
Abstract
A growing body of literature has begun to delineate the unique and potent biological properties of vitamin E involved in cardiovascular disease, cancer, chronic inflammation, and neurodegeneration. Vitamin E was initally proposed as a dietary factor essential in preventing embryonic mortality and soon after considered as lipid-soluble antioxidant that inhibits lipid peroxidation by scavenging reactive oxygen species. Recent mechanistic studies indicate that vitamin E possesses functions that are independent of its antioxidant ability and mainly modulate cell signal transduction and gene expression. Despite overt vitamin E deficiency is rare, most commonly it can be found in children with inherited abnormalities that prevent the absorption or maintenance of normal blood concentration of vitamin E. This review summarizes the genetic disorders associated with congenital vitamin E deficiency. In parallel, it provides a brief overview on the historical, metabolic, and functional aspects of vitamin E.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Since its discovery, research recognized potential ability of vitamin E to prevent chronic diseases and clinical syndromes. Particularly this was supported by a number of clinical conditions believed to have an oxidative stress component such as diabetes (Evans et al. 2002), cardiovascular (Rimm et al. 1993), neurodegenerative diseases (Butterfield et al. 2002), and cancer (London et al. 1992) (Fig. 1). Nonetheless, the role of this vitamin in other conditions is still debated (Rossato and Mariotti 2014).

Summary of vitamin E activities
In the recent years, it has become increasingly evident that not all actions of vitamin E are dependent on its antioxidant properties, but may involve nonantioxidant activities, especially those connected with cell signal transduction, protein expression, enzyme activities, and gene expression regulation (Zingg and Azzi 2004).
The purpose of this review is to describe the congenital disorders associated with vitamin E deficiency. In addition, we sought to provide a concise summary of the history, metabolism, and regulatory functions of vitamin E.
History, Metabolism, and Function of Vitamin E: General Overview
Historical Background
Vitamin E was first discovered in 1922 by Evans and Bishop as a dietary factor essential for reproduction in rats. They found that laboratory rats failed to reproduce when lard was their only source of food fat. According to the researchers, there was a compound in both wheat germ and lettuce that corrected the problem and they decided to call it “Factor X” and the “antisterility factor” (Evans and Bishop 1922). Similar results were observed by Sure and he called it “Vitamin E,” as by then vitamins A, B, C, and D were already known (Sure 1924). In 1936, Evans et al. isolated a pure compound from the nonsaponifiable fraction of wheat-germ oil having the properties of vitamin E. This active compound was called alpha-tocopherol (α-tocopherol), from the ancient Greek word phero meaning “to bring” and tocos, meaning “childbirth” (Evans et al. 1936).
Natural vitamin E now encompasses a family of eight fat-soluble isoforms that exhibit the biological activity of α-tocopherol: the α-, β-, γ-, and δ-tocopherols and the α-, β-, γ-, and δ-tocotrienols, which are synthesized by plants from homogentisic acid (Rimbach et al. 2002).
Tocopherols and tocotrienols both include a chromanol ring and differ in their side chains. Saturated phytyl side chain is involved in the structure of tocopherols, while unsaturated geranylgeranyl side chain with three double bonds is involved in the structure of tocotrienols (Kamal-Eldin and Appelqvist 1996). Tocopherols and tocotrienols are classified as α-, β-, γ-, and δ according to the methyl group on the chromanol ring. Among the eight naturally occurring forms of vitamin E family, α-tocopherol is considered the most common form in human tissues followed by γ-tocopherol, while tocotrienols are usually not detected in tissues (Frank et al. 2012). Tocopherols are exclusively synthesized by photosynthetic organisms, and plant-derived oils are the major sources of vitamin E in the human diet. The most common form of tocopherol in the North American diet is γ-tocopherol, the predominant form of vitamin E in corn oils, while the most common form in European diets is α-tocopherol, found in olive and sunflower oils (Dutta and Dutta 2003). Tocotrienols are found in palm oil, barley, oats, and rice bran, and have higher antioxidant activity than tocopherols.
Metabolism
The vitamin Es present in ingested food, either as a free molecule or esterified, leave the stomach to be hydrolyzed in the duodenal lumen by the pancreatic lipases and subsequently absorbed through the brush border membrane of the enterocytes (Borel et al. 2001; Hacquebard and Carpentier 2005). After their uptake, vitamin E isomers reach the basolateral side of enterocytes to be equally incorporated into chylomicrons together with triacylglycerol, phospholipids, and cholesterol (Hacquebard and Carpentier 2005).The chylomicron-bound vitamin E forms are transported via lymphatic system into the circulation where the triaclyglycerol components of the chylomicrons are hydrolyzed by lipoprotein lipase of the capillary endothelium and adipose tissue forming lipid-depleted chylomicrons components (Hacquebard and Carpentier 2005). The chylomicrons remnants containing most of the absorbed tocopherols and tocotrienols are then taken up by the liver (via the endocytic receptors, such as the low-density lipoprotein (LDL) receptor and heparin sulfate proteoglycans) which sorts out α-tocopherol and preferentially secretes it within very low-density lipoprotein (VLDL) and high-density lipoprotein (HDL) into the bloodstream for distribution in the body (Traber et al. 1993). On the other hand, excess α-tocopherol and the other tocopherols and tocotrienol analogues are either excreted unchanged or metabolized before elimination in urine and bile (Traber et al. 1993). This selective accumulation of α-tocopherol is mediated by hepatic cytosolic protein, α-tocopherol-transfer protein (α-TTP) which preferentially binds to α-tocopherol over other vitamers (Eggermont 2006) (Fig. 2). While α-TTP presents high affinity to α-tocopherol (100%), it has much lower affinity toward other vitamin E forms, e.g., 38%, 9%, or 1% affinity to β-, γ-, and δ- tocopherols, respectively (Eggermont 2006). In addition, it was proposed that non-α-tocopherol forms of vitamin E are catabolized in liver into carboxyethyl hydroxychromanol (CEHC) metabolites via cytochrome P450 (CYP4F2) initiated ω-hydroxylation and oxidation followed by β-oxidation of the phytyl chain (Birringer et al. 2002; Sontag and Parker 2007).

