
Abstract
Cellular immunotherapy is currently one of the most exciting and promising new cancer treatments to reach the clinic. Expanded knowledge of T cell biology and new developments in synthetic biology have led to an ability to engineer T cells to express antibody-like recognition molecules linked to T cell-activating domains (chimeric antigen receptors, CARs) and tumor-specific T cell receptors (TCRs) which redirect patient-derived T cells toward their cancers. In this chapter, we will review the biology of T cell receptors, describe the rationale for generating affinity-enhanced TCRs with particular emphasis on the development of the affinity-enhanced NY-ESO-1-specific TCR. We also review clinical trials using TCR-engineered autologous T cells and highlight some important toxicities that have been observed in these studies. Finally, we provide an overview of some of the opportunities and challenges for wider application of this technology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Lymphoma
- Leukemia
- Myeloma
- Chimeric antigen receptor
- Modified T cells
- Immunotherapy
- Tumor-associated antigens
- Tumor-infiltrating lymphocytes
- TCR-engineered T cells
1 Introduction
The potential for cellular immunotherapy to offer significant therapeutic benefit to patients suffering from advanced forms of cancer was highlighted by the success of allogeneic blood and marrow hematopoietic cell transplants (HCTs) for hematologic malignancies and tumor-infiltrating lymphocytes (TIL) therapy for solid tumors, especially melanoma. These forms of therapy carry risks and technical challenges including potentially lethal graft-versus-host disease (GvHD) in the case of allogeneic HCTs and the inability to isolate and successfully prepare TIL for up to 50% of patients (Tran et al. 2008). Furthermore while the high frequency of clinical responses to TIL therapy amply demonstrated that autologous T cell therapy of cancer was possible, the cure rates from TIL were very low and likely limited by the prevalence of tumor-reactive T cells bearing low-affinity TCRs for tumor-associated antigens (TAA) as a result of thymic selection as well as the reestablishment of immunosuppressive mechanisms. For tumors characterized by genomic instability which display higher levels of patient tumor-specific neoantigens, the therapeutic potential for TIL may be greater (Maby et al. 2015). The identification of key immune checkpoints (e.g., CTLA-4/CD80 and PD-1/PDL-1) and the development of antibodies (e.g., ipilimumab, nivolumab, pembrolizumab) which block these inhibitory receptors and pathways have led to durable systemic responses in patients with a variety of advanced solid tumors including melanoma, lung cancer, and head and neck cancer—an unprecedented occurrence in the history of cancer therapy (Hodi et al. 2010; Brahmer et al. 2015; Seiwert et al. 2016). Nonetheless, the overall impact of such therapies may still be limited by the restricted repertoire and low affinity of the naturally occurring endogenous T cells present in the patient. Therefore, there remains a strong rationale for the engineering of tumor specificity into a patient’s own T cells, in order to overcome these limitations.
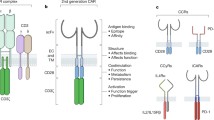
Progress in the understanding of TCR structure and function and advancements in synthetic biology and cell transduction methodologies have converged to make it feasible to engineer patient-derived T cells to express novel receptors or receptor constructs which can redirect the T cells to known tumor targets. Eshhar and colleagues were among the first to demonstrate that T cells could be redirected to tumors by introducing tumor-specific “T-body” or “CAR (chimeric antigen receptor)” constructs by gene modification (Gross et al. 1989). Through a series of elegant experimental advancements, these and other investigators have generated second- and third-generation CARs against the B cell antigen CD19 which exert a high frequency of durable responses in patients with advanced and refractory chronic lymphocytic leukemia, B cell lymphoma, and perhaps most impressively relapsed and resistant pediatric and adult acute lymphoblastic leukemia (ALL) (Maus et al. 2014; Porter et al. 2011; Grupp et al. 2013; Kochenderfer et al. 2015). While the CAR approach offers a cellular immunotherapy strategy that is not HLA-restricted and thereby expands the scope of patients who can be treated with each construct while avoiding the problem of MHC downregulation in tumors (Gross and Eshhar 1992), several limitations and clinical challenges exist: The tumor antigens for CAR-engineered T cells must be expressed on the cell surface and absent from critical non-tumor target organs and tissues. Also, the “non-physiologic” signaling mechanisms for CARs which are composed of the CD3-zeta cytoplasmic domain and one or more costimulatory domains (e.g., CD28 or 41BB or both) may lead to rapid proliferation and cytokine release syndrome (CRS) which requires careful clinical management and sometimes can be fatal (Barrett et al. 2014). However other factors including the density and accessibility of target antigen and the distribution of tumor cells may also contribute to the magnitude of T cell proliferation and the occurrence of CRS following adoptive transfer of both CAR T cells and TCR-engineered T cells.
Immunotherapy using TCR-engineered T cells involves the transfer of gene constructs encoding TCR alpha and beta chains which recognize 8–10 amino acid peptides processed from TAA and expressed in the context of HLA molecules. To date, the most common HLA-restricted TCRs tested in humans have been specific for HLA-A*02:01 which is found in about 50% of Caucasians, about 40% of Hispanics, and about 20–24% of African-Americans (Ellis et al. 2000; Gonzalez-Galarza et al. 2015). The frequency of HLA-A*02:01 is about 22% among Japanese, about 18% among US Asians, and highly variable in Chinese (0–24%). Because these peptides can be drawn from both the intracellular and extracellular proteins, the range of TAA which can be recognized by TCR-engineered T cells is estimated to be five- to tenfold higher than that for CARs based in part on the fact that only about 28% of all cellular proteins are expressed on the cell membrane in whole or in part (Uhlen et al. 2015). Furthermore, these TCR-engineered T cells typically employ physiological signaling pathways which may offer a safety advantage with respect to CRS. In this chapter, we will discuss the development of tumor-specific TCRs, their clinical applications and safety considerations, and future directions.
