
Abstract
Epigenetic programming of the pathogen and the host can have a marked influence on the development and progression of acute and chronic disease. Bacterial pathogenesis may be viewed as a developmental program similar to that of cell differentiation and development in eukaryotes. Bacterial epigenetic programming is imparted by DNA methylation, whereby the virulence traits expressed by a pathogen may depend on the cumulative interactions between the microbe and its environment. Such bacterial “memory” provides a means for adaptation to the varied subsequent microenvironments encountered during the infective process. DNA methylation can affect DNA–protein interactions and resultant gene expression by altering DNA thermodynamic stability and curvature and by methyl-group-mediated steric hindrance. Some of these epigenetic interactions can form heritable DNA methylation patterns in the microbial genome that control gene expression in their progeny cells. Microbes can also stimulate heritable changes in the host epigenome via infection-associated alterations to host epigenetic determinants including DNA methylation, histone modifications, chromatin-associated complexes, and noncoding RNA-mediated silencing. The resultant changes in host chromatin remodeling and gene expression may be localized and/or systemic due to direct microbe-to-host cell communication or via dissemination of microbial-host signaling. Thus, the role of epigenetics in host–microbe interactions may be the nexus of many pathological syndromes even though there may be no apparent direct link between infection and disease, providing the basis for the development of novel therapeutics and diagnostic tests for diseases with epigenomic determinants.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Epigenetics
- DNA methylation
- Epigenetic disease
- Infectious disease
- Epigenetic programming
- Epigenome
- Bacterial memory
- Epigenetic host-microbe interactions
- Epigenetic memory
5.1 Introduction
Deciphering the mechanisms that govern epigenetic programming in the pathogen and the host is crucial to the development of new therapeutic approaches to control acute and chronic disease. For instance, pathogenic Escherichia coli utilize heritable DNA methylation patterns to control pili production via a phase variation mechanism, whereby individual cells either express pili (phase-ON) or not (phase-OFF), resulting in periods of attachment and detachment that are critical for progression of an ascending urinary tract infection (Low and Casadesús 2008; Marinus and Casadesús 2009). Microbial infection can also stimulate epigenetic changes in the host epigenome, potentially leading to a variety of human diseases including cancer and autoimmune disorders (Bierne et al. 2012; Dawson and Kouzarides 2012; Elinav et al. 2013; Feinberg and Tycko 2004; Stein 2011). Despite this knowledge, the role of epigenetic modifications on pathogen virulence is poorly understood, and the role of host epigenetic modifications that contribute to, or result from, infectious diseases are only just beginning to be elucidated.
5.2 DNA Methylation
DNA methylation is a fundamental epigenetic process that provides a means to impart additional information to the genomic sequence. In bacteria, DNA methylation occurs at the N6 position of adenine (6mA) and the C5 or N4 positions of cytosine (5mC; 4mC), and these modifications are catalyzed by DNA methyltransferases (Noyer-Weidner and Trautner 1992; Palmer and Marinus 1994; Sánchez-Romero et al. 2015; Wion and Casadesús 2006). Such epigenetic information can alter the timing and targeting of cellular events including transcription, transposition, chromosomal replication, and DNA repair. The most common DNA modification in eukaryotes is 5mC, which is involved in a variety of processes including gene regulation, genomic imprinting, X-chromosome inactivation, and epigenetic memory maintenance (Jones 2012; Jones and Takai 2001; Smith and Meissner 2013). Additionally, 6mA has been recently reported as a possible epigenetic mark in eukaryotes that plays a potential role in transcription and epigenetic inheritance (Luo et al. 2015).
5.2.1 Bacterial Restriction Modification Systems
DNA methylation is a standard means by which restriction-modification (R-M) systems serve to protect bacterial cells from foreign DNA (viruses, transposons, plasmids) (Kobayashi et al. 1999; Meselson et al. 1972; Roberts and Macelis 2001). In most R-M systems, base methylation by a methyltransferase (on adenine or cytosine) prevents DNA cleavage of host DNA by cognate restriction enzymes. Recent evidence suggests that the role of R-M-associated methyltransferases is not restricted to protecting host genomes as the lack of certain R-M systems alters the gene expression pattern of the cell, suggesting a role in epigenetic control of gene expression (Fang et al. 2012; Furuta et al. 2014; Sánchez-Romero et al. 2015; Vasu and Nagaraja 2013). E. coli O104:H4 is a hemolytic uremic syndrome (HUS)-linked outbreak strain that harbors multiple active adenine methyltransferases—some of which are associated with R-M systems (Fang et al. 2012). E. coli O104:H4 also contains a lysogenic lambdoid phage, fStx104, which encodes Shiga toxin (the cause of HUS), and an R-M system that engenders both the production of Shiga toxin and alteration of the bacterium’s transcriptome. These findings indicate that DNA methyltransferases associated with R-M systems can make considerable contributions to bacterial virulence (discussed further below).
5.2.2 Solitary DNA Methyltransferases
Some bacterial DNA methyltransferases lack cognate restriction enzymes and thus are not part of R-M systems. These solitary methyltransferases play roles in cellular regulatory events including those that control bacterial gene regulation, cell-cycle events, and virulence (Low et al. 2001; Marinus and Casadesús 2009; Reisenauer et al. 1999).
5.2.2.1 Dam Methylase
DNA adenine methylase (Dam) is a solitary methyltransferase of Gammaproteobacteria (e.g., E. coli and Salmonella) that methylates the N6 position of adenine in the sequence “GATC” of the bacterial genome and plays a role in the timing and targeting of many cellular events by influencing the interactions of regulatory proteins with DNA (Casadesús and Low 2006; Løbner-Olesen et al. 2005; Low and Casadesús 2008; Low et al. 2001; Marinus and Casadesús 2009). DNA adenine methylation can affect DNA–protein interactions at GATC sequences by altering DNA thermodynamic stability and curvature and by methyl-group-mediated steric hindrance (Wion and Casadesús 2006). There are about 130 molecules of Dam per cell in E. coli, a level that allows sufficient time for some DNA–protein binding between DNA synthesis and the methylation of GATC sequences within newly synthesized DNA (Boye et al. 1992). Competition between Dam and DNA-binding proteins resulted in the formation of ~35 nonmethylated GATC sequences in the E. coli genome (Hale et al. 1994; Ringquist and Smith 1992; Tavazoie and Church 1998; Wang and Church 1992). The actual number of nonmethylated sites at any one time is dependent on bacterial growth rate and growth phase, supporting the hypothesis that the DNA-binding proteins are in competition with Dam at these sites to control the timing and targeting of cellular regulatory events.
5.2.2.2 Cytosine Methylases
Although the role of cytosine methylases has been generally associated with R-M systems, recent evidence suggests that this view may need to be broadened (Marinus and Casadesús 2009; Sánchez-Romero et al. 2015). DNA cytosine methylase (Dcm) is a solitary methyltransferase of Gammaproteobacteria that methylates the internal cytosine in the CCA/TGG motif at the C5 position (5mC) (Bigger et al. 1973; Kahramanoglou et al. 2012). E. coli dcm mutants display increased expression of the stress response sigma factor, RpoS, suggesting cytosine methylation may be involved in gene expression and the stress response (Kahramanoglou et al. 2012). Further, the absence of a solitary cytosine methyltransferase (5mC), HpyA-VIBM, in Helicobacter pylori alters the expression of genes involved in motility, adhesion, and virulence (Kumar et al. 2012).
5.2.2.3 CcrM Methylase
The role of DNA adenine methylation in cell-cycle-related events has been extensively studied in Caulobacter crescentus, serving as a model organism for bacterial cell-cycle regulation and development (Gonzalez et al. 2014; Marczynski and Shapiro 2002; McAdams and Shapiro 2003; Reisenauer et al. 1999). C. crescentus is a member of the Alphaproteobacteria, which includes Brucella abortus (brucellosis), Agrobacterium tumefaciens (crown gall disease), and Sinorhizobium meliloti (nitrogen-fixation). It has a dimorphic life cycle, spending part of its life cycle as a non-replicating motile swarmer cell and the other as a replicating sessile stalked cell. Many of the cellular events leading to differentiation into these morphological stages are modulated by the solitary cell-cycle regulated methyltransferase, CcrM, which methylates the N6 position of adenine in the sequence GANTC (Marczynski and Shapiro 2002; McAdams and Shapiro 2003; Reisenauer et al. 1999). The C. crescentus chromosomal methylation state (unmethylated, hemimethylated, fully methylated) controls a regulatory cascade that couples DNA replication and the expression of cell-cycle master regulators, which facilitate progression of the Caulobacter cell cycle (Collier et al. 2006).