Absorption, transport, and metabolism of vitamin E. All forms of vitamin E are absorbed equally. Intestinal enzymatic digestion is followed by the distribution to the liver and nonhepatic tissues. Discrimination between the different forms of vitamin E in favor of α-tocopherol occurs mainly in the liver by α-TTP, which protects α-tocopherol from excessive degradation and excretion (With permission from Eggermont 2006)
Molecular Function of Vitamin E
Antioxidant Activity
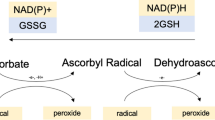
Vitamin E is widely accepted as one of the most potent antioxidant. Biochemically, tocopherols and tocotrienols are potent lipophilic antioxidants by scavenging lipid peroxyl radicals via donating hydrogen from the phenolic group on the chromanol ring and thus neutralize free radicals or reactive oxygen species (ROS) (Jiang et al. 2001). At equal molar concentrations in vitro, because of possessing similar phenolic moiety, all vitamin E forms are considered to have potent antioxidant activities. It was reported that the α-tocopherol and γ-tocopherol isoforms and the tocotrienol forms have relatively similar capacity to scavenge ROS during lipid oxidation (Yoshida et al. 2007). In vivo, there is likely more ROS scavenging by α-tocopherol than γ- tocopherol since it is at a 10-fold higher concentration within tissues. In addition to scavenging ROS, γ-tocopherol, in contrast to α-tocopherol, also reacts with nitrogen species such as peroxynitrite forming 5-nitro-γ-tocopherol (Wolf 1997).
Nonantioxidant Activity
As already mentioned in the introduction, it has been reported that vitamin E exhibits some properties that cannot be assigned to its antioxidant capacity. Here below we summarize some of those nonantioxidant functions.
Effects on Enzyme Inhibition
Protein kinase C (PKC) is a serine/threonine kinase that utilizes the cofactors phosphatidylserine, diacylglycerol, and calcium for activation. It is considered one of the major cellular transduction systems triggered by various ligands as hormones, neurotransmitters, and growth factors (Azzi et al. 1992). PKC is one of the pathways used by α-tocopherol (Boscoboinik et al. 1991) where it exerts a specific inhibitory activity compared to β-tocopherol (Tasinato et al. 1995). In monocytes this leads to the inhibition of phosphorylation and translocation of the cytosolic factor p47 (phox) and to an impaired assembly of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and of superoxide production (Cachia et al. 1998). Moreover, α-tocopherol exerted an antiproliferative effect on vascular smooth muscle cell model by inhibiting PKC pathway (Özer et al. 1998). However, these inhibitory effects were not observed by its isomer β-tocopherol or another antioxidant (probucol) (Özer et al. 1998). Additionally, α-tocopherol protects against endothelial damage by regulating the endothelial cell PKC (Abdala-Valencia et al. 2012). Remaining within this context, vitamin E has shown to increase the production of vasodilator prostanoids by suppressing cyclooxygenase (COX) activity in cellular environments and therefore effectively contribute to the anti-inflammatory process (Wu et al. 2005).
Effects on Gene Expression
It has been reported that vitamin E regulates the expression of genes involved in oxidative stress, proliferation, inflammation, and apoptosis. Some of these gene classes modulated by vitamin E include α-TTP, scavenger receptors (CD36, SR-BI, SR-AI/II), P450-Cytochromes, transcriptional factors (NF-κB, AP-1), genes involved in the modulation of extracellular proteins (tropomyosin, collagen-alpha-1, matrix metalloproteinases (MMP-1, -19), and connective tissue growth factor) (Azzi et al. 2004; Brigelius-Flohé 2009).
Moreover, genes connected to adhesion and inflammation (E-selectin, Intercellular Adhesion Molecule 1, integrins, glycoprotein IIb, IL-2, IL-4, IL-1b, and transforming growth factor-beta), lipid metabolism (Apolipoprotein E, peroxisome proliferator-activated receptor gamma, LDL- receptor), and cell cycle regulation are also affected by α-tocopherol at the transcriptional level (Brigelius-Flohé 2009).
Despite well-documented antioxidant and other beneficial effects as well as negative association between α-tocopherol intake and chronic diseases, supplementation of α-tocopherol has failed to offer consistent benefits to prevention of chronic diseases including cancer and cardiovascular diseases in many large clinical intervention studies (Moya-Camarena and Jiang 2012; Myung et al. 2013; Papaioannou et al. 2011; Dolara et al. 2013). On the other hand, accumulating evidence suggests that metabolites mainly deriving from non-α-tocopherol vitamers have unique properties that are superior to α-tocopherols.
For instance, short-chain carboxychromanols like CEHCs generated by CYP4F2-initiated ω- oxidation of the side chain have been shown to have natriuretic activities (Wechter et al. 1996). In hemodialysis patients, supplementation of γ-tocopherol but not α-tocopherol is associated with a consistent increase of serum CEHC and a decrease in pro-inflammatory IL-6 and C-reactive protein (Himmelfarb et al. 2003). Moreover, long-chain carboxychromanols, including 13′-carboxychromanol, exhibit a potent anti-inflammatory (Jiang et al. 2008) and anticancer (Birringer et al. 2010) effects that may provide new insights into physiological role of less tissue-preserved forms of vitamin E.