2 TCR Structure and Function
Physiologic T cell responses are primarily dependent upon the intermolecular interaction between the clonotypic alpha-beta TCR and the cognate peptide-MHC (major histocompatibility complex). This interaction is stabilized by CD4 and CD8 which bind to specific constant regions of the MHC class II and class I molecules, respectively, as well as by interactions between CD2 and CTLA-4 and CD28 on the T cells and CD58 (for CD2) and CD80 (for CTLA-4 and CD28) on the antigen-presenting or tumor target cells (Bridgeman et al. 2012). The interface between the TCR and the peptide-MHC complex (pMHC) involves the pMHC surface and the three hypervariable complementarity determining regions (CDRs) of the alpha and beta chains (Rudolph et al. 2006). The CDR1α and CDR2α regions are encoded by one of 47 TCR-α germline variable genes, while the CDR1β and CDR2β regions are encoded by one of 57 TCR-β germline variable genes. The CDR2 loops mainly contact the MHC molecule, while the CDR1 loops can contact both the MHC and the peptide. On the other hand, the CDR3α and CDR3β loops of the alpha and beta chains are encoded by variable (V), diversity (D, β chains only), and joining (J) segments and further diversified enzymatically by random nucleotide insertions at the junctional regions of the V-D-J gene segments. As predicted, the hypervariable CDR3α and CDR3β loops mainly contact the antigen peptide. Altogether the TCR generation mechanism can produce ~1015 to 1020 unique alpha and beta pairs able to recognize an enormous range of antigenic structures (Miles et al. 2011). TCRs on the surface of T cells bind to the pMHC with a certain affinity which is determined during T cell ontogeny. During T cell development and maturation in the thymus, T cells bearing TCRs that bind to cognate antigens too strongly are eliminated by negative selection, while T cells that bind too weakly are eliminated by apoptosis (Jameson et al. 1995). More specifically, early-stage double-positive (CD4+/CD8+) T cells are first positively selected through interaction with peptide-MHC molecules expressed by thymic cortical epithelial cells, while in the medulla of the thymus, single-positive (CD4+/CD8− or CD4−/CD8+) T cells encounter medullary epithelial cells which express an abundance of peptides derived from genes expressed in all somatic tissues (Nitta et al. 2010; Groettrup et al. 2010). As a result of this interaction, T cells which bear TCRs which exhibit high-affinity interactions with self-peptides are negatively selected. This affinity maturation process results in a vast repertoire of TCRs that bind to their cognate antigens in the context of the MHC, strongly if derived from nonself (e.g., microbial) antigens but weakly if derived from self-antigens (dissociation constant or K D ~ 0.1 to 500 μM or greater). Of note, antibodies typically bind their cognate antigens with K D values in the nM or pM range (van der Merwe and Davis 2003).
This editing mechanism significantly reduces the risk of autoimmune disease but conversely also limits the ability of the native immune system to recognize TAA since in most cases these are peptides that are derived from self-proteins that have been re-expressed or overexpressed in the tumor. This model of T cell ontogeny helps to explain the lack of success of cancer vaccination strategies which have thus far yielded limited clinical benefits with an objective response rate of 3.3% among more than 1300 patients who received a variety of cancer vaccines both at the NIH Surgery Branch and in the published literature (Rosenberg et al. 1994, 2004). It also explains the differential affinity ranges that have been observed for virus and cancer-specific TCRs with markedly higher affinities for TCRs that bind viral antigens than cancer-related antigens (Aleksic et al. 2012). In general the binding affinity of TCRs for cancer-related antigens that are “self”-antigens is about tenfold lower than the binding affinity of TCRs for nonself and microbial antigens.
Another important principle is that the avidity of a TCR for its cognate pMHC and the kinetics of the interaction are major determinants of T cell activation (Zoete et al. 2013; Davis et al. 1998; Irving et al. 2012; Stone et al. 2009). Using techniques such as surface plasmon resonance (SPR) and isothermal titration calorimetry, the biophysical characteristics have been calculated for dozens of TCR-pMHC interactions (Bridgeman et al. 2012). On the basis of this work, two major models of T cell activation have been developed: the “affinity model” which proposes that the level of T cell activation depends on the total number of TCRs bound to peptide-MHC complexes and the “half-life model” whereby optimal T cell activation requires that the TCR engage the peptide-MHC complex with sufficient binding strength and time to induce signaling (Zoete et al. 2013). It follows that strategies which generate TCRs with higher affinity for cognate tumor antigens could lead to superior therapeutic effect especially when combined with forced expression of those affinity-enhanced TCRs on cytotoxic T cells. Indeed multiple investigators have shown that affinity enhancement of TCRs for cognate peptide-MHC complex within the physiologic TCR affinity range (K D ~ 200 μM to 1 μM) and in the presence of low levels of peptide-MHC complexes which is typical for most tumors results in improved T cell function (Robbins et al. 2008; Zhao et al. 2007). On the other hand, affinity enhancement into the supraphysiologic range (K D < 1 μM) often led to functional impairment due in part to diminished expression of costimulatory molecules accompanied by increased PD-1 expression and upregulation of SHP-1 and SHP-2 phosphatases which serve to downregulate T cell function (Irving et al. 2012; Hebeisen et al. 2013, 2015). Further enhancement of affinity to K D levels of <1 nM may lead to cross-reactivity with other peptide-MHC complexes. Thus an overarching principle has emerged that optimal function (including T cell activation and target specificity) of a given TCR occurs within a certain window of binding affinities. Within the physiologic range of TCR affinity mentioned above, the specific affinity for optimal T cell function is not possible to predict for each TCR-p-MHC complex and must be empirically determined in each case.
3 Isolation of Therapeutic TCRs and Strategies for Functional Enhancement
Candidate TCRs for therapeutic applications have generally been identified initially by isolating TIL from patient tumors. To illustrate the process of generating a TCR for therapeutic application, the steps taken to generate an affinity-enhanced TCR for a naturally processed peptide that is derived from the cancer-testis antigens (CTAgs) NY-ESO-1 and LAGE-1 is described in some detail. This process involved both experimental iteration and serendipity. Among the various classes of TAA which could be targeted by T cells, the CTAgs or cancer-germline antigens (CGs) are attractive because of their relatively clean expression profiles, being mainly restricted to germ cells and cancers (Ilyas and Yang 2015; Kvistborg et al. 2013). In addition, these antigens are shared by a variable but significant proportion of patients who develop specific cancers, and some may promote cancer cell survival and confer chemotherapy resistance (Monte et al. 2006), thus increasing the potential clinical benefit from targeting these antigens with TCR-modified T cells. Other classes of TAA including tissue differentiation or lineage-specific antigens and neoantigens derived from patient-specific mutations may be less desirable targets for TCR-engineered T cells due to “off-tumor” expression leading to damage of normal tissues and application to very small numbers of patients, respectively.
