
Abstract
Radiation therapy for skull base tumors requires knowledge, skill, and experience by the radiation oncologist. Tumors often require high target dosages, which can conflict with the competing goals of normal organ dose limitation for risk reduction of long-term functional impairments and anatomic disfigurement in the surviving children and adolescents. Protons are well suited to meet those challenges, specifically by employing active scanning technology. Chordomas represent the most challenging histology. Postoperative or definitive proton therapy with target doses ranging between 72–76–80GyRBE for small to large tumor volumes, respectively, has resulted in local control rates similar to the adult patient cohort. Due to the rarity of the disease no prospective randomized data are available, but comparison with published data using alternative conformal and/or stereotactic photon modalities strongly indicates a more favorable outcome using protons. Considering the anatomically close proximity of tumors to critical normal structures in the skull base, high target dosages require also relatively high normal tissue dose constraints compared to general radiation oncology practices. Likely due to favorable dose–volume effects of protons, i.e., its sharp dose fall-off within defined organs, the rates of severe adverse events appear acceptable. This chapter reviews the principles and practice of proton therapy for skull base chordomas.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1 Introduction
Skull base tumors can arise either directly from bony, cartilaginous, or soft tissues composing the skull base or by direct extension from either extracranial or intracranial origins. Examples for the former are chordoma, chondrosarcoma, and osteogenic malignancies like osteosarcoma and Ewing’s sarcoma of the skull base. The latter mainly comprises extracranial parameningeal rhabdomyosarcoma and intracranial meningiomas. Rhabdomyosarcomas constitute the most common histology and those children and adolescents are typically enrolled in national or international clinical protocols specifying radiotherapy parameters. This chapter focuses on the management of skull base chordomas that due to their radioresistant nature require tumoricidal radiation doses exceeding tolerance of surrounding normal tissues and organs. Target contouring and dose prescriptions based on knowledge about the natural history of chordomas, their propensity and likelihood of spread and involvement of specific anatomic compartments as well as definition of normal organ at risk tolerance dose levels are the key components to give this vulnerable group of patients the optimum chance of survival with the least risks of permanent, treatment-related impairments.
Chordomas are rare tumors, and the overall incidence across all age groups is approximately 0.1 per year per 1,000,000 population (Rombi et al. 2013). Only 5% of cases have been described in the pediatric age. A recent review of the Surveillance, Epidemiology, and End Result (SEER) database identified that in a period of 38 years (1973–2011) only 86 pediatric patients ≤19 years of age within a cohort of 1358 primary chordoma patients (Lau et al. 2016).
Chordomas arise from remnants in the notochord thus are midline structures occurring in the clivus superiorly down to the sacrococcygeal region inferiorly. Its distribution in the adult population along the axial skeleton is approximately 50% in skull base and cervical spine, i.e., occipito-cervical tumors, 30% in the sacrococcygeal region, and 20% throughout the T- and L-spine vertebral bodies.
An important treatment management question is, if chordomas in pediatric patients have a different pathophysiological profile, i.e., natural history, compared to their adult counterparts. Indeed, on occasion a child will present with a short clinical history of rapidly evolving symptoms accompanied by fast tumor growth. Indeed, the SEER database review confirmed that despite the majority of primary chordomas present with loco-regional involvement (90.4%), pediatric patients had more distant disease (14.8 vs. 9.2%, p < 0.05) (Lau et al. 2016). Despite ongoing efforts to better characterize these tumors, no reliably reproducible prognostic factor has been identified (Hoch et al. 2006).