Serum Vitamin E Assessment
The adequacy of the mean range of α-tocopherol intake is difficult to define being strongly influenced by concentration of circulating lipids, and does not accurately reflect tissue vitamin levels. Serum α-tocopherol concentrations less than 12 μmol/L were defined by the Institute of Medicine (IOM) to be in the deficient/inadequate range for healthy adults (Institute of Medicine 2000). It is recognized that vitamin E inadequacy is associated with increased erythrocyte fragility. Therefore, results from hydrogen peroxide-induced erythrocyte lysis test were used to define vitamin E status and consequently to assess vitamin E supplementation (Institute of Medicine 2000). As will be discussed in the next section, most commonly low α-tocopherol concentrations are caused by the combination of consumption of diets low in vitamin E, along with inadequate intakes or absorption of fat, protein, and calories. These latter dietary components are necessary for fat absorption and thus for vitamin E absorption and its lipoprotein transport.
Congenital Disorders with Vitamin E Deficiency
Vitamin E deficiency is seldom found in adults but more frequently can be found in children, likely as a result of genetic defects that lead to fat-malabsorption syndromes or a rapid depletion of plasma α-tocopherol. In this section we highlight the pathophysiology, clinical presentation, and management of the main genetic disorders associated with vitamin E deficiency (Table 1).
Ataxia with Vitamin E Deficiency
Human vitamin E defici ency symptoms began to be reported in the 1960s in various case studies of patients with lipoprotein abnormalities. Since these patients had malabsorption of other nutrients, it was not clear the extent to which various symptoms could be attributed to lack of vitamin E (Kayden et al. 1965). In the early 1980s, studies of humans with vitamin E deficiency symptoms without fat malabsorption began to appear in the literature (Burck et al. 1981). This form of vitamin E deficiency was later called AVED, or familial isolated vitamin E deficiency (Doerflinger et al. 1995). AVED (MIM 277460) is a rare genetic neurodegenerative disease transmitted in an autosomal recessive mode, and caused by mutations in the α-tocopherol transfer protein gene (TTPA) located on chromosome 8q13 (Doerflinger et al. 1995). This defect impairs incorporation of vitamin E into plasma VLDL and thus cannot reach peripheral circulation.
The concomitant presence of specific neurological phenotype and low plasma levels of vitamin E, in the absence of other clinical conditions associated with fat malabsorption, can guide the diagnosis for AVED (Anheim et al. 2010). The onset of neurologic features in AVED is between 4 and 18 years of age with a phenotype that resembles patients with Friedreich’s ataxia, a genetic disorder caused by a mutation in the gene for frataxin, a mitochondrial iron-binding protein (Bradley et al. 2000; Yokota et al. 1997).
The signs and symptoms are usually devastating and progressive and mainly including truncal and extremity ataxia, loss of deep tendon reflexes, disturbances in proprioceptive and vibratory sensations, dysarthria, and positive Babinski sign (El Euch-Fayache et al. 2014). Head titubation, retinopathy, and dystonia are more common in patients with AVED while cardiomyopathy, glucose intolerance, scoliosis, and foot deformities in Friedreich ataxia (Cavalier et al. 1998; Benomar et al. 2002).
The molecular mechanism that underlies the neurological damage in patients with AVED is not yet known in detail, but oxidative stress due to reduced delivery of vitamin E to the central nervous system is likely to play a major role (Copp et al. 1999). Neuropathological findings derived from human and animal models with vitamin E deficiency documented the presence of severe dying back-type degeneration of the posterior column and massive accumulation of lipofuscin in neurons including dorsal root ganglion cells (Yokota et al. 2000).
Different mutations have been described in different ethnic groups. In North Africa populations, the most frequent mutation is 744delA on chromosome 8q13 (Cavalier et al. 1998; Gabsi et al. 2001). The mutation 513insTT predominates in AVED families of North European origin, 175 C4T (R59W) on exon 1 and 437delT on exon 3 in the case reported from Netherland as well as G552A on exon 3 in Japan (Yokota et al. 1997). In Mediterranean region, both the 744delA and 513insTT account for approximately 80% of the TTPA mutated alleles in Italian AVED (Doerflinger et al 1998, Cavalier et al. 1998).
Several studies described a correlation between the type of mutation and the function of the α-tocopherol protein and thereby vitamin E serum level and the severity of the neurological signs (Cavalier et al. 1998; Mariotti et al. 2004). Different mutations have been reported, including missense, nonsense, frameshift, and splice site mutations, and may affect the severity of the disease, presumably via residual protein activity with certain mutations (Cavalier et al. 1998).
Today experts agree that there is no specific instrumental approach for establishing the diagnosis of AVED (Harding et al. 1982). However, the presence of sensory neuropathy with normal motor conduction and absent or markedly reduced sensory nerve action potentials (SNAPs) is considered a neurophysiologic hallmark of this disease. Additionally, it has been described a distal motor neuropathy with normal sensory conduction AVED patient (Fusco et al. 2008).
Individuals with AVED are treated with life-long vitamin E supplementation. Remarkably, the administration vitamin E in early stages of the disease seems to prevent the progression of neurological impairment, atherosclerosis, and retinopathy in these patients (Marzouki et al. 2005) and can mildly improve cerebellar ataxia (Gabsi et al 2004). A mouse model has been developed that shows late-onset head tremor, ataxia, and retinal degeneration, the neurological aspects of which resolve with supplementation of vitamin E (Yokota et al. 2001).
