
Abstract
Calcium (Ca2+) is one of the main intracellular signals used by the cell to transmit and translate extracellular inputs into specific function activation. A complex web, formed by different interconnected cellular structures, and able to take up and release the cation, is present throughout the cell and controls Ca2+ dynamics under physiological and pathological conditions. Among different organelles, mitochondria represent a central hub of this net and make diverse physical and functional couplings with several other intracellular structures, fundamental not only for Ca2+ signaling but also for multiple pathways regulating the cell fate. In this chapter, we update mitochondria-organelles connections, with a special attention at Ca2+ crosstalk, from different points of view: the intracellular conditions that allow the mitochondrion to be one of the most important Ca2+ modulator, the different connections undertaken by mitochondria with several organelles and the functional consequences of these couplings, the molecules involved in the formation/modulation of these inter-organelles structures, and the most studied diseases in which alterations of these relationships have been reported to play a pathogenic role.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Calcium (Ca2+)
- Mitochondria
- Inter-organelle contacts
- Endoplasmic reticulum (ER)
- Tethering
- Mitochondria-associated membranes (MAMs)
1 The Mitochondrial Network as a Fundamental Structure in Cellular Ca2+ Homeostasis
The capacity of mitochondria to take up Ca2+ was first documented in the 1960s (Deluca and Engstrom 1961). Mitchell’s chemiosmotic theory, proposed in the same years (Mitchell and Moyle 1967), early provided the ultimate thermodynamic basis for the entry of Ca2+ (i.e., a positively charged ion) into the mitochondrial matrix, thanks to the generation, by the respiratory chain, of an electrochemical gradient (Δѱ) across the inner mitochondrial membrane (IMM), negative on the side of the matrix (−180 mV). However, the molecular mechanisms and the significance of this accumulation have started to be elucidated much more recently. Since the pivotal studies on isolated mitochondria, it was clear that the outer mitochondrial membrane (OMM) is largely permeable to ions and small solutes (at least in part due to the presence of different isoforms of the voltage-dependent anion channels, VDACs; see Chaps. 7, 8, 9, 10, and 11), and thus it is not a limiting step for mitochondrial Ca2+ accumulation. On the contrary, the inner mitochondrial membrane (IMM) is highly impermeable to ions and requires specialized transport mechanisms (Bragadin et al. 1979). The molecular identity of the underlying proteins has been only recently revealed (Baughman et al. 2011) (see also Chap. 2), but their activity was functionally characterized in the 1970s. The mitochondrial high-capacity mechanism to take up Ca2+ was called “mitochondrial Ca2+ uniporter” (MCU) and demonstrated to exhibit dependence on Δѱ, sensitivity to the inhibitor ruthenium red, low affinity for Ca2+ (Kd ~ 20 μM), and Ca2+ cooperativity (Carafoli 2003). In addition, the existence of antiporters that export Ca2+ from the matrix in exchange for Na+ (especially in excitable tissues, such as the brain and heart) or H+ (in non-excitable tissues, such as liver) further controls and limits mitochondrial Ca2+ accumulation (Nicholls and Crompton 1980).
The initial enthusiasm for mitochondria in the cellular Ca2+ scenario temporarily vanished in the 1980s. Indeed, the development of fluorescent indicators and the discovery that inositol 1,4,5-triphosphate (IP3, generated upon stimulation of plasma-membrane (PM) receptors coupled to phospholipase C (PLC)) induces the release of Ca2+ into the cytosol from a “non-mitochondrial intracellular store” (Streb et al. 1983) clearly demonstrated that physiological cytosolic [Ca2+] oscillations (ranging from 50 to 100 nM in basal conditions, to peaks of 1–3 μM) are not compatible with the activation of MCU, whose Kd is around 20 μM (Patron et al. 2013). Thus, the initial idea of mitochondria as key organelles in the modulation of intracellular Ca2+ homeostasis was overcome by the general consensus that, only under pathological conditions with a massive cytosolic Ca2+ overload, an appreciable mitochondrial Ca2+ uptake would occur.
The introduction of genetically encoded Ca2+ probes targeted to the mitochondrial matrix (firstly aequorin (Rizzuto et al. 1992) and then GFP-based fluorescent probes (Miyawaki et al. 1997)) showed, however, that, upon cell stimulation, mitochondria promptly take up Ca2+ in a fashion that largely exceeds what is expected on the basis of the Kd of MCU for Ca2+, thus actively taking part in the regulation of the intracellular Ca2+ dynamics. The contradiction between these data was only apparent and was solved by the demonstration that mitochondria are strategically positioned close to the regions of Ca2+ release from the intracellular stores (mainly the endoplasmic reticulum, ER (Rizzuto et al. 1998)), or Ca2+ entry from the PM. Indeed, in these areas, the opening of specific Ca2+ channels generates, close to their mouths, transient microdomains of high [Ca2+] that are experienced by nearby mitochondria, allowing the overcoming of the low affinity of MCU and resulting in a rapid mitochondrial Ca2+ accumulation that follows the cytosolic Ca2+ rise. Below, a brief summary on Ca2+ microdomain generation is provided.
1.1 Molecular Determinants of Ca2+ Microdomains
A microdomain can be defined as a localized region, within a cell, that differs in composition from the surrounding areas. Usually, the term refers to the presence, for specific molecules, of appreciable concentration gradients in a given environment, which can be relatively long lasting or, on the contrary, promptly drop out within a few ms. The importance of the existence of microdomains is immediately clear when referred to those of Ca2+. Indeed, the versatility of Ca2+ as a key intracellular second messenger, regulating different physiological processes, is achieved by combining the possibility to transmit Ca2+ signals as temporally distinct oscillations and by confining these events within spatially defined regions (not necessarily membrane enclosed), allowing a fine and localized regulation of specific activities (Berridge et al. 2000). The generation of a Ca2+ microdomain typically begins with the opening of few Ca2+ channels located in the membrane of intracellular Ca2+ stores (endoplasmic/sarcoplasmic reticulum or Golgi apparatus) or in the PM. In the simpler model, immediately after their opening, Ca2+ flows from the mouth of the channels and spreads into the cytosol (where its concentration is lower), following Fick’s diffusion laws (for a recent review, see Filadi and Pozzan (2015)). Accordingly, its flux in a given point is inversely proportional to the distance from the channel. However, the presence of different Ca2+ buffers (typically proteins, such as parvalbumin/calbindin or the Ca2+-modulated calmodulin/calcineurin, but also other molecules, such as ATP and negatively charged phospholipids), the high viscosity of the cytosol, the existence of organelles endowed with pumps/exchangers/channels that can promptly remove or further release Ca2+, and the possibility to tune the frequency and the time of channel opening are all players that actively shape the microdomain (Filadi and Pozzan 2015).
Elegant mathematical models (Naraghi and Neher 1997) and, more recently, Ca2+-imaging experiments with sufficient temporal and spatial resolutions (Tadross et al. 2013) allowed to define the shape of a Ca2+ microdomain. Upon opening of a channel, within a few hundreds of nm, a Ca2+ microdomain is formed and reaches the steady state in less than 1 ms. When the channel closes, the microdomain vanishes immediately. A Ca2+ gradient was calculated to extend up to 50–70 nm from the mouth of a channel (Naraghi and Neher 1997), but clearly the amplitude of a Ca2+ microdomain in the cellular context depends on a multitude of parameters. Among them, particularly important are the flux of Ca2+ from a given channel determined by its current (the higher the flux, the higher the [Ca2+] reached in the microdomain), the concentration/affinity/diffusion constant of the buffers that surround the channel (the higher these parameters are, the higher the capacity of a given buffer to damp the microdomain), the presence of isolated or clustered channels (the more channels opening, the higher the flux of Ca2+), and the abundance of the Ca2+ reservoir (virtually infinite in the case of the extracellular milieu, depletable in the case of intracellular Ca2+ stores).
The generation of Ca2+ microdomains on the surface of mitochondria has variable and important consequences for the cell fate, from activation of mitochondrial metabolism to control of cell death, from regulating autophagy to sustain tumor growth (see below).
1.2 Generation of Ca2+ Microdomains on the Mitochondria Surface: The Importance of Being Coupled with Other Cell Compartments
As discussed above (and see also Chap. 2), the process of mitochondrial Ca2+ uptake largely takes advantage from the exposure of mitochondria to high [Ca2+] microdomains on their surface, thanks to the location of these organelles (or, more precisely, of part of them) near the mouths of Ca2+ channels. On this aspect, the close proximity of mitochondria to the ER and, to a lesser extent, to PM, has been extensively investigated (Table 1.1 and see also next section).
As to the former, the first evidence that, upon an IP3-dependent Ca2+ release from the ER, mitochondria experienced on their surface an averaged Ca2+ concentration higher than that of the bulk cytosol was obtained thanks to the targeting of the photoprotein aequorin to the mitochondrial inter-membrane space (MIMS) (Rizzuto et al. 1998). Regions of close proximity between mitochondria and ER were observed in living cells, supporting the idea that mitochondrial Ca2+ uptake may be favored by their juxtaposition to the sites of Ca2+ release from the ER (Rizzuto et al. 1998). A multitude of evidence in support of this notion were provided, for instance, an increased heterogeneity in the mitochondrial Ca2+ peaks upon a dynamin-related protein 1 (Drp1)-overexpression-dependent mitochondria fragmentation (Szabadkai et al. 2004). These results, obtained in single-cell Ca2+ imaging experiments employing mitochondrial GFP-based Ca2+ probes, further suggested a physiological origin of the mitochondrial Ca2+ rises from localized regions that then spread along the mitochondrial network. Finally, using FRET-based Ca2+ probes spanning on the cytosolic side of the OMM, it was directly demonstrated that, during an IP3-dependent ER Ca2+ release, microdomains of high [Ca2+] (10–30 μM, compared to peaks of up to 3 μM in the bulk cytosol) are promptly generated on discrete sites on the OMM (Csordas et al. 2010; Giacomello et al. 2010) (Fig. 1.1).