2 Presenting Symptoms
The majority of chordomas is relatively slowly growing thus can reach considerable size before creating symptoms. In general, symptoms are due to compression, displacement, or pressure of normal structures (Borba et al. 1996; Hug et al. 1999). Chordomas typically remain extraaxial and almost never invade brain parenchyma, spinal cord, or cranial nerves. Rather, symptoms are a function of compression of normal structures in the region of the tumor. Diplopia from CN-VI compression in the cavernous sinus is the predominant symptom from upper skull base tumors. Suprasellar tumor extension will frequently cause bitemporal hemianopsia via pressure on the optic chiasm. Trigeminal nerve dysfunction may result in involvement of Meckel’s cave. Headaches may be associated with intracranial extension and displacement or compression of normal brain parenchyma. Since chordomas do not invade a brain parenchyma itself, seizures are rarely observed. Involvement of the lower clivus will cause symptoms depending on region of growth. Destruction of clivus and subsequent impairment of various neural foramina in the inferior skull base, impairment of the lower cranial nerves may be a first sign of inferior skull base tumors causing slurred speech due to XII nerve dysfunction with hemiatrophy of the tongue, or dysphagia or potential aspiration. Impairment of swallowing may result from either anterior chordoma growth into the retropharyngeal soft tissues or by involvement of the lower cranial nerve function. Posterior clival intracranial extension can result in brainstem compression and its accompanied symptoms. Further inferior extension involving the occipito-cervical junction with potential erosion of C1 and/or massa lateralis may cause severe headaches, mechanical instability of the occipito-cervical junction, and a foramen magnum syndrome with upper C-spine compression.
3 The Role of Surgery
The role of surgery in chordomas is undisputed. It is at least as important in children as in adult patients according to a recent review of 86 pediatric chordoma patient included in the SEER Database. For the pediatric patient cohort having surgery, only survival was significantly longer than for adults (22.5 vs. 14.3 years, p < 0.001) (Lau et al. 2016). Munzenrider et al. reported that gross total or near total resection—if achievable with acceptable risks—is associated with the highest chance of permanent local control in both adult and pediatric patients (Munzenrider and Liebsch 1999). In children, this requires often two-stage surgical procedures, each addressing a different portion of tumor resection. Postoperative radiotherapy should preferably be integral part of the initial treatment strategy. Thus, these children benefit significantly from an interdisciplinary management team as outlined previously in a case report by Dr. Habrand (Habrand et al. 2016). In general, with possibly few exceptions in the infant age group, this should be followed by postoperative high-dose radiation. Proton radiotherapy has been shown to have advantages in dose delivery and normal tissue sparing in this situation. It is not clear, at this time, whether it is better to proceed with immediate postoperative high-dose radiation therapy after gross total resection or to delay radiation therapy in young children with chordoma. In case of radiographically confirmed near total or subtotal resection, however, there is little doubt that postoperative high-dose radiation treatment should be recommended unless specific extenuating circumstances exist. In cases of large tumors where gross total resection is not feasible, a strategic debulking procedure probably also has significant merits including decompression of critical normal structures that are dose-limiting (e.g., optic chiasm, optic nerves, brainstem, or cervical spine) for symptom control and optimal radiation planning. The total tumor dose required for gross residual disease generally exceeds the tolerance of adjacent organs at risk (OAR). When the tumor abuts and/or compresses these OARs, tumor dose in that area will be limited to respect the maximum permissible OAR dose. Hug and Munzenrider reported a relationship between an increased incidence of local failure in cases where the chordoma abuts or compresses OARs (Hug et al. 1999; Munzenrider and Liebsch 1999).
The advent of endoscopic minimally invasive surgery has revolutionized the surgery of skull base tumors. Children with small anterior or mid skull base tumors are now spared from large open craniotomies; however, in patients with large chordomas with bilateral extension towards the brainstem and/or massive extension into the lower skull base, upper cervical spine, or parapharyngeal/paravertebral soft tissues, major two-stage approaches with or without occipito-cervical fusion might still be required. We have presently no indication that subtotal resection with significant residual tumor and high-dose particle therapy is equivalent in outcome compared to a near total resection with high-dose particle therapy. This emphasizes the need to maximize surgery as long as it is feasible in the individual patient.