Currently, vitamin E is manufactured as the acetate esters of RRR-α-tocopherol by using plant materials and all-racemic-α-tocopherol by chemical synthetic methods. Even though, α-TTP preferentially packages RRR-α-tocopherol into nascent VLDL, however, some AVED individuals lacking α-TTP or with a marked defect in the RRR-α-tocopherol binding site cannot discriminate between α-tocopherol stereoisomers (Traber et al. 1993; Cavalier et al. 1998).
Cavalier et al. suggested that administration of 800 mg RRR α-tocopherol twice daily, with meals that contain fat, results in plasma α-tocopherol levels at or above the normal range (Cavalier et al. 1998).
Abetalipoproteinemia
Abetalipoproteinemia (ABL; MIM 200100) or Bassen-Kornzweig syndrome is a rare autosomal-recessive disease that is characterized by very low plasma concentrations of triglyceride and cholesterol (under 30 mg/dl) and undetectable levels of LDL and apolipoprotein (apo) B (Berriot-Varoqueaux et al. 2000). It is caused by a mutation in microsomal triglyceride transfer protein (MTP) gene on chromosome 4q22-24 (Wang and Hegele 2000). MTP, physiologically expressed on the luminal side of the endoplasmic reticulum in intestine, is involved in the assembly of chylomicrons in the enterocytes and of VLDL particles in hepatocytes (Wang and Hegele 2000).
Despite the gold standard diagnostic test would be by sequencing the MTP gene, and a clinical diagnosis can be made for ABL based on lipid profile, blood smear, and clinical symptoms. Key clinical features in ABL patients in the first years of life are steatorrhea due to fat malabsorption, and failure to thrive. This is often accompanied by digestive symptoms, such as diarrhea, vomiting, and abdominal distension (Burnett et al. 2012).
Additionally, neurological disorders which may appear later in childhood due to profound vitamin E deficiency often have the greatest impact on quality of life especially if there has been no therapeutic intervention. Absent tendon reflexes is an early clinical sign, followed by deep sensory loss in the lower limbs and then a spinocerebellar syndrome with an ataxic gait, dysmetria, and dysarthria (Berriot-Varoqueaux et al. 2000; Kane and Havel 2001). Ophthalmological findings may include atypical retinitis pigmentosa where the presence of lipofuscin pigment in the retina suggests that vitamin E deficiency plays a central role in this retinopathy (Berriot-Varoqueaux et al. 2000, Kane and Havel 2001).
Among laboratory investigations, a blood smear may show acanthocytosis that results from either vitamin E deficiency or an altered membrane lipid composition (Kane and Havel 2001). As mentioned previously, lipid profile shows decrease of plasma levels of cholesterol and triglycerides and almost undetectable levels of apoB-containing lipoproteins including chylomicrons, VLDL, and LDL (Berriot-Varoqueaux et al. 2000; Wang and Hegele 2000). The current standard treatment consists of strict adherence to specialized diets and oral vitamin E supplementation. Total fat intake should be restricted to less than 30% of the total caloric intake which will eliminate steatorrhea and allow absorption of nutrients essential for growth and development (Berriot-Varoqueaux et al. 2000; Wang and Hegele 2000; Zamel et al. 2008).
High-dose oral vitamin E supplementation (100–300 mg/kg/day) is recommended to halt the progression of the neurological deterioration (Zamel et al. 2008). Combined oral vitamin E and A is usually administered to attenuate the severity of retinal degeneration (Zamel et al. 2008).
Hypobetalipoproteinemia
Hypobetalipoproteinemia (HBL) represents a rare co-dominant condition characterized by low plasma levels of total cholesterol, low-density lipoprotein-cholesterol (LDL-C), and apoB below the 5th percentile of the general population (Tarugi et al. 2007). Familial hypobetalipoproteinemia (FHBL; MIM 107730) is the most frequent monogenic form of HBL. It may be due to loss-of-function mutations in apoB gene (APOB-linked FHBL) or, less frequently, in PCSK9 (PCSK9-linked FHBL) (Tarugi et al. 2007; Schonfeld 2003). However, in many subjects the genetic basis of FHBL remains unexplained (Orphan FHBL) (Tarugi et al. 2007). Most APOB gene mutations lead to the formation of truncated apoB protein of various sizes (Tarugi et al. 2007, Schonfeld 2003). Missense nontruncating mutations of the APOB gene can also cause FHBL (Burnett et al. 2003).
The best-characterized form of FHBL occurs with a dominant mode of inheritance and it has been linked to heterozygous pathogenic mutations in the APOB gene that are generally asymptomatic but often develop liver steatosis (Tarugi et al. 2007). Patients with the clinical diagnosis of homozygous FHBL are rare and they can be either carriers of homozygous or compound heterozygous mutations in APOB gene (Tarugi et al. 2007). The clinical manifestations of homozygous FHBL show great variability. They may have a similar biochemical and clinical phenotype to patients affected by ABL due to fat malabsorption and fat-soluble vitamin deficiency, particularly vitamins A and E (Lee and Hegele 2014).
Chylomicron Retention Disease
Chylomicron retention disease (CMRD ; MIM 246700), also called Anderson disease, is an autosomal recessive disorder condition characterized by the accumulation of lipid droplets within the enterocytes and the selective absence of apoB-48-containing particles from plasma (Boldrini et al. 2001). CMRD is caused by homozigous and compound heterozygous mutations in SAR1B, a gene encoding Sar1b protein, which is involved in chylomicron trafficking from the endoplasmic reticulum to the Golgi apparatus (Jones et al. 2003). Until now, missense mutations have represented the majority of SAR1B mutations.