Mitochondria-ER tethering is fundamental for their Ca2+ crosstalk. Left panel: EM image of a MEF cell showing several close appositions between ER (orange) and OMM (purple) (The EM image was acquired by G. Turacchio, Institute of Protein Biochemistry (CNR), Naples). Scale bar: 100 nm. The juxtaposition between the two opposing membranes allows the generations of high [Ca2+] on OMM upon Ca2+ release from ER Ca2+ channels. Upper right panel: an SH-SY5Y cell transiently expressing the OMM-targeted cameleon Ca2+ probe N33-D1cpv. Yellow-to-red spikes indicate the generation of high [Ca2+] microdomains in discrete regions of OMM upon bradykinin cell stimulation (100 nM) and IP3-mediated ER Ca2+ release. Scale bar: 5 μm (Image modified from Filadi et al. 2012). Lower right panel: simulations of [Ca2+] changes over time upon IP3-mediated ER Ca2+ release in the bulk cytoplasm (black, dotted trace), at regions of ER-mitochondria interface (black, continuous trace) and in mitochondrial matrix (red trace). Note the different scales and that mitochondrial Ca2+ uptake is slightly delayed compared to the almost immediate generation of Ca2+ hot spots on mitochondrial surface
Regarding the distance between ER and mitochondria for an efficient Ca2+ transfer, it has been demonstrated, by the use of artificial linkers, that a decrease in the distance, or an increase in the surface of apposition, between the two organelles, correlates with an increase in the efficiency of Ca2+ transfer (Csordas et al. 2006, 2010). However, below 7 nm in the distance between the two opposing membranes, the process resulted to be substantially impaired, likely because the IP3Rs span for ~10 nm from the ER membrane and thus cannot be accommodated when the width of the cleft is lower. However, the physiological thickness of ER-mitochondria appositions observed by EM (10–15 up to 25–30 nm in the case of smooth ER and 25–30 up to 50–80 nm in the case of rough ER) seems to be largely compatible with the process. Moreover, it must be stressed that, as discussed above, a Ca2+ microdomain is predicted to extend from the mouth of a channel for at least 50 nm (Naraghi and Neher 1997). In addition, given that IP3Rs form clusters (Foskett et al. 2007) and that they can open together, potentially engaging also the opening of Ryanodine receptors (RyRs) in the process of the Ca2+-induced Ca2+ release (CICR), further increasing the amount of Ca2+ locally released from the ER, it appears reasonable that, under certain conditions, microdomains of high [Ca2+] can reach an extension of up to 100–150 nm. Thus, even mitochondria located at distances of ~100 nm may be exposed to [Ca2+] sufficiently high to induce an efficient uptake of Ca2+. Importantly, regions in which ER membranes (especially those of the rough ER) are juxtaposed and follow the profile of the OMM at distances of 50–100 nm have been reported (Filadi et al. 2015; Giacomello and Pellegrini 2016); whether they are physiologically relevant is not clear. Recently, a mathematical model proposed a distance between IP3Rs and MCU of ~30–85 nm for an optimal ER-mitochondria Ca2+ transfer (Qi et al. 2015). Considering that the distance between the OMM and the IMM (where MCU is located) is at least of 10–20 nm (Reichert and Neupert 2002), the optimal gap between the ER and the OMM is predicted to be in the range of 10–60 nm. Surely, the fact that the thickness of the cleft is dynamically regulated may suggest that different types of contact could have different functions or represent dormant states of contact, ready to be recruited and reactivated upon a change in the metabolic state of the cells (Sood et al. 2014; Giacomello and Pellegrini 2016).
In addition to IP3Rs, the ER of different cell types and the sarcoplasmic reticulum (SR) of the striated muscles are endowed with RyRs. Though in principle the situation is similar to that of IP3Rs (i.e., a Ca2+ microdomain near the mouth of RyRs can be formed upon their opening and be experienced by juxtaposed mitochondria), the peculiar organization and ordered distribution of these Ca2+ channels in striated muscles make the case of SR-mitochondria coupling unique. Indeed, in this tissue, the situation is peculiar; while regions of close contact (mediated by proteinaceous tethers of 10–15 nm (Franzini-Armstrong 2007; Boncompagni et al. 2009)) between SR and mitochondria have been clearly documented, RyRs are specifically clustered in the dyadic cleft, i.e., the portion of SR (junctional SR) that faces the T-tubules (for a recent review, see Filadi and Pozzan (2015)). Importantly, the space between T-tubules and SR is too narrow (~10 nm) to allow the accommodation of mitochondria, which have been reported to be at a distance of ~130 nm in skeletal muscle (Boncompagni et al. 2009) and ~35 nm in cardiomyocytes (Sharma et al. 2000), where the SR cisternae are flatter. However, as discussed above, the presence of RyR clusters and the fact that the two opposing membranes in the dyadic cleft represent a barrier (that only allows the lateral diffusion of Ca2+) are factors that shape the Ca2+ microdomain and laterally spread it, so that mitochondria surrounding the Ca2+ releasing unit (CRU) are exposed to [Ca2+] sufficiently high to induce an appreciable uptake, as measured in different experimental models (Pacher et al. 2000, 2002; Szalai et al. 2000; Robert et al. 2001; Rudolf et al. 2004; Drago et al. 2012). Though in adult cardiomyocytes it is still a matter of debate whether mitochondria could efficiently take up Ca2+ during the fast physiological Ca2+ transients in a beat-to-beat manner, in neonatal cardiomyocytes, such oscillations in mitochondrial Ca2+ have been measured (Robert et al. 2001; Pacher et al. 2002; Drago et al. 2012) and demonstrated to actively impact the amplitude of cytosolic Ca2+ rises (Drago et al. 2012).
Finally, as far as Ca2+ crosstalk is concerned, it is important to stress that the existence of regions of close apposition between mitochondria and ER/SR is physiologically relevant not only for the process of mitochondrial Ca2+ uptake but also for ER Ca2+ handling. First of all, the physical presence of mitochondria near ER membranes limits the free Ca2+ diffusion upon release from the ER Ca2+ channels, potentially sustaining the local [Ca2+] and favoring its reuptake by SERCA pumps. Moreover, the local high [Ca2+] may have opposite effects: on the one hand, affecting the process of CICR (recruiting and opening more Ca2+ releasing channels, RyRs and IP3Rs), and, on the other, negatively regulating the opening of IP3Rs when the local [Ca2+] reaches a certain threshold (Landolfi et al. 1998; Foskett et al. 2007). On the contrary, upon MCU activation and rapid mitochondrial Ca2+ accumulation, mitochondria can act as Ca2+ buffers that dampen not only bulk cytosolic Ca2+ peaks but also, locally, Ca2+ microdomains (Qi et al. 2015). However, given that mitochondria do not store Ca2+ (with the only exception of Ca2+ phosphates precipitation, favored by the alkaline pH; (Nicholls and Chalmers 2004)) and, after Ca2+ uptake, they release it back to the cytosol, the latter phenomenon could facilitate Ca2+ reuptake into the ER by the SERCA. In addition, mitochondria have been reported to locally sustain SERCA activity by fueling it with ATP (De Marchi et al. 2011), further indicating the existence of a functional, bidirectional crosstalk.
Much less investigated has been the role of mitochondria-PM contacts in the process of mitochondrial Ca2+ uptake. In many excitable cell types, a fast mitochondrial Ca2+ rise has been measured upon voltage-operated channel (VOC)’ s opening and Ca2+ entry from the extracellular environment, subsequent to PM depolarization (see Giacomello et al. 2010 and, for a review, Rizzuto and Pozzan 2006). Thus, regions of high [Ca2+] are likely to be generated on the surface of sub-PM located mitochondria when Ca2+ enters through VOC’s . On the contrary, more debated is the generation of such Ca2+ microdomains on the OMM upon capacitative Ca2+ entry (CCE)). Indeed, evidence that mitochondria are exposed to CCE-generated Ca2+ microdomains has been provided (Quintana et al. 2006; Watson and Parekh 2012), and mitochondrial membrane potential, as well as functional mitochondrial Na+/Ca2+ exchanger, has been shown to be necessary to sustain CCE, likely because of mitochondria capacity to buffer sub-PM local Ca2+ increases and maintain CCE activation (Naghdi et al. 2010). However, mitochondrial depolarization has also been described to negatively impact on CCE by a different Mfn2-dependent mechanism, impairing migration of the ER Ca2+ sensor stromal interaction molecule 1 (STIM-1) protein to form PM-located ER punctae, critical for CCE induction (Singaravelu et al. 2011). Notably, during CEE, the close apposition between ER and PM is narrow enough to exclude mitochondria from the mouth of PM-located Orai1 channels (calcium release-activated calcium channel protein 1), thus arguing against the possibility that mitochondria could be exposed to high [Ca2+] during CCE. Moreover, data recently obtained in our lab (Giacomello et al. 2010; Filadi et al. unpublished results) did not support an appreciable contribution of CCE to mitochondrial Ca2+ uptake. Probably, these contrasting results depend on the specific cell type employed in different studies and, consequently, on the amplitude of the Ca2+ influx and on the presence, or not, of mitochondria in the regions surrounding the ER-PM platform generated during CCE. Further investigations will be required to address this issue. Interestingly, however, in HeLa cells, the presence of sub-PM mitochondria has been suggested to indirectly modulate the activity of both PM Ca2+ ATPases (PMCA) and store-operated Ca2+ channels (Frieden et al. 2005). The phenomenon was proposed to be due, again, to a possible Ca2+ buffering effect exerted by sub-PM mitochondria on the entry of the ion through the PM located channels (Frieden et al. 2005). Thus, it appears clear how the interrelationship between mitochondria and PM, as for the ER, may be bidirectional.
2 Functional Consequences of Mitochondria-ER Ca2+ Cross-Talk
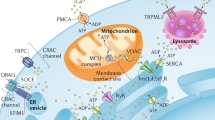
Ca2+ rises on the cytosolic surface of both the ER and mitochondria can have important regulatory roles. For example, [Ca2+] in the mitochondrial inter-membrane space modulates the activity of metabolite carriers or dehydrogenases located on the outer surface of the IMM (see below); furthermore, as mentioned above, local mitochondrial Ca2+ sequestration has profound effects on the allosteric modulation of ER Ca2+-releasing channels. Regarding the [Ca2+] within mitochondria, two possible and opposite effects can be achieved: activation of mitochondrial metabolism by stimulating the Krebs cycle, NADH formation, and the respiratory chain activity, with the result of an increase in ATP production, or sensitization toward cell death pathways, due to mitochondrial Ca2+ overload, opening of the mitochondrial permeability transition pore (mPTP), and release of cytochrome C. Moreover, an important role for ER-mitochondria Ca2+ transfer in modulating autophagy/mitophagy and tumor progression has been recently shown. While a more detailed discussion on the role of ER-mitochondria Ca2+ transfer in regulating cell death is provided elsewhere (see Chap. 6 and Orrenius et al. 2015 for a recent review), below a brief summary on activation of mitochondrial metabolism, autophagy, and cancer growth modulation by ER-mitochondria Ca2+ crosstalk is provided (Fig. 1.2).

Mitochondria interact with different organelles/compartments within the cell. The cartoon represents multiple mitochondria interactions with other organelles, as detailed in the text. Red-dotted squares underline specific functions sustained by the indicated inter-organelle interaction, with the representation of the main proteins involved; yellow-dotted squares are those only identified in yeast; in the black-dotted square are represented proteins that have been implicated in the structure of the inter-organelle interface. See text for all the abbreviations used and the description of the specific functions. In italic, yeast proteins
2.1 Mitochondrial Metabolism
Bioenergetics homeostasis and mitochondrial functionality are two essential features that provide the correct energy to the cell accordingly to its requirements; in particular, in excitable cells, such as neurons, cardiac, and skeletal muscle cells, ATP produced through the glycolysis process is not enough to guarantee the multiplicity of activities of these cells, and thus mitochondria become their energy suppliers.
Ca2+ is a specific signal for activating mitochondria metabolism: under resting conditions, the mitochondrial matrix Ca2+ level is in equilibrium with that of the cytosol, i.e., around 100 nM. Upon cytosolic Ca2+ rises, due to Ca2+ release from the intracellular stores, mainly the ER, or to Ca2+ entry from the extracellular space, and the generation of high Ca2+ microdomains (see above), mitochondria can take up Ca2+ thus increasing its concentration in the matrix, reaching, in some types of cells, values above 100 μM (Rizzuto et al. 1992; Pozzan and Rizzuto 2000). It has been known for a long time that a rise in Ca2+ levels in the matrix, due to an increase in the workload or to a specific cell stimulation, increases NADH levels, mitochondrial metabolism, and ATP synthesis (Jouaville et al. 1999; Pitter et al. 2002). On the other hand, it has been demonstrated that the buffering of Ca2+ rises, obtained upon cell stimulations, in the cytosol or in the mitochondrial matrix, limits the increase in mitochondrial metabolism (Wiederkehr et al. 2011). More recently, it has been demonstrated that also at resting conditions, the ER-mitochondria Ca2+ transfer is important for mitochondrial energy metabolism: the inhibition, or the absence, of the transfer causes a decrease in ATP levels, an increase in AMPK phosphorylation, and, eventually, a strong autophagy activation (Cardenas et al. 2010) (see below).