4 Target Delineation
Target delineation should consider the anatomic compartments at risk of harboring microscopic disease. Treating only radiographically visible disease seen on diagnostic MR or CT scans is insufficient and will likely result in poor local control probability. Studies of radiosurgery to the gross residual disease without a margin for microscopic disease report inferior local control rates. In contrast, treatment contouring for tumor delineation for patients with chordoma can be separated into three approaches: One for gross residual disease (gross tumor volume, GTV), one for high-risk macroscopic disease (GTV), and one for low- to intermediate-risk microscopic disease (clinical target volume, CTV). In clinical practice, an oncologic en bloc resection is not possible for skull base tumors, and tumors are resected in a piecemeal fashion.
In practice, it is best to define the GTV as a combination of demonstrated, unequivocal gross disease as well as areas of high-risk microscopic disease. A GTV should include the immediate surgical bony resection margins in the skull base, the area surrounding major blood vessels, or the entire cavernous sinus even if partially involved on MRI. Frequently, there can be discrepancy between the surgeon’s report of a gross total resection and postoperative imaging suggesting small area of residual disease or equivocal findings. In general, any area that has persistent abnormal imaging findings postoperatively should be considered as being at high risk for macroscopic disease.
The CTV is defined as volume/anatomic compartments considered to be of low to intermediate risk for microscopic disease. Examples would be the prepontine space if the resected tumor involved the prepontine space, or the surgical access route and tumor bed with anatomic margin. A uniform CTV margin is not recommended since the CTV is often highly irregular and may vary in different dimensions. For example, a 1–2 cm margin would be appropriate when extracranial tumor extension into the posterior pharyngeal soft tissues is noted but a 2–3 mm margin only is needed into the brain parenchyma. Chordomas in general do not invade the brain parenchyma. Therefore, even in cases of significant initial compression of the temporal lobes causing edema within the temporal lobes, the edematous portion of the temporal lobe does not require inclusion in the CTV. Rather, in case of re-expansion of the temporal lobe following surgery a small rim of 2–3 mm typically suffices.
An approximate 5% risk of seeding into the surgical axis route has been reported in the adult chordoma population (Munzenrider and Liebsch 1999). In children, in case of an anterior transnasal transsphenoidal approach, it is useful to extend the CTV indeed into the nasal cavity or paranasal sinuses as shown in Fig. 20.1. In case of a transcranial approach, however, a strategy to routinely include the entire surgical access would result in a majority of children in significant permanent side effects; thus, this approach has not been favored in transcranial approaches.

Target contouring of a large skull base chordoma with extensive extracranial extension following subtotal, transnasal/transsphenoidal resection. Gross tumor volume (GTV) includes residual gross disease, equivocal areas as per imaging, and potential high-risk microscopic disease (red). Clinical target volume (CTV) includes GTV, potential microscopic areas but respecting anatomic barriers and considerations of infiltrative potential, as well as nasal surgical route. Note: brain parenchyma typically not included in the CTV except an outer “rim”
5 Tumor and Target Volume Prescription Doses
Most available data in clinical practice are based on 1.8–2.0 GyRBE dose fraction regimen, assuming a relative biologic effectiveness (RBE) factor of 1.1 of protons versus photons.
Although no formal dose escalation study has been conducted, there appears to be a radiation dose–response curve for chordomas. This conclusion is supported by the inferior results associated with photon therapy with doses of 66–68 Gy in contrast to the vast majority of proton data, where GTV prescription doses are in excess of 72 GyRBE. It appears that the minimum dose for very small GTV targets should be 72 GyRBE, for small to medium size tumors approximately 75–76 GyRBE and for large targets up to 78–80 GyRBE.