5.3 Dam Methylation Modulates the Timing and Targeting of Cellular Processes
Dam plays a role in the timing and targeting of many cellular processes including DNA repair, DNA replication, transposition, conjugation, as well as those specifically involved in bacterial virulence (Løbner-Olesen et al. 2005; Low and Casadesús 2008; Low et al. 2001; Marinus and Casadesús 2009; Sánchez-Romero et al. 2015).
5.3.1 Dam Controls DNA Repair and Replication
Errors that occur during replication are corrected by methyl-directed mismatch repair that can distinguish base mismatches on the newly synthesized strand. Such DNA strand discrimination is accomplished using hemimethylated DNA that arises after passage of the replication fork, whereby the parental strand is methylated at Dam-target sequences (GATC sites) and the newly synthesized strand is non-methylated (Pukkila et al. 1983). DNA base mismatches on newly synthesized DNA are recognized and removed by the MutHLS DNA mismatch repair proteins, and the errors are corrected using the parental strand as a template (Iyer et al. 2006). Subsequently during the cell cycle, the newly synthesized strand is methylated by Dam at GATC sites resulting in fully methylated DNA. Dam levels are controlled primarily at the transcriptional level (Løbner-Olesen et al. 2003) and the absence, or overproduction, of Dam leads to an increase in spontaneous mutation frequency due to the lack of hemimethylated DNA needed for strand discrimination during DNA mismatch repair (Heithoff et al. 2007; Herman and Modrich 1982; Marinus and Morris 1974).
The timing of DNA replication is controlled by a competition between Dam and DNA-binding proteins that recognize hemimethylated DNA. SeqA binds specifically to several hemimethylated GATC sites at and near the origin of replication (oriC), delaying their methylation by Dam (Kang et al. 1999; Lu et al. 1994). The sequestration of these hemimethylated sites by SeqA delays further replication fork initiation since it represses transcription of the replication initiator (dnaA) and inhibits DnaA binding at oriC as both processes operate optimally at fully methylated GATC sites (Marinus and Casadesús 2009). Additionally, SeqA acts at hemimethylated sites to play a role in nucleoid structure, organization, and partitioning into daughter cells (Bach et al. 2003; Helgesen et al. 2015; Joshi et al. 2013; Skarstad and Katayama 2013). Thus, competition between Dam and DNA-binding proteins controls the timing and targeting of many cellular events that are critical to the cell cycle.
5.3.2 Dam Controls Bacterial Gene Expression
Dam methylation of GATC sites can control gene expression via altering the affinity of DNA-binding proteins to regulatory sequences, as described in the following examples (Casadesús and Low 2006; Løbner-Olesen et al. 2005; Low and Casadesús 2008; Low et al. 2001; Marinus and Casadesús 2009; Sánchez-Romero et al. 2015).
Initiation of DNA Replication
Sequestration of hemimethylated GATC sites by SeqA at the oriC region delays replication fork initiation via dnaA transcriptional repression and DnaA-binding inhibition at oriC (discussed above). Implications: Maintenance of hemimethylated DNA near the oriC region limits the number of replication forks that can initiate before cell division.
Transposition
Tn10 transposition occurs upon the generation of hemimethylated GATC sites in the transposase promoter (Roberts et al. 1985). The transposase promoter is only active when the transposase-coding strand is methylated and the noncoding strand is not methylated. Implications: Transposition is repressed through most of the cell cycle, preventing high-level transposition that would otherwise cause detrimental effects to the genome. Transposition is limited to one copy while the other copy remains in the original location.
Conjugal Plasmid Transfer
Stimulation of the tra operon for conjugal transfer of the Salmonella virulence plasmid occurs upon generation of hemimethylated GATC sites within the upstream regulatory sequences for traJ expression, a transcriptional activator of the tra operon. Methylation of the noncoding strand (but not the coding strand) stimulates binding of the leucine-responsive regulatory protein (Lrp), with resultant traJ transcription, and conjugal transfer of the methylated noncoding single-stranded DNA into the recipient bacterium (Camacho and Casadesús 2002; Camacho and Casadesús 2005). Implications: Conjugal transfer is repressed through most of the cell cycle via a traJ epigenetic switch, thereby modulating the considerable metabolic and energetic cost of mating functions to the cell. Recipient cells are competent for conjugation since the noncoding, methylated strand serves as a template for DNA replication, reproducing the DNA methylation pattern that permits Lrp binding.
Cell Invasion
Salmonella invasion of human epithelial cells is impaired in the absence of Dam methylation (Garcia-Del Portillo et al. 1999). Binding of the HdfR regulatory protein to unmethylated GATC sites in regulatory sequences for the std fimbrial operon stimulates StdEF-mediated repression of invasion determinants encoded on Salmonella Pathogenicity Island I (SPI-1) (Jakomin et al. 2008; López-Garrido and Casadesús 2012). Implications: Methylation state of invasion-associated regulatory sequences ensures bacterial invasion of only appropriate cells/cellular compartments that contribute to the onset and progression of infection.
Pili Phase Variation
Dam methylation controls the production of E. coli pyelonephritis-associated pili (pap) via a phase-variation mechanism that results in cells that either express or do not express the pili. Dam is in competition with two transcriptional activators (Lrp, PapI) for GATC sites in upstream regulatory sequences for pap expression, forming DNA methylation patterns that can be inherited in progeny populations similar to that observed in eukaryotes (Low and Casadesús 2008; Marinus and Casadesús 2009). Implications: Phase variation (ON-OFF) control of pili adherence via Dam methylation results in periods of bacterial attachment and detachment, facilitating uropathogenic E. coli progression from the bladder to kidney, resulting in pyelonephritis.
5.4 DNA Methylation Plays an Essential Role in Bacterial Virulence
DNA methylation has been shown to play a role or has been implicated in the virulence of many bacterial pathogens (Casadesús and Low 2006; Heusipp et al. 2007; Low et al. 2001; Marinus and Casadesús 2009; Sánchez-Romero et al. 2015). Representative examples are discussed below, including pathogens that utilize solitary or R-M methyltransferases to modulate bacterial virulence.
5.4.1 DNA Methylation Controls Bacterial Pathogenesis
Salmonella spp.
Nontyphoidal Salmonella (NTS) is the greatest foodborne-disease burden in the United States, with greater than one million illnesses annually (Gilliss et al. 2011; Scallan et al. 2011). Salmonella enterica infection can result in any of four disease syndromes: enterocolitis/diarrhea, bacteremia, typhoid fever, and chronic asymptomatic carriage (Coburn et al. 2007). Many serovars infect both humans and animals, with the particular syndrome a function of the serovar (serotypic variant), strain virulence, and host susceptibility (Coburn et al. 2007; Heithoff et al. 2012). Dam methylation plays an essential role in Salmonella virulence (Garcia-Del Portillo et al. 1999; Heithoff et al. 1999). The lack or overproduction of Dam confers significant virulence attenuation (10,000-fold) in murine models of typhoid fever. Dam methylation is involved in the invasion of nonphagocytic cells, M-cell cytotoxicity, bile resistance, envelope stability, cell motility, fimbrial, O-antigen and cytotoxin production, systemic dissemination, and the elicitation of host innate and adaptive immune responses (Badie et al. 2007; Garcia-Del Portillo et al. 1999; Heithoff et al. 1999, 2001, 2007, 2008; López-Garrido and Casadesús 2010; Pucciarelli et al. 2002; Sarnacki et al. 2009; Shtrichman et al. 2002; Simon et al. 2007). Implications: Dam methylation controls the production of many factors underlying microbial virulence (adhesins, invasins, toxins) and impacts host–pathogen interactions that compromise host immunity. Salmonella dam mutants are capable of eliciting cross-protection against a diversity of salmonellae and are well-tolerated when applied as modified live vaccines in mice (Heithoff et al. 2001, 2008, 2015), poultry (Dueger et al. 2001, 2003a), sheep (Mohler et al. 2011) and calves (Dueger et al. 2003b; Mohler et al. 2006, 2008). Induction of immunity is rapid, and the vaccine can be delivered in drinking water for low-cost and low-stress vaccination of livestock populations (Mohler et al. 2011, 2012).
Yersinia spp.
