the PDH flux as a function of both ratios, when either glucose (via pyruvate) is changing at constant FA input (via PCoA ) or vice versa. Overall, and as expected from the behavior of isolated PDH, its flux decreases as a function of increasing AcCoA/CoA (compare Figs. 13.4a and 13.5a). However, PDH flux as a function of NADH/NAD displays a different behavior than its activity when isolated, i.e., increasing rather than decreasing as a function of the ratio (compare Figs. 13.4b and 13.5b). When integrated, the PDH flux is the result of the instantaneous composition of all regulatory effectors. This interpretation was confirmed by the model’s ability to reproduce the trajectory of PDH flux in the integrated system, across the family of PDH activity curves corresponding to the isolated enzyme (Fig. 13.6), which is obtained when using the steady-state values of all regulatory effectors to parameterize the PDH rate expression (e.g., see Eqs. 13.1 and 13.2). When PCoA is changing at constant Pyr, we observe an overall similar qualitative behavior although for a more restricted range of variation in both ratios and activity (Fig. 13.5). Together, these results suggest that AcCoA/CoA regulation prevails over NADH/NAD in the integrated system, where the flux through the PDH complex as a systemic property results from the instantaneous levels of all regulatory effectors.

Steady-state PDH flux in the integrated model when either pyruvate (Pyr) or palmitoyl-CoA (PCoA ) was varied (PCoA/Pyr ratio). A computed simulation of the complete model was run until all state variables reached steady state (i.e., their time derivatives were <1 × 10−10). Pyr was adjusted in the range from 4 × 10−3 to 0.01 mM (indicated in blue lines) while keeping PCoA constant at 0.04 mM. In the steady states indicated with a red line PCoA was varied from 0.01 to 0.06 mM, while Pyr remained constant at 0.009 mM. In these plots the AcCoA/CoA and NADH/NAD ratios are computed from their steady-state values occurring upon variations in the relative proportion of substrates Pyr and PCoA. Consequently, all axes are representing state variables thus explaining the trend observed in the PDH flux value as a function of NADH/NAD, since the values of every regulatory effector of PDH activity are simultaneously changing in these simulations

Modular study of the PDH activity under conditions reproducing the steady states obtained with different PCoA /Pyr ratios . The concentrations of substrates and effectors other than the ones in the x-axis were fixed at the values corresponding to the steady states represented in the curves in Fig. 13.5. The PDH activity was calculated as a function of the ratio AcCoA/CoA and NADH/NAD under conditions mimicking variations in Pyr (top panels) or PCoA (bottom panels). The squares indicate the exact conditions for all substrates and effectors that were represented in Fig. 13.5
Respiration and H2O2 emission both increase as a function of PCoA /Pyr ratio when either one of them is changing in constant proportion to the other, suggesting that ROS generation is matched by the ROS scavenging systems under these conditions (Fig. 13.7).

Steady-state respiration and H2O2 emission fluxes as a function of PCoA /Pyr ratios. The O2 consumption (solid lines) and H2O2 emission (dashed lines) fluxes were obtained in the same simulations presented in Fig. 13.5 when Pyr (in blue or gray) or PCoA (in red or magenta) was changed. All simulation conditions correspond to those specified in the legend of Fig. 13.5
To address the question of substrate selection , we quantified the fluxes driven by Pyr or PCoA as a function of their ratio to simulate changing nutrient availability and the sensitivity of the pathways’ flux to both substrates. Figure 13.8a shows the fluxes through β-oxidation via carnitine palmitoyl transferase 1 (CPT1) and PDH as a function of the ratio PCoA/Pyr. When Pyr increases at constant PCoA, i.e., decreasing PCoA/Pyr ratio, the flux through PDH increases 3.6-fold, whereas that of CPT1 decreased 15%. On the other hand, increasing PCoA at constant Pyr produced ~40% increase in flux through CPT1 as compared to ~11% decrease through PDH. Together, these results suggest that the flux from Pyr is much more sensitive to nutrient availability than from PCoA , thus making substrate selection toward Pyr more sensitive, at least under these conditions (Fig. 13.8b). Quantitatively speaking, the flux values through PDH and CPT1 determined with our model agree very well with published results, respectively, of glucose oxidation in whole heart (3–10 vs. 5–7 μM s−1) (Buchanan et al. 2005; Cortassa et al. 2015; Kashiwaya et al. 1994) and with palmitate oxidation in cardiomyocytes (0.4–0.6 vs. 0.4 μM s−1) (Luiken et al. 2009) or respiration in the presence of both palmitate and glucose in cardiomyocytes (0.12 vs. 0.4 μM s−1) (Wang et al. 2011).