NY-ESO-1 was originally identified as a putative human tumor antigen by a method called serological expression cloning of recombinant cDNA libraries from human tumors (SEREX) using tissue obtained from a patient with squamous cell carcinoma of the esophagus (Chen et al. 1997). The function of NY-ESO-1 is unknown. NY-ESO-1 expression is detected in testis, ovary, and weakly in uterus specimens, but no mRNA can be detected by reverse transcription-polymerase chain reaction (RT-PCR) in any other normal tissue. NY-ESO-1 (CTAG-1B) is an immunogenic cancer-testis antigen (CTA) associated with spontaneous and vaccine-induced immunity that can lead to clinical cancer responses (Hunder et al. 2008; Yuan et al. 2008). Up to 60% of advanced myelomas have been reported to express NY-ESO-1, a feature correlated to tumor proliferation and high-risk features (van Baren et al. 1999; Jungbluth et al. 2005; Condomines et al. 2007; Atanackovic et al. 2007; van Rhee et al. 2005).
In addition to myeloma, multiple solid tumors express NY-ESO-1 at rates of up to 50% including melanoma and cancers of the bladder, lung, ovary, uterus, and esophagus (Chen et al. 1997). Reported expression rates vary between different studies; RT-PCR is more sensitive than immunohistochemistry (IHC) and tends to give higher figures for NY-ESO-1 expression. Figures derived from IHC are more reliable, since this technique detects protein rather than RNA. A feature of CTAgs like NY-ESO-1 is that they can have heterogenous expression in the tumor and so it is informative to measure both aspects in tissue sections by RNA- or protein-based approaches. CTLs recognizing the HLA-A*0201-restricted epitope NY-ESO157–165 (SLLMWITQC) have been grown from the blood and lymph nodes of myeloma patients by several different groups (Atanackovic et al. 2007; van Rhee et al. 2005). LAGE-1, a highly homologous TAA with a very similar expression pattern as NY-ESO-1, also shares the same epitope, and T cell clones specific for this epitope also kill antigen-positive tumor cells (van Rhee et al. 2005).
TCR gene cDNA sequences were isolated from the NY-ESO-1 HLA-A*0201-SLLMWITQC-restricted T cell clone 1G4 (Jager et al. 1998). This CTL clone was cultured from a metastatic lymph node derived from an 81-year-old woman with melanoma who exhibited both strong serologic and cytolytic reactivities against autologous tumor cells. The CTL clone (1G4) was found to recognize the SLLMWITQC peptide corresponding to amino acids 157–165 of NY-ESO-1 in an HLA-A*02:01-restricted manner. Note that the SLLMWITQC peptide sequence is the identical sequence derived and expressed from the LAGE-1 antigen, and therefore LAGE-1 antigen-positive tumors are also targeted by the 1G4 T cell clone.
The cDNA coding sequences for the mature extracellular regions of the α and β chain TCR proteins were cloned into separate E. coli plasmid vectors and expressed as protein inclusion bodies. These inclusion bodies were purified, solubilized, and then refolded as soluble α/β heterodimeric TCR proteins (sTCR). Both TCR chains were genetically truncated at the C terminus immediately before the native intra-chain cysteine residues and joined together by means of an artificial disulfide bond engineered between the α and β chain TCR constant regions. The 1G4 sTCR protein was purified, and its HLA-peptide antigen-binding kinetics were analyzed by surface plasmon resonance (SPR) using a BIAcore 3000. The 1G4 gene sequences also served as a platform to generate variants with enhanced antigen-binding affinity using bacteriophage display of large numbers of mutated 1G4 TCR proteins.
The HLA-A*0201-SLLMWITQC-peptide antigen complex was required for validation of antigen binding of the 1G4 T cell clone and the soluble version, as well as for testing and selection of affinity-enhanced variants generated by phage display. This complex was made by cloning the HLA-A*0201 protein and β2 microglobulin into E. coli expression vectors. These proteins were then expressed separately as protein inclusion bodies prior to solubilization, mixing with the SLLMWITQC peptide and refolding. The refolded pMHC antigen complex was then purified by ion exchange and size exclusion chromatography.
A 1G4 sTCR phage-display library was constructed with mutations covering the hypervariable complementarity determining region (CDR3 region) of the β chain. Three rounds of selection/enrichment for high-affinity TCR clones were performed. Competition ELISA assays for high-affinity mutant TCR phage identified several candidate TCR β-chain CDR3 mutations. These high-affinity β chain mutants then formed the basis of a library where the CDR3 α chain was also mutated in a similar manner (Li et al. 2005). This complex library was then used to isolate still higher affinities. Later, mutations were introduced into the CDR2 regions of both chains, and these libraries were then reselected (Dunn et al. 2006).
4 Biochemical Validation and Efficacy Testing of the Affinity-Enhanced NY-ESO-1 sTCR Clones
High-affinity mutant TCR alpha and beta chain genes were cloned separately into E. coli expression vectors. These mutant TCR chains were expressed and refolded in various paired combinations including with wild-type chains. They were then purified and analyzed for binding to HLA-A*0201-SLLMWITQC antigen by SPR. As mentioned, earlier studies using T cells transfected with high-affinity TCRs indicated that TCRs with very high affinities could exhibit diminished function and altered target specificity suggesting that TCRs with moderately increased affinity should be preferentially evaluated (Zhao et al. 2007). Thus, the high-affinity mutant CDR3α chain TCR sequences (Li et al. 2005) and mutant CDR2β chain TCR sequences (Dunn et al. 2006) were partially back-mutated to the wild-type 1G4 TCR sequence. A panel of these phage-derived 1G4 TCR mutants was then assessed in TCR-transfected T cells (Robbins et al. 2008). From these data TCRs with single or dual amino acid substitutions in the antigen-binding region and which were anticipated to have optimal cellular properties were selected for comparison in lentiviral T cell transduction and functional studies. These studies consisted of cytokine release assays and cytotoxicity assays against a panel of NY-ESO-1 positive and negative tumor cell lines. From these studies, an affinity-enhanced NY-ESO-1-TCR construct emerged as the “winner” based on enhanced binding properties, enhanced cytokine release, and target cell killing as well as retention of antigen specificity. This construct consisted of an alpha-chain variant (c259) carrying amino acid substitutions at positions 95 (threonine → leucine) and 96 (serine → tyrosine) of the CDR3 region of the 1G4 NY-ESO-1 TCR clone combined with the wild-type beta chain of the 1G4 NY-ESO-1 TCR clone. This affinity-enhanced TCR had a T 1/2 of 19 s and K D of 730 nM vs 2.2 seconds and 9.3 μM for the wild-type 1G4 TCR, indicating about tenfold higher “dwell” time and binding strength for the 1G4 NY-ESO-1 TCR variant known as c259 (Robbins et al. 2008).