Yersinia pseudotuberculosis and enterocolitica are zoonotic foodborne pathogens that can cause severe disease in humans including gastroenteritis, mesenteric lymphadenitis, and septicemia (Galindo et al. 2011; Tauxe 2015). Many pathogenic strains infect both humans and animals whereby the particular syndrome is a function of the serotype, strain virulence, and host susceptibility. The dam gene is essential in certain strains of Yersinia species, and the lack or overproduction of Dam in Y. pseudotuberculosis leads to severe virulence attenuation in murine models of bacteremia (Julio et al. 2001; Kubicek-Sutherland et al. 2014; Taylor et al. 2005) and confers protection to heterologous Y. pseudotuberculosis or Y. pestis challenge (Julio et al. 2001; Kubicek-Sutherland et al. 2014; Taylor et al. 2005). Dam overproducing Y. pseudotuberculosis ectopically secrete several Yersinia outer proteins (e.g., YopE cytotoxin) as well as LcrV, a low-calcium-responsive virulence factor normally involved in Yop synthesis, localization, and suppression of host inflammatory activities (Badie et al. 2004; Julio et al. 2001, 2002). Dam overproducing Y. enterocolitica confer altered invasion, motility, and composition of the lipopolysaccharide (LPS) O-antigen and also display ectopic Yop secretion via increased ClpP protease degradation of the LcrG regulatory protein that normally blocks Yop secretion (Fälker et al. 2005, 2006, 2007). Implications: Dam methylation controls the strict environmental regulation of Yersinia virulence function synthesis and localization, serving to modulate bacterial pathogenesis and host inflammatory activities.
Enterohemorrhagic E. coli
EHEC are a subgroup of Shiga toxin-producing E. coli (STEC) that can cause severe intestinal disease [i.e., hemorrhagic colitis [HC] and hemolytic uremic syndrome [HUS] (Hartland and Leong 2013; Mahan et al. 2013)]. EHEC intestinal adherence requires the delivery of the type III secretion system (TTSS) effector proteins Tir and EspFU into the host cell and expression of the bacterial outer membrane adhesin, intimin. Increased adherence exhibited by dam mutant EHEC was correlated with increased protein levels of Tir, EspFU, and intimin (Campellone et al. 2007). Dam also controls the maintenance of lysogeny for a bacteriophage (933W) that encodes Shiga toxin (Stx-2), which inhibits protein synthesis (via ribosomal inactivation) and leads to renal toxicity in HUS patients (Murphy et al. 2008). Implications: Dam methylation modulates EHEC intestinal adherence and Shiga toxin production during infection.
Brucella abortus
B. abortus is an intracellular pathogen and the causative agent of brucellosis, a zoonotic disease that causes abortions and stillbirths in livestock and acute febrile illness in humans, which may progress to chronically debilitating disease (World Health Organization 2006). It is also designated as a select agent with the potential for bioterrorism due to the chronic nature of disease in livestock and humans and its ability to undergo aerosolization (Centers for Disease Control and Prevention 2015). B. abortus is a member of the Alphaproteobacteria which have defined morphological stages that are modulated by the solitary cell cycle-regulated DNA methyltransferase, CcrM (Marczynski and Shapiro 2002). CcrM is essential for viability in B. abortus, and its overexpression attenuates replication within murine macrophages (Robertson et al. 2000). Implications: CcrM methylation may play a role in intracellular replication of the bacterium within phagocytes, a key virulence characteristic for both acute and chronic cases of brucellosis.
Mycobacterium tuberculosis
M. tuberculosis infections cause nine million active cases and 1.5 million tuberculosis deaths annually, with one-third of the world’s population having latent infections (World Health Organization 2014a). Although there are no predicted dam homologues, the M. tuberculosis solitary DNA methyltransferase, MamA, plays a role in gene expression and fitness during hypoxia, and different methyltransferases are observed in different lineages of M. tuberculosis (Shell et al. 2013). Implications: DNA methylation may play a role in M. tuberculosis lineage-specific differences in preferences for distinct host environments and different disease courses in humans.
Haemophilus influenzae
Non-typeable H. influenzae (NTHi) is a major cause of middle ear (otitis media) infections in children (Haggard 2008). NTHi contains R-M systems comprised of a methyltransferase (mod) and a restriction endonuclease (res) (Srikhanta et al. 2010). Phase variable (ON-OFF) switching of mod alleles (due to the presence of tandem repeats in the corresponding mod genes) regulates the expression of multiple proteins that are involved in antibiotic resistance, biofilm formation, and immune evasion. Recent studies indicate that mod switching to the ON orientation was highly selected in a chinchilla model of otitis media, and ON phase-variants formed more robust biofilms in vitro (Atack et al. 2015). These findings suggest that mod is involved in bacterial virulence, immune evasion, and niche adaptation. Several other human pathogens contain phase-variable R-M systems, including H. pylori (atrophic gastritis), Neisseria meningitidis (meningitis), N. gonorrhoeae (gonorrhea), and Moraxella catarrhalis (otitis media) (Atack et al. 2015; Srikhanta et al. 2010). Implications: Phase-variable R-M systems that modulate microbial virulence traits may be shared across the microbial realm.
5.5 Perspectives: Epigenetic Programming of the Pathogen and Disease Susceptibility
Bacterial pathogenesis can be regarded as a developmental program (Casadesús and D’Ari 2002; Mahan et al. 2010) similar to eukaryotic cell differentiation and development (Bird 2002, 2007; Jaenisch and Bird 2003). Bacterial epigenetic programming is imparted by DNA methylation, whereby the virulence traits expressed are dependent on the aggregate of interactions between the microbe and its environment. Thus, the bacterial epigenome provides a means for bacterial “memory,” engendering the capacity for adaption to the disparate microenvironments encountered as the infection proceeds due to dissemination to new host sites, tissue breakdown, inflammation, and immune clearance mechanisms. Thus, a microbial population may comprise a spectrum of genotypically identical cells with significant phenotypic differences in virulence traits since pathogenicity may be a reflection of cumulative exposure to selective pressures within host(s) and environments experienced during the microbial life cycle. Epigenetic programming may provide insights into the virulence disparities of closely related strains that exhibit marked differences with regard to pathogenicity, host range, and preferences for distinct host environments and different disease courses in humans.
5.6 Microbial Infection, Epigenetic Reprogramming, and Human Disease
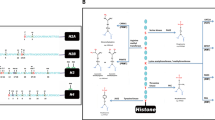
Microbe-associated changes in the host epigenome can play a significant role in human disease via chromatin remodeling and resultant transcriptional reprogramming driven by host DNA methylation, histone modifications, chromatin-associated complexes, and noncoding RNA-mediated silencing (Bannister and Kouzarides 2011; Bierne et al. 2012; Dawson and Kouzarides 2012; Herceg et al. 2013; Paschos and Allday 2010). DNA methylation occurs at the 5′ position of cytosines within CpG dinucleotides, and can recruit protein complexes that can alter chromatin structure or affect the binding of transcription factors with resultant gene silencing (Bird 2002, 2007; Dawson and Kouzarides 2012; Jones and Takai 2001). Histone modifications (e.g., methylation, acetylation, phosphorylation, ubiquitination) can alter chromatin structure and affect gene expression (Bannister and Kouzarides 2011; Dawson and Kouzarides 2012). Noncoding RNA-mediated silencing involves microRNAs (noncoding, 18–25 nucleotides) that target mRNAs and negatively control gene expression (He and Hannon 2004; Sato et al. 2011).
Such transcriptional reprogramming can alter host defense genes involved in TLR (Toll-like receptor), MAPK (mitogen-activated protein kinase), interferon (IFN), and NF-kB signaling pathways (Gómez-Díaz et al. 2012; Paschos and Allday 2010; Stein 2011). For instance, M. tuberculosis inhibits IFN-γ-induced chromatin remodeling by TLR2 and MAPK signaling (via inhibition of histone acetylation), leading to reduced expression of several immune genes and resultant persistence of chronic infections (Pennini et al. 2006). Influenza virus suppresses the antiviral response via the production of a histone mimic that serves as a sink for a host transcription factor (hPAF1) involved in antiviral gene expression (Marazzi et al. 2012). Conversely, some microbe-associated epigenome changes are protective. Following acute viral infection, chromosome remodeling is implicated in the formation of memory CD8+ T cells that provide the host with long-term protective immunity against the pathogen (Youngblood et al. 2010).
5.6.1 Microbial Infection and Cancer
Microbe-associated cancers account for a significant proportion (>20%) of all human cancers (Moore and Chang 2010; zur Hausen 2009). The molecular basis involves microbe-stimulated changes in the host epigenome, with resultant changes in host chromatin remodeling, gene expression, and metabolism (Dawson and Kouzarides 2012; Esteller 2008; Feinberg and Tycko 2004; Herceg et al. 2013; Stein 2011).