Steady-state fluxes through PDH and CPT1 upon changing proportions between pyruvate and PCoA . The fluxes through the transport step of FAs CPT1 (solid lines) and regulatory PDH (dashed lines) obtained in the simulations in Fig. 13.5 were represented as a function of the ratio of PCoA over pyruvate. Blue and gray lines indicate when Pyr was varied at constant PCoA, whereas red and magenta ones represent changes in PCoA at constant Pyr. The scheme in panel B is a graphical representation of the sensitivity of flux regulation through the PDH complex and CPT1 in response to changing proportion of glucose- and FA -derived substrates
Taken together, the results obtained agree with the idea that the crucial regulatory role played by PDH in substrate selection in the short term depends on glucose and FAs ’ availability and on the resulting composition of metabolite levels that activate or inhibit the kinases and phosphatases from the PDH complex. Within this complex regulatory picture, our simulations also indicate that, under these conditions, the phosphorylated form of PDH predominates over the non-phosphorylated one, as can be judged by the low αPDH values (~1.2 × 10−3, when either pyruvate or PCoA is varying; see also Figs. 13.2, 13.4, and 13.5). Accordingly, a higher response of pyruvate flux through PDH as compared to FAs through CPT1 was determined, indicating a higher sensitivity of selection toward carbohydrates (Fig. 13.8). Judging from the agreement between the PDH behaviors whether isolated from or integrated with mitochondrial metabolism, simulations also show a predominant regulatory impact by the AcCoA/CoA ratio as compared to NADH/NAD (compare Figs. 13.4 and 13.5).
6 Modulation of Substrate Selection and Metabolic Remodeling in Health, Disease, and Aging
(Patho)physiological situations involve differential selection of substrate fuel which can exert a significant impact on the organism or cell behavior. For instance, obese and type 2 diabetic patients exhibit greater rates of FA oxidation and insulin resistance unlike lean healthy individuals (the latter whom, under insulin stimulation, are able to switch from predominantly FA oxidation to elevation of glucose uptake, oxidation, and storage). This capacity to adjust fuel selection as a function of nutrient availability has been termed metabolic flexibility (Kelley and Mandarino 2000), in which the mitochondrial PDH complex plays a crucial regulatory role (Randle 1986; Roche et al. 2001; Roche and Hiromasa 2007; Sugden et al. 1998; Zhang et al. 2014). PDH regulation involves short-term (e.g., allosteric inhibition/activation) as well as long-term (e.g., gene expression, transcriptional, posttranslational) mechanisms (Randle 1998; Roche et al. 2001; Roche and Hiromasa 2007; Sugden et al. 1997, 1998) which, in turn, are subjected to circadian regulation (Bellet and Sassone-Corsi 2010). The temporal regulatory dimension of PDH activity is of great importance because both acute (e.g., fast-feed transition) and chronic (e.g., metabolic disorder) nutritional conditions demand flexible, or generate inflexible, metabolic responses. For example, long-term consumption of a high-saturated fat diet may cause hyperglycemia, hyperinsulinemia, glucose intolerance, and obesity. In skeletal muscle , consumption of a high-fat diet leads to the use of lipid-derived fuels as respiratory substrates, a switch modulated, in part, by upregulation of PDK, the kinase activity associated with PDH (Zhang et al. 2014). Another level of regulation is given by nutrient availability-sensitive posttranslational modifications in the presence or absence of sirtuin3 (SIRT3) that reshape the mitochondrial acetylome, potentially affecting multiple enzymatic activities (Finkel 2015; Foster et al. 2013).
Glucose stores are important for their utilization as preferred substrate by the central nervous system. Inactivation of PDH along with greatly diminished glucose oxidation may happen in muscle, liver, and fat cells due to diabetes , starvation , long-term feeding of high-fat diet, or obesity. As the studies of Randle and coworkers first demonstrated (Randle 1986; Randle et al. 1963), during starvation and the diabetic state, the acute decreases in the PDH activity of these tissues are engendered by a marked induction of kinase activity (Randle 1998; Roche et al. 2001). In starvation and hibernation, enhanced PDK activity is beneficial for preventing loss of body carbohydrate while favoring the use of the more abundant lipid fuels. Along with increased kinase levels, hormonally controlled increase in FA and ketone body oxidation elevates AcCoA and NADH , which, in turn, stimulate the activity of certain PDK isoforms.