Mitochondria produce ATP for the cell through two main processes, the tricarboxylic acid (TCA) or Krebs cycle and the respiration by the electron transport chain (ETC). These two processes need pyruvate, the product of glycolysis, to move in the mitochondrial matrix where it is converted in acetyl-CoA, which enters in the TCA cycle. From each molecule of acetyl-CoA, mitochondria produce three NADH and one FADH2. These are two high-energy molecules that are essential for the ETC activity. Since 1970, it is known that four mitochondrial dehydrogenases are regulated by Ca2+. One of these, the FAD-glycerol phosphate dehydrogenase (GPDH), is located in the IMM and senses the cation concentration in the intermembrane space, while the other three enzymes catalyze reactions of the TCA cycle (or immediately upstream) and sense mitochondrial matrix [Ca2+]: pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and oxoglutarate dehydrogenase (OGDH) (Denton 2009). GPDH transfers, through a redox reaction, reducing equivalents from NADH, produced by glycolysis, to the ETC as FADH2; it presents a Ca2+-binding motif that lies in the intermembrane space and has a Kd for Ca2+ of 0.1 μM; moreover, its activation increases cellular ATP levels (Garrib and McMurray 1986). PDH plays a crucial role in mitochondrial metabolism since it converts pyruvate into acetyl-CoA, allowing the TCA cycle activation. Its activity is regulated by a kinase/phosphatase cycle: when phosphorylated it is inactive, while a dephosphorylation event activates it. Ca2+ binds (Kd ~ 1 μM) both the dehydrogenase itself and its phosphatase, increasing the active form of the enzyme (Turkan et al. 2004). On the other hand, ICDH and OGDH catalyze two reactions of the TCA cycle: they are both regulated by Ca2+ through the direct binding of the cation; the latter event induces an increase in the affinity of the enzymes for their substrates (Kd of 20–30 μM and 1 μM, respectively; (Denton et al. 1978)).
More recently, in isolated mitochondria, it has been reported that Ca2+ can regulate directly the ETC and the F1F0 ATP synthase activity, indicating that mitochondrial matrix Ca2+ concentration is a key factor not only for TCA cycle enzymes but it can directly stimulate, independently from dehydrogenase functionalities, ATP production, and the activity of the respiratory chain complexes (Territo et al. 2000; Glancy et al. 2013).
As mentioned above, Ca2+ can regulate mitochondrial functions without reaching their matrix, modulating the shuttle of nucleotides, metabolites, and cofactors inside the organelles. Specific mitochondrial carriers (MCs), localized in the IMM, exchange nucleotides, substrates, and metabolites between cytosol and mitochondria. Among them, there are two Ca2+-binding MCs (CaMCs): the L-CaMCs (long Ca2+-dependent MCs) and the S-CaMCs (short Ca2+-dependent MCs). Both molecules sense Ca2+ in the intermembrane space by the presence of EF-hand Ca2+-binding domains localized in their N-terminal fragments facing the intermembrane space (del Arco and Satrustegui 2004; Satrustegui et al. 2007).
L-CaMCs are the aspartate/glutamate carriers (AGC) , and they belong to the malate-aspartate shuttle (MAS). They catalyze the exchange of a glutamate and an H+ (from the cytosol) for an aspartate (from mitochondria) and, being part of the MAS, the entry into mitochondria of a NADH molecule that contributes to mitochondrial metabolism (Palmieri et al. 2001). The role of AGC in substrates transport has been known for several years but only recently, thank to structural studies, it has been shown its Ca2+ dependence by the presence of an EF-hand domain in its N-terminal fragment. Moreover, AGC is activated by low [Ca2+] (its Kd is around 300 nM), well below the concentration needed for MCU activation. Thus, also low cytosolic Ca2+ rises can induce the entrance into mitochondria of NADH and metabolites, stimulating ETC activity and ATP production (Pardo et al. 2006; Thangaratnarajah et al. 2014).
S-CaMCs, instead, are ATP-Mg/Pi carriers that catalyze the exchange of ATP and ADP for one phosphate across the IMM, modulating the levels of adenine nucleotides (AdN: AMP+ADP+ATP) inside mitochondria; due to this function, these carriers can modulate several cell functionalities, such as mitochondrial metabolism and oxidative phosphorylation. A defect in these carriers, or their absence, in different tissues, can induce an impairment in energy production required for several functionalities (Anunciado-Koza et al. 2011; Amigo et al. 2013). The activity of ATP-Mg/Pi carriers is regulated by Ca2 +, and they also present EF-hand Ca2+ binding domains, homologous to calmodulin, in their N-terminal parts facing the intermembrane space; their Kd is around 1.5–3 μM, requiring a higher increase in cytosolic [Ca2+], than L-CaMCs, for their activation and the regulation of AdN levels within mitochondria (Haynes et al. 1986; Nosek et al. 1990).
2.2 Autophagy/Mitophagy
Ca2+ plays a complex role in autophagy regulation and the first link between autophagy, and intracellularly stored Ca2+ was described in rat hepatocytes in 1993 (Gordon et al. 1993). More recently, both inhibitory and activatory effects of Ca2+ on autophagy induction have been reported (see La Rovere et al. (2016) for a recent review).
As far as Ca2+ transfer from the ER to mitochondria is concerned, it has been shown that, under basal conditions, the constitutive, low level, IP3R-mediated Ca2+ release and subsequent Ca2+ uptake by mitochondria, throughout the MCU complex, are essential for the maintenance of optimal cellular bioenergetics (see above). As a consequence of this energetic balance, autophagy is maintained at low levels in healthy cells. On the other hand, disturbances in ER-mitochondria Ca2+ transfer trigger autophagy by increasing the AMP/ATP ratio (due to a decrease in mitochondria bioenergetics) and activating AMPK, a highly sensitive indicator of cellular energy status whose activity increases under conditions of metabolic stress (Cardenas et al. 2010). The importance of the ER-mitochondria Ca2+ crosstalk in autophagy induction was also confirmed by molecular (upon MCU overexpression) or pharmacological (upon treatments with drugs that increase the uptake of Ca2+ by mitochondria) approaches finalized to correct dysfunctional mitochondrial Ca2+ uptake in human fibroblasts from mitochondrial disorder patients, resulting, in these cells, in a rescue of autophagy levels similar to those observed in control cells (Granatiero et al. 2016). Consistently, stable knockdown of MCU and MCUR1, key components of the mitochondrial Ca2+ uptake machinery (Murgia and Rizzuto 2015) (and see also Chap. 2), reduced cellular oxygen consumption rate, activated AMPK, and induced autophagy (Mallilankaraman et al. 2012).
Finally, other alterations in mitochondrial Ca2+ signaling can lead to mitophagy. PINK-1, a protein involved in mitophagy initiation, regulates Ca2+ efflux from mitochondria through the Na+/Ca2+ exchanger, and its loss may lead to mitochondrial Ca2+ accumulation (Gandhi et al. 2009), leading to mPTP opening and release of pro-apoptotic factors. In addition, elevation of local Ca2+ concentrations in the vicinity of mitochondria, a phenomenon very much depending on ER-mitochondria tethering (see above), with impaired membrane potential, promotes the activation of the mitochondrial shaping molecule Drp1, triggering mitochondrial fission and subsequent mitophagy (Sandebring et al. 2009).
More recently, it has been demonstrated that the overexpression of Parkin, the cytosolic E3 ubiquitin ligase involved in mitophagy, favors, in normal conditions, ER-mitochondria tethering and their Ca2+ crosstalk, sustaining mitochondria morphology and ATP production and consequently limiting ubiquitination of mitochondrial targets and mitophagy (Cali et al. 2013a). Upon mitochondrial damage (such as organelle depolarization), the protective effect of Parkin is however insufficient, and, after its recruitment to mitochondria, the mitophagic process is activated.
2.3 Cancer
Among multiple mechanisms, cancer growth relies mainly on dysregulation on cell proliferation and cell death, and Ca2+ and mitochondria are crucial actors in modulating both events. In particular, while the correct mitochondrial Ca2+uptake sustains the increased energy demand of proliferating cells, mitochondrial Ca2+ overload induces organelles morphology alterations, permeabilization, and swelling, with the subsequent release of pro-apoptotic factors. Both Ca2+ -dependent pathways are very much linked to mitochondria-ER coupling.
Indeed, enhanced resistance to apoptosis of cancer cells involves also dysregulation of the ER-mitochondria Ca2+ axis and several tumor suppressor and oncogenic proteins, mutated or deleted in various types of human cancers, act at the interface between the two organelles and modify their Ca2+ crosstalk. The oncogene AKT, for example, has been reported to phosphorylate two key targets, hexokinase 2 (HK2), promoting its binding with the mitochondrial channel VDAC1 (Majewski et al. 2004), and the ER Ca2+-releasing channel IP3R3 (Marchi et al. 2012), which in turn negatively affects the Ca2+-dependent apoptotic response. On the contrary, the tumor suppressor protein phosphatase and tensin homolog (PTEN), commonly lost or mutated in human cancers, have been shown to directly interact with IP3R, counteracting, by its enzymatic activity, the reduced IP3R-dependent Ca2+ release mediated by AKT phosphorylation (Bononi et al. 2013). The tumor suppressor PML has been shown to act on this ER-mitochondria Ca2+ pathway as well: it promotes the formation of a multiprotein complex containing IP3R3, AKT, and the protein phosphatase PP2a, which regulates ER-mitochondria Ca2+ transfer (Giorgi et al. 2010). Finally, other two examples that further indicate the ER-mitochondria Ca2+ crosstalk as a critical hub in cancer growth are the tumor suppressor proteins BRCA1 and p53: the first was found to be recruited to the ER during apoptosis in an IP3R-dependent manner, sensitizing the IP3R to its ligand, thus favoring the Ca2+-dependent apoptotic response (Hedgepeth et al. 2015); the second, in an extra-nuclear fraction, has been shown to interact with the ER Ca2+-ATPase SERCA, modulating ER Ca2+ content and mitochondrial Ca2+ uptake, organelle swelling, and apoptosis induction (Giorgi et al. 2015). Of note, recently, the cancer-testis antigen FATE1 has been reported to uncouple the two organelles and to protect from Ca2+- and drug-induced cell death (Doghman-Bouguerra et al. 2016), highlighting the importance of the modulation of ER-mitochondria interface in cancer cells.
3 Molecular Components Involved in Mitochondria-Organelles Contacts
The first electron microscopy (EM) studies of cellular ultrastructure reporting that mitochondria are in physical contact with other organelles, in particular with the ER, dated back to the 1950s (Robertson 1960). In these identified regions of juxtaposition, the two opposing organelles do not fuse but maintain their identity (Fig. 1.1 and Table 1.1).
Contacts with the ER are conserved between different species (Rowland and Voeltz 2012) and have been well documented in yeast, mammals, and, recently, also in plants (Mueller and Reski 2015). Different types of contact have been reported, depending on the fact that mitochondria can be engaged in juxtaposition with both smooth ER and rough ER and that distances between ER membranes and OMM can be extremely variable, ranging from ~10 nm up to 80–100 nm (for a recent review, see (Giacomello and Pellegrini 2016)). However, while in the case of the classical close contacts (below 25–30 nm), OMM and ER membranes appear to be clearly tethered by electron-dense filamentous structures, proteinaceous in their nature (Csordas et al. 2006); such structures, to the best of our knowledge, have never been observed for the more distant areas of apposition. A complete list of the proteins that has been involved in the modulation of ER-mitochondria coupling is far away from the scope of the present chapter. However, below, a brief summary of both yeast and mammalian tethers is provided (Fig. 1.2).