Hepatitis B Virus (HBV)
Liver cancer is the second leading cause of cancer death worldwide (World Health Organization 2014b, 2015). The risk for liver cancer is increased 100-fold in individuals with chronic HBV infection (Fernandez et al. 2009), and recent estimates indicate that ~250 million individuals in the human population are chronically infected with HBV (Schweitzer et al. 2015). HBV persists in host cells by the nuclear accumulation of covalently closed circular DNA (cccDNAs) that serve as a template for transcription of all viral mRNAs and are organized into minichromosomes by histones and nonhistone viral and cellular proteins (Grimm et al. 2011; Protzer 2015). High viral loads in patients with chronic hepatitis correlate with hyperacetylation of histone H3 and H4 bound to cccDNA in liver biopsy samples (Pollicino et al. 2006), allowing access of the HBV cccDNA chromatin-like structure to liver-specific transcription factors and subsequent replication (Quasdorff et al. 2008). The HBx regulatory protein (Kew 2011) relieves chromatin-mediated transcriptional repression of HBV cccDNA that involves the histone methyltransferase, SETDB1 (Rivière et al. 2015). HBx also upregulates several DNA methyltransferases (DNMTs), resulting in increased promoter methylation and repression of tumor-suppressor genes encoding p16, a cyclin-dependent kinase inhibitor that functions in cell-cycle arrest and cellular senescence, and E-cadherin, a cell–cell adhesion molecule that affects tumor invasiveness (Fernandez and Esteller 2010; Jung et al. 2007; Tian et al. 2013). Patient samples from various stages of HBV infection show increased methylation of the HBV genome as an acute infection transitions to a chronic infection and during the subsequent progression to premalignant lesions and cancer (Fernandez and Esteller 2010; Fernandez et al. 2009; Stein 2011). Additionally, microRNA (miR-152), whose normal function is to downregulate DNMT1, is downregulated in patients with HBV-associated liver cancer, thus causing DNA hypermethylation (Huang et al. 2010; Saito et al. 2014). These findings suggest a tumor-suppressive role of miR-152, and therapeutic use of this microRNA may reduce aberrant DNA methylation.
Human Papillomavirus (HPV)
Genital human papillomavirus (HPV) is the most commonly diagnosed sexually transmitted infection in the United States and is associated with 95% of cervical and anal cancers and 60% of oropharyngeal cancers. (Centers for Disease Control and Prevention 2012; Gilmer 2015). Through preexisting lesions, HPV infects the basal (lower) layer of the stratified cervical epithelium, and viral genomes are maintained as episomal DNA in the nuclei of infected cells (Kajitani et al. 2012). HPV oncoproteins E6 and E7 inactivate p53 and retinoblastoma (pRb) tumor-suppressing proteins, respectively, resulting in aberrant proliferation and delayed differentiation of infected host cells (Münger et al. 2004). The productive phase of the lifecycle (genome amplification, virion assembly/release) occurs in upper layers of the cervical epithelium that are terminally differentiated. In infections with HPV “high-risk” invasive serotypes (16 and 18), progression of the disease is associated with increased methylation of the HPV genome and considerable suppression of E-cadherin (Anayannis et al. 2015; Fernandez and Esteller 2010; Fernandez et al. 2009; Sun et al. 2011; Wilson et al. 2013). E-cadherin is utilized by Langerhans cells (antigen processing/presentation) to move through stratified epithelium, and its reduction may impact HPV clearance and the length of persistent infections. In a keratinocyte cell line, the HPV E7 oncoprotein is necessary for E-cadherin downregulation via augmentation of host DNMT1 levels and resultant E-cadherin repression (Laurson et al. 2010). DNMT inhibition (via 5-aza-deoxycytidine administration) restored E-cadherin levels, suggesting that epigenetic intervention may have utility in combating persistent infections via restoring influx of Langerhans cells to infected tissue. Further, epigenetic alterations to the viral genome via methylation of viral promoter regions have been implicated in HPV E6 and E7 expression during a transforming infection (Steenbergen et al. 2014). The overall consequence of deregulated expression of E6 and E7 in proliferating cells is chromosomal instability, leading to accumulation of lesions in host cell cancer genes and subsequent progression toward cancer (Korzeniewski et al. 2011).
Epstein–Barr Virus (EBV)
EBV is carried in the vast majority (>90%) of the human population as an asymptomatic lifelong infection, yet it is also correlated with several nonmalignant and malignant diseases (Odumade et al. 2011; Rickinson et al. 2014; Thompson and Kurzrock 2004; Thorley-Lawson 2015). EBV causes mononucleosis and many human tumors of B cell, T cell, and epithelial origin such as Burkett’s lymphoma, Hodgkin’s disease, gastric carcinoma, nasopharyngeal carcinoma, and lymphoproliferative tumors in immunocompromised individuals. The EBV lifecycle involves infection of oropharyngeal cells; host colonization through growth-transforming latent infection of B cells within oropharyngeal lymphoid tissues; long-term persistence within recirculating memory B cells as a silent latent infection; and reactivation to the viral lytic phase and subsequent infection of naïve host cells (Rickinson et al. 2014). Many of these events are driven by epigenetic reprogramming of the pathogen and host, whereby B cell growth transformation is facilitated by several latent proteins, including EBV nuclear antigens (EBNAs) and latent membrane proteins (LMPs), followed by regulated shutdown of latent protein expression that ultimately results in latency in recirculating B cells (Hammerschmidt 2015; Paschos and Allday 2010). Increased methylation of the EBV genome occurs as an acute infection transitions to chronic infection and during subsequent development and progression of cancer (Fernandez and Esteller 2010; Fernandez et al. 2009), and infection of B lymphocytes or nasopharyngeal carcinoma cell lines results in the expression of several DNMTs (Schmeinck 2011; Tsai et al. 2006). During the latent phase, EBV lytic genes are transcriptionally silenced by histone methyltransferase EZH2, a component of the Polycomb Repressive Complex 2, PRC2; and these “histone marks” are erased upon lytic phase induction (Hammerschmidt 2015; Woellmer et al. 2012). These findings indicate that epigenetic modifications of viral DNA determine viral latency.
Helicobacter pylori
Stomach cancer is the third leading cause of cancer death worldwide (World Health Organization 2014b, 2015). H. pylori is a gastric pathogen that colonizes approximately 50% of the world’s population (Wroblewski et al. 2010), associated with 65% of gastric cancers, and classified as a class I carcinogen (Polk and Peek 2010; World Health Organization 2014b). In patients infected with H. pylori, aberrant methylation and repression of tumor-suppressor genes (E-cadherin, p16) was linked with increased gastric cancer risk (Kaise et al. 2008; Maekita et al. 2006; Nakajima et al. 2006; Yoshida et al. 2013). In a gerbil model of gastric cancer, H. pylori infection was shown to be causally involved in the induction of aberrant methylation in the host epigenome, which was associated with the upregulation of several inflammation-related genes (CXCL2, IL-1β, NOS2, TNF-α) (Niwa et al. 2010). Methylation decreased upon bacterial clearance but remained significantly higher than that observed in uninfected control animals. Suppressing inflammation with the immunosuppressive drug, cyclosporin A, prevented aberrant methylation without affecting colonization, indicating that epigenetic modifications occurred as a consequence of inflammation rather than the infection itself. These studies revealed an “epigenetic field defect” whereby increased DNA methylation that arises as a result of infection marks a region with higher risk for transformation (Niwa et al. 2010; Stein 2011). Thus, DNA methylation has potential clinical utility as a biomarker for the risk of malignant transformation for a number of cancers, offering new therapeutic opportunities that target and monitor epigenetic changes (discussed below).
5.7 Concluding Remarks: Microbial Infection and Its Impact on the Host Epigenome and Disease
The origin of some diseases may have a microbial component even though there may be no apparent direct link between infection and disease. How does this occur and what are the possible implications? Microbial infection can trigger heritable changes in the host epigenome that lead to profound differences in disease susceptibility, host cell metabolism, inflammation, and immune responses, and some of these responses may be maintained long after microbial clearance (Bierne et al. 2012; Davis et al. 2011; Stein 2011). The primary challenge toward establishing a causal link between infection-associated changes in the host epigenome and disease origin is the considerable interplay between epigenetic, genetic (mutational), and nonmicrobial (e.g., carcinogen) risk factors that cloud the assignment of primary versus secondary events leading to disease development and progression (Fig. 5.1). Microbes cause cancer directly via harboring oncogenes that contribute to cell transformation or indirectly through chronic inflammation whereby ultimately carcinogenic mutations are generated in host cells (Moore and Chang 2010; Parsonnet 1999; zur Hausen 2001). Additionally, microbial and nonmicrobial associated alterations in host epigenetic determinants influence many biological processes that are fundamental to the development of cancer including the inactivation of tumor-suppressor genes and/or DNA-repair genes, which predispose the genome to mutation (Baylin and Herman 2000; Dawson and Kouzarides 2012; Herceg et al. 2013; Moore and Chang 2010; Paschos and Allday 2010; Romani et al. 2015; Stein 2011).