Severe limitation of glucose use by PDH inactivation raises blood glucose in the diabetic state, while glucose clearance is impaired making a major contribution to the pathology of diabetes . High blood glucose damages vascular cells (Brownlee 2001; Choi et al. 2008; Giacco and Brownlee 2010; Laakso 1999; Lasker 1993) and the myocardium leading to progressive vascular damage and heart dysfunction in both type 1 (insulin-deficient) and type 2 (insulin-resistant) diabetes (Aon et al. 2015; Bhatt et al. 2015; Tocchetti et al. 2012, 2015), which amount to ~4% and 96%, respectively, of the diabetic population according to the 2012 American Diabetes Association statistics. Diabetes is characterized by increased circulating concentrations of glucose and FAs . Irrespective of hyperglycemia , the heart from diabetics relies heavily on FA utilization with a concomitant decrease in glucose oxidation (Boudina and Abel 2010; Carley and Severson 2005). Historically, the glucose-FA cycle, also known as the Randle cycle (Randle 1998; Randle et al. 1963), has played a relevant role as the biochemical mechanism explaining the control and functional impact of fuel selection, FA over glucose oxidation (Hue and Taegtmeyer 2009). However, on its own, hyperglycemia induces cellular damage that involves the increase of the flux of glucose and other sugars through the polyol, hexosamine, advanced glycation end products (AGEs), and diacylglycerol (DAG) pathways, the latter leading to protein kinase C (PKC) activation (Brownlee 1995, 2001; Giacco and Brownlee 2010). These changes are also implicated in the hyperglycemia-mediated modifications and impairments of cell redox assets (Aon et al. 2015; Brownlee 2001; Tocchetti et al. 2012, 2015; Williamson et al. 1993).
Under starvation and diabetic conditions, the PDK4 isoform (less sensitive to pyruvate inhibition vs. other isoforms) is overexpressed in several tissues, particularly in heart and skeletal muscle under conditions of limited consumption of carbohydrate (Holness et al. 2000; Roche et al. 2001; Sugden et al. 1998, 1999, 2000; Wu et al. 1998, 2000). Besides prolonged starvation , feeding a high-fat, low-carbohydrate, diet increases PDK4 in both slow- and fast-twitch muscles . Refeeding or insulin treatment reverses the effects of starvation or diabetes , respectively. In rat liver, PDK2 and PDK4 are both overexpressed under conditions of starvation , diabetes , or feeding a high-fat, low-carbohydrate diet or in response to artificial elevation of cAMP or 3,5,3′-triiodothyronine (Denyer et al. 1986; Jones et al. 1992; Sugden et al. 1998; Sugden and Holness 1994; Wu et al. 2000). These effects can be also reversed by insulin or refeeding with a carbohydrate-rich diet (Holness et al. 1988). Selective elevation of PDK2, following maintenance on a high-fat diet, could be prevented or reversed by a diet supplemented with long-chain ω-3-fatty acids (Sugden et al. 1998).
A healthy heart can utilize various substrates (glucose, FAs , ketones, lactate) to satisfy its continuous energy requirements, although under postabsorptive or fasting conditions it preferentially uses FAs (Kolwicz and Tian 2009; Lionetti et al. 2011). The heart’s high adaptability can also be found throughout its life cycle where at the fetal stage it relies on carbohydrate substrates, whereas at more mature stages it predominantly consumes FAs as fuel. The FA dependence of the heart can be enhanced by diabetes (Belke et al. 1999; Lopaschuk 2002). In general, aging and disease drive substantial metabolic remodeling that includes changes in mitochondrial function, impaired metabolic flexibility, and reduced insulin sensitivity (Finkel 2015). In the aged heart, the capacity for glucose utilization prevails over FA oxidation (Hansford 1983; Lesnefsky et al. 2016; Van Bilsen et al. 2009), although tending to be insufficient for sustaining energy supply under stress (Kolwicz and Tian 2009). Unlike the reported decline of protein levels from mitochondrial metabolism (including respiratory complexes, TCA cycle , FA, and amino acid metabolisms), a significant increase in glycolytic and extracellular structural proteins happens with age (Tocchi et al. 2015). Aging impairs mitochondrial OxPhos , particularly so in interfibrillar mitochondria, affecting the activity of complexes III and IV, which accounts in large measure for the known decrease in respiration (Lesnefsky et al. 2016). Phenotypically, the metabolic profile of the aging heart bears some similarity to that of heart failure. Pathological hypertrophy is associated with reversion to a fetal gene expression pattern and an increased reliance on carbohydrate fuel rather than FAs which in turn are less consumed (Kolwicz and Tian 2009).