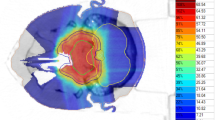
For the clinical target volume (CTV), doses analogous to general sarcoma principles for low-risk microscopic disease are used, i.e., doses between 50 and 54 GyRBE. Figures 20.2 and 20.3 depict typical isodose distributions that can be accomplished with spot scanning technology for locally advanced skull base chordomas.

A 13-year-old boy with extensive chordoma of the clivus with extracranial extension. Status post 2 subtotal resections. Proton therapy of GTV to 75.7 GyRBE and CTV to 54 GyRBE at 1.8 GyRBE dose per fraction

Chordoma in a 19-year-old male. Status post-transnasal subtital resection. Contour display of PTV of CTV (green shaded volume) and GTV (red volume). Proton therapy prescription of CTV to 54 GyRBE (green shaded volume = PTV of CTV). GTV to 73.8 Gy (RBE) at 1.8 GyRBE dose per fraction
Although chondrosarcomas are very rare in children compared to chordomas, it is worthwhile to briefly outline some differences, as they have been established in young adults and adults in general: No prospective trials have been undertaken between chordomas and chondrosarcomas, it appears that higher local control rates can be obtained in chondrosarcomas with total prescription dosage ranging between 70 and 76 GyRBE. Most published data indicate actuarial local control rates >80% and up to 95% at 5-years and even 10-year local control rates >80% for chondrosarcomas, which is even more remarkable considering the higher percentage of chondrosarcoma patients compared to chordoma patients with medium- to large-size disease since they are relatively unresectable compared to chordoma due the inherent calcified nature. There is presently no analysis available that indicates a dose differential between smaller size and larger size chondrosarcomas since local control rates appear equally high. In clinical practice, the GTV dose ranges between 70 GyRBE for small residual disease and 75 GyRBE for large-volume disease. If the patient is referred postoperatively for no residual disease, then presumably high-risk areas could be treated to approximately 66–68 GyRBE only.
6 Organs at Risk, Special Dose Considerations
A matter of significant debate remains the critical OAR constraints since the maximum permissible dosages used in some proton centers exceed the typical dose constraints used in conventional photon therapy. The general low risk of high-grade adverse events reported after proton therapy with high critical OAR constraints is likely supported by a dose–volume effect, i.e., the sharp dose fall-off within an OAR. This is mentioned in the QUANTEC report for OAR recommendations where it is explicitly stated that “for dosages at the level of 60 Gy no data are presently available on the dose volume effect” (Marks et al. 2010).
The ALARA (As Low As Reasonably Achievable) rule of dose exposure applies to each patient. If a tumor is not in immediate vicinity, abutting or compressing an OAR, then every effort should be made to keep this OAR at lower dose levels compared to situations with tumor abutment. The following OAR constraints are presently used by the author and reflect general guidelines only that need to be carefully adjusted to the individual patient and situation. The author allows a maximum dose to 0.01 cm3 up to 60 GyRBE to either optic chiasm or one of the optic nerves. Dose levels up to 60 GyRBE to single optic structures have resulted in the majority of publications in severe adverse event rates, i.e., grade 3 or higher toxicity, of optic structures of less than 1% (Munzenrider and Liebsch 1999). One should consider that inherently the dose tolerance of optic structure is likely lower if the patient has preexisting visual impairment. In addition, OAR constraints to optic structures should be reduced if the patient has monocular vision. In the author’s opinion, a significant risk of complete blindness is unacceptable and renders the patient unsuitable for proton therapy. Ethical considerations apply to the use of high-dose radiation analogous to surgical resection. If complete tumor resection would require resection of the optic chiasm, such tumor is deemed “unresectable” by the neurosurgeons. Radiation dose to the optic structures that are associated with a high risk of complete blindness should not be considered.