In yeast, the ER-mitochondria encounter structure (ERMES), formed by the cytosolic protein Mdm12, the ER membrane protein Mmm1, and the OMM proteins Mdm34 and Mdm10, was identified by a genetic screening (Kornmann et al. 2009). ERMES has been implicated in ER-mitochondria lipid transport (AhYoung et al. 2015), though different groups failed to find defects in lipid metabolism in cells lacking the complex (reviewed in Murley and Nunnari 2016). Importantly, ERMES homologs have not been identified in mammals.
Recently, a proteomic analysis identified the ER transmembrane protein Ltc1/Lam6 as a potential additional tether in yeast, thanks to its interaction with the OMM proteins TOM70/TOM71 (Murley et al. 2015).
ER-mitochondria contacts , in both yeast and mammals, mark sites of mitochondrial division (Friedman et al. 2011), and, in yeast, the additional ERMES-associated subunit Gem1 (a GTPase whose mammalian homolog is Miro-1) regulates ER-mitochondria connections (Kornmann et al. 2011) and ER-dependent mitochondrial division (Murley et al. 2013).
In metazoan cells, while the functional significance of the ER-mitochondria juxtaposition has been established in a number of different studies, the nature of the proteins involved in the physical tethering between the two organelles remains much less understood. Different proteins have been demonstrated to modulate this parameter, but the formal demonstration that the lack of a given protein abolish close contacts has never been provided. Notably, the possibility that different and independent tethering complexes may exist, and compensate one for the lack of the others, increases the complexity of the above analysis.
Among the proteins that have been associated with ER-mitochondria tethering, the OMM resident protein Mitofusin-2 (Mfn2) has received a lot of attention. Mfn2 was initially proposed to be a tether, given the presence of a fraction of the protein in ER membranes (particularly in the mitochondria associated membranes, MAMs) and its ability to form homotypic interactions with the OMM resident counterpart (de Brito and Scorrano 2008). The demonstration that the lack of this protein deeply reduces ER-mitochondria co-localization, visualized by confocal microscopy techniques, as well as a number of indirect evidence involving Mfn2 in organelles’ coupling (Schneeberger et al. 2013), initially created a consensus on the fact that Mfn2 could be a tether. However, we (Filadi et al. 2015, 2016; Leal et al. 2016) and others (Cosson et al. 2012; Wang et al. 2015) have shown, by a number of different approaches, that Mfn2 depletion is actually associated to an increased ER-mitochondria physical and functional coupling, challenging the initial idea of Mfn2 as a tether. Interestingly, a Gp78 (an E3-ubiquitin ligase)-dependent Mfn2 degradation has been correlated with an increased association of rough ER to mitochondria, while Mitofusin-1 has been shown to negatively affect the tethering with the smooth ER, thus suggesting that the coupling with mitochondria of rough and smooth ER may be differently regulated (Wang et al. 2015).
In addition to Mfn2 , others proteins have been reported to be involved in the regulation of ER-mitochondria juxtaposition. PACS-2, a cytosolic multi-sorting protein, have been shown to control their apposition in a BAP31 (an ER cargo receptor)-dependent manner (Simmen et al. 2005), though its role as a molecular scaffold or as a simple regulator is not fully clarified.
Interestingly, a physical interaction between the ER resident IP3Rs (especially the MAM-enriched IP3R3), the cytosolic fraction of the mitochondrial chaperone Grp75, and the OMM located VDAC1 has been reported and demonstrated to be functionally involved in the efficacy of mitochondrial Ca2+ uptake (Szabadkai et al. 2006). However, the interpretation that sees the IP3Rs-Grp75-VDAC1 complex as a potential tether between the two opposing organelles is, in our opinion, unlikely, because in DT40 cells knockout (KO) for the three IP3Rs isoforms, it has been demonstrated, by EM analysis, that ER-mitochondria physical association is not modified (Csordas et al. 2006), thus arguing against a role of the IP3Rs as tethers.
The familial Alzheimer’s disease (FAD)- related protein Presenilin-2 (PS2) has been shown by us to favor the physical and functional ER-mitochondria coupling, with FAD-linked PS2 mutants more effective than the wt counterpart in the modulation of these parameters (Zampese et al. 2011; Kipanyula et al. 2012). Recently, we have demonstrated that PS2 is able to increase the number of ER-mitochondria close contacts in a Mfn2-dependent manner, by sequestering the latter protein and thus removing its negative effect on organelles tethering (Filadi et al. 2015, 2016) (see also below).
An interaction between the ER resident protein VAPB and the OMM protein PTPIP51 has also been demonstrated to positively correlate with the physical and functional ER-mitochondrial coupling (De Vos et al. 2012).
Finally, the ER-stress related protein kinase RNA-like ER kinase (PERK; see also below) has also been shown to modulate ER-mitochondria contact sites, independently from its enzymatic activity and canonical role during ER stress (Verfaillie et al. 2012; van Vliet and Agostinis 2016). As to the contacts with the PM, their functions and molecular composition have been much less investigated, and contrasting results have been obtained. For instance, in HeLa cells, it has been reported that ~10% of the PM surface co-localized with mitochondria and that sub-PM mitochondria are important for the modulation of PM Ca2+ ATPases (Frieden et al. 2005). However, given the optical resolution of confocal microscopy (at best, ~0.5 μm along the z-axis), this value may be overestimated and include also mitochondria that are in proximity but not really associated to PM. Indeed, in another study performed in RBL-2H3 and H9c2 cells, just a few mitochondria-PM contact points were observed, and a large fraction of peri-PM mitochondria was found to lack a direct contact with the PM (Csordas et al. 2010). Often, an interleaving ER-stack was observed to be located between PM and mitochondria, hindering a direct association between them (Csordas et al. 2010). Moreover, it has been shown that, upon Drp1/Fis1 overexpression-induced mitochondrial fragmentation, the majority of mitochondria loose the co-localization with the PM, suggesting that just few tethering points between the two structures may exist (Frieden et al. 2005).
The molecular nature of the junctions between mitochondria and the PM is mysterious. Recently, by employing a proteomic approach in murine liver, it has been reported that the connexin protein Cx32, enriched at gap junctions, but retrieved also in the IMM, physically interacts with the OMM resident fraction of syderoflexin-1 (SFXN-1), thus suggesting a putative role for the Cx32-SFXN1 axis as a PM-mitochondria tether (Fowler et al. 2013). In yeast, the interaction between the PM protein Num1 (Klecker et al. 2013), the OMM-associated adaptor protein Mdm36 (Lackner et al. 2013), and still unknown additional partners has been demonstrated to be important for the association of mitochondria with the cell cortex and for the retention of part of them in the mother cell after cell division (reviewed in Klecker et al. 2014). Thus, PM-mitochondria contacts may be essential for the partitioning of mitochondria between the mother cell and the bud.
Recently, different types of contact have been also described between mitochondria and other cellular organelles, such as peroxisomes, through the binding of the peroxisomal protein Pex11 and the ERMES subunit Mdm34 in yeast (Mattiazzi Usaj et al. 2015), and melanosomes, through undefined Mfn2-dependent molecular tether structures (Daniele et al. 2014) (see also below). Moreover, tight contacts (vCLAMPs, ~10 nm the distance between the opposing membranes and ~100 nm the length of the juxtaposition) have been reported between vacuoles and mitochondria in yeast (Elbaz-Alon et al. 2014). These contacts depend on the vacuolar proteins Vps39 and the Rab GTPase Ypt7 and have a role in lipid metabolism/exchange between these organelles (see below).
4 Mitochondria Inter-organelle Contacts : Not Only Matter of Ca2+ Signaling
4.1 Lipid Synthesis and Trafficking
The maintenance of the lipid composition of cellular membranes involves a tight communication between organelles (Lebiedzinska et al. 2009). The more extensively studied, and probably the more important interorganellar interaction for this function, is between mitochondria and the ER. Indeed, lipid transfer is one of the most prominent feature of ER-mitochondria contact domains, even though the underlying molecular mechanisms are mostly unknown (Murley and Nunnari 2016).
The ER is the master organelle for lipid synthesis. Nevertheless, the specific domains associated to mitochondria (MAMs) have been shown to be enriched, compared to the bulk ER membranes, in enzymes involved in lipid metabolism, such as membrane-anchoring proteins, cholesterol metabolism, and triglycerides/phospholipids synthesis enzymes, including the two phosphatidylserine (PS) synthases (Stone and Vance 2000). Interestingly, certain phospholipids synthetic pathways require enzymes that are located at both the ER and the mitochondria: for example, biosynthesis of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) follows a complete circuit, starting from PS synthesis at the ER side of MAM by PS synthase (Zborowski et al. 1983; Stone and Vance 2000) to its decarboxylation into PE at the IMM and back to the ER for conversion into PC (Vance 1990; Voelker 2000; Osman et al. 2011).
Beyond synthesis, there is a bidirectional and extensive lipid exchange between membranes of the two organelles, although the factors involved in lipid transport remain elusive (Rowland and Voeltz 2012). PC is the most abundant mitochondrial phospholipid, but it is synthesized only in elements of the ER. PS and PC must then be imported from the ER to the mitochondria, through yet uncharacterized mechanisms but likely involving ER membranes and OMM close juxtapositions (Vance 2014).
The importance of mitochondria-ER lipid exchanges is also illustrated by the fact that the precursor for the mitochondrial synthesis of cardiolipin, the phosphatidic acid, comes from the ER, through a yet unknown mechanism (Vance 2014). Moreover, sterols and sphingolipids are minor but potentially functionally significant constituents of mitochondrial membranes. MAMs have been reported to be enriched in cholesterol, compared to the bulk ER, and studies have suggested that depletion of cholesterol from MAMs promotes the association of the two organelles (Fujimoto et al. 2012). Thus, mitochondria-ER contacts could contain microdomains that are enriched in cholesterol and gangliosides, modified sphingolipids, similarly to the detergent-resistant lipid rafts at the PM (Hayashi and Fujimoto 2010). Interestingly, in yeast, sterols have been suggested to be directly transported from the ER to the mitochondria through Ltc1 (also known as Lam6), an ER-located sterol transporter enriched in MAMs (Murley et al. 2015) which interacts with the ERMES complex (Gatta et al. 2015). Finally, ceramides, like other sphingolipids, constitute a quantitatively minor, but functionally significant, constituent of mitochondria, since they can induce OMM permeabilization-mediated apoptosis. Interestingly, ceramides synthesis has been shown to occur at MAMs (Stiban et al. 2008).
Even though the interplay between mitochondria and ER seems undeniable in the maintenance of cellular lipid homeostasis, a clear identification of molecular actors implicated in the process in mammalian cells is still missing. Insights for molecular mechanisms come almost exclusively from yeast. Indeed, an exciting clue arised from the identification in yeast of the members of the ERMES complex (see above) as highly hydrophobic proteins involved in lipid trafficking (Kopec et al. 2010). More precisely, recent studies showed that synaptotagmin-like mitochondrial lipid-binding protein (SMP) domains, present in the ERMES complex, are able to assemble in heterotetramer domains to form tubular structures, as hydrophobic tunnels for lipid transfer (AhYoung et al. 2015). Nevertheless, the fact that these SMP domains display a strong preference for PC (AhYoung et al. 2015) suggests that other glycerol-phospholipids transfer might be mediated by different parts of the ERMES complex or by other molecules (Murley and Nunnari 2016). Evidence has pointed toward a possible implication of some yeast members of the oxysterol-binding homology (Osh) protein family that display high affinity for PS and are crucial for PS homeostasis (Maeda et al. 2013), as well as for the ER membrane complex (EMC) proteins (Lahiri et al. 2014), for increasing the rate of transfer of PS from the ER to mitochondria. All these elements suggest that defining the molecular mechanism of lipid transfer between ER and mitochondria could imply the discovery of mammalian homologues of ERMES to allow phospholipids transfer between organelles.