It should be noted that other investigators have taken a structural approach starting with crystallographic structures of TCR-peptide-MHC complexes in order to elucidate points of contact. Amino acid substitutions can then be made logically rather than randomly based on structure compatibilities in order to achieve affinity enhancements (Zoete et al. 2013; Haidar et al. 2009; Malecek et al. 2014). This approach has also led to development of affinity-enhanced variants of the HLA-A*02:01-restricted TCR for NY-ESO-1157–165 (Schmid et al. 2010). Furthermore, a high-throughput TCR gene-capture methodology has also been developed to more rapidly isolate and identify tumor antigen-specific TCR sequences from human tumor tissue both with and without prior knowledge of antigen specificities. This methodology was used to develop a library of CTAg-specific TCRs and may also facilitate TCR-engineered T cell immunotherapy against private neoantigens which are expressed by individual patient tumors (Linnemann et al. 2013).
The cytotoxic effects of T cells transduced with the affinity-enhanced variant of the HLA-A*02:01-restricted TCR for NY-ESO-1157–165 were then evaluated in the immunodeficient NSG (NOD/scid/γc null) mouse model using the human B cell precursor acute lymphoblastic leukemia cell line (NALM-6) as the tumor target. The immunodeficient NOD/scid/γc null (NOG) mouse is an excellent xenotransplantation model to measure the in vivo repopulation of human CD4 T cells (Ito et al. 2009). Following engraftment, the human hematopoietic cells can be maintained in NSG mice for at least 2 months or until fatal xenogeneic GvHD. Intravenous injection of NALM-6 into NSG mice provides a systemic tumor model with rapid evolution toward animal death within 20–23 days. Parental NALM-6 cells express both HLA-A1 and HLA-A2 molecules and also low levels of certain cancer-testis antigens including MAGE A3, but no NY-ESO-1 antigen. To achieve higher expression of this antigen, NALM-6 cells were transduced with lentiviruses expressing NY-ESO-1 proteins in conjunction with the green fluorescent (GFP) protein (NALM6-GFP-NY-ESO1). As a control cell line, NALM-6 cells were transduced with GFP only (NALM6-GFP). In previous experiments, the NALM6-GFP-NY-ESO1 cells induced mouse death within 23 days, similar to the parental NALM-6 cells.
The efficacy study used parental and transduced NALM-6 cell lines and evaluated the impact of CD4 and CD8 T cells which were genetically modified by lentiviral transfection to express NY-ESO-1 TCR on animal survival. The infused study cell number was 5 × 106 CD4 and CD8 T cells. This dose was chosen based on pilot data in the NALM-6 model which indicated that this is the effective dose required to observe an antitumor effect. Since a human is on average 3000-fold larger than an average mouse, a cell dose of 5 × 106 cells in a mouse roughly corresponds to a human dose of ~10 billion cells. The starting dose in human trials was expected to be about 1 × 109, and so the dose evaluated in the preclinical murine experiments represented about ten times greater than the human dose that would be administered in phase I trials.
As expected, all the control mice (injected with saline and mock/untransduced T cells) died between day 19 and 23. Also, the high-affinity NY-ESO1 TCR (c259)-transduced T cells did not give the mice any survival advantage when mice were inoculated with the NY-ESO-1-negative NALM6 tumor cells. However when mice carrying the NALM6-NY-ESO-1 positive tumor cells were treated with the NY-ESO-1-TCR-transduced T cells, a significant survival advantage was seen regardless of the TCR affinity (wt or the affinity-enhanced c259 TCR variant). These data suggested that both NY-ESO-1 TCRs were effective against tumor cells expressing the cognate antigen.
5 Clinical Translation of the Affinity-Enhanced NY-ESO-1 (c259) TCR
In the course of translating the affinity-enhanced NY-ESO-1 (c259) TCR to the bedside, several additional considerations merited attention including optimization of function by the transduced T cells and preclinical testing to minimize the risk of off-tumor effects. Two potential impediments to tumor targeting and killing by TCR-modified T cells include the potential problem of “mispairing” of transduced alpha and beta chains with the native T cell alpha and beta chains as well as the limited availability of components of the CD3 signaling complex. Second, both mispairing and the physiological redundancy of any TCR to potentially recognize a large number different peptides in the context of HLA molecules can lead to “off-target” recognition and off-tumor toxicities. The formation and surface expression of a functional TCR requires pairing of the new alpha and beta chains followed by association with the four invariant chains of the CD3 complex, namely, CD3γ, CD3δ, CD3ε (two subunits), and CD3ζ (two subunits). Furthermore, the availability of the CD3 subunits particularly CD3ζ is limited and partially formed TCR-CD3 complexes that undergo degradation in the endoplasmic reticulum (Minami et al. 1987, Mallabiabarrena et al. 1992). Ex vivo studies have shown that introduction of a novel TCR into a lymphocyte which expresses its native TCR results in mispairing between exogenous alpha and beta chains and endogenous beta and alpha chains sufficient to generate neoreactivities which are either HLA class I or class II restricted and directed against both allogeneic and autologous targets (van Loenen et al. 2010). Furthermore, such mispairing with generation of autoreactive T cells was thought to contribute to lethal GvHD in a mouse model which utilized an unusually intensive conditioning regimen (Bendle et al. 2010). It should be noted that to date, no known immunotoxicity has been demonstrated to occur in humans on the basis of mispairing between native and exogenous TCR chains. Potential strategies for reducing mispairing and enhancing the surface expression and function of transduced therapeutic TCRs include the optimization of equimolar translation of the introduced alpha and beta chains using the internal ribosome entry site (IRES) sequence of the encephalomyocarditis virus or more recently the “self-cleaving” 2A sequences from picornaviruses or porcine teschovirus which allows the ribosome to skip from one chain sequence to an adjacent one in order to achieve nearly equivalent translation and production of each peptide chain (Furler et al. 2001; Szymczak et al. 2004). Two other strategies to augment preferential pairing of the exogenous TCR chains include introduction of a second cysteine residue to generate an extra disulfide bond between the introduced alpha and beta chains and use of murine TCR sequences to facilitate species-specific pairing (van Loenen et al. 2010; Cohen et al. 2006, 2007). The molecular techniques which promote equimolar synthesis of the exogenous TCR alpha and beta chains have already been employed in the development of therapeutic TCRs for human studies.