CMRD presents shortly after birth with malabsorptive diarrhea and failure to thrive, with vomiting and abdominal distension often present (Peretti et al. 2010). Vitamin E is the most affected among the liposoluble vitamins in CMRD, because its transport is highly dependent on apo B-containing lipoproteins (Berriot-Varoqueaux et al. 2000). Hepatomegaly and hepatic steatosis may develop in some patients, but in contrast to ABL and FHBL, liver cirrhosis has not been reported in CMRD (Peretti et al. 2010). The neurological complications are usually an alarm sign of vitamin E deficiency in these subjects and include hyporeflexia and loss of proprioception in adolescents and ataxia, myopathy, and sensory neuropathy in adults (Peretti et al. 2010).
In CMRD, total cholesterol, LDL cholesterol, and HDL cholesterol concentrations are low, but triglyceride levels are generally normal. An increased plasma creatine kinase concentration of up to five times the normal level may be observed from infancy (Peretti et al. 2010).
Regarding the management of CMRD patients, there are no specific recommendations for the follow-up or treatment of CMRD, even if low-fat diet regimens have shown to improve digestive symptoms in these patients (Peretti et al. 2010).
Cystic Fibrosis
Cystic Fibrosis (CF; MIM 219700) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, located on chromosome 7. The CFTR protein is a chloride ion channel and is expressed in many different organs (e.g., the pancreas, airways, lungs, liver, salivary glands, sweat and reproductive tract glands) (Gibson et al. 2003). CFTR protein dysfunction leads to defective ion transport across epithelial surfaces, which causes impaired mucociliary clearance. The most common manifestations in newborns and children include sinopulmonary symptoms, failure to thrive, steatorrhea, and meconium ileus (Gibson et al. 2003). However, intestinal malabsorption mediated by pancreatic insufficiency, deranged bile acid function, and enzyme inactivation by hyperacidity remains the most common feature in CF subjects (Peretti et al. 2005). Vitamin E status in these individuals has shown to be associated with hemolytic anemia (Wilfond et al. 1994), cognitive impairment (Koscik et al. 2005), and increased rate of pulmonary exacerbations (Hakim et al. 2007). Today, it has been recommended to include a dosage of 100 UI to 400 UI per day for all subjects with CF (Sinaasappel et al. 2002).
Conclusion
Vitamin E deficiency is quite rare in adult humans. It is more frequently found in children with an inherited condition that impairs their ability to absorb this vitamin.
It has been shown that vitamin E plays a vital role in various disorders through its oxidative properties, although it has also nonoxidative effects in humans. Therefore, further studies should be addressed beyond the free radical-scavenging properties of vitamin E and its metabolites.
In spite of the promising potential, the experimental analysis of tocotrienols accounts for only a small portion of vitamin E research. Hence, the current state of knowledge deserves further investigation into this lesser known form of vitamin E.
It must be emphasized that early supplementation of vitamin E could halt the disease progress and a fast progression of the disease should be avoided. However, it is well known that this vitamin is transported with lipoproteins, and therefore its plasma concentration is not indicative of whole body and peripheral tissue stores. In this context, additional markers of inadequate vitamin E status especially in newborns are needed. This would also promote the evaluation of vitamin E supplementation outcomes in terms of beneficial effects and safety.
Policies and protocols
In this chapter we described the genetic disorders associated with vitamin E deficiency in humans. Despite vitamin E deficiency is rare, it can occur as a result of genetic abnormalities in α-tocopherol-transfer protein or as a result of fat malabsorption syndromes (abetalipoproteinemia, hypobetalipoproteinemia, chylomicron retention disease, and cystic fibrosis).
-
Generally, vitamin E deficiency manifests in early childhood by progressive neurologic damage and serious clinical consequences. Therefore, policies focused on the prompt recognition of individuals with vitamin E deficiency should be considered.
-
The physiological role and the health consequences of vitamin E deficiency should get the desired attention in international micronutrient recommendations. In addition, public health authorities should encourage large-scale research studies to determine optimal vitamin E dosage and to evaluate the outcomes.
-
As described previously, hydrogen peroxide-induced erythrocyte lysis test is used to define vitamin E status and consequently to assess vitamin E supplementation. However, when interpreting the results it is important to consider serum lipid profile components in these individuals.
-
A full clinical investigation is recommended in individuals diagnosed by AVED. In addition to laboratory testing, assessment must include neurologic, ophthalmologic, and cardiac examination. Moreover, it is appropriate to perform a predictive genetic testing in at-risk families for the purpose of early diagnosis and treatment.
Dictionary of Terms
-
Tocopherols and tocotrienols – Two main families of vitamin E. Each composed of four forms with different chemical structure.
-
Reactive oxygen species – Highly reactive molecules produced by physiologic and nonphysiologic processes and may cause damage to cell membrane or DNA molecules.
-
Antioxidant – Is a molecule that protects against harmful chemical reactions inside the organism.
-
Malabsorption – Impairment in absorption of food nutrients from intestinal tract.
-
Congenital disorder – When a baby has a disease that is present from birth. It can be inherited from mother and/or father (genetic disorder) or caused by environmental factors.
Summary
-
Vitamin E is a fat-soluble vitamin that refers to a group of compounds that include both tocopherols and tocotrienols.
-
Vitamin E is a potent antioxidant that acts as a peroxyl radical scavenger and can play a role in regulating cell signaling and modulating gene transcription through a nonantioxidant activity.
-
Vitamin E has been shown to play a role in cardiovascular disease, cancer, chronic inflammation, and neurodegeneration.
-
In humans severe vitamin E deficiency occurs as a result of genetic defects in the α-tocopherol transfer protein gene or in presence fat-malabsorption syndromes.