Recent studies, however, have provided the first possible molecular identity in a different protein implicated in ER-mitochondria steroid transfer. Indeed, the translocator protein (TSPO), located in the OMM, is implicated in the import and processing of steroids into mitochondria (Flis and Daum 2013), and many clues point toward a possible presence of TSPO in MAMs, particularly through its interaction with VDAC (Guilarte et al. 2016).
The ER is not the only partner with which mitochondria interact to maintain cellular lipid homeostasis. Indeed, experimental disruption of ERMES has been shown to be compensated by the expansion of mitochondrial contacts with vacuoles/lysosomes, called vacuolar and mitochondrial patches (vCLAMPs), to maintain lipid homeostasis, and vice versa. Moreover, the molecular ratio between ERMES and vCLAMP components in yeast is dependent on the metabolic state of the cell. Ltc1 (see above) could also be expressed at vCLAMPs, in conditions of expansion, and is necessary for the compensation of ERMES loss (Honscher et al. 2014 and reviewed in Murley and Nunnari 2016).
Peroxisomes have been also shown to interact with mitochondria (see also below). Interestingly, in yeast, they make contacts at sites of mitochondria-ER juxtapositions, through the interaction between the peroxisomal protein Pex11 and the ERMES subunit Mdm34 (Mattiazzi Usaj et al. 2015). Interestingly, in these areas, the mitochondrial matrix enzyme PDH, responsible for the production of acetyl-CoA (see above), is also always present. Thus, acetyl-coA, which is a metabolite at the crossroad between glucose and lipids metabolism, is produced at a cellular hub of organelles’ interactions (Cohen et al. 2014). These observations could lead to exciting speculations about a nutrient-sensing system based on the distribution of mitochondria/ER/vacuoles and peroxisome contacts, as well as on lipid transporters composition, to adapt lipid homeostasis and metabolism to cellular environment (Murley and Nunnari 2016).
It is important to underlie, however, that even if close contacts have the strongest relevance in inter-organelle lipid trafficking, other mechanisms might exist. In particular, soluble cytosolic lipid transfer proteins have been identified, especially for PC and PI (Vance 2015).
4.2 Autophagy
The process of autophagy is considerably dependent on intracellular organelles’ membranes and their possible interactions (Yang and Klionsky 2009).
The major intracellular source from which the initial isolation membrane (also known as “phagophore”) can form has been reported to be the ER, both in yeast (Suzuki and Ohsumi 2010) and mammals (Axe et al. 2008; Hayashi-Nishino et al. 2009; Itakura and Mizushima 2010; Matsunaga et al. 2010). More recently, multiple pieces of evidence have been reported for mitochondria-ER contact sites as sources of membranes for autophagosome biogenesis. Depletion of ER-mitochondria tethering regulators, such as Mfn2, has been shown to impair autophagosome formation (Hailey et al. 2010).
In addition, a milestone following study (Hamasaki et al. 2013) clearly demonstrated that autophagosomes can form at mitochondria-ER contact sites: not only key components of the autophagic process (Atg5, Atg14, Beclin-1, Vps15, Vps34) were found to be localized in MAMs under starved conditions (and this association at mitochondria-ER contact sites was stable throughout the autophagosomes formation process) but also alterations of this association led to decreased autophagy. Moreover, it has been shown that MAM raft-like microdomains, containing the raft marker ganglioside-3 (GD3), were pivotal in autophagosome formation: under starved conditions, calnexin, a Ca2 +-binding chaperone protein enriched in MAMs, strongly associates with GD3 and crucial upstream regulators of autophagy, such as AMBRA1 and WIPI1, allowing autophagy to proceed (Garofalo et al. 2016). The reduction in GD3 levels, by knocking down a member of the glycosyltransferase family responsible for its synthesis, led to impaired starvation-induced association of core complex molecules at MAMs and defective autophagy, highlighting the key role played by the MAM platform in driving the process.
Finally, by scanning electron microscopy and electron tomography, a morphological analysis confirmed that phagophores can assemble at the mitochondria-ER contact sites (Biazik et al. 2015). This observation, together with the finding that lipids transported from the ER to the mitochondria are subsequently translocated into autophagosomes under starved conditions (Hailey et al. 2010), strongly indicates mitochondria-ER communications as fundamental for resupplying lipids to mitochondria to compensate for their transport into autophagosomes.
Very recently, FUNDC1, the integral OMM protein that functions as LC3 receptor during hypoxia-induced mitophagy (Liu et al. 2012), has been found to be enriched in MAMs and interact with calnexin, DRP1 and OPA1, mediating either mitochondria elongation in response to hypoxic stimuli or mitochondria fragmentation and mitophagy, depending on its phosphorylation pattern (Wu et al. 2016). Thus, FUNDC1, localized at mitochondria-ER contact sites, functions as an adaptor protein coordinating mitochondrial dynamics and quality control in response to different signaling pathways and mitochondrial stresses.
4.3 The Unfolded Protein Response (UPR)
Several physiological and pathological stress conditions can alter ER homeostasis, decreasing the amount of Ca2+ accumulated within the ER or compromising the folding ER capacity, leading to accumulation of unfolded proteins in the ER lumen, and causing the ER stress response known as “unfolded proteins response” (UPR) (Verfaillie et al. 2013).
Within the cell, the mitochondria-ER axis is a critical player in the complex modulation of the UPR; indeed, both organelles have a key role during UPR, and they are both modulated by it in their functionality and interrelationship. For example, the early phase of UPR is linked to changes in ER morphology and redistribution of mitochondria, which move toward the ER structure, strengthening ER-mitochondria contact sites. The increased ER-mitochondria interaction sustained a higher Ca2+ transfer between them, rising mitochondrial metabolism, oxygen consumption, and ATP production. The outcome can help the cell to rescue the stress state, supplying ATP to chaperone proteins, such as Bip, involved during UPR in the folding process (Bravo et al. 2011).
As mentioned above, if, however, the stress and the damage are too severe, the pro-survival pathway, mediated by the three sensors, turns into a pro-death response through the regulation of pro-apoptotic proteins. In this case, the stronger ER-mitochondria interaction favors a deregulated Ca2+ transfer, leading to mitochondrial Ca2+ overload, mPTP opening, and apoptotic protein activation. In particular, among the proteins that can trigger apoptosis, the truncated form of SERCA (S1T) has been shown to be involved in the late ER stress response: highly expressed during ER stress by the PERK-ATF4 pathway, the truncated pump increases ER-mitochondria tethering and blocks mitochondrial movements, causing an increased ER-mitochondrial Ca2+ transfer, mitochondrial Ca2+ overload, and eventually cell death (Chami et al. 2008).
4.4 Organelle Activity and Biogenesis
A peculiar molecular and functional relationship between mitochondria and peroxisomes has been known for a long time. Indeed, these organelles not only share some components of their fission machinery (such as Drp1, Fis1, and Mff in mammals and Dnm1 and Fis1 in yeast) but also cooperate in some metabolic pathways, such as β-oxidation of fatty acids (reviewed in Schrader et al. 2015). Moreover, the mitochondrial biogenesis transcriptional co-activator PGC-1α has been found to directly increase peroxisomes number (Bagattin et al. 2010).
Mitochondria and peroxisomes have been reported to be closely associated in ultrastructural studies in mammalian cells (Hicks and Fahimi 1977), and multiple evidences indicate physical connections between mitochondria and peroxisomes in yeast (Rosenberger et al. 2009), although their contribution to metabolite exchange between the two organelles is unclear. Instead, mitochondrion-derived vesicles (MDVs) were suggested to allow the latter feature (Neuspiel et al. 2008) and appear, together with vesicles derived from the ER, to be important for peroxisomal biogenesis (Mohanty and McBride 2013). As to the functions of mitochondria-peroxisomes contacts, they have been suggested to be relevant for peroxisome fission in yeast (Mao et al. 2014), as well as for the innate immune response against viral and bacterial infections (reviewed in Schrader et al. 2015 and see also Horner et al. 2011, 2015) and for redox homeostasis (Nordgren and Fransen 2014) (reviewed in Schrader et al. 2015). Moreover, the same molecular fission complex (see above) is reported to be recruited during the autophagic degradation of the two organelles, mitophagy (Mao et al. 2013), and pexophagy (Mao et al. 2014), respectively. Moreover, peroxisome division, occurring early in the process, has been shown to take place at mitochondria-peroxisome contact sites (Mao et al. 2014), highlighting a functional interplay of the two organelles in autophagy.
Mitochondria make also contacts with other organelles, although the significance and molecular characterization of these sites are not defined. For example, in yeast the acidic vacuole is functionally linked to mitochondria, and any dysregulation of each organelle is negatively reflected to the other (Hughes and Gottschling 2012). In mammals, a vesicular transport pathway has been described from mitochondria to lysosomes, as an early response to oxidative stress, independent from mitophagy and preventing mitochondrial dysfunctionality (Soubannier et al. 2012).
Finally, it has been recently demonstrated that ~7% of melanosomes are in close contact with mitochondria, thanks to the presence of proteinaceous bridges similar to those observed in ER-mitochondria regions of apposition (Daniele et al. 2014). This association has been reported to be important for melanosomes biogenesis, likely through a local ATP supply from mitochondria to melanosomes or through an exchange of other metabolites, such as Ca2+. Interestingly, though its abundance in the sites of interorganellar apposition is strongly reduced compared to the bulk OMM (as revealed by immunogold analysis), Mfn2 has been suggested to be important for these contacts, because its downregulation decreases their number (Daniele et al. 2014).
4.5 Other Cell Functions
4.5.1 Mitochondria Inheritance
In yeast, it has been demonstrated that active nucleoids (the nucleoprotein structures responsible for mtDNA inheritance) are associated with a protein complex that spans the OMM and the IMM (Meeusen and Nunnari 2003). Components of the ERMES complex (involved in mitochondria-ER contacts in yeast, see above) have been observed to be spatially associated to active nucleoids (Murley et al. 2013). Moreover, Mmr1, an OMM protein specific of bud’s but not of mother cell’s mitochondria, physically interacts with Myo2 (Itoh et al. 2004), a class V myosin that mediates the transport of mitochondria to the bud; of note, Mmr1 has been observed to be enriched at mitochondria-ER contact sites (Swayne et al. 2011), thus suggesting that these regions of contact may be important for mitochondria inheritance.
Very recently, in mammalian cells, it has been reported that mtDNA synthesis is spatially associated to a subset of mitochondria-ER contacts involved in mitochondrial division (Lewis et al. 2016). Importantly, mitochondria-ER tether formation precedes and is necessary (but not sufficient) for mtDNA replication and mitochondrial division, thus playing a crucial role in the proper inheritance of mtDNA between dividing mitochondria. It is tempting to speculate that the existence of a contact with the ER creates a specialized platform on OMM and IMM, characterized by a specific lipid/protein composition (lipid rafts-like), important for the successive recruitment of proteins involved in specific activities, such as mtDNA replication and mitochondrial division.
4.5.2 Inflammation and Antiviral Response
The activation of innate immune response is another pathway in which the mitochondria-ER axis seems to be actively involved (reviewed in Marchi et al. 2014; van Vliet et al. 2014; Schrader et al. 2015). The cytosolic pathogen receptor RIG-1, responsible for the production of pro-inflammatory cytokines, has been demonstrated to be recruited for proper assembly by the OMM adaptor protein MAVS (mitochondria antiviral signaling protein; Belgnaoui et al. 2011), which has been recently reported to be specifically located at MAMs, where it regulates mitochondrial and peroxisomal signaling events during viral infections (Horner et al. 2011). A proteomic analysis revealed, in these latter conditions, a dynamic change in MAM protein composition, with an increase in MAVS interactors (Horner et al. 2015).