7 Concluding Remarks
Mitochondrial metabolism and PDH activity are central players in substrate selection, a process that underlies metabolic remodeling and flexibility in healthy, diseased, and aged states. Within the complex regulatory picture of the PDH activity, with dedicated kinases and phosphatases as key targets, the mitochondrial AcCoA/CoA ratio assumes a predominant role compared to NADH/NAD. Under conditions of different nutrient availability, the higher sensitivity of PDH toward glucose-derived substrates (i.e., pyruvate) is in agreement with the crucial role played by this enzymatic complex in preserving glucose stores for brain function that, in turn, determines the overall balance between nutrient utilization, storage, and turnover in the organism’s function. By accounting for all major effectors of the PDH complex, modeling and results presented herein now enable a more realistic and detailed understanding of the regulation of selection between glucose- and FA -derived substrates in the presence of all major redox-energetic mitochondrial functions.
References
Aggarwal NT, Makielski JC (2013) Redox control of cardiac excitability. Antioxid Redox Signal 18:432–468
Akar FG, Aon MA, Tomaselli GF, O’Rourke B (2005) The mitochondrial origin of postischemic arrhythmias. J Clin Invest 115:3527–3535
Alleman RJ, Katunga LA, Nelson MA, Brown DA, Anderson EJ (2014) The “goldilocks zone” from a redox perspective-adaptive vs. deleterious responses to oxidative stress in striated muscle. Front Physiol 5:358
Alleman RJ, Tsang AM, Ryan TE, Patteson DJ, Mcclung JM, Spangenburg EE, Shaikh SR, Neufer PD, Brown DA (2016) Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol 310:H1360–H1370
Aon MA (2013) Complex systems biology of networks: the riddle and the challenge. In: Aon MA, Saks V, Schlattner U (eds) Systems biology of metabolic and signaling networks. energy, mass and information transfer, 1st edn. Springer-Verlag Berlin Heidelberg, Heidelberg
Aon MA, Cortassa S, O’rourke B (2004) Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A 101:4447–4452
Aon MA, Cortassa S, Maack C, O’Rourke B (2007) Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem 282:21889–21900
Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O’rourke B (2009) From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol 41:1940–1948
Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O’Rourke B, Paolocci N, Cortassa S (2012) Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol 139:479–491
Aon MA, Bhatt N, Cortassa S (2014) Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol 5:1–13
Aon MA, Tocchetti CG, Bhatt N, Paolocci N, Cortassa S (2015) Protective mechanisms of mitochondria and heart function in diabetes. Antioxid Redox Signal 22:1563–1586
Aon MA, Cortassa S, Juhaszova M, Sollott SJ (2016) Mitochondrial health, the epigenome and healthspan. Clin Sci (Lond) 130:1285–1305
Bao H, Kasten SA, Yan X, Hiromasa Y, Roche TE (2004a) Pyruvate dehydrogenase kinase isoform 2 activity stimulated by speeding up the rate of dissociation of ADP. Biochemistry 43:13442–13451
Bao H, Kasten SA, Yan X, Roche TE (2004b) Pyruvate dehydrogenase kinase isoform 2 activity limited and further inhibited by slowing down the rate of dissociation of ADP. Biochemistry 43:13432–13441
Batenburg JJ, Olson MS (1976) Regulation of pyruvate dehydrogenase by fatty acid in isolated rat liver mitochondria. J Biol Chem 251:1364–1370
Baxter MA, Coore HG (1978) The mode of regulation of pyruvate dehydrogenase of lactating rat mammary gland. Effects of starvation and insulin. Biochem J 174:553–561
Belke DD, Larsen TS, Lopaschuk GD, Severson DL (1999) Glucose and fatty acid metabolism in the isolated working mouse heart. Am J Phys 277:R1210–R1217
Bellet MM, Sassone-Corsi P (2010) Mammalian circadian clock and metabolism – the epigenetic link. J Cell Sci 123:3837–3848
Bhatt NM, Aon MA, Tocchetti CG, Shen X, Dey S, Ramirez-Correa G, O’Rourke B, Gao WD, Cortassa S (2015) Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. Am J Physiol Heart Circ Physiol 308:H291–H302
Boudina S, Abel ED (2010) Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11:31–39
Bowker-Kinley MM, Davis WI, Wu P, Harris. RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329(Pt 1):191–196
Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337:96–100
Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B (2010) Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol 48:673–679
Brownlee M (1995) Advanced protein glycosylation in diabetes and aging. Annu Rev Med 46:223–234
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820
Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED (2005) Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146:5341–5349