The issues of “on-target, off-tumor” and “off-target, off-tumor” toxicities whereby adoptively transferred TCR gene-modified T cells recognize cognate peptide antigen on non-tumor cells and elicit damage to normal tissues or recognize unintended, “cross-reactive” peptides or HLA molecules on non-tumor cells and elicit normal tissue injury are likely more common causes of TCR-engineered T cell toxicities. These toxicities will also be discussed further in the context of the clinical studies. Steps taken during preclinical development to mitigate against these toxicities include the following: (i) alanine substitution scanning to identify which amino acid residues in the antigen peptide are critical for TCR binding and thereby identify structurally similar peptide sequences in other genes which the TCR could potentially recognize, followed by binding assays to test for potential cross-reactivity; (ii) testing of the TCR against an “alloreactivity panel” to search for cross-reactivity with other HLA molecules in addition to the one that the TCR is known to bind; (iii) testing of the TCR against a large panel of primary cells and tissues; and (iv) careful assessment of the true distribution of the target antigen to minimize the risk of “on-target/off- tumor” effects. As mentioned, on-target/off-tumor toxicity and off-target/off-tumor toxicity will also be discussed further in the context of the clinical studies.
6 Early Clinical Studies of TCR-Engineered Lymphocytes
The first clinical trial of TCR-engineered T cells for cancer immunotherapy focused on the melanocyte differentiation antigen MART-1 which is expressed in 80–90% melanoma cases and frequently recognized by melanoma TIL. Using a TCR isolated from a melanoma TIL which was HLA-A*02:01 restricted and recognized the MART-1:27-35 epitope AAGIGILTV, investigators cloned a cDNA for this TCR into a retrovirus and then transduced autologous peripheral blood lymphocytes from 15 HLA-A*02:01+ patients with advanced melanoma (Morgan et al. 2006). After myeloablative therapy to enhance the impact of the T cell immunotherapy through various mechanisms, these patients received the transduced T cells followed by maintenance therapy using IL-2. Of the 15 patients who were treated, 1 had a complete regression that lasted for 23 months, while a second patient exhibited a complete regression of an axillary mass and a 90% reduction in the size of a liver lesion which was resected 10 months later, and the patient remained disease-free 9 years after treatment.
To augment response rates, a higher-affinity TCR for the MART was identified and tested. TCRs for the MART-1:27-35 AAGIGILTV were isolated from TIL derived from 24 melanoma patients and tested for avidity for the HLA-peptide complex and IFN-γ production by transduced lymphocytes. A specific TCR termed DMF5 exhibited the strongest response and was selected for further clinical development (Johnson et al. 2006). Similar preclinical studies for another melanocyte differentiation antigen gp100 led to development and selection of a relatively high-affinity TCR directed against the gp100:154-162 epitope KTWGQYWQV which was isolated from a T cell clone generated from an HLA-A*02:01+-transgenic mouse that had been immunized with the gp100:154-162 epitope. Treatment of patients with autologous T cells engineered to express the high-affinity MART-1 TCR (DMF5) led to objective responses in 6 of 20 patients (30%) with advanced melanoma and in 3 of 16 patients (17%) who received autologous lymphocytes transduced with the high-affinity gp100 TCR (Johnson et al. 2009). Of the 36 total patients treated in these two studies, 34 eventually relapsed, while 1 patient was an ongoing partial responder nearly 8 years after receiving treatment with the MART-1:27-35 TCR, and a second patient had an ongoing complete response nearly 8 years after receiving treatment with the gp100:154-162 TCR.
Importantly, severe “on-target, off-tumor” toxicities were observed in most of the patients who were treated with cells engineered to express both the MART-1 and the gp100 TCRs including skin rash in 29 of 36 patients which culminated in loss of the majority of epidermal melanocytes (Robbins 2015). In addition, uveitis developed in 11/20 patients who received the MART-1 TCR-modified T cells and 4/16 patients who received the gp100 TCR gene-modified T cells; 13 of the 15 affected patients were successfully treated with steroid eye drops. Acute hearing loss developed in 10/20 patients who received MART-1 TCR-expressing T cells and 5/16 patients treated with gp100 TCR-expressing T cells, while 9 of the 36 total patients developed vertigo presumably due to engineered T cell attack on inner ear melanocytes; all patients responded to intratympanic steroid injections (Robbins 2015). The higher objective response rates and also the on-target/off-tumor toxicity rates in these latter studies suggest that affinity enhancement may increase the potency of TCR-modified T cells.
Another clinical trial was conducted involving the adoptive transfer of T cells engineered to express an affinity-enhanced TCR for an HLA-A*02:01 restricted immunogenic peptide composed of amino acids 691–699 from the carcinoembryonic antigen (CEA) found on most colon cancers but also normal colonic epithelial cells found in the crypts. This TCR also contained a serine → threonine substitution at codon 112 in the CDR3 region of the TCR-α which seemed to augment recognition of the CEA peptide on colon cancer cell lines (Parkhurst et al. 2009). Treatment of HLA-A*02:01+ patients with metastatic colon cancer who had high levels of circulating CEA using CEA691–699-directed TCR-expressing T cells elicited a 6-month partial response in one of three patients but predictably led to severe inflammatory colitis and grade 1 diarrhea in one patient but grade 3 diarrhea in two patients requiring administration of oral corticosteroids in two of the three patients (Robbins 2015; Parkhurst et al. 2009). While these autoimmune toxicities resolved in 4–6 weeks, the trial was terminated early. The clinical experience with autologous T cells engineered to express TCRs directed against lineage-specific or “differentiation” antigens which are expressed on tumors but also normal tissue counterparts suggests that occasional clinical responses can be obtained which are sometimes durable, but the benefits seem more than offset by “on-target, off-tumor” immunotoxicities. The lack of selectivity against tumor tissue may be explained in part by the presence of immuno-inhibitory factors which are operative in the tumor bed but not in the normal tissue. This has led to studies using T cells engineered to carry TCRs which are directed against antigens that are mainly or exclusively expressed by tumors including the cancer-testis antigens (CTAs) or cancer-germ line antigens (CGs), most notably NY-ESO-1.