-
Early supplementation of vitamin E may prevent and reverse clinical complications of vitamin E deficiency.
-
Future research should focus on finding biomarkers of inadequate vitamin E status to optimize both diagnostic and therapeutic approaches in individuals with vitamin E deficiency.
Abbreviations
- ABL:
-
Abetalipoproteinemia
- AD:
-
Autosomal dominant
- apo:
-
Apolipoprotein
- AR:
-
Autosomal recessive
- AVED:
-
Ataxia with vitamin E deficiency
- CEHC:
-
Carboxyethyl hydroxychromanol
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- CMRD:
-
Chylomicron retention disease
- COX:
-
Cyclooxygenase
- FFA:
-
Free fatty acids
- FHBL:
-
Familial hypobetalipoproteinemia
- HBL:
-
Hypobetalipoproteinemia
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- LDL-C:
-
Low-density lipoprotein-cholesterol
- MIM:
-
Mendelian Inheritance in Man
- MMPs:
-
Matrix metalloproteinases
- MTP:
-
Microsomal triglyceride transfer protein
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- PKC:
-
Protein kinase C
- ROS:
-
Reactive oxygen species
- TTPA:
-
α-tocopherol transfer protein gene
- VLDL:
-
Very low-density lipoprotein
- α-TTP:
-
α-Tocopherol-transfer protein
References
Abdala-Valencia H, Berdnikovs S, Cook-Mills JM (2012) Vitamin E isoforms differentially regulate intercellular adhesion molecule-1 activation of PKCα in human microvascular endothelial cells. PLoS One 7:e41054
Anheim M, Fleury M, Monga B, Laugel V, Chaigne D, Rodier G, Ginglinger E, Boulay C, Courtois S, Drouot N et al (2010) Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics 11:1–12
Azzi A, Boscoboinik D, Hensey C (1992) The protein kinase C family. Eur J Biochem 208:547–557
Azzi A, Gysin R, Kempná P, Munteanu A, Negis Y, Villacorta L, Visarius T, Zingg JM (2004) Vitamin E mediates cell signaling and regulation of gene expression. Ann N Y Acad Sci 1031:86–95
Benomar A, Yahyaoui M, Meggouh F, Bouhouche A, Boutchich M, Bouslam N, Zaim A, Schmitt M, Belaidi H, Ouazzani R et al (2002) Clinical comparison between AVED patients with 744 del A mutation and Friedreich ataxia with GAA expansion in 15 Moroccan families. J Neurol Sci 198:25–29
Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR (2000) The role of the microsomal triglygeride transfer protein in abetalipoproteinemia. Annu Rev Nutr 20:663–697
Birringer M, Pfluger P, Kluth D, Landes N, Brigelius-Flohé R (2002) Identities and differences in the metabolism of tocotrienols and tocopherols in HepG2 cells. J Nutr 132:3113–3118
Birringer M, Lington D, Vertuani S, Manfredini S, Scharlau D, Glei M, Ristow M (2010) Proapoptotic effects of long-chain vitamin E metabolites in HepG2 cells are mediated by oxidative stress. Free Radic Biol Med 49:1315–1322
Boldrini R, Biselli R, Bosman C (2001) Chylomicron retention disease – the role of ultrastructural examination in differential diagnosis. Pathol Res Pract 197:753–757
Borel P, Pasquier B, Armand M, Tyssandier V, Grolier P, Alexandre-Gouabau MC, Andre M, Senft M, Peyrot J, Jaussan V et al (2001) Processing of vitamin A and E in the human gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 280:G95–G103
Boscoboinik D, Szewczyk A, Hensey C, Azzi A (1991) Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J Biol Chem 266:6188–6194
Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH (2000) Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum Mol Genet 9:275–282
Brigelius-Flohé R (2009) Vitamin E: the shrew waiting to be tamed. Free Radic Biol Med 46:543–554
Burck U, Goebel HH, Kuhlendahl HD, Meier C, Goebel KM (1981) Neuromyopathy and vitamin E deficiency in man. Neuropediatrics 12:267–278
Burnett JR, Shan J, Miskie BA, Whitfield AJ, Yuan J, Tran K, McKnight CJ, Hegele R, Yao Z (2003) A novel nontruncating APOB gene mutation, R463W, causes familial hypobetalipoproteinemia. J Biol Chem 278:13442–13452
Burnett JR, Bell DA, Hooper AJ, Hegele RA (2012) Clinical utility gene card for: Familial hypobetalipoproteinaemia (APOB). Eur J Hum Genet 20(8). https://doi.org/10.1038/ejhg.2012.85
Butterfield DA, Castegna A, Drake J, Scapagnini G, Calabrese V (2002) Vitamin E and neurodegenerative disorders associated with oxidative stress. Nutr Neurosci 5:229–239
Cachia O, Benna JE, Pedruzzi E, Descomps B, Gougerot-Pocidalo MA, Leger CL (1998) alpha-tocopherol inhibits the respiratory burst in human monocytes. Attenuation of p47(phox) membrane translocation and phosphorylation. J Biol Chem 273:32801–32805
Cavalier L, Ouahchi K, Kayden HJ, Di Donato S, Reutenauer L, Mandel JL, Koenig M (1998) Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet 62:301–310
Copp RP, Wisniewski T, Hentati F, Larnaout A, Ben Hamida M, Kayden HJ (1999) Localization of alpha-tocopherol transfer protein in the brains of patients with ataxia with vitamin E deficiency and other oxidative stress related neurodegenerative disorders. Brain Res 822:80–87
Doerflinger N, Linder C, Ouahchi K, Gyapay G, Weissenbach J, Le Paslier D, Rigault P, Belal S, Ben Hamida C, Hentati F et al (1995) Ataxia with vitamin E deficiency: refinement of genetic localization and analysis of linkage disequilibrium by using new markers in 14 families. Am J Hum Genet 56:1116–1124