Burgoyne JR, Mongue-Din H, Eaton P, Shah AM (2012) Redox signaling in cardiac physiology and pathology. Circ Res 111:1091–1106
Carley AN, Severson DL (2005) Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta 1734:112–126
Cascante M, Marin S (2008) Metabolomics and fluxomics approaches. Essays Biochem 45:67–81
Cate RL, Roche TE (1978) A unifying mechanism for stimulation of mammalian pyruvate dehydrogenase(a) kinase by reduced nicotinamide adenine dinucleotide, dihydrolipoamide, acetyl coenzyme A, or pyruvate. J Biol Chem 253:496–503
Choi SW, Benzie IF, Ma SW, Strain JJ, Hannigan BM (2008) Acute hyperglycemia and oxidative stress: direct cause and effect? Free Radic Biol Med 44:1217–1231
Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15:536–550
Christians ES, Benjamin IJ (2012) Proteostasis and REDOX state in the heart. Am J Physiol Heart Circ Physiol 302:H24–H37
Cortassa S, O’Rourke B, Aon MA (2014) Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta 1837:287–295
Cortassa S, Caceres V, Bell LN, O’Rourke B, Paolocci N, Aon MA (2015) From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys J 108:163–172
Crane D, Haussinger D, Graf P, Sies H (1983) Decreased flux through pyruvate dehydrogenase by thiol oxidation during t-butyl hydroperoxide metabolism in perfused rat liver. Hoppe Seylers Z Physiol Chem 364:977–987
D’autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8:813–824
Dedkova EN, Blatter LA (2008) Mitochondrial Ca2+ and the heart. Cell Calcium 44:77–91
Denton RM, Mccormack JG, Thomas AP (1986) Mechanisms whereby insulin and other hormones binding to cell surface receptors influence metabolic pathways within the inner membrane of mitochondria. Ann N Y Acad Sci 488:370–384
Denton RM, Mccormack JG, Rutter GA, Burnett P, Edgell NJ, Moule SK, Diggle TA (1996) The hormonal regulation of pyruvate dehydrogenase complex. Adv Enzym Regul 36:183–198
Denyer GS, Kerbey AL, Randle PJ (1986) Kinase activator protein mediates longer-term effects of starvation on activity of pyruvate dehydrogenase kinase in rat liver mitochondria. Biochem J 239:347–354
Dey S, Sidor A, O’Rourke B (2016) Compartment-specific control of reactive oxygen species scavenging by antioxidant pathway enzymes. J Biol Chem 291:11185–11197
Donohoe DR, Bultman SJ (2012) Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol 227:3169–3177
Eaton S (2002) Control of mitochondrial beta-oxidation flux. Prog Lipid Res 41:197–239
Eaton S, Bartlett K, Pourfarzam M (1996) Mammalian mitochondrial beta-oxidation. Biochem J 320(Pt 2):345–357
Egan B, Zierath JR (2013) Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17:162–184
Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R, Katz A, Bruton JD, Westerblad H (2007) Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes 56:1136–1142
Finkel T (2015) The metabolic regulation of aging. Nat Med 21:1416–1423
Fisher-Wellman KH, Neufer PD (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab 23:142–153
Foster DW (2012) Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest 122:1958–1959
Foster DB, Liu T, Rucker J, O’Meally RN, Devine LR, Cole RN, O’Rourke B (2013) The cardiac acetyl-lysine proteome. PLoS One 8:e67513
Gauthier LD, Greenstein JL, Cortassa S, O’Rourke B, Winslow RL (2013) A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys J 105:1045–1056
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107:1058–1070
Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, Lane EE, Dohlman A, Zepeda-Orozco D, Xie J, Rutter J, Norris AW, Cox JE, Burgess SC, Potthoff MJ, Taylor EB (2015) Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab 22:669–681
Hafstad AD, Boardman N, Aasum E (2015) How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid Redox Signal 22:1587–1605
Hansford RG (1976) Studies on the effects of coenzyme A-SH: acetyl coenzyme A, nicotinamide adenine dinucleotide: reduced nicotinamide adenine dinucleotide, and adenosine diphosphate: adenosine triphosphate ratios on the interconversion of active and inactive pyruvate dehydrogenase in isolated rat heart mitochondria. J Biol Chem 251:5483–5489
Hansford RG (1983) Bioenergetics in aging. Biochim Biophys Acta 726:41–80
Hoek JB, Rydstrom J (1988) Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem J 254:1–10
Holness MJ, Sugden MC (1990) Glucose utilization in heart, diaphragm and skeletal muscle during the fed-to-starved transition. Biochem J 270:245–249
Holness MJ, Maclennan PA, Palmer TN, Sugden MC (1988) The disposition of carbohydrate between glycogenesis, lipogenesis and oxidation in liver during the starved-to-fed transition. Biochem J 252:325–330
Holness MJ, Kraus A, Harris RA, Sugden MC (2000) Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 49:775–781
Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM (1998) Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem 273:17680–17688
Hue L, Taegtmeyer H (2009) The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297:E578–E591
Jeong EM, Liu M, Sturdy M, Gao G, Varghese ST, Sovari AA, Dudley SC Jr (2012) Metabolic stress, reactive oxygen species, and arrhythmia. J Mol Cell Cardiol 52:454–463
Jones DP (2002) Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 348:93–112
Jones DP, Go YM (2010) Redox compartmentalization and cellular stress. Diabetes Obes Metab 12(Suppl 2):116–125
Jones DP, Sies H (2015) The redox code. Antioxid Redox Signal 23:734–746
Jones BS, Yeaman SJ, Sugden MC, Holness MJ (1992) Hepatic pyruvate dehydrogenase kinase activities during the starved-to-fed transition. Biochim Biophys Acta 1134:164–168
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ (2004) Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113:1535–1549
Kaludercic N, Deshwal S, Di Lisa F (2014) Reactive oxygen species and redox compartmentalization. Front Physiol 5:285
Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV (1994) Control of glucose utilization in working perfused rat heart. J Biol Chem 269:25502–25514
Keating ST, El-Osta A (2015) Epigenetics and metabolism. Circ Res 116:715–736
Kelley DE, Mandarino LJ (2000) Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49:677–683
Kembro JM, Aon MA, Winslow RL, O’Rourke B, Cortassa S (2013) Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys J 104:332–343
Kembro JM, Cortassa S, Aon MA (2014) Complex oscillatory redox dynamics with signaling potential at the edge between normal and pathological mitochondrial function. Front Physiol 5:257
Kienesberger PC, Pulinilkunnil T, Nagendran J, Dyck JR (2013) Myocardial triacylglycerol metabolism. J Mol Cell Cardiol 55:101–110
Kolwicz SC Jr, Tian R (2009) Metabolic therapy at the crossroad: how to optimize myocardial substrate utilization? Trends Cardiovasc Med 19:201–207
Kurz FT, Aon MA, O’Rourke B, Armoundas AA (2010) Spatio-temporal oscillations of individual mitochondria in cardiac myocytes reveal modulation of synchronized mitochondrial clusters. Proc Natl Acad Sci U S A 107:14315–14320
Kurz FT, Aon MA, O’Rourke B, Armoundas AA (2014) Cardiac mitochondria exhibit dynamic functional clustering. Front Physiol 5:329
Kurz FT, Derungs T, Aon MA, O’Rourke B, Armoundas AA (2015) Mitochondrial networks in cardiac myocytes reveal dynamic coupling behavior. Biophys J 108:1922–1933
Kurz FT, Kembro JM, Flesia AG, Armoundas AA, Cortassa S, Aon MA, Lloyd D (2016) Network dynamics: quantitative analysis of complex behavior in metabolism, organelles and cells, from experiments to models and back. Wiley Interdiscip Rev Syst Biol Med. doi:10.1002/wsbm.1352
Laakso M (1999) Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 48:937–942
Lanpher B, Brunetti-Pierri N, Lee B (2006) Inborn errors of metabolism: the flux from Mendelian to complex diseases. Nat Rev Genet 7:449–460
Lasker RD (1993) The diabetes control and complications trial. Implications for policy and practice. N Engl J Med 329:1035–1036
Lee J, Homma T, Kurahashi T, Kang ES, Fujii J (2015) Oxidative stress triggers lipid droplet accumulation in primary cultured hepatocytes by activating fatty acid synthesis. Biochem Biophys Res Commun 464:229–235
Lesnefsky EJ, Chen Q, Hoppel CL (2016) Mitochondrial metabolism in aging heart. Circ Res 118:1593–1611
Lionetti V, Stanley WC, Recchia FA (2011) Modulating fatty acid oxidation in heart failure. Cardiovasc Res 90:202–209
Liu T, Takimoto E, Dimaano VL, Demazumder D, Kettlewell S, Smith G, Sidor A, Abraham TP, O’Rourke B (2014) Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res 115:44–54
Lloyd D, Cortassa S, O’Rourke B, Aon MA (2012) What yeast and cardiomyocytes share: ultradian oscillatory redox mechanisms of cellular coherence and survival. Integr Biol (Camb) 4:65–74
Lopaschuk GD (2002) Metabolic abnormalities in the diabetic heart. Heart Fail Rev 7:149–159
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010) Myocardial fatty acid metabolism in health and disease. Physiol Rev 90:207–258
Luiken JJ, Niessen HE, Coort SL, Hoebers N, Coumans WA, Schwenk RW, Bonen A, Glatz JF (2009) Etomoxir-induced partial carnitine palmitoyltransferase-I (CPT-I) inhibition in vivo does not alter cardiac long-chain fatty acid uptake and oxidation rates. Biochem J 419:447–455
Mailloux RJ (2015) Still at the center of it all: novel functions of the oxidative krebs cycle. Bioenergetics 4:122
Malloch GD, Munday LA, Olson MS, Clark JB (1986) Comparative development of the pyruvate dehydrogenase complex and citrate synthase in rat brain mitochondria. Biochem J 238:729–736