With regard to the brainstem, the author uses a surface dose constraint of 64 GyRBE and a brainstem center dose of 53 GyRBE, as they have been developed early on by the group at the Harvard Cyclotron Laboratory and Massachusetts General Hospital (MGH/HCL). The surface dose can be defined as meaningful volume (e.g., 0.5 cm3) and as the isodose line that is repeatedly permitted to touch the brainstem surface. The low brainstem center dose ascertains a sharp dose fall-off inside the brainstem. Based on the publication in 1999 by Debus who reported the MGH/HCL experience (Debus et al. 1999), this approach has resulted in exceedingly low rate of brainstem toxicity in adults and children with skull base chordomas and chondrosarcomas. At most there are only anecdotal reports of severe brainstem injuries in other large series.
Grade 3 or higher brain parenchymal toxicities remain the highest single organ severe adverse event rate reported in high-dose skull base tumor treatments. Several publications have reported rates of approximately 4–6% for adult patients. Despite significant efforts of predictive analysis, at present no generally agreed upon OAR constraints have proven to reduce this rate significantly. A constraint of 72 GyRBE to <2 cm3 of volume per individual lobe appears practical. The medial temporal lobes are at the highest risk when treating superior skull base tumors, which may be due to the competing adjacent OARs. In order to avoid the optic chiasm, optic nerves and brainstem require use of lateral fields that either range out in the contralateral medial temporal lobe with passive scattering technology or place repeated highly weighted spots in the medial temporal lobes with active spot scanning. These issues illustrate the need of additional fields besides lateral beams for upper skull base lesions to minimize the risk of temporal lobe necrosis especially when the PTV extends into this area.
7 Normal Tissue Constraints Versus Target Coverage
GTV prescription doses for either residual chordoma or chondrosarcoma exceed the majority of normal tissue tolerances; hence, the specification of OAR constraints significantly impacts on the general ability to cover the GTV volume and defines the steepness of the shoulder region of the target dose–volume histograms. Figure 20.4 illustrates a patient treated to a total target dose of 73.6 Gy (RBE) for relatively small residual disease chordoma of the upper clivus with no brainstem compression and no optic apparatus abutment—a rather favorable situation. Treatment plans were generated using identical contours and prescription dose with either the author’s OAR constraints of 60 and 64 GyRBE or the more common limits of 55 and 55 GyRBE to the optic chiasm and brainstem surface, respectively. The resulting GTV DVHs are displayed in Fig. 20.5. The use of lower OAR constraints alone resulted in a significant reduction of therapeutic dose for this small and favorable GTV. Although the exact coverage reduction is dependent on institution specific equipment, it exemplifies qualitatively the importance for each treating physician to carefully consider their OAR constraints and integrate published proton center constraints into their plans. Using low OAR constraints might allow a lower risk of high-grade complications but might also considerably reduce the patient’s chances of local control and potentially eradicate any gain in tumor control probability by protons over conventional photon therapy.

Influence of decreasing OAR constraints on resulting target coverage. Prescription to GTV 74 Gy(RBE). OAR constraint for optic nerves and chiasm of 60 Gy(RBE) (Panel a) versus 54 Gy(RBE) (Panel b). OAR constraint for brainstem surface of 64 Gy(RBE) (Panel c) versus 58 Gy(RBE) (Panel d). Isodose display ranging from 54 (orange) to 64 (blue) to 74 Gy(RBE) (dark red)

Influence of OAR dose constraints on target coverage according to dose–volume histogram
8 Prognostic Factors
In general for skull base tumors, chordomas need to be distinguished from chondrosarcoma since they are two distinctly different histologies with different prognosis. Low-grade chondrosarcomas by and large have a better prognosis following high-dose proton therapy than chordomas. Since the majority of skull base chondrosarcomas can be controlled long term with proton therapy, little information is available on prognostic factors. There is no reliable information available if large size, unresectable skull base chondrosarcomas have a lesser prognosis compared to small residual disease following high-dose proton therapy.
In contrast, for low-grade chordomas various prognostic factors have been identified in the adult population that determines the likelihood of accomplishing local control and long-term survival. Although those have not been specifically validated for the pediatric population, this still serve as helpful guidelines.