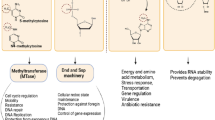
Epigenetic programming of the pathogen and the host can stimulate the development and progression of acute and chronic disease. (a) The epigenome of pathogenic microbes can be modified to stimulate the production of virulence determinants (via Dam; host DNMTs). Pathogenic bacteria can modify the host epigenome (dark circles) via DNA methylation, histone modifications, chromatin-associated complexes, and noncoding RNA mediated silencing. (b) Environmental inputs can alter disease susceptibility by stimulating genetic (mutational; red circles) or epigenetic changes (nonmutational; dark circles) in the host genome via carcinogen exposure, cell signaling, and inflammation. (c) Chronic disease (e.g., cancer) can be stimulated directly by genetic changes in the host genome caused by exposure to carcinogens, microbial oncogenes, and chronic inflammation or indirectly via epigenetic changes in the host genome by inactivation of tumor-suppressor genes and/or DNA-repair genes, which predispose the genome to mutation. Such complex interactions between genetic, epigenetic, and environmental inputs result in host gene dysregulation and human disease
Despite these challenges, significant advances have been made toward establishing a direct link between microbe-associated changes in the host epigenome and cancer, and it remains a possibility that certain disorders are a consequence of chronic inflammation with microbial origin (Bierne et al. 2012; Costenbader et al. 2012; Elinav et al. 2013; Feinberg and Tycko 2004; Grivennikov et al. 2010; Herceg et al. 2013; Liu et al. 2008; Portela and Esteller 2010; Schett et al. 2013; Ushijima and Hattori 2012; Van Vliet et al. 2007; Wilson 2008). For example, the gut microbiome (the largest reservoir of microbes in the body) stimulates host epigenome changes that are linked to inflammatory bowel disease (Khor et al. 2011; Knights et al. 2013; Kostic et al. 2014; Ventham et al. 2013). Since there are ~100 trillion microbial cells in the gastrointestinal tract—roughly ten times more than the cells in the human body—the gut microbiome has the capacity to produce a variety of compounds that can impact host genomic/epigenomic processes and metabolism (Bianconi et al. 2013; Garagnani et al. 2013; Shenderov 2012; Stilling et al. 2014). Examples include microbial structural components and metabolites (e.g., peptides, polysaccharides, endotoxins, short-chain fatty acids, co-factors) that are potential epigenomic modifiers, which can affect gene expression and metabolism in the host via transcriptional reprogramming of host signaling pathways (Gómez-Díaz et al. 2012; Knights et al. 2013; Paschos and Allday 2010; Stein 2011). Notably, host–gut microbe interactions can lead to considerable systemic signaling, involving many organs and organ systems, including the central nervous system (Stilling et al. 2014). Thus, the role of epigenetics in host–microbe interactions leading to pathological syndromes—with the potential of the disruption of homeostasis due to pathogen exposure—provides the foundation for the development of new medicines and diagnostic tests for diseases with epigenomic determinants.
The significant challenge of epigenetic therapies lies in the lack of specificity—and the global hypomethylation achieved by DNMT inhibitors—which may be detrimental to developing an effective treatment. Notwithstanding, cancer treatment applications include administration of small molecules that inhibit epigenetic factors (Dawson and Kouzarides 2012; Romani et al. 2015), risk assessments that link the degree of aberrant DNA methylation to the likelihood of cell transformation (Niwa et al. 2010; Stein 2011), and gene therapy targeting epigenetic factors (Yao et al. 2015). The use of “epigenetic modifier drugs” may extend beyond cancer to other epigenetically based diseases as evidenced by their current testing in noncancer clinical trials (e.g., irritable bowel syndrome, Alzheimer disease, cardiovascular disease, thalassemia, psoriasis) (Romani et al. 2015). Additionally, combinational therapies—comprising epigenetic modifier drugs and antimicrobials—may prove useful in combating infectious diseases and associated disease manifestations such as blood clotting and inflammation that can cause severe tissue damage and organ failure leading to death (Grewal et al. 2013; Herceg et al. 2013; Moore and Chang 2010; Schleithoff et al. 2012; Yang et al. 2015).
References
Anayannis N, Schlecht N, Belbin T (2015) Epigenetic mechanisms of human papillomavirus-associated head and neck cancer. Arch Pathol Lab Med. doi:10.5858/arpa.2014-0554-RA
Atack J, Srikhanta Y, Fox K, Jurcisek J, Brockman K, Clark T, Boitano M, Power P, Jen F-C, McEwan A (2015) A biphasic epigenetic switch controls immunoevasion, virulence and niche adaptation in non-typeable Haemophilus influenzae. Nat Commun 6:7828. doi:10.1038./ncomms8828
Bach T, Krekling M, Skarstad K (2003) Excess SeqA prolongs sequestration of oriC and delays nucleoid segregation and cell division. EMBO J 22:315–323
Badie G, Heithoff D, Mahan M (2004) LcrV synthesis is altered by DNA adenine methylase overproduction in Yersinia pseudotuberculosis and is required to confer immunity in vaccinated hosts. Infect Immun 72:6707–6710
Badie G, Heithoff D, Sinsheimer R, Mahan M (2007) Altered levels of Salmonella DNA adenine methylase are associated with defects in gene expression, motility, flagellar synthesis, and bile resistance in the pathogenic strain 14028 but not in the laboratory strain LT2. J Bacteriol 189:1556–1564
Bannister A, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Baylin S, Herman J (2000) DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet 16:168–174
Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri M, Tassani S, Piva F (2013) An estimation of the number of cells in the human body. Ann Hum Biol 40:463–471
Bierne H, Hamon M, Cossart P (2012) Epigenetics and bacterial infections. Cold Spring Harb Perspect Med 2:a010272
Bigger C, Murray K, Murray N (1973) Recognition sequence of a restriction enzyme. Nature 244:7–10
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bird A (2007) Perceptions of epigenetics. Nature 447:396–398
Boye E, Marinus M, Løbner-Olesen A (1992) Quantitation of Dam methyltransferase in Escherichia coli. J Bacteriol 174:1682–1685
Camacho E, Casadesús J (2002) Conjugal transfer of the virulence plasmid of Salmonella enterica is regulated by the leucine-responsive regulatory protein and DNA adenine methylation. Mol Microbiol 44:1589–1598
Camacho E, Casadesús J (2005) Regulation of traJ transcription in the Salmonella virulence plasmid by strand-specific DNA adenine hemimethylation. Mol Microbiol 57:1700–1718
Campellone K, Roe A, Løbner-Olesen A, Murphy K, Magoun L, Brady M, Donohue-Rolfe A, Tzipori S, Gally D, Leong J (2007) Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157: H7. Mol Microbiol 63:1468–1481
Casadesús J, D’Ari R (2002) Memory in bacteria and phage. Bioessays 24:512–518
Casadesús J, Low D (2006) Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev 70:830–856
Centers for Disease Control and Prevention (2012) Human papillomavirus-associated cancers-United States, 2004–2008. Morb Mortal Wkly Rep 61:258
Centers for Disease Control and Prevention (2015) Humans and Brucella species. http://www.cdc.gov/brucellosis/clinicians/brucella-species.html
Coburn B, Grassl G, Finlay B (2007) Salmonella, the host and disease: a brief review. Immunol Cell Biol 85:112–118
Collier J, Murray SR, Shapiro L (2006) DnaA couples DNA replication and the expression of two cell cycle master regulators. EMBO J 25:346–356
Costenbader K, Gay S, Alarcón-Riquelme M, Iaccarino L, Doria A (2012) Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun Rev 11:604–609
Davis B, Wen H, Ting J (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707
Dawson M, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
Dueger EL, House JK, Heithoff DM, Mahan MJ (2001) Salmonella DNA adenine methylase mutants elicit protective immune responses to homologous and heterologous serovars in chickens. Infect Immun 69:7950–7954