Marchington DR, Kerbey AL, Jones AE, Randle PJ (1987) Insulin reverses effects of starvation on the activity of pyruvate dehydrogenase kinase in cultured hepatocytes. Biochem J 246:233–236
Mcknight SL (2010) On getting there from here. Science 330:1338–1339
Mitchell SJ, Madrigal-Matute J, Scheibye-Knudsen M, Fang E, Aon M, Gonzalez-Reyes JA, Cortassa S, Kaushik S, Gonzalez-Freire M, Patel B, Wahl D, Ali A, Calvo-Rubio M, Buron MI, Guiterrez V, Ward TM, Palacios HH, Cai H, Frederick DW, Hine C, Broeskamp F, Habering L, Dawson J, Beasley TM, Wan J, Ikeno Y, Hubbard G, Becker KG, Zhang Y, Bohr VA, Longo DL, Navas P, Ferrucci L, Sinclair DA, Cohen P, Egan JM, Mitchell JR, Baur JA, Allison DB, Anson RM, Villalba JM, Madeo F, Cuervo AM, Pearson KJ, Ingram DK, Bernier M, De Cabo R (2016) Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23:1093–1112
Muoio DM, Neufer PD (2012) Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metab 15:595–605
Neely JR, Bowman RH, Morgan HE (1969) Effects of ventricular pressure development and palmitate on glucose transport. Am J Phys 216:804–811
Nelson DL, Cox MM (2013) Lehninger principles of biochemistry. W. H. Freeman and Company, New York
Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284:13291–13295
Nickel A, Kohlhaas M, Maack C (2014) Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol 73:26–33
Nickel AG, von Hardenberg A, Hohl M, Loffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kastner L, Lipp P, Zeller T, Muller C, Knopp A, Laufs U, Bohm M, Hoth M, Maack C (2015) Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 22:472–484
Nulton-Persson AC, Starke DW, Mieyal JJ, Szweda LI (2003) Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry 42:4235–4242
Oram JF, Bennetch SL, Neely JR (1973) Regulation of fatty acid utilization in isolated perfused rat hearts. J Biol Chem 248:5299–5309
Pettit FH, Pelley JW, Reed LJ (1975) Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun 65:575–582
Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem 289:8312–8325
Randle PJ (1986) Fuel selection in animals. Biochem Soc Trans 14:799–806
Randle PJ (1998) Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14:263–283
Randle PJ, Garland PB, Hales CN, Newsholme EA (1963) The glucose fatty-acid cycle Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1:785–789
Ravindran S, Radke GA, Guest JR, Roche TE (1996) Lipoyl domain-based mechanism for the integrated feedback control of the pyruvate dehydrogenase complex by enhancement of pyruvate dehydrogenase kinase activity. J Biol Chem 271:653–662
Roche TE, Hiromasa Y (2007) Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci 64:830–849
Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA (2001) Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol 70:33–75
Roul D, Recchia FA (2015) Metabolic alterations induce oxidative stress in diabetic and failing hearts: different pathways, same outcome. Antioxid Redox Signal 22:1502–1514
Rydstrom J (2006) Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta 1757:721–726
Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A (2014) Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal 26:1598–1603
Schafer FQ, Buettner GR (2001) Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30:1191–1212
Schonfeld P, Wojtczak L (2008) Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic Biol Med 45:231–241
Sies H (2015) Oxidative stress: a concept in redox biology and medicine. Redox Biol 4:180–183
Singh R, Cuervo AM (2012) Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012:282041
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Stanley BA, Sivakumaran V, Shi S, Mcdonald I, Lloyd D, Watson WH, Aon MA, Paolocci N (2011) Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem 286:33669–33677
Steussy CN, Popov KM, Bowker-Kinley MM, Sloan RB Jr, Harris RA, Hamilton JA (2001) Structure of pyruvate dehydrogenase kinase. Novel folding pattern for a serine protein kinase. J Biol Chem 276:37443–37450
Sugden MC, Holness MJ (1994) Interactive regulation of the pyruvate dehydrogenase complex and the carnitine palmitoyltransferase system. FASEB J 8:54–61
Sugden MC, Fryer LG, Holness MJ (1996) Regulation of hepatic pyruvate dehydrogenase kinase by insulin and dietary manipulation in vivo. Studies with the euglycaemic-hyperinsulinaemic clamp. Biochim Biophys Acta 1316:114–120
Sugden MC, Orfali KA, Fryer LG, Holness MJ, Priestman DA (1997) Molecular mechanisms underlying the long-term impact of dietary fat to increase cardiac pyruvate dehydrogenase kinase: regulation by insulin, cyclic AMP and pyruvate. J Mol Cell Cardiol 29:1867–1875
Sugden MC, Fryer LG, Orfali KA, Priestman DA, Donald E, Holness MJ (1998) Studies of the long-term regulation of hepatic pyruvate dehydrogenase kinase. Biochem J 329(Pt 1):89–94