The inflammasome NOD-like receptor NLRP3 is a multiprotein complex that works like a sensor of damage (intracellular pattern-recognition receptors that recognize pathogen-associated molecular patterns, PAMPs) activating the pro-inflammatory response through specific pathways. Normally located at ER membranes, after mitochondrial-ROS-induced inflammasome activation, NLRP3 redistributes to MAMs (Zhou et al. 2011), thus highlighting the importance of this cell sub-compartment in ROS signaling and activation of inflammatory response. Importantly, the activity of VDACs, which are OMM proteins and important regulators of mitochondrial metabolic activity (see Chaps. 7, 8, 9, 10, 11, and 12 for extensive descriptions), is crucial for NLRP3 activation. Moreover, in cystic fibrosis cell models, it has been recently shown that MCU-dependent mitochondrial Ca2+ uptake integrates pro-inflammatory signals initiated by pathogen-associated molecules and relays the information to NLRP3 (Rimessi et al. 2015), acting as a synergic stress stimulus in triggering exacerbated inflammatory response. Thus, not only mitochondria play a central role in the orchestration of the inflammatory response but also constitute the signal-integrating organelle for inflammasome activation.
4.5.3 Mitochondria Dynamics and Transport
Mitochondria are dynamic organelles, continuously undergoing fusion and fission. These opposite processes maintain the shape, size, and number of mitochondria as well as their physiological functionality (Youle and van der Bliek 2012). Numerous studies have suggested a role for mitochondria-ER interactions in mitochondrial fission and fusion (see Marchi et al. 2014 for a recent review), through mechanisms involving physical and functional (Ca2+) coupling. Moreover, many of the proteins involved in MAM structural integrity are mitochondria dynamics-related proteins.
Mitochondrial fission, in particular, seems to be highly dependent on ER proximity. The fragmentation process relies on the recruitment, by specific receptors at the mitochondrial surface, of dynamin-related proteins, forming helical oligomers that wrap around the organelles and divide them in a GTP-dependent manner (Klecker et al. 2014). Interestingly, these fission complexes assemble at specific sites where ER tubules circumscribe mitochondria to form constriction sites both in yeast and metazoan cells (Smirnova et al. 2001). In these latter, mitochondrial division mediated by ER constrictions involves actin polymerization to tighten the noose formed by Drp1, through the action of the ER protein inverted formin-2 (INF2), a promoter of actin polymerization and depolarization (Prudent and McBride 2016). Moreover, the mitochondrial protein Miro1, at the OMM, has been shown to sense [Ca2+], stop mitochondria motility, and induce fission in a Drp1-dependent manner (Saotome et al. 2008). Interestingly, its yeast homolog Gem1, which presents similar features (Frederick et al. 2004), interacts with the MAM complex ERMES at sites of mitochondrial division (Nguyen et al. 2012). This interaction is relying on the Gem1 EF-hand Ca2+-binding domain, suggesting that the association of the protein with ERMES could be Ca2+ dependent (Kornmann et al. 2011). Moreover, reduced mitochondria-ER interactions have been associated with mitochondrial fragmentation during glucose sensing in the liver (Theurey et al. 2016).
Apart from fission, mitochondria-ER interactions have been shown to be crucial for fusion processes as well. The main actors in the control of mitochondrial fusion are Mfn1 and Mfn2, two homologous GTPase proteins present, both, at OMM, but, the second one, also at ER membranes. In particular, Mfn2 coordinates the interactions between different mitochondria, leading to the stabilization of the whole mitochondrial network, but is also crucial for tuning their tethering to the ER, modulating MAM formation (see above) (Koshiba et al. 2004).
Only mitochondria-ER contacts have been clearly demonstrated to have a central role, with the described mechanisms, in mitochondrial shaping events. Nevertheless, clues suggest that mitochondria-peroxisome interactions might also play a part. For instance, induction of oxidative stress by expression of KillerRed in peroxisomes induces mitochondrial fragmentation, even though only functional interplay through ROS has been implicated in the phenomenon (Ivashchenko et al. 2011). Interestingly, both organelles undergo similar dynamics processes and share components of their division machinery, such as Drp1 and Fis1 (Schrader et al. 2016) (see above).
To this day, the only link observed between mitochondrial dynamics and mitochondria-PM interaction is the observation that organelles’ fragmentation mediated by Fis1 or Drp1 overexpression is associated with their decreased interactions with the cell membrane. However, it is more likely that this element underlines the possibility that a small number of anchor points exist at the mitochondria-PM interface rather than a real functional interplay between mitochondrial dynamics and PM (Frieden et al. 2005).
5 Mitochondria-ER Contacts and Diseases
As detailed above, the communication between mitochondria and ER regulates a multitude of physiological processes, ranging from intracellular Ca2+ homeostasis to lipid metabolism and control of cell death. Thus, the emergence of alterations in organelles’ physical and functional tethering as a common hallmark in different pathologies, such as neurodegenerative diseases, diabetes, obesity, and cancer is not surprising. A comprehensive and detailed review of all these findings is far away from the scope of the present chapter. Here, we limit ourselves to a brief summary of the main studies involving alterations in the mitochondria-ER axis as a key event in three neurodegenerative diseases: Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). The interested reader is also referred to additional reviews on this topic (Cali et al. 2013b).
5.1 Alzheimer’s Disease
In Alzheimer’s disease (AD), the pathogenicity of the altered production/accumulation of amyloid β peptide (Aβ) has been well established (Goedert and Spillantini 2006). Additionally, however, AD patients display early intracellular alterations (such as those in Ca2+ homeostasis, lipid metabolism, axonal transport, ROS production, autophagy, and mitochondrial dynamics) that have been much less investigated as potential therapeutic targets (Agostini and Fasolato 2016). Importantly, many of these altered processes are known to take place at MAMs. Furthermore, Presenilin 1 and 2 (PS1 and PS2), the two alternative core proteins of the γ-secretase complex responsible for Aβ production, mutated in the familial forms of the disease, as well as γ-secretase activity, have also been found to be enriched in MAMs (Area-Gomez et al. 2009; Schreiner et al. 2015). In addition, it has been originally demonstrated that wt PS2 and, more potently, familial AD (FAD)-PS2 mutants, but not PS1 neither FAD-PS1, favor ER-mitochondrial Ca2+ transfer by increasing the physical association between the two organelles (Zampese et al. 2011). Though an increased interorganellar apposition in the presence of FAD-PS2 mutations has been later confirmed by others in human fibroblasts from FAD patients (Area-Gomez et al. 2012), an increased tethering with an impact on phospholipids and cholesterol metabolism has been additionally observed in fibroblasts from FAD-PS1 and sporadic AD patients (Area-Gomez et al. 2012). In this paper, however, the authors reported that both endogenous PS1 and PS2 negatively affect ER-mitochondria coupling, while their FAD mutants favor the apposition. It is, however, unclear how sporadic AD cases may converge on MAMs alterations similarly to those mechanistically caused by PSs mutations, as it has been proposed. Moreover, in another independent study, FAD-PS1 expression in a neuronal cell model was found to be associated to a decreased, and not to an increased, ER-mitochondria physical and functional coupling (Sepulveda-Falla et al. 2014).
Very recently, we confirmed that only PS2 increases ER-mitochondria communications, by binding and sequestering Mfn2 (Filadi et al. 2016) (see also above). It is tempting to speculate that, while PS2 directly affect ER-mitochondria tethering, in the cases of FAD-PS1 and sporadic AD, other pathways may indirectly, and over a longer period of time, converge on ER-mitochondria association, though the molecular mechanism is not clear. On this line, an altered coupling between these two organelles has been reported in different AD models (Kipanyula et al. 2012; Hedskog et al. 2013), and an acute Aβ oligomer exposure has been demonstrated to increase ER-mitochondria Ca2+ transfer (Hedskog et al. 2013). Finally, it has been observed that increasing ER-mitochondria contacts (through Mfn2 down-regulation) deeply affects the process of Aβ production, by altering the maturation of γ-secretase (Leal et al. 2016), further suggesting that the ER-mitochondria platform may represent a novel target for AD pharmacological research.
5.1.1 Parkinson’s Disease
The association between Parkinson’s disease (PD) and mitochondrial alterations is particularly immediate. Indeed, some of the genes mutated in the familial form of the disease encode for proteins that are targeted or are transiently associated, to mitochondria, such as PINK-1 and Parkin (reviewed in Cali et al. 2011). As to the contacts with the ER, overexpression of wt Parkin has been reported to favor ER-mitochondria physical and functional coupling, increasing mitochondrial Ca2+ uptake and sustaining ATP production, a function exerted also by endogenous Parkin, whose downregulation results in mitochondrial fragmentation (Cali et al. 2013). However, a recent report described an increased ER-mitochondria apposition in fibroblasts from Parkin (PARK-2) KO mice, as well as from PD patients harboring PARK-2 mutations (Gautier et al. 2016). This increase has been associated to higher levels of the Parkin target Mfn2, proposed as an ER-mitochondria tether. However, the complete list of the possible Parkin targets still lacks, and thus whether the effects of Parkin-KO on the tethering depend or not from its effects on Mfn2 levels is not clear. Overexpression of wt DJ-1 (another protein whose mutations are associated with PD familial forms) has been also shown to increase ER-mitochondria communication, by counteracting the negative effects of the tumor suppressor protein p53 on this parameter (Ottolini et al. 2013). Interestingly, a small fraction of DJ-1 has been recently demonstrated to translocate into mitochondrial matrix upon nutrient deprivation, a process important to sustain ATP levels, which is lost in the presence of pathogenic DJ-1 mutants (Cali et al. 2015). α-Synuclein has been also reported to be involved in the modulation of ER-mitochondrial physical tethering, enhancing mitochondrial Ca2+ uptake (Cali et al. 2012), and has been recently demonstrated to be localized at MAMs (Guardia-Laguarta et al. 2014), with its disease-linked mutants less associated to MAMs and resulting in mitochondria-ER uncoupling. Finally, in drosophila PINK1/Parkin mutants, it has been found that defective mitochondria activate the PERK branch of UPR, a neurotoxic process that seems to be potentially modulated by Mfn2 (Celardo et al. 2016).
5.1.2 Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease and is clinically and genetically associated to frontotemporal dementia (FTD) (Ling et al. 2013). In the nucleus and cytosol of neurons and glia of patients, inclusions of TDP-43-, FUS-, and C9ORF72-derived protein can be retrieved. Importantly, mutations in the genes encoding for these proteins are responsible for ~50% of the familial forms of the disease (Ling et al. 2013). Recently, increasing evidence has been provided for a key role of the mitochondria-ER axis in ALS/FTD onset/progression (reviewed in Manfredi and Kawamata 2016). In particular, overexpression of both wt and mutated TDP-43 decreases ER-mitochondria contacts and Ca2+ transfer (Stoica et al. 2014), by disrupting the VAPB-PTPIP51 tethering complex through activation of GSK-3β (see above). Similarly, FUS, wt, and ALS-linked mutant overexpression has been reported to disrupt ER-mitochondria juxtaposition, Ca2+ shuttling, and ATP production (Stoica et al. 2016), by affecting, in a GSK-3β-dependent manner, the VAPB-PTPIP51 interaction. Importantly, VAPB mutations are responsible for some familial forms of ALS type-8, and VAPB-P56S expression increases mitochondrial Ca2+ uptake after its release from the ER (De Vos et al. 2012). In mouse embryonic motor neurons, overexpression of G93A hSOD1, as an ALS animal model, has been shown to affect ER and mitochondrial Ca2+ homeostasis (Lautenschlager et al. 2013). Recently, the loss of the MAM protein SIGMA1-R (a condition associated to some familial forms of ALS/FTD) has been reported to impair mitochondria-ER crosstalk (Bernard-Marissal et al. 2015), and, similarly, SIGMA1-R loss-of-function mutations (associated to distal hereditary motor neuropathies, dHMNs) have been shown to disrupt this interorganellar communication (Gregianin et al. 2016). Thus, different studies convincingly suggested that ER-mitochondria axis could be an interesting platform to exploit as a therapeutic target in ALS/FTD.