-
(a)
Size of residual disease plays an important role: Small-volume disease (defined as approximately 25–30 cm3) appears to be correlated with an excellent long-term prognosis of >90% at 5 years. The chances drop significantly from small-volume to mid-size (approximately 50 cm3) to larger volumes.
-
(b)
Compression or tumor abutment of dose-limiting OARs at the time of proton therapy is correlated with reduced chances of obtaining permanent local control—likely due to the dose gradient between maximum permissible OAR dose and tumoricidal prescription dose.
-
(c)
High-grade as compared to low-grade histology correlates as well with survival.
There is no consistent evidence in the published literature that patients with recurrent disease will fare worse compared to patients receiving proton therapy at the time of initial diagnosis. It remains a matter of discussion if postoperative proton therapy can be delayed in very young children after near total resection. A policy of watchful waiting with frequent MRI scans has to be carefully weighted against the possible disadvantages of recurrent disease requiring additional surgery at time of detection. No clear recommendations can be made and since the data support either side. The natural history of low versus fast tumor growth plays an important role in the decision process.
9 Proton Therapy Results
Historically, proton therapy results have been consistently superior to conventional proton therapy. In 1999, the MGH publication by Munzenrider established the excellent prognosis for chondrosarcomas following high-dose proton therapy and established the mixed prognosis for chordoma patients depending on the previously stated prognostic factors (Munzenrider and Liebsch 1999). At the time, however, the chordoma data were clearly superior to any available photon data. At present, the best available data have shown 5-year-old local control rates even of long term for favorable chordomas of approximately 80% and 5 years and 80% even at 10 years considering that for small chordomas this may well be up in the 90% range even long time but less favorable for large-sized chordomas with compression of OARs. Conventional photon data have historically and consistently produced inferior results but most recent publication by the Canadian Group (Saghal 2015) has raised the possibility of excellent long-term data even by use from high-dose, conformal photon therapy. Undoubtedly, it would be very beneficial that prospective trials will establish best practice, also establish and confirm outcomes data according to prognostic factors before ultimately proceeding with a direct photon-to-proton comparison. In addition, we presently face with a dramatic transition of proton therapy from passive scattering technology to spot scanning technology and within the spot scanning technology from first-generation equipment to more advanced delivery technology (Lomax et al. 2004).
Few retrospective series have been published outside case reports. Rombi for the group at Paul Scherrer Institute, Switzerland, reported on pediatric patients treated with spot scanning-based proton therapy delivery technique (Rombi et al. 2013). Twenty-six pediatric patients ranging in age between 3.7 and 20.8 years (mean age 13.2 years) were treated at the PSI for chordoma or chondrosarcoma. Nineteen chordoma and 7 chondrosarcoma patients with tumors originated from the skullbase (17) and the axial skeleton (9). The mean prescribed dose was 74 GyRBE (range, 73.8–75.6) for chordoma and 66 GyRBE (range, 54–72) for chondrosarcoma, at 1.8–2.0 GyRBE dose per fraction. With a mean follow-up period of 46 months (range, 4.5–126.5), actuarial 5-year local control rates were 81% for chordoma and 80% for chondrosarcoma and actuarial 5-year overall survival rates were 89% for chordoma and 75% for chondrosarcoma, respectively. With relatively limited follow-up, the authors did not report any grade 3 or higher late toxicities. Benk et al. reported the MGH/HCL experience of 18 pediatric patients with chordomas located at either skull base (15) or C-spine (3) and treated with mixed photons and protons to a median dose of 69 GyRBE (Benk et al. 1995). At a median follow-up of 72 months (range, 19–120), the actuarial local control and overall survival at 5 years was 63% and 68%, respectively. Hug et al. for the group at Loma Linda University Medical Center (LLUMC) analyzed 13 skull base pediatric patients (10 chordoma and 3 chondrosarcoma), treated with protons (6 patients) or mixed protons and photons (7 patients) to a median prescribed dose of 73.7 and 70 GyRBE for chordoma and chondrosarcoma, respectively (Hug et al. 1999). After mean follow-up of 40 months (range, 13–92 months), 5-year local control and 5-year overall survival was 60%, and 100% chordoma and chondrosarcoma, respectively. Table 20.1 summarizes the results of the few larger retrospective studies reported thus far.