Dueger EL, House JK, Heithoff DM, Mahan MJ (2003a) Salmonella DNA adenine methylase mutants prevent colonization of newly hatched chickens by homologous and heterologous serovars. Int J Food Microbiol 80:153–159
Dueger EL, House JK, Heithoff DM, Mahan MJ (2003b) Salmonella DNA adenine methylase mutants elicit early and late onset protective immune responses in calves. Vaccine 21:3249–3258
Elinav E, Nowarski R, Thaiss C, Hu B, Jin C, Flavell R (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159
Fälker S, Schilling J, Schmidt M, Heusipp G (2007) Overproduction of DNA adenine methyltransferase alters motility, invasion, and the lipopolysaccharide O-antigen composition of Yersinia enterocolitica. Infect Immun 75:4990–4997
Fälker S, Schmidt M, Heusipp G (2005) DNA methylation in Yersinia enterocolitica: role of the DNA adenine methyltransferase in mismatch repair and regulation of virulence factors. Microbiology 151:2291–2299
Fälker S, Schmidt M, Heusipp G (2006) Altered Ca2+ regulation of Yop secretion in Yersinia enterocolitica after DNA adenine methyltransferase overproduction is mediated by Clp-dependent degradation of LcrG. J Bacteriol 188:7072
Fang G, Munera D, Friedman D, Mandlik A, Chao M, Banerjee O, Feng Z, Losic B, Mahajan M, Jabado O (2012) Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol 30:1232–1239
Feinberg A, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4:143–153
Fernandez A, Esteller M (2010) Viral epigenomes in human tumorigenesis. Oncogene 29:1405–1420
Fernandez A, Rosales C, Lopez-Nieva P, Graña O, Ballestar E, Ropero S, Espada J, Melo S, Lujambio A, Fraga M (2009) The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res 19:438–451
Furuta Y, Namba-Fukuyo H, Shibata T, Nishiyama T, Shigenobu S, Suzuki Y, Sugano S, Hasebe M, Kobayashi I (2014) Methylome diversification through changes in DNA methyltransferase sequence specificity. PLoS Genet 10:e1004272
Galindo C, Rosenzweig J, Kirtley M, Chopra A (2011) Pathogenesis of Y. enterocolitica and Y. pseudotuberculosis in human yersiniosis. J Pathog 2011:1–16
Garagnani P, Pirazzini C, Franceschi C (2013) Colorectal cancer microenvironment: among nutrition, gut microbiota, inflammation and epigenetics. Curr Pharm Des 19:765–778
Garcia-Del Portillo F, Pucciarelli MG, Casadesus J (1999) DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc Natl Acad Sci U S A 96:11578–11583
Gilliss D, Cronquist A, Cartter M, Tobin-D’Angelo M, Blythe D, Smith K, Lathrop S, Birkhead G, Cieslak P (2011) Vital signs: incidence and trends of infection with pathogens transmitted commonly through food—foodborne diseases active surveillance Network, 10 U.S. Sites, 1996–2010. Morb Mortal Wkly Rep 60:749–755
Gilmer L (2015) Human papillomavirus vaccine update. Prim Care Clin Off Pract 42:17–32
Gómez-Díaz E, Jordà M, Peinado M, Rivero A (2012) Epigenetics of host–pathogen interactions: the road ahead and the road behind. PLoS Pathog 8:e1003007
Gonzalez D, Kozdon J, McAdams H, Shapiro L, Collier J (2014) The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res 42:3720–3735
Grewal P, Aziz P, Uchiyama S, Rubio G, Lardone R, Le D, Varki N, Nizet V, Marth J (2013) Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc Natl Acad Sci U S A 110:20218–20223
Grimm D, Thimme R, Blum H (2011) HBV life cycle and novel drug targets. Hepatol Int 5:644–653
Grivennikov S, Greten F, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Haggard M (2008) Otitis media: prospects for prevention. Vaccine 26:G20–G24
Hale W, Van der Woude M, Low D (1994) Analysis of nonmethylated GATC sites in the Escherichia coli chromosome and identification of sites that are differentially methylated in response to environmental stimuli. J Bacteriol 176:3438–3441
Hammerschmidt W (2015) The epigenetic life cycle of Epstein–Barr virus. Curr Top Microbiol Immunol 390(Pt 1):103–117
Hartland E, Leong J (2013) Enteropathogenic and enterohemorrhagic E. coli: ecology, pathogenesis, and evolution. Front Cell Infect Microbiol 3:15–20
He L, Hannon G (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531
Heithoff D, Badie G, Julio S, Enioutina E, Daynes R, Sinsheimer R, Mahan M (2007) In vivo-selected mutations in methyl-directed mismatch repair suppress the virulence attenuation of Salmonella dam mutant strains following intraperitoneal, but not oral, infection of naïve mice. J Bacteriol 189:4708–4717
Heithoff DM, Enioutina EY, Bareyan D, Daynes RA, Mahan MJ (2008) Conditions that diminish myeloid-derived suppressor cell activities stimulate cross-protective immunity. Infect Immun 76:5191–5199
Heithoff DM, Enioutina EY, Daynes RA, Sinsheimer RL, Low DA, Mahan MJ (2001) Salmonella DNA adenine methylase mutants confer cross-protective immunity. Infect Immun 69:6725–6730
Heithoff D, House J, Thomson P, Mahan M (2015) Development of a Salmonella cross-protective vaccine for food animal production systems. Vaccine 33:100–107
Heithoff DM, Shimp WR, House JK, Xie Y, Weimer BC, Sinsheimer RL, Mahan MJ (2012) Intraspecies variation in the emergence of hyperinfectious bacterial strains in nature. PLoS Pathog 8:e1002647
Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ (1999) An essential role for DNA adenine methylation in bacterial virulence. Science 284:967–970
Helgesen E, Fossum-Raunehaug S, Sætre F, Schink K, Skarstad K (2015) Dynamic Escherichia coli SeqA complexes organize the newly replicated DNA at a considerable distance from the replisome. Nucleic Acids Res 43:2730–2743
Herceg Z, Lambert M-P, van Veldhoven K, Demetriou C, Vineis P, Smith M, Straif K, Wild C (2013) Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis 34:1955–1967
Herman G, Modrich P (1982) Escherichia coli dam methylase. Physical and catalytic properties of the homogeneous enzyme. J Biol Chem 257:2605–2612
Heusipp G, Fälker S, Schmidt M (2007) DNA adenine methylation and bacterial pathogenesis. Int J Med Microbiol 297:1–7
Huang J, Wang Y, Guo Y, Sun S (2010) Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus–related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology 52:60–70
Iyer R, Pluciennik A, Burdett V, Modrich P (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106:302–323
Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254
Jakomin M, Chessa D, Bäumler A, Casadesús J (2008) Regulation of the Salmonella enterica std fimbrial operon by DNA adenine methylation, SeqA, and HdfR. J Bacteriol 190:7406–7413
Jones P (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492
Jones P, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science 293:1068–1070
Joshi M, Magnan D, Montminy T, Lies M, Stepankiw N, Bates D (2013) Regulation of sister chromosome cohesion by the replication fork tracking protein SeqA. PLoS Genet 9:e1003673
Julio S, Heithoff D, Provenzano D, Klose K, Sinsheimer R, Low D, Mahan M (2001) DNA Adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect Immun 69:7610–7615
Julio S, Heithoff D, Sinsheimer R, Low D, Mahan M (2002) DNA adenine methylase overproduction in Yersinia pseudotuberculosis alters YopE expression and secretion and host immune responses to infection. Infect Immun 70:1006–1009
Jung J, Arora P, Pagano J, Jang K (2007) Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1 pathway. Cancer Res 67:5771–5778
Kahramanoglou C, Prieto A, Khedkar S, Haase B, Gupta A, Benes V, Fraser G, Luscombe N, Seshasayee A (2012) Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat Comm 3:886
Kaise M, Yamasaki T, Yonezawa J, Miwa J, Ohta Y, Tajiri H (2008) CpG island hypermethylation of tumor-tuppressor genes in H. pylori-infected non-Neoplastic gastric mucosa is linked with gastric cancer risk. Helicobacter 13:35–41
Kajitani N, Satsuka A, Kawate A, Sakai H (2012) Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front Microbiol 3:1–12
Kang S, Lee H, Han J, Hwang D (1999) Interaction of SeqA and Dam methylase on the hemimethylated origin of Escherichia coli chromosomal DNA replication. J Biol Chem 274:11463–11468
Kew MC (2011) Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 26:144–152