References
Agostini M, Fasolato C (2016) When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s disease. Cell Calcium 60(5):289–298
AhYoung AP, Jiang J, Zhang J et al (2015) Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci U S A 112(25):E3179–E3188
Amigo I, Traba J, Gonzalez-Barroso MM et al (2013) Glucagon regulation of oxidative phosphorylation requires an increase in matrix adenine nucleotide content through Ca2+ activation of the mitochondrial ATP-Mg/Pi carrier SCaMC-3. J Biol Chem 288(11):7791–7802
Anunciado-Koza RP, Zhang J, Ukropec J et al (2011) Inactivation of the mitochondrial carrier SLC25A25 (ATP-Mg2+/Pi transporter) reduces physical endurance and metabolic efficiency in mice. J Biol Chem 286(13):11659–11671
Area-Gomez E, de Groof AJ, Boldogh I et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175(5):1810–1816
Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD et al (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31(21):4106–4123
Arruda AP, Pers BM, Parlakgul G et al (2014) Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20(12):1427–1435
Axe EL, Walker SA, Manifava M et al (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182(4):685–701
Bagattin A, Hugendubler L, Mueller E (2010) Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci U S A 107(47):20376–20381
Baughman JM, Perocchi F, Girgis HS et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476(7360):341–345
Belgnaoui SM, Paz S, Hiscott J (2011) Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol 23(5):564–572
Bernard-Marissal N, Medard JJ, Azzedine H, Chrast R (2015) Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138(Pt 4):875–890
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1(1):11–21
Biazik J, Yla-Anttila P, Vihinen H et al (2015) Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 11(3):439–451
Boncompagni S, Rossi AE, Micaroni M et al (2009) Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell 20(3):1058–1067
Bononi A, Bonora M, Marchi S et al (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ 20(12):1631–1643
Bragadin M, Pozzan T, Azzone GF (1979) Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry 18(26):5972–5978
Bravo R, Vicencio JM, Parra V et al (2011) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124(Pt 13):2143–2152
Cali T, Ottolini D, Brini M (2011) Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 37(3):228–240
Cali T, Ottolini D, Negro A, Brini M (2012) Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem 287(22):17914–17929
Cali T, Ottolini D, Negro A, Brini M (2013a) Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta 1832(4):495–508
Cali T, Ottolini D, Brini M (2013b) Calcium and endoplasmic reticulum-mitochondria tethering in neurodegeneration. DNA Cell Biol 32(4):140–146
Cali T, Ottolini D, Soriano ME, Brini M (2015) A new split-GFP-based probe reveals DJ-1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Hum Mol Genet 24(4):1045–1060
Carafoli E (2003) Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci 28(4):175–181
Cardenas C, Miller RA, Smith I et al (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142(2):270–283
Celardo I, Costa AC, Lehmann S et al (2016) Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson's disease. Cell Death Dis 7(6):e2271
Chami M, Oules B, Szabadkai G et al (2008) Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell 32(5):641–651
Cohen Y, Klug YA, Dimitrov L et al (2014) Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol BioSyst 10(7):1742–1748
Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One 7(9):e46293
Csordas G, Renken C, Varnai P et al (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174(7):915–921
Csordas G, Varnai P, Golenar T et al (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39(1):121–132
Daniele T, Hurbain I, Vago R et al (2014) Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr Biol 24(4):393–403
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456(7222):605–610
De Marchi U, Castelbou C, Demaurex N (2011) Uncoupling protein 3 (UCP3) modulates the activity of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J Biol Chem 286(37):32533–32541
De Vos KJ, Morotz GM, Stoica R et al (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21(6):1299–1311
del Arco A, Satrustegui J (2004) Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J Biol Chem 279(23):24701–24713
Deluca HF, Engstrom GW (1961) Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A 47:1744–1750
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787(11):1309–1316
Denton RM, Richards DA, Chin JG (1978) Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J 176(3):899–906
Doghman-Bouguerra M, Granatiero V, Sbiera S et al (2016) FATE1 antagonizes calcium- and drug-induced apoptosis by uncoupling ER and mitochondria. EMBO Rep 17(9):1264–1280
Drago I, De Stefani D, Rizzuto R, Pozzan T (2012) Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A 109(32):12986–12991
Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V et al (2014) A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 30(1):95–102
Filadi R, Pozzan T (2015) Generation and functions of second messengers microdomains. Cell Calcium 58(4):405–414
Filadi et al (2012) In: Agostinis P, Samali A (eds) Endoplasmic reticulum stress in health and disease. Springer Science+Business Media, Dordrecht
Filadi R, Greotti E, Turacchio G et al (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A 112(17):E2174–E2181
Filadi R, Greotti E, Turacchio G et al (2016) Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep 15(10):2226–2238
Flis VV, Daum G (2013) Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb Perspect Biol 5(6):a013235
Fonteriz RI, de la Fuente S, Moreno A et al (2010) Monitoring mitochondrial [Ca2+] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium 48(1):61–69
Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87(2):593–658
Fowler SL, Akins M, Zhou H et al (2013) The liver connexin32 interactome is a novel plasma membrane-mitochondrial signaling nexus. J Proteome Res 12(6):2597–2610
Franzini-Armstrong C (2007) ER-mitochondria communication. How privileged? Physiology (Bethesda) 22:261–268
Frederick RL, McCaffery JM, Cunningham KW et al (2004) Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol 167(1):87–98
Frieden M, Arnaudeau S, Castelbou C, Demaurex N (2005) Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem 280(52):43198–43208
Friedman JR, Lackner LL, West M et al (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362
Fujimoto M, Hayashi T, Su TP (2012) The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem Biophys Res Commun 417(1):635–639
Gandhi S, Wood-Kaczmar A, Yao Z et al (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33(5):627–638
Garofalo T, Matarrese P, Manganelli V et al (2016) Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy 12(6):917–935
Garrib A, McMurray WC (1986) Purification and characterization of glycerol-3-phosphate dehydrogenase (flavin-linked) from rat liver mitochondria. J Biol Chem 261(17):8042–8048
Gatta AT, Wong LH, Sere YY et al (2015) A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. elife 4:e07253
Gautier CA, Erpapazoglou Z, Mouton-Liger F et al (2016) The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet 25:2972. pii: ddw148
Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ 23(9):1417–1427
Giacomello M, Drago I, Bortolozzi M et al (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38(2):280–290
Giorgi C, Ito K, Lin HK et al (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330(6008):1247–1251
Giorgi C, Bonora M, Sorrentino G et al (2015) p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A 112(6):1779–1784
Glancy B, Willis WT, Chess DJ, Balaban RS (2013) Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52(16):2793–2809
Goedert M, Spillantini MG (2006) A century of Alzheimer’s disease. Science 314(5800):777–781
Gordon PB, Holen I, Fosse M et al (1993) Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 268(35):26107–26112
Granatiero V, Giorgio V, Cali T et al (2016) Reduced mitochondrial Ca(2+) transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ 23(2):231–241
Gregianin E, Pallafacchina G, Zanin S et al (2016) Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum Mol Genet 25:3741
Guardia-Laguarta C, Area-Gomez E, Rub C et al (2014) Alpha-synuclein is localized to mitochondria-associated ER membranes. J Neurosci 34(1):249–259
Guilarte TR, Loth MK, Guariglia SR (2016) TSPO finds NOX2 in microglia for redox homeostasis. Trends Pharmacol Sci 37(5):334–343
Hailey DW, Rambold AS, Satpute-Krishnan P et al (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667
Hamasaki M, Furuta N, Matsuda A et al (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495(7441):389–393
Hayashi T, Fujimoto M (2010) Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol 77(4):517–528
Hayashi-Nishino M, Fujita N, Noda T et al (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11(12):1433–1437
Haynes RC Jr, Picking RA, Zaks WJ (1986) Control of mitochondrial content of adenine nucleotides by submicromolar calcium concentrations and its relationship to hormonal effects. J Biol Chem 261(34):16121–16125
Hedgepeth SC, Garcia MI, Wagner LE 2nd et al (2015) The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem 290(11):7304–7313
Hedskog L, Pinho CM, Filadi R et al (2013) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc Natl Acad Sci U S A 110(19):7916–7921
Hicks L, Fahimi HD (1977) Peroxisomes (microbodies) in the myocardium of rodents and primates. A comparative ultrastructural cytochemical study. Cell Tissue Res 175(4):467–481
Honscher C, Mari M, Auffarth K et al (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30(1):86–94
Horner SM, Liu HM, Park HS et al (2011) Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A 108(35):14590–14595
Horner SM, Wilkins C, Badil S et al (2015) Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS One 10(3):e0117963
Hughes AL, Gottschling DE (2012) An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 492(7428):261–265
Islinger M, Luers GH, Zischka H et al (2006) Insights into the membrane proteome of rat liver peroxisomes: microsomal glutathione-S-transferase is shared by both subcellular compartments. Proteomics 6(3):804–816
Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6(6):764–776
Itoh T, Toh EA, Matsui Y (2004) Mmr1p is a mitochondrial factor for Myo2p-dependent inheritance of mitochondria in the budding yeast. EMBO J 23(13):2520–2530
Ivashchenko O, Van Veldhoven PP, Brees C et al (2011) Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar cross-talk. Mol Biol Cell 22(9):1440–1451
Jouaville LS, Pinton P, Bastianutto C et al (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A 96(24):13807–13812
Kipanyula MJ, Contreras L, Zampese E et al (2012) Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 11(5):885–893
Klecker T, Scholz D, Fortsch J, Westermann B (2013) The yeast cell cortical protein Num1 integrates mitochondrial dynamics into cellular architecture. J Cell Sci 126(Pt 13):2924–2930
Klecker T, Bockler S, Westermann B (2014) Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol 24(9):537–545
Kopec KO, Alva V, Lupas AN (2010) Homology of SMP domains to the TULIP superfamily of lipid-binding proteins provides a structural basis for lipid exchange between ER and mitochondria. Bioinformatics 26(16):1927–1931
Kornmann B, Currie E, Collins SR et al (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325(5939):477–481
Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A 108(34):14151–14156
Koshiba T, Detmer SA, Kaiser JT et al (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305(5685):858–862
La Rovere RM, Roest G, Bultynck G, Parys JB (2016) Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 60(2):74–87
Lackner LL, Ping H, Graef M et al (2013) Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc Natl Acad Sci U S A 110(6):E458–E467
Lahiri S, Chao JT, Tavassoli S et al (2014) A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol 12(10):e1001969
Landolfi B, Curci S, Debellis L et al (1998) Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured in situ in intact cells. J Cell Biol 142:1235–1243
Lautenschlager J, Prell T, Ruhmer J et al (2013) Overexpression of human mutated G93A SOD1 changes dynamics of the ER mitochondria calcium cycle specifically in mouse embryonic motor neurons. Exp Neurol 247:91–100
Leal NS, Schreiner B, Pinho CM et al (2016) Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J Cell Mol Med 20(9):1686–1695
Lebiedzinska M, Szabadkai G, Jones AW et al (2009) Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int J Biochem Cell Biol 41(10):1805–1816
Lewis SC, Uchiyama LF, Nunnari J (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353(6296):aaf5549
Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79(3):416–438
Liu L, Feng D, Chen G et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14(2):177–185