10 Systemic Therapy
The effect of conventional chemotherapy agents has been limited in the management of chordoma. Some young children have been managed with upfront chemotherapy, due to extensive loco-regionally or distant presentation, but results have been of limited success except some delay in disease progression measured in months.
Newer approaches using small molecule-targeted agents, such as imatinib and sorafenib, have resulted in some disease stability (Stacchiotti and Casali 2011; Stacchiotti et al. 2012; Bompas et al. 2015; Di Maio et al. 2015; Hindi et al. 2015). Brachyury, a nuclear transcription factor, is a strong mediator of the epithelial to mesenchymal transition and is over-expressed in chordoma. A recombinant vaccine encoding brachyury that activates human T cells has been developed. It has yielded encouraging preliminary results in a phase 1 study for patients with progressive chordoma (Heery et al. 2015). These therapies, however, have been restricted to small adult clinical trials and require further evaluation for both adults and children.
11 Summary
Proton therapy offers excellent chances of permanent local control survival and for many children patients with skull base chordomas. The outcomes data for chondrosarcomas are already excellent for essentially all patient groups. They are good for pediatric chordoma patients with good prognostic factors, namely small residual disease. For unfavorable prognostic factors, further improvements are needed that should be best established by enrolling patients in prospective clinical trials. The role of systemic therapy needs further refinements. Late effects data to elucidate on the long-term effects of high-dose proton therapy are presently still missing, but high-grade toxicities within the first 3–5 years after therapy are within acceptable range.
References
Benk V, Liebsch NJ, Munzenrider JE, Efird J, McManus P, Suit H (1995) Base of skull and cervical spine chordomas in children treated by high-dose irradiation. Int J Radiat Oncol Biol Phys 31(3):577–581. doi:10.1016/0360-3016(94)00395-2
Bompas E, Le Cesne A, Tresch-Bruneel E, Lebellec L, Laurence V, Collard O, Saada-Bouzid E, Isambert N, Blay JY, Amela EY, Salas S, Chevreau C, Bertucci F, Italiano A, Clisant S, Penel N (2015) Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO)dagger. Ann Oncol 26:2168–2173. doi:10.1093/annonc/mdv300
Borba LA, Al-Mefty O, Mrak RE, Suen J (1996) Cranial chordomas in children and adolescents. J Neurosurg 84(4):584–591. doi:10.3171/jns.1996.84.4.0584
Debus J, Hug EB, Liebsch NJ, O’Farrel D, Finkelstein D, Efird J, Munzenrider JE (1999) Dose-volume tolerance of the brainstem after high-dose radiotherapy. Front Radiat Ther Oncol 33:305–314
Di Maio S, Yip S, Al Zhrani GA, Alotaibi FE, Al Turki A, Kong E, Rostomily RC (2015) Novel targeted therapies in chordoma: an update. Ther Clin Risk Manag 11:873–883. doi:10.2147/TCRM.S50526
Habrand JL, Datchary J, Bolle S, Beaudre A, de Marzi L, Beccaria K, Stefan D, Grill J, Dendale R (2016) Chordoma in children: case-report and review of literature. Rep Pract Oncol Radiother 21(1):1–7. doi:10.1016/j.rpor.2015.10.007