Khor B, Gardet A, Xavier R (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474:307–317
Knights D, Lassen K, Xavier R (2013) Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut 62:1505–1510
Kobayashi I, Nobusato A, Kobayashi-Takahashi N, Uchiyama I (1999) Shaping the genome–restriction–modification systems as mobile genetic elements. Curr Opin Genet Dev 9:649–656
Korzeniewski N, Spardy N, Duensing A, Duensing S (2011) Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett 305:113–122
Kostic A, Xavier R, Gevers D (2014) The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146:1489–1499
Kubicek-Sutherland J, Heithoff D, Ersoy S, Shimp W, Mahan M (2014) Immunization with a DNA adenine methylase over-producing Yersinia pseudotuberculosis vaccine confers robust cross-protection against heterologous pathogenic serotypes. Vaccine 32:1451–1459
Kumar R, Mukhopadhyay A, Ghosh P, Rao D (2012) Comparative transcriptomics of H. pylori strains AM5, SS1 and their hpyAVIBM deletion mutants: possible roles of cytosine methylation. PLoS One 7(8):e42303
Laurson J, Khan S, Chung R, Cross K, Raj K (2010) Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 31:918–926
Liu L, Li Y, Tollefsbol T (2008) Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol 10:25
Løbner-Olesen A, Marinus M, Hansen F (2003) Role of SeqA and Dam in Escherichia coli gene expression: a global/microarray analysis. Proc Natl Acad Sci U S A 100:4672–4677
Løbner-Olesen A, Skovgaard O, Marinus M (2005) Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8:154–160
López-Garrido J, Casadesús J (2010) Regulation of Salmonella enterica pathogenicity island 1 by DNA adenine methylation. Genetics 184:637–649
López-Garrido J, Casadesús J (2012) Crosstalk between virulence loci: regulation of Salmonella enterica pathogenicity island 1 (SPI-1) by products of the std fimbrial operon. PLoS One 7:e30499
Low D, Casadesús J (2008) Clocks and switches: bacterial gene regulation by DNA adenine methylation. Curr Opin Microbiol 11:106–112
Low D, Weyand N, Mahan M (2001) Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun 69:7197–7204
Lu M, Campbell J, Boye E, Kleckner N (1994) SeqA: a negative modulator of replication initiation in E. coli. Cell 77:413–426
Luo G-Z, Blanco M, Greer E, He C, Shi Y (2015) DNA N6-methyladenine: a new epigenetic mark in eukaryotes? Nat Rev Mol Cell Biol 16:705–710
Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M (2006) High levels of aberrant DNA methylation in Helicobacter pylori–infected gastric mucosae and its possible association with gastric cancer risk. Clin Can Res 12:989–995
Mahan MJ, Kubicek-Sutherland JZ, Heithoff DM (2013) Rise of the microbes. Virulence 4:213–222
Mahan M, Sinsheimer R, Shimp W, Heithoff D (2010) Covert operations: the adaptable plan of attack deployed by pathogenic bacteria. In: Maloy S, Hughes K, Casadesús J (eds) The lure of bacterial genetics: a tribute to John Roth. American Society for Microbiology Press, Washington, DC, pp 185–200
Marazzi I, Ho J, Kim J, Manicassamy B, Dewell S, Albrecht R, Seibert C, Schaefer U, Jeffrey K, Prinjha R (2012) Suppression of the antiviral response by an influenza histone mimic. Nature 483:428–433
Marczynski G, Shapiro L (2002) Control of chromosome replication in Caulobacter crescentus. Annu Rev Microbiol 56:625–656
Marinus M, Casadesús J (2009) Roles of DNA adenine methylation in host–pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev 33:488–503
Marinus M, Morris N (1974) Biological function for 6-methyladenine residues in the DNA of Escherichia coli K12. J Mol Biol 85:309–322
McAdams H, Shapiro L (2003) A bacterial cell-cycle regulatory network operating in time and space. Science 301:1874–1877
Meselson M, Yuan R, Heywood J (1972) Restriction and modification of DNA. Annu Rev Biochem 41:447–466
Mohler V, Heithoff D, Mahan M, Hornitzky M, Thomson P, House J (2012) Development of a novel in-water vaccination protocol for DNA adenine methylase deficient Salmonella enterica serovar Typhimurium vaccine in adult sheep. Vaccine 30:1481–1491
Mohler VL, Heithoff DM, Mahan MJ, Walker KH, Hornitzky MA, Gabor L, Thomson PC, Thompson A, House JK (2011) Protective immunity conferred by a DNA adenine methylase deficient Salmonella enterica serovar Typhimurium vaccine when delivered in-water to sheep challenged with Salmonella enterica serovar Typhimurium. Vaccine 29:3571–3582
Mohler V, Heithoff D, Mahan M, Walker K, Hornitzky M, McConnell C, Shum L, House J (2006) Cross-protective immunity in calves conferred by a DNA adenine methylase deficient Salmonella enterica serovar Typhimurium vaccine. Vaccine 24:1339
Mohler V, Heithoff D, Mahan M, Walker K, Hornitzky M, Shum L, Makin K, House J (2008) Cross-protective immunity conferred by a DNA adenine methylase deficient Salmonella enterica serovar Typhimurium vaccine in calves challenged with Salmonella serovar Newport. Vaccine 26:1751–1758
Moore P, Chang Y (2010) Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer 10:878–889
Münger K, Baldwin A, Edwards K, Hayakawa H, Nguyen C, Owens M, Grace M, Huh K (2004) Mechanisms of human papillomavirus-induced oncogenesis. J Virol 78:11451–11460
Murphy K, Ritchie J, Waldor M, Løbner-Olesen A, Marinus M (2008) Dam methyltransferase is required for stable lysogeny of the Shiga toxin (Stx2)-encoding bacteriophage 933W of enterohemorrhagic Escherichia coli O157: H7. J Bacteriol 190:438–441
Nakajima T, Maekita T, Oda I, Gotoda T, Yamamoto S, Umemura S, Ichinose M, Sugimura T, Ushijima T, Saito D (2006) Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev 15:2317–2321
Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T (2010) Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 70:1430–1440
Noyer-Weidner M, Trautner T (1992) Methylation of DNA in prokaryotes. In: Jost JP, Saluz H (eds) DNA methylation: molecular biology and biological significance. Birkhauser Basel, Switzerland, pp 39–108
Odumade O, Hogquist K, Balfour H (2011) Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev 24:193–209
Palmer B, Marinus M (1994) The dam and dcm strains of Escherichia coli—a review. Gene 143:1–12
Parsonnet J (1999) Microbes and malignancy: infection as a cause of human cancers. Oxford University Press, USA
Paschos K, Allday M (2010) Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol 18:439–447
Pennini M, Pai R, Schultz D, Boom W, Harding C (2006) Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-γ-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 176:4323–4330
Polk D, Peek R (2010) Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer 10:403–414
Pollicino T, Belloni L, Raffa G, Pediconi N, Squadrito G, Raimondo G, Levrero M (2006) Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 130:823–837
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28:1057–1068
Protzer U (2015) Hepatitis: epigenetic control of HBV by HBx protein-releasing the break? Nat Rev Gastroenterol Hepatol 12. doi:10.1016/j.jhep.2015.1006.1023
Pucciarelli M, Prieto A, Casadesús J, Garcıa-del Portillo F (2002) Envelope instability in DNA adenine methylase mutants of Salmonella enterica. Microbiology 148:1171–1182
Pukkila P, Peterson J, Herman G, Modrich P, Meselson M (1983) Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 104:571–582
Quasdorff M, Hösel M, Odenthal M, Zedler U, Bohne F, Gripon P, Dienes HP, Drebber U, Stippel D, Goeser T (2008) A concerted action of HNF4α and HNF1α links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol 10:1478–1490
Reisenauer A, Kahng L, McCollum S, Shapiro L (1999) Bacterial DNA methylation: a cell cycle regulator? J Bacteriol 181:5135–5139
Rickinson A, Long H, Palendira U, Münz C, Hislop A (2014) Cellular immune controls over Epstein–Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol 35:159–169
Ringquist S, Smith C (1992) The Escherichia coli chromosome contains specific, unmethylated dam and dcm sites. Proc Natl Acad Sci U S A 89:4539–4543
Rivière L, Gerossier L, Ducroux A, Dion S, Deng Q, Michel M-L, Buendia M-A, Hantz O, Neuveut C (2015) HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol 63:1093–1102