7 Clinical Studies of NY-ESO-1 TCR-Expressing T Cells in Solid Tumors
The NY-ESO-1 cancer-testis or cancer-germline antigen is widely expressed in solid tumors including melanoma, lung, breast, ovarian, prostate, and bladder tumors where the expression frequency ranges between 10 and 50% (Chen et al. 1997). NY-ESO-1 is abundantly and even more frequently expressed in synovial cell sarcomas where it is found in approximately 60–70% of tumors (Jungbluth et al. 2001). Based on the higher frequency and selectivity of expression in synovial cell sarcoma and melanoma, a clinical trial was conducted using autologous T cells engineered to express the affinity-enhanced HLA-A*02:01-restricted (c259) variant of the 1G4 NY-ESO-1 TCR carrying amino acid substitutions at positions 95 (threonine → leucine) and 96 (serine → tyrosine) of the alpha-chain CDR3 region combined with the wild-type beta chain of the 1G4 NY-ESO-1 TCR clone. After lymphodepleting chemotherapy consisting of fludarabine and cyclophosphamide, patients received retrovirally transduced autologous T cells carrying the affinity-enhanced NY-ESO-1 TCR followed by IL-2 maintenance therapy (Robbins et al. 2011). Four of six HLA-A*02:01+ patients with synovial cell sarcoma and five of eleven advanced melanoma patients exhibited clinical responses by RECIST criteria; 2/11 melanoma patients had complete responses that persisted for more than 1 year, while a partial response in one patient with synovial sarcoma lasted for 18 months. A recent report using a similar experimental design with expanded cohorts of patients and longer follow-up included 18 patients with progressive synovial cell sarcoma, 11 of whom had objective responses, and 20 patients with advanced melanoma, 11 of whom had objective responses (Robbins et al. 2015). The projected 3- and 5-year overall survival rates for the synovial cell sarcoma patients were 38% and 14%, respectively, while the projected survivals for the melanoma patients were 33% at both timepoints. Using this larger study, the investigators sought to identify predictors of response. Although some patients in this study were also immunized with a peptide vaccine derived from the NY-ESO-1157–165 epitope, receipt of this vaccine did not correlate with response, while higher T cell numbers and one measure of enhanced functionality (IFNγ production in response to peptide-pulsed EBV-transformed lymphocyte targets) did seem to correlate to better clinical response. Importantly, no toxicities attributable to the transduced T cells were observed in accordance with the restricted expression of NY-ESO-1 to tumors and germ cells.
8 Clinical Studies of NY-ESO-1 TCR-Expressing T Cells in Myeloma
Up to 60% of advanced myelomas have also been reported to express NY-ESO-1, a feature which is correlated to enhanced tumor proliferation and other high-risk features including relapsed and extramedullary disease (van Baren et al. 1999; Jungbluth et al. 2005; Condomines et al. 2007; Atanackovic et al. 2007; van Rhee et al. 2005). High-dose chemotherapy followed by autologous stem cell transplantation (AHCT) has been a mainstay of therapy for myeloma and better clinical outcomes following AHCT for myeloma and other hematologic neoplasms that may be associated with rapid post-transplant lymphocyte recovery (Porrata et al. 2001; Porrata and Markovic 2004). In addition, tumor-reactive T cells are present at low frequencies in the marrow and blood of myeloma patients which may target myeloma cells upon activation (Dhodapkar et al. 2002; Noonan et al. 2005). Thus autologous immune-mediated control of myeloma may be possible.
We and other investigators have studied whether cancer vaccines administered post-AHCT could be immunogenic and improve outcomes. Our studies involved combined cellular and vaccine strategies under the hypothesis that transfers of ex vivo costimulated autologous T cells will improve functional T cell recovery, thereby providing a platform for enhanced vaccine-directed immune responses. For these studies autologous T cells were stimulated by coculture with immunomagnetic beads conjugated with anti-CD3 and anti-CD28 monoclonal antibodies to prevent T cell anergy through combined CD3 and CD28 signaling (Li et al. 1999; Boussiotis et al. 2000). Using microbial vaccines including a pneumococcal conjugate vaccine (Prevnar®) and an influenza vaccine as well as a cancer antigen vaccine based on peptides derived from hTERT and survivin, these studies showed that an early post-transplant infusion of 1–5 × 1010 in vivo vaccine-primed and ex vivo costimulated T cells followed by booster immunizations led to protective antimicrobial antibody responses in a majority of patients and cancer vaccine-directed T cell responses in about 1/3 of patients (Rapoport et al. 2005, 2011; Stadtmauer et al. 2011). The addition of novel adjuvants such as the toll-like receptor-3 (TLR3) agonist Poly-ICLC (Hiltonol®) to a MAGE-A3 tumor antigen vaccine led to functional T cell responses in more than 2/3 of patients and a marginally significant better EFS for patients who developed IFN-γ responses on both CD4+ and CD8+ T cells (Rapoport et al. 2014). Furthermore, a pattern of schedule-dependent T cell expansion was observed, whereby the most robust CD4+ and CD8+ T cell recoveries post-AHCT occurred when the ex vivo costimulated T cells were infused very early after high-dose chemotherapy (on day +2 after autologous stem cell transplantation rather than day +14 or day +100) presumably as a result of the homeostatic expansion mechanisms (e.g., unbound IL-15) that prevail after lymphodepleting chemotherapy (Rapoport et al. 2009). Using this platform of high-dose, lymphodepleting chemotherapy followed by AHCT and then adoptive transfer of ex vivo costimulated autologous T cells, we conducted and reported a phase I/II clinical trial (NCT01352286) designed to evaluate the safety and activity of autologous T cells genetically engineered to express the affinity-enhanced TCR (NY-ESOc259) that recognizes the NY-ESO-1/LAGE-1 peptide complex HLA-A*0201-SLLMWITQC (NY-ESO-1157–165) and infused post-AHCT (Rapoport et al. 2015). Patients with high-risk or relapsed multiple myeloma (MM), who were HLA-A*0201 positive and whose myeloma was positive for NY-ESO-1 and/or LAGE-1 by quantitative qRT-PCR, were eligible. Figure 7.1 shows a flow diagram for this clinical trial. Briefly, autologous CD25-depleted CD4 and CD8 T cells were activated and expanded using anti-CD3/anti-CD28 antibody-conjugated microbeads, and genetically modified with a self-inactivating (SIN) lentiviral vector encoding the affinity-enhanced NY-ESO/LAGE-1 TCR. Engineered T cells were administered 4 days after high-dose melphalan and 2 days following auto-HCT (day +2 of AHCT), at a dose range of 1–10 billion total cells. We hypothesized that adoptive transfer of NY-ESOc259 TCR-engineered T cells would improve the duration and depth of post-AHCT clinical responses in HLA-A201-positive patients with advanced NY-ESO-1/LAGE-1-expressing MM.