Sugden MC, Holness MJ, Donald E, Lall H (1999) Substrate interactions in the short- and long-term regulation of renal glucose oxidation. Metabolism 48:707–715
Sugden MC, Kraus A, Harris RA, Holness MJ (2000) Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem J 346(Pt 3):651–657
Sung MM, Hamza SM, Dyck JR (2015) Myocardial metabolism in diabetic cardiomyopathy: potential therapeutic targets. Antioxid Redox Signal 22:1606–1630
Swain L, Kesemeyer A, Meyer-Roxlau S, Vettel C, Zieseniss A, Guntsch A, Jatho A, Becker A, Nanadikar MS, Morgan B, Dennerlein S, Shah AM, El-Armouche A, Nikolaev VO, Katschinski DM (2016) Redox imaging using cardiac myocyte specific transgenic biosensor mice. Circ Res 119:1004–1016
Thomas AP, Denton RM (1986) Use of toluene-permeabilized mitochondria to study the regulation of adipose tissue pyruvate dehydrogenase in situ. Further evidence that insulin acts through stimulation of pyruvate dehydrogenase phosphate phosphatase. Biochem J 238:93–101
Thomas AP, Diggle TA, Denton RM (1986) Sensitivity of pyruvate dehydrogenase phosphate phosphatase to magnesium ions. Similar effects of spermine and insulin. Biochem J 238:83–91
Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O’rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, Aon MA (2012) GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 61:3094–3105
Tocchetti CG, Stanley BA, Sivakumaran V, Bedja D, O’Rourke B, Paolocci N, Cortassa S, Aon MA (2015) Impaired mitochondrial energy supply coupled to increased H2O2 emission under energy/redox stress leads to myocardial dysfunction during Type I diabetes. Clin Sci (Lond) 129:561–574
Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS (2015) Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta 1847:1424–1433
Van Bilsen M, Van Nieuwenhoven FA, Van Der Vusse GJ (2009) Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res 81:420–428
Van Eunen K, Simons SM, Gerding A, Bleeker A, Den Besten G, Touw CM, Houten SM, Groen BK, Krab K, Reijngoud DJ, Bakker BM (2013) Biochemical competition makes fatty-acid beta-oxidation vulnerable to substrate overload. PLoS Comput Biol 9:e1003186
Wallace DC (2010) The epigenome and the mitochondrion: bioenergetics and the environment [corrected]. Genes Dev 24:1571–1573
Wallace DC, Fan W (2010) Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10:12–31
Walther TC, Farese RV Jr (2009) The life of lipid droplets. Biochim Biophys Acta 1791:459–466
Walther TC, Farese RV Jr (2012) Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81:687–714
Wang H, Sreenevasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52:2159–2168
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080
Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, Van Den Enden M, KILO C, Tilton RG (1993) Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42:801–813
Wojtczak L, Schonfeld P (1993) Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1183:41–57
Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA (1998) Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem J 329(Pt 1):197–201
Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA (2000) Starvation increases the amount of pyruvate dehydrogenase kinase in several mammalian tissues. Arch Biochem Biophys 381:1–7
Xie C, Biary N, Tocchetti CG, Aon MA, Paolocci N, Kauffman J, Akar FG (2013) Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am J Physiol Heart Circ Physiol 304:H916–H926
Yan J, Lawson JE, Reed LJ (1996) Role of the regulatory subunit of bovine pyruvate dehydrogenase phosphatase. Proc Natl Acad Sci U S A 93:4953–4956
Yang D, Gong X, Yakhnin A, Roche TE (1998) Requirements for the adaptor protein role of dihydrolipoyl acetyltransferase in the up-regulated function of the pyruvate dehydrogenase kinase and pyruvate dehydrogenase phosphatase. J Biol Chem 273:14130–14137
Zhang S, Hulver MW, Mcmillan RP, Cline MA, Gilbert ER (2014) The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond) 11:10
Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B (2010) A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol 6:e1000657
Zima AV, Blatter LA (2006) Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71:310–321
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Cortassa, S., Sollott, S.J., Aon, M.A. (2017). Substrate Selection and Its Impact on Mitochondrial Respiration and Redox. In: Rostovtseva, T. (eds) Molecular Basis for Mitochondrial Signaling. Biological and Medical Physics, Biomedical Engineering. Springer, Cham. https://doi.org/10.1007/978-3-319-55539-3_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-55539-3_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55537-9
Online ISBN: 978-3-319-55539-3
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)