Dolara P, Bigagli E, Collins A (2013) Antioxidant vitamins and mineral supplementation, life span expansion and cancer incidence: a critical commentary. Eur J Nutr 51:769–781
Dutta A, Dutta SK (2003) Vitamin E and its role in the prevention of atherosclerosis and carcinogenesis: a review. J Am Coll Nutr 22:258–268
Eggermont E (2006) Recent advances in vitamin E metabolism and deficiency. Eur J Pediatr 165:429–434
El Euch-Fayache G, Bouhlal Y, Amouri R, Feki M, Hentati F (2014) Molecular, clinical and peripheral neuropathy study of Tunisian patient with ataxia with vitamin E deficiency. Brain 137:402–410
Evans HM, Bishop KS (1922) On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 56:650–651
Evans HM, Emerson OH, Emerson GA (1936) The isolation from wheat germ oil of an alcohol, α-tocopherol, having the properties of vitamin E. J Biol Chem 113:319–332
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23:599–622
Frank J, Chin XW, Schrader C, Eckert GP, Rimbach G (2012) Do tocotrienols have potential as neuroprotective dietary factors? Ageing Res Rev 11:163–180
Fusco C, Frattini D, Pisani F, Gellera C, Della Giustina E (2008) Isolated vitamin E deficiency mimicking distal hereditary motor neuropathy in a 13-year-old boy. J Child Neurol 23:1328–1330
Gabsi S, Gouider-Khouja N, Belal S, Fki M, Kefi M, Turki I, Ben Hamida M, Kayden H, Mebazaa R, Hentati F (2001) Effect of vitamin E supplementation in patients with ataxia with vitamin E deficiency. Eur J Neurol 8:477–481
Gibson RL, Burns JL, Ramsey BW (2003) Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951
Hacquebard M, Carpentier YA (2005) Vitamin E: absorption, plasma transport and cell uptake. Curr Opin Clin Nutr Metab Care 8:133–138
Hakim F, Kerem E, Rivlin J, Bentur L, Stankiewicz H, Bdolach-Abram T, Wilschanski M (2007) Vitamins A and E and pulmonary exacerbations in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 45:347–353
Harding AE, Muller DP, Thomas PK, Willison HJ (1982) Spinocerebellar degeneration secondary to chronic intestinal malabsorption: a vitamin E deficiency syndrome. Ann Neurol 12:419–424
Himmelfarb J, Kane J, McMonagle E, Zaltas E, Bobzin S, Boddupalli S, Phinney S, Miller G (2003) Alpha and gamma tocopherol metabolism in healthy subjects and patients with end-stage renal disease. Kidney Int 64:978–991
Institute of Medicine (2000) Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. The National Academies Press, Washington, DC
Jiang Q, Christen S, Shigenaga MK, Ames BN (2001) gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr 74:714–722
Jiang Q, Yin X, Lill MA, Danielson ML, Freiser H, Huang J (2008) Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc Natl Acad Sci USA 105:20464–20469
Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S et al (2003) Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet 34:29–31
Kamal-Eldin A, Appelqvist LA (1996) The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 31:671–701
Kane JP, Havel RJ (2001) Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly WS (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp 2717–2752
Kayden HJ, Silber R, Kossmann CE (1965) The role of vitamin E deficiency in the abnormal autohemolysis of acanthocytosis. Trans Assoc Am Phys 78:334–342
Koscik RL, Lai HJ, Laxova A, Zaremba KM, Kosorok MR, Douglas JA, Rock MJ, Splaingard ML, Farrell PM (2005) Preventing early, prolonged vitamin E deficiency: an opportunity for better cognitive outcomes via early diagnosis through neonatal screening. J Pediatr 147:S51–S56
Lee J, Hegele RA (2014) Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis 37:333–339
London SJ, Stein EA, Henderson IC, Stampfer MJ, Wood WC, Remine S, Dmochowski JR, Robert NJ, Willett WC (1992) Carotenoids, retinol, and vitamin E and risk of proliferative benign breast disease and breast cancer. Cancer Causes Control 3:503–512
Mariotti C, Gellera C, Rimoldi M, Mineri R, Uziel G, Zorzi G, Pareyson D, Piccolo G, Gambi D, Piacentini S et al (2004) Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci 25:130–137
Marzouki N, Benomar A, Yahyaoui M, Birouk N, Elouazzani M, Chkili T, Benlemlih M (2005) Vitamin E deficiency ataxia with (744 del A) mutation on alpha-TTP gene: genetic and clinical peculiarities in Moroccan patients. Eur J Med Genet 48:21–28
Moya-Camarena SY, Jiang Q (2012) The role of vitamin E forms in cancer prevention and therapy-Studies in human intervention trials and animal models. In: Sarkar FH (ed) Nutraceuticals and Cancer. Springer, New York, pp 323–354. ISBN:978-94-007-2629-1
Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ (2013) Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ 346:f10
Özer NK, Şirikçi Ö, Taha S, Engin KN, Boscobionik D, Clement S, Stocker A, Azzi A (1998) Prevention of atherosclerosis by α-tocopherol in smooth muscle cells by a mechanism involving signal transduction modulation. In: Özben T (ed) Free radicals, oxidative stress and antioxidants. Springer, New York, pp 333–342. ISBN:978-1-4419-3292-1