Autologous leukapheresis is performed. CD25-depleted CD4 and CD8 T cells are activated and expanded using anti-CD3/anti-CD28 antibody-conjugated microbeads, and genetically modified with a self-inactivating (SIN) lentiviral vector encoding the affinity-enhanced NY-ESO/LAGE-1 TCR. Engineered T cells are administered 4 days after high-dose melphalan or 2 days following auto-HCT (day +2 of AHCT), at a dose range of 1–10 billion total cells
Prior to enrollment on study, patients had received a median of three prior therapies (range 1–4) including five with prior AHCT. Sixty percent (12/20) of tumors contained cytogenetic abnormalities most of which were considered high risk. Twenty patients (median age of 58, range 44–72 years) received a mean of 8 billion total CD3 T cells (range 1–10 billion) which were genetically modified at an average of 33% (range 18–49%, study minimum was 10%). Thus a mean of 2.4 billion transduced CD3 T cells (range 0.45–3.9 billion) were infused. Infusions were well-tolerated, and no clinically apparent CRS was detected although significant elevations of serum IL-6 were detected in all patients (median 22-fold increase; range 8- to 2272-fold) within 7–28 days post infusion which overlapped with the period of maximum T cell expansion. A subset of responding patients with high levels of engineered T cells were evaluated by flow cytometry for cytokine production (IFN-γ) and cytotoxic potential (granzyme B production and CD107α surface expression) in response to peptide-loaded targets. The data showed that polyfunctional T cells which were generated during manufacturing engrafted in the patients where they remained functional in the peripheral blood for up to a year after infusion.
The majority of adverse events were related to the high-dose melphalan. Importantly, there were no treatment-related fatalities. All serious adverse events (SAEs) (5) were resolved, and the 17 adverse events which were at least probably related to the treatment were grade 3 or lower. Notably a skin rash with lymphocytosis occurred in 3/20 patients, and some patients had a diarrheal syndrome that occurred later than expected for melphalan-induced mucositis, and three cases were confirmed by biopsy to be autologous graft-versus-host disease (autoGvHD). However, analysis of engineered T cells in inflamed and normal colonic tissue and peripheral blood was performed in patients who developed autoGvHD, and while engineered T cells were present in inflamed tissue, they were diluted at sites of inflammation compared to adjacent non-inflamed tissues, suggesting that they were not driving the event. Also we previously observed acute GvHD (aGvHD) involving the GI tract after adoptive transfer of activated but non-gene-modified T cells (Rapoport et al. 2009).
The median progression-free survival (PFS) of this high-risk cohort was 19.1 months (the lower bound of the 95% CI was 8.5 months, upper bound has not been reached yet), while 15/20 (75%) patients remained alive at the time of the initial report. Engineered T cells were found to expand, traffick to marrow, and persist for at least 6 months in all but one patient as determined by Q-PCR and/or flow cytometry. Engineered cells were detected in the blood or marrow by flow cytometry for as long as 2 years after infusion in two patients. This length of persistence is unusual for TCR gene-modified T cells and for gene-modified T cells in general and may reflect adoptive transfer in the setting of AHCT (after intensive lymphodepleting chemotherapy), the use of ex vivo costimulation of the transduced T cells and/or properties of the NY-ESO-1-TCR, and expression vector. Evidence for specific targeting of antigen-positive myeloma cells came from several directions: Compared to enrollment levels, loss of NY-ESO-1 and LAGE-1 transcripts by qRT-PCR analysis on marrow specimens was observed in 12/15 evaluable patients at day 100 and in 11/13 evaluable patients at day 180. Conversely, the 3/15 patients who had detectable levels of NY-ESO-1 and LAGE-1 transcripts in the marrow at day 100 also had very low or undetectable levels of engineered T cells in the peripheral blood which was followed by a relapse in two patients. Four patients had an increase in CD138 transcripts (as a measure of plasma cells in general) in the absence of NY-ESO-1/LAGE-1 transcripts, suggesting that pressure from the immune response was potentially selecting for tumor escape subclones that lacked target tumor antigen. Two patients with prolonged persistence of gene-modified T cells developed durable partial responses associated with residual NY-ESO-1/LAGE-1-negative myeloma cells also consistent with the phenomenon of antigen-negative tumor escape. Furthermore, on a statistical basis, between days 0 through 180 post-transplant, the persistence of gene-modified T cells in peripheral blood was inversely correlated with the level of NY-ESO-1 expression in the marrow (p = 0.022) and possibly with LAGE-1 (p = 0.098). In contrast, there was no relationship over time between T cell persistence in blood and CD138 expression (reflecting total plasma cells) in the marrow. Altogether, these data suggest induction of a robust and tumor antigen-specific memory immune response.
9 Risks of TCR-Engineered T Cell Therapy: The MAGE-A3 and MART-1 Experiences
The three major categories of toxicity resulting from TCR-modified T cell therapy include (1) on-target/off-tumor toxicity, (2) off-target/off-tumor toxicity, (3) and CRS presumably resulting from excessive T cell activation. The clinical studies using affinity-enhanced MART-1 (DMF5) and gp100 TCR-engineered T cells that were described earlier amply illustrated examples of “on-target/off-tumor toxicity” resulting from T cell attack on normal melanocytes in the skin and inner ear. These studies suggest that further development of TCR immunotherapy against lineage-specific or tissue differentiation antigens may be difficult. The following studies provide examples of “off-target/off-tumor toxicity.”