Roberts D, Hoopes B, McClure W, Kleckner N (1985) IS10 transposition is regulated by DNA adenine methylation. Cell 43:117–130
Roberts R, Macelis D (2001) REBASE—restriction enzymes and methylases. Nucleic Acids Res 29:268–269
Robertson G, Reisenauer A, Wright R, Jensen R, Jensen A, Shapiro L, Roop R (2000) The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J Bacteriol 182:3482–3489
Romani M, Pistillo M, Banelli B (2015) Environmental epigenetics: crossroad between public health, lifestyle, and cancer prevention. BioMed Res Int 2015:587983
Saito Y, Hibino S, Saito H (2014) Alterations of epigenetics and microRNA in hepatocellular carcinoma. Hepatol Res 44:31–42
Sánchez-Romero M, Cota I, Casadesús J (2015) DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol 25:9–16
Sarnacki S, Marolda C, Llana M, Giacomodonato M, Valvano M, Cerquetti M (2009) Dam methylation controls O-antigen chain length in Salmonella enterica serovar enteritidis by regulating the expression of Wzz protein. J Bacteriol 191:6694–6700
Sato F, Tsuchiya S, Meltzer S, Shimizu K (2011) MicroRNAs and epigenetics. Febs J 278:1598–1609
Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM (2011) Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15
Schett G, Elewaut D, McInnes I, Dayer J-M, Neurath M (2013) How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med 19:822–824
Schleithoff C, Völter-Mahlknecht S, Dahmke I, Mahlknecht U (2012) On the epigenetics of vascular regulation and disease. Clin Epigenetics 4. http://www.clinicalepigeneticsjournal.com/content/4/1/7
Schmeinck A (2011) Acquisition and loss of chromatin modifications during an Epstein-Barr Virus infection. Dissertaion, Ludwig Maximilians-University, Munich
Schweitzer A, Horn J, Mikolajczyk R, Krause G, Ott JJ (2015) Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 246:1546–1555
Shell S, Prestwich E, Baek S-H, Shah R, Sassetti C, Dedon P, Fortune S (2013) DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLoS Pathog 9:e1003419
Shenderov B (2012) Gut indigenous microbiota and epigenetics. Microb Ecol Health Dis 23. doi:10.3402/mehd.v23i0.17195
Shtrichman R, Heithoff D, Mahan M, Samuel C (2002) Tissue selectivity of interferon-stimulated gene expression in mice infected with Dam+ versus Dam− Salmonella enterica serovar Typhimurium strains. Infect Immun 70:5579–5588
Simon R, Heithoff D, Mahan M, Samuel C (2007) Comparison of tissue-selective proinflammatory gene induction in mice infected with wild-type, DNA adenine methylase-deficient, and flagellin-deficient Salmonella enterica. Infect immun 75:5627–5639
Skarstad K, Katayama T (2013) Regulating DNA replication in bacteria. Cold Spring Harb Perspect Biol 5:a012922
Smith Z, Meissner A (2013) DNA methylation: roles in mammalian development. Nat Rev Genet 14:204–220
Srikhanta Y, Fox K, Jennings M (2010) The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat Rev Microbiol 8:196–206
Steenbergen R, Snijders P, Heideman D, Meijer C (2014) Clinical implications of (epi) genetic changes in HPV-induced cervical precancerous lesions. Nat Rev Cancer 14:395–405
Stein R (2011) Epigenetics—the link between infectious diseases and cancer. JAMA 305:1484–1485
Stilling R, Dinan T, Cryan J (2014) Microbial genes, brain & behaviour–epigenetic regulation of the gut–brain axis. Genes Brain Behav 13:69–86
Sun C, Reimers L, Burk R (2011) Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol 121:59–63
Tauxe R (2015) Epidemiology of yersiniosis. In: Calderwood SB, section editor, Bloom A, deputy editor (eds) http://www.uptodate.com/contents/epidemiology-of-yersiniosis. Accessed 14 Aug 2015
Tavazoie S, Church G (1998) Quantitative whole-genome analysis of DNA-protein interactions by in vivo methylase protection in E. coli. Nat Biotechnol 16:566–571
Taylor V, Titball R, Oyston P (2005) Oral immunization with a dam mutant of Yersinia pseudotuberculosis protects against plague. Microbiology 151:1919–1926
Thompson P, Kurzrock R (2004) Epstein-Barr virus and cancer. Clin Can Res 10:803–821
Thorley-Lawson D (2015) EBV persistence—Introducing the virus. In: Münz C (ed) Epstein Barr Virus Volume 1. Springer, Switzerland, pp 151–209
Tian Y, Yang W, Song J, Wu Y, Ni B (2013) Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol 33:2810–2816
Tsai C-L, Li H-P, Lu Y-J, Hsueh C, Liang Y, Chen C-L, Tsao S, Tse K-P, Yu J-S, Chang Y-S (2006) Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH2-terminal kinase signaling. Cancer Res 66:11668–11676
Ushijima T, Hattori N (2012) Molecular pathways: involvement of helicobacter pylori–triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Can Res 18:923–929
Van Vliet J, Oates N, Whitelaw E (2007) Epigenetic mechanisms in the context of complex diseases. Cell Mol Life Sci 64:1531–1538
Vasu K, Nagaraja V (2013) Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol Mol Biol Rev 77:53–72
Ventham N, Kennedy N, Nimmo E, Satsangi J (2013) Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology 145:293–308
Wang M, Church G (1992) A whole genome approach to in vivo DNA-protein interactions in E. coli. Nature 360:606–610
Wilson A (2008) Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J Periodontol 79:1514–1519
Wilson G, Lechner M, Köferle A, Caren H, Butcher L, Feber A, Fenton T, Jay A, Boshoff C, Beck S (2013) Integrated virus-host methylome analysis in head and neck squamous cell carcinoma. Epigenetics 8:953–961
Wion D, Casadesús J (2006) N6-methyl-adenine: an epigenetic signal for DNA–protein interactions. Nat Rev Microbiol 4:183–192
Woellmer A, Arteaga-Salas J, Hammerschmidt W (2012) BZLF1 governs CpG-methylated chromatin of Epstein-Barr virus reversing epigenetic repression. PLoS Pathog 8:e1002902
World Health Organization (2006) http://www.who.int/csr/resources/publications/Brucellosis.pdf. In: Corbel MJ, Elberg SS, Cosivi O (eds) Geneva, pp 1–102
World Health Organization (2014a) Global tuberculosis report 2014. http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?ua=1. WHO Press, Geneva, pp 1–171
World Health Organization (2014b) World cancer report, 2014. In: Stewart B, Wild C (eds) WHO Report. WHO Press, Geneva
World Health Organization (2015) Cancer. Fact sheet No. 297. WHO Press, Geneva
Wroblewski L, Peek R, Wilson K (2010) Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23:713–739
Yang W, Aziz P, Heithoff D, Mahan M, Smith J, Marth J (2015) An intrinsic mechanism of secreted protein aging and turnover. Proc Natl Acad Sci U S A 112:13657–13662
Yao S, He Z, Chen C (2015) CRISPR/Cas9-mediated genome editing of epigenetic factors for cancer therapy. Hum Gene Ther 26:463–471
Yoshida T, Kato J, Maekita T, Yamashita S, Enomoto S, Ando T, Niwa T, Deguchi H, Ueda K, Inoue I (2013) Altered mucosal DNA methylation in parallel with highly active Helicobacter pylori-related gastritis. Gastric Cancer 16:488–497
Youngblood B, Davis C, Ahmed R (2010) Making memories that last a lifetime: heritable functions of self-renewing memory CD8 T cells. Int Immunol. doi:10.1093/intimm/dxq437
zur Hausen H (2001) Oncogenic DNA viruses. Oncogene 20:7820–7823
zur Hausen H (2009) The search for infectious causes of human cancers: where and why. Virology 392:1–10
Acknowledgments
The scope of this chapter and its space limitations have unfortunately resulted in the inability to separately cite many of the original publications that have contributed substantially to the field. We sincerely apologize to the authors of these publications. This work was supported by G. Harold & Leila Y. Mathers Foundation and Santa Barbara Cottage Hospital Research Program (to M.J.M).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Mahan, M.J., Heithoff, D.M., Barnes V, L., Sinsheimer, R.L. (2017). Epigenetic Programming by Microbial Pathogens and Impacts on Acute and Chronic Disease. In: Doerfler, W., Casadesús, J. (eds) Epigenetics of Infectious Diseases. Epigenetics and Human Health. Springer, Cham. https://doi.org/10.1007/978-3-319-55021-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-55021-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55019-0
Online ISBN: 978-3-319-55021-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)