Papaioannou D, Cooper KL, Carroll C, Hind D, Squires H, Tappenden P, Logan RF (2011) Antioxidants in the chemoprevention of colorectal cancer and colorectal adenomas in the general population: a systematic review and meta analysis. Color Dis 13:1085–1099
Peretti N, Marcil V, Drouin E, Levy E (2005) Mechanisms of lipid malabsorption in Cystic Fibrosis: the impact of essential fatty acids deficiency. Nutr Metab (Lond) 2:11
Peretti N, Sassolas A, Roy CC, Deslandres C, Charcosset M, Castagnetti J, Pugnet-Chardon L, Moulin P, Labarge S, Bouthillier L et al (2010) Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J Rare Dis 5:24
Rimbach G, Minihane AM, Majewicz J, Fischer A, Pallauf J, Virgli F, Weinberg PD (2002) Regulation of cell signalling by vitamin E. Proc Nutr Soc 61:415–425
Rimm EB, Stampfer MJ, Ascherio A, Giovannucci E, Colditz GA, Willett WC (1993) Vitamin E consumption and the risk of coronary heart disease in men. N Engl J Med 328:1450–1456
Rossato M, Mariotti C (2014) Normal spermatogenesis and sperm function in a subject affected by cerebellar ataxia due to congenital vitamin E deficiency. Andrologia 46:322–324
Schonfeld G (2003) Familial hypobetalipoproteinemia: a review. J Lipid Res 44:878–883
Sinaasappel M, Stern M, Littlewood J, Wolfe S, Steinkamp G, Heijerman HG, Robberecht E, Döring G (2002) Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibros 1:51–75
Sontag TJ, Parker RS (2007) Influence of major structural features of tocopherols and tocotrienols on their omega-oxidation by tocopherol-omega-hydroxylase. J Lipid Res 48:1090–1098
Sure B (1924) Dietary requirements for reproduction. II. The existence of a specific vitamin for reproduction. J Biol Chem 58:693–709
Tarugi P, Averna M, Di Leo E, Cefalù AB, Noto D, Magnolo L, Cattin L, Bertolini S, Calandra S (2007) Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis 195:e19–e27
Tasinato A, Boscoboinik D, Bartoli GM, Maroni P, Azzi A (1995) d-alpha-tocopherol inhibition of vascular smooth muscle cell proliferation occurs at physiological concentrations, correlates with protein kinase C inhibition, and is independent of its antioxidant properties. Proc Natl Acad Sci USA 92:12190–12194
Traber MG, Sokol RJ, Kohlschütter A, Yokota T, Muller DP, Dufour R, Kayden HJ (1993) Impaired discrimination between stereoisomers of alpha-tocopherol in patients with familial isolated vitamin E deficiency. J Lipid Res 34:201–210
Wang J, Hegele RA (2000) Microsomal triglyceride transfer protein (MTP) gene mutations in Canadian subjects with abetalipoproteinemia. Hum Mutat 15:294–295
Wechter WJ, Kantoci D, Murray ED Jr, D'Amico DC, Jung ME, Wang WH (1996) A new endogenous natriuretic factor: LLU-alpha. Proc Natl Acad Sci USA 93:6002–6007
Wilfond BS, Farrell PM, Laxova A, Mischler E (1994) Severe hemolytic anemia associated with vitamin E deficiency in infants with cystic fibrosis. Implications for neonatal screening. Clin Pediatr (Phila) 33:2–7
Wolf G (1997) gamma-Tocopherol: an efficient protector of lipids against nitric oxide-initiated peroxidative damage. Nutr Rev 55:376–378
Wu D, Liu L, Meydani M, Meydani SN (2005) Vitamin E increases production of vasodilator prostanoids in human aortic endothelial cells through opposing effects on cyclooxygenase-2 and phospholipase A2. J Nutr 135:1847–1853
Yokota T, Shiojiri T, Gotoda T, Arita M, Arai H, Ohga T, Kanda T, Suzuki J, Imai T, Matsumoto H et al (1997) Friedreich-like ataxia with retinitis pigmentosa caused by the His101Gln mutation of the alpha-tocopherol transfer protein gene. Ann Neurol 41:826–832
Yokota T, Uchihara T, Kumagai J, Shiojiri T, Pang JJ, Arita M, Arai H, Hayashi M, Kiyosawa M, Okeda R et al (2000) Postmortem study of ataxia with retinitis pigmentosa by mutation of the alpha-tocopherol transfer protein gene. J Neurol Neurosurg Psychiatry 68:521–525
Yokota T, Igarashi K, Uchihara T, Jishage K, Tomita H, Inaba A, Li Y, Arita M, Suzuki H, Mizusawa H et al (2001) Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proc Natl Acad Sci USA 98:15185–15190
Yoshida Y, Saito Y, Jones LS, Shigeri Y (2007) Chemical reactivities and physical effects in comparison between tocopherols and tocotrienols: physiological significance and prospects as antioxidants. J Biosci Bioeng 104:439–445
Zamel R, Khan R, Pollex RL, Hegele RA (2008) Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis 3:19
Zingg JM, Azzi A (2004) Non-antioxidant activities of vitamin E. Curr Med Chem 11:1113–1133
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
El Hadi, H., Vettor, R., Rossato, M. (2019). Congenital Vitamin E Deficiency. In: Preedy, V., Patel, V. (eds) Handbook of Famine, Starvation, and Nutrient Deprivation. Springer, Cham. https://doi.org/10.1007/978-3-319-55387-0_86
Download citation
DOI: https://doi.org/10.1007/978-3-319-55387-0_86
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55386-3
Online ISBN: 978-3-319-55387-0
eBook Packages: MedicineReference Module Medicine