Maeda K, Anand K, Chiapparino A et al (2013) Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature 501(7466):257–261
Majewski N, Nogueira V, Bhaskar P et al (2004) Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 16(5):819–830
Mallilankaraman K, Cardenas C, Doonan PJ et al (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14(12):1336–1343
Manfredi G, Kawamata H (2016) Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol Dis 90:35–42
Mao K, Wang K, Liu X, Klionsky DJ (2013) The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell 26(1):9–18
Mao K, Liu X, Feng Y, Klionsky DJ (2014) The progression of peroxisomal degradation through autophagy requires peroxisomal division. Autophagy 10(4):652–661
Marchi S, Marinello M, Bononi A et al (2012) Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis 3:e304
Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837(4):461–469
Matsunaga K, Morita E, Saitoh T et al (2010) Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190(4):511–521
Mattiazzi Usaj M, Brloznik M, Kaferle P et al (2015) Genome-wide localization study of yeast Pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J Mol Biol 427(11):2072–2087
Meeusen S, Nunnari J (2003) Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J Cell Biol 163(3):503–510
Mitchell P, Moyle J (1967) Chemiosmotic hypothesis of oxidative phosphorylation. Nature 213(72):137–139
Miyawaki A, Llopis J, Heim R et al (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388:882–887
Mohanty A, McBride HM (2013) Emerging roles of mitochondria in the evolution, biogenesis, and function of peroxisomes. Front Physiol 4:268
Mueller SJ, Reski R (2015) Mitochondrial dynamics and the ER: the plant perspective. Front Cell Dev Biol 3:78
Murgia M, Rizzuto R (2015) Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium 58(1):11–17
Murley A, Nunnari J (2016) The emerging network of mitochondria-organelle contacts. Mol Cell 61(5):648–653
Murley A, Lackner LL, Osman C et al (2013) ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. elife 2:e00422
Murley A, Sarsam RD, Toulmay A et al (2015) Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol 209(4):539–548
Naghdi S, Waldeck-Weiermair M, Fertschai I et al (2010) Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J Cell Sci 123(Pt 15):2553–2564
Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17(18):6961–6973
Neuspiel M, Schauss AC, Braschi E et al (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18(2):102–108
Nguyen TT, Lewandowska A, Choi JY et al (2012) Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13(6):880–890
Nicholls DG, Chalmers S (2004) The integration of mitochondrial calcium transport and storage. J Bioenerg Biomembr 36(4):277–281
Nicholls DG, Crompton M (1980) Mitochondrial calcium transport. FEBS Lett 111(2):261–268
Nordgren M, Fransen M (2014) Peroxisomal metabolism and oxidative stress. Biochimie 98:56–62
Nosek MT, Dransfield DT, Aprille JR (1990) Calcium stimulates ATP-Mg/Pi carrier activity in rat liver mitochondria. J Biol Chem 265(15):8444–8450
Orrenius S, Gogvadze V, Zhivotovsky B (2015) Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun 460(1):72–81
Osman C, Voelker DR, Langer T (2011) Making heads or tails of phospholipids in mitochondria. J Cell Biol 192(1):7–16
Ottolini D, Cali T, Negro A, Brini M (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 22(11):2152–2168
Pacher P, Csordas P, Schneider T, Hajnoczky G (2000) Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol 529(Pt 3):553–564
Pacher P, Thomas AP, Hajnoczky G (2002) Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc Natl Acad Sci U S A 99(4):2380–2385
Palmieri L, Pardo B, Lasorsa FM et al (2001) Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria. EMBO J 20(18):5060–5069
Pardo B, Contreras L, Serrano A et al (2006) Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem 281(2):1039–1047
Patron M, Raffaello A, Granatiero V et al (2013) The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem 288(15):10750–10758
Perkins GA, Tjong J, Brown JM et al (2010) The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J Neurosci 30(3):1015–1026
Pitter JG, Maechler P, Wollheim CB, Spat A (2002) Mitochondria respond to Ca2+ already in the submicromolar range: correlation with redox state. Cell Calcium 31(2):97–104
Pozzan T, Rizzuto R (2000) The renaissance of mitochondrial calcium transport. Eur J Biochem 267:5269–5273
Prudent J, McBride HM (2016) Mitochondrial dynamics: ER actin tightens the Drp1 noose. Curr Biol 26(5):R207–R209
Qi H, Li L, Shuai J (2015) Optimal microdomain crosstalk between endoplasmic reticulum and mitochondria for Ca2+ oscillations. Sci Rep 5:7984
Quintana A, Schwarz EC, Schwindling C et al (2006) Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J Biol Chem 281(52):40302–40309
Rapizzi E, Pinton P, Szabadkai G et al (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159(4):613–624
Reichert A, Neupert W (2002) Contact sites between the outer and inner membrane of mitochondria-role in protein transport. Biochim Biophys Acta 1592(1):41–49
Rimessi A, Bezzerri V, Patergnani S et al (2015) Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun 6:6201
Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86(1):369–408
Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358(6384):325–327
Rizzuto R, Pinton P, Carrington W et al (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280(5370):1763–1766
Robert V, Gurlini P, Tosello V et al (2001) Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J 20(17):4998–5007
Robertson JD (1960) The molecular structure and contact relationships of cell membranes. Prog Biophys Mol Biol 10:343–418
Rosenberger S, Connerth M, Zellnig G, Daum G (2009) Phosphatidylethanolamine synthesized by three different pathways is supplied to peroxisomes of the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1791(5):379–387
Rowland AA, Voeltz GK (2012) Endoplasmic reticulum-mitochondria contacts: function of the junction. Nature reviews. Mol Cell Biol 13(10):607–625
Rudolf R, Mongillo M, Magalhães PJ, Pozzan T (2004) In vivo monitoring of Ca2+ uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol 166(4):527–536
Sandebring A, Thomas KJ, Beilina A et al (2009) Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One 4(5):e5701
Saotome M, Safiulina D, Szabadkai G et al (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 105(52):20728–20733
Satrustegui J, Pardo B, Del Arco A (2007) Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev 87(1):29–67
Schneeberger M, Dietrich MO, Sebastian D et al (2013) Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155(1):172–187
Schrader M, Godinho LF, Costello JL, Islinger M (2015) The different facets of organelle interplay-an overview of organelle interactions. Front Cell Dev Biol 3:56
Schrader M, Costello JL, Godinho LF et al (2016) Proliferation and fission of peroxisomes – an update. Biochim Biophys Acta 1863(5):971–983
Schreiner B, Hedskog L, Wiehager B, Ankarcrona M (2015) Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 43(2):369–374
Sepulveda-Falla D, Barrera-Ocampo A, Hagel C et al (2014) Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J Clin Invest 124(4):1552–1567
Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS (2000) Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr 32(1):97–104
Simmen T, Aslan JE, Blagoveshchenskaya AD et al (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24(4):717–729
Singaravelu K, Nelson C, Bakowski D et al (2011) Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J Biol Chem 286(14):12189–12201
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12(8):2245–2256
Sood A, Jeyaraju DV, Prudent J et al (2014) A mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc Natl Acad Sci U S A 111(45):16017–16022
Soubannier V, McLelland GL, Zunino R et al (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22(2):135–141
Stiban J, Caputo L, Colombini M (2008) Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J Lipid Res 49(3):625–634
Stoica R, De Vos KJ, Paillusson S et al (2014) ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5:3996
Stoica R, Paillusson S, Gomez-Suaga P et al. (2016) ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep (17)9: 1326–1342.
Stone SJ, Vance JE (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275(44):34534–34540
Streb H, Irvine RF, Berridge MJ, Schulz I (1983) Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306:67–68
Suski JM, Lebiedzinska M, Wojtala A et al (2014) Isolation of plasma membrane-associated membranes from rat liver. Nat Protoc 9(2):312–322
Suzuki K, Ohsumi Y (2010) Current knowledge of the pre-autophagosomal structure (PAS). FEBS Lett 584(7):1280–1286
Swayne TC, Zhou C, Boldogh IR et al (2011) Role for cER and Mmr1p in anchorage of mitochondria at sites of polarized surface growth in budding yeast. Curr Biol 21(23):1994–1999
Szabadkai G, Simoni AM, Chami M et al (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16(1):59–68
Szabadkai G, Bianchi K, Varnai P et al (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175(6):901–911
Szalai G, Csordas G, Hantash BM et al (2000) Calcium signal transmission between ryanodine receptors and mitochondria. J Biol Chem 275(20):15305–15313
Tadross MR, Tsien RW, Yue DT (2013) Ca2+ channel nanodomains boost local Ca2+ amplitude. Proc Natl Acad Sci U S A 110(39):15794–15799
Territo PR, Mootha VK, French SA, Balaban RS (2000) Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol 278(2):C423–C435
Thangaratnarajah C, Ruprecht JJ, Kunji ER (2014) Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat Commun 5:5491
Theurey P, Tubbs E, Vial G et al (2016) Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J Mol Cell Biol 8(2):129–143
Tubbs E, Theurey P, Vial G et al (2014) Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63(10):3279–3294
Turkan A, Hiromasa Y, Roche TE (2004) Formation of a complex of the catalytic subunit of pyruvate dehydrogenase phosphatase isoform 1 (PDP1c) and the L2 domain forms a Ca2+ binding site and captures PDP1c as a monomer. Biochemistry 43(47):15073–15085
van Vliet AR, Agostinis P (2016) When under pressure, get closer: PERKing up membrane contact sites during ER stress. Biochem Soc Trans 44(2):499–504
van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843(10):2253–2262
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265(13):7248–7256
Vance JE (2014) MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841(4):595–609
Vance JE (2015) Phospholipid synthesis and transport in mammalian cells. Traffic 16(1):1–18
Verfaillie T, Rubio N, Garg AD et al (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19(11):1880–1891
Verfaillie T, Garg AD, Agostinis P (2013) Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett 332(2):249–264
Voelker DR (2000) Interorganelle transport of aminoglycerophospholipids. Biochim Biophys Acta 1486(1):97–107
Wang PT, Garcin PO, Fu M et al (2015) Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J Cell Sci 128(15):2759–2765
Watson R, Parekh AB (2012) Mitochondrial regulation of CRAC channel-driven cellular responses. Cell Calcium 52(1):52–56
Wieckowski MR, Giorgi C, Lebiedzinska M et al (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4(11):1582–1590
Wiederkehr A, Szanda G, Akhmedov D et al (2011) Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab 13(5):601–611
Wu W, Lin C, Wu K et al (2016) FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J 35(13):1368–1384
Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 335:1–32
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337(6098):1062–1065
Zampese E, Fasolato C, Kipanyula MJ et al (2011) Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A 108(7):2777–2782
Zborowski J, Dygas A, Wojtczak L (1983) Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett 157(1):179–182
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225
Acknowledgments
The authors thank grants from the University of Padova, the Italian Ministry of University and Scientific Research, Fondazione Cassa di Risparmio di Padova e Rovigo (CARIPARO Foundation; Progetti d’eccellenza 2011/2012), and EU Joint Programme in Neurodegenerative Disease, 2015–2018, “Cellular Bioenergetics in Neurodegenerative Diseases: A system-based pathway and target analysis,” for their support of the research work performed in their lab. P.T. is research fellow of the EU Joint Programme – Neurodegenerative Disease.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Filadi, R., Theurey, P., Rossi, A., Fedeli, C., Pizzo, P. (2017). Mitochondrial Ca2+ Handling and Behind: The Importance of Being in Contact with Other Organelles. In: Rostovtseva, T. (eds) Molecular Basis for Mitochondrial Signaling. Biological and Medical Physics, Biomedical Engineering. Springer, Cham. https://doi.org/10.1007/978-3-319-55539-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-55539-3_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55537-9
Online ISBN: 978-3-319-55539-3
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)