Heery CR, Singh BH, Rauckhorst M, Marte JL, Donahue RN, Grenga I, Rodell TC, Dahut W, Arlen PM, Madan RA, Schlom J, Gulley JL (2015) Phase I trial of a yeast-based therapeutic cancer vaccine (GI-6301) targeting the transcription factor brachyury. Cancer Immunol Res 3(11):1248–1256. doi:10.1158/2326-6066.CIR-15-0119
Hindi N, Casali PG, Morosi C, Messina A, Palassini E, Pilotti S, Tamborini E, Radaelli S, Gronchi A, Stacchiotti S (2015) Imatinib in advanced chordoma: a retrospective case series analysis. Eur J Cancer 51(17):2609–2614. doi:10.1016/j.ejca.2015.07.038
Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE (2006) Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol 30(7):811–818. doi:10.1097/01.pas.0000209828.39477.ab
Hug EB, Loredo LN, Slater JD, DeVries A, Grove RI, Schaefer RA, Rosenberg AE, Slater JM (1999) Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg 91(3):432–439. doi:10.3171/jns.1999.91.3.0432
Lau CS, Mahendraraj K, Ward A, Chamberlain RS (2016) Pediatric chordomas: a population-based clinical outcome study involving 86 patients from the Surveillance, Epidemiology, and End Result (SEER) Database (1973-2011). Pediatr Neurosurg 51(3):127–136. doi:10.1159/000442990
Lomax AJ, Bohringer T, Bolsi A, Coray D, Emert F, Goitein G, Jermann M, Lin S, Pedroni E, Rutz H, Stadelmann O, Timmermann B, Verwey J, Weber DC (2004) Treatment planning and verification of proton therapy using spot scanning: initial experiences. Med Phys 31(11):3150–3157. doi:10.1118/1.1779371
Marks LB, Yorke ED, Jackson A, Ten Haken RK, Constine LS, Eisbruch A, Bentzen SM, Nam J, Deasy JO (2010) Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys 76(3 Suppl):S10–S19. doi:10.1016/j.ijrobp.2009.07.1754
Munzenrider JE, Liebsch NJ (1999) Proton therapy for tumors of the skull base. Strahlenther Onkol 175(Suppl 2):57–63
Rombi B, Ares C, Hug EB, Schneider R, Goitein G, Staab A, Albertini F, Bolsi A, Lomax AJ, Timmermann B (2013) Spot-scanning proton radiation therapy for pediatric chordoma and chondrosarcoma: clinical outcome of 26 patients treated at paul scherrer institute. Int J Radiat Oncol Biol Phys 86(3):578–584. doi:10.1016/j.ijrobp.2013.02.026
Sahgal A, Chan MW, Atenafu EG, Masson-Cote L, Bahl G, Yu E, Millar BA, Chung C, Catton C, O’Sullivan B, Irish JC, Gilbert R, Zadeh G, Cusimano M, Gentili F, Laperriere NJ (2015) Image-guided, intensity-modulated radiation therapy (IG-IMRT) for skull base chordoma and chondrosarcoma: preliminary outcomes. Neuro Oncol 17(6):889–894.
Stacchiotti S, Casali PG (2011) Systemic therapy options for unresectable and metastatic chordomas. Curr Oncol Rep 13(4):323–330. doi:10.1007/s11912-011-0176-x
Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, Bertuzzi A, Tamborini E, Pilotti S, Messina A, Spreafico C, Gronchi A, Amore P, Vinaccia V, Casali PG (2012) Phase II study of imatinib in advanced chordoma. J Clin Oncol 30(9):914–920. doi:10.1200/JCO.2011.35.3656
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Hug, E.B. (2018). Proton Therapy for Skull Base Chordomas in the Pediatric and Adolescent Patient. In: Mahajan, A., Paulino, A. (eds) Radiation Oncology for Pediatric CNS Tumors. Springer, Cham. https://doi.org/10.1007/978-3-319-55430-3_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-55430-3_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55428-0
Online ISBN: 978-3-319-55430-3
eBook Packages: MedicineMedicine (R0)