A companion myeloma trial involving post-AHCT adoptive transfers of autologous T cells engineered to express an affinity-enhanced TCR for the HLA-A*01-restricted MAGE-A3 peptide (EVDPIGHLY) complex was also developed and implemented. This high-affinity MAGE-A3 TCR (MAGE-A3a3a TCR) carried four substitutions in the alpha chain of the CDR2 region, while the beta chain remained wild type. As with the NY-ESO-1 affinity-enhanced TCR, this MAGE-A3 TCR underwent extensive preclinical development involving synthetic biology, biophysical and immunological testing, and extensive screening of normal tissues and cells. After high-dose melphalan (day 2), AHCT (day 0), and T cell infusion (day +2), the first patient treated on this study developed cardiogenic shock accompanied by fever, hypoxia, and hypotension on days +3 to +5 and died 5 days after T cell transfer (day +7) (Linette et al. 2013). PCR analysis of the peripheral blood for the MAGE-A3a3a TCR sequences revealed robust in vivo gene-modified T cell expansion which was >400 cells/μl just 3 days after T cell transfer. By the time of the patient’s death, the transduced T cells were estimated to have expanded ~200-fold in vivo. The transduced T cells localized to the bone marrow, lung, heart, and liver, but the highest concentrations were in the blood and pericardial fluid. At autopsy, there was extensive myocardial necrosis with a striking CD3+ lymphoid cellular infiltration in the myocardium; similar infiltration was not observed in the skeletal muscle or other examined organs. Cytokine analysis of the blood and pericardial fluid was consistent with immune cell activation (including ~100-fold increases in IFN-γ and ~1000-fold increases in IL-6). A similar clinical course and cardiac histopathology were observed in a second patient who had melanoma and received MAGE-A3a3a TCR-transduced T cells after conditioning with high-dose cyclophosphamide (Linette et al. 2013). Elegant post-SAEs in vitro studies using an alanine-scanning methodology to delineate critical TCR-binding residues in the MAGE-A3 peptide EVDPIGHLY ultimately identified a peptide (ESDPIVAQY) derived from the very large (3-megadalton) cardiac muscle protein titin as the likely target of off-tumor, off-target TCR cross-reactivity (Cameron et al. 2013). Interestingly extensive preclinical testing revealed no concerns for off-target activity, and post-SAEs testing of 38 cardiac-derived primary cells (including 10 which were HLA-A*01+) showed no evidence for activation of MAGE-A3a3a TCR-transduced T cells by IFN-γ ELISpot analysis. The only model which demonstrated robust reactivity with the affinity-enhanced TCR was an iCell cardiomyocyte culture system which was derived from induced pluripotent stem cells and included a mixture of spontaneously electrically active atrial, nodal, and ventricular-like myocytes which may be more representative of normal heart tissue.
In another clinical trial using an affinity-enhanced HLA-A*2:01 TCR against the MAGEA3: 112-120 peptide (KVAELVHFL), two of seven melanoma patients who received chemotherapy conditioning followed by adoptive transfer of transduced autologous T cells along with IL-2 had complete responses, one lasting more than 4 years, and two additional patients had objective partial responses (Morgan et al. 2013). A single patient with synovial cell sarcoma also had a partial response which lasted for 5 months. However severe neurological toxicity occurred in three patients characterized by mental status changes in all, seizures in two, and white matter vacuolation in one. A possible explanation was cross-reactivity of the affinity-enhanced TCR against a peptide derived from MAGE-A12 which is expressed at low levels in brain tissue. Nonetheless it is unclear why this neurological toxicity was observed in only a subset of treated patients. These studies highlight both the clinical potency of affinity-enhanced TCR-engineered T cells as well as the potential danger of serious and even fatal off-tumor and off-target toxicities. Improvements in the margin of safety for these reagents may require more complex preclinical testing using (i) amino acid (alanine) scanning methodologies to identify critical binding residues and thereby expand the search for potentially cross-reactive peptides in the human genome as well as (ii) more relevant human tissue models such as organ-like structures derived from induced pluripotent stem cells.
Although not thought to be as common as in CAR T cell trials, a fatality apparently due to CRS from excessive T cell activation was recently reported in a study of autologous T cells engineered to express a TCR for the HLA-A*0201-restricted 26-35 epitope of MART-1, which was not affinity enhanced (van den Berg et al. 2015). A patient with very bulky and widely metastatic melanoma (including an 18 cm retroperitoneal mass, a 16 cm pelvic mass, malignant abdominal ascites, and brain and pulmonary metastases) was treated with MART-1 TCR-modified T cells after conditioning with cyclophosphamide and fludarabine. Six days after T cell infusion, the patient had seizures, cerebral hemorrhage, and cardiac arrest and died several days later with multiorgan failure and irreversible brain injury. Following T cell infusion, levels of IL-6, IFN-γ, C-reactive protein (CRP), and procalcitonin were extremely elevated suggestive of CRS or excessive T cell activation. Although the gene-modified T cells were widely distributed in known tumor sites as well as in multiple vital organs including the heart, there was no evidence for any cross-reactivity with any experimental models of cardiac tissue including beating heart cardiomyocyte cultures.
10 Current Clinical Trials Using TCR Gene-Modified T Cells
A number of clinical trials have demonstrated the feasibility and efficacy of genetically modified TCR therapies for different types of cancer as reported above, with clinical activity including tumor regression being reported in a significant subset of patients. These early studies have spawned an outgrowth of promising TCR trials directed against an increasing array of tumor-associated targets. Table 7.1 below lists current clinical trials that are registered with the National Cancer Institute. Notably, some of these new trials also target epitopes of TAA such as WT1 that are expressed at low levels in normal hematopoietic stem cells but at much higher levels in leukemia.
11 Expert Point of View and Future Directions
A therapeutic “proof of principle” for affinity-enhanced TCR-engineered autologous T cells has been amply demonstrated for both certain hematological malignancies (e.g., myeloma) and solid tumors (e.g., melanoma, synovial sarcoma). Several challenges remain before this form of therapy can be more widely applicable. High-affinity TCRs will need to be developed against a greater variety of new targets including peptides derived from established tumor-specific genes such as the widely expressed cancer-testis or cancer-germline antigens. Other candidates could be peptides derived from recurring cancer-specific mutations (e.g., IDH-1/IDH-2 in acute leukemia or KRAS in solid tumors) or the common breakpoint regions of cancer-specific fusion genes (e.g., BCR-ABL in CML) since these mutations may also generate “shared” tumor-associated antigenic peptides. Investigators will need to address the inherent problem of HLA-restriction by offering TCRs that reach a wider range of HLA molecules beyond the traditional and common targets of HLA-A*2:01 and HLA-A*01. As noted above preclinical testing will need to expand significantly beyond studying cell lines and normal tissue arrays to ensure a greater margin of safety for these powerful cell-based therapies. Additional barriers to successful cellular immunotherapy using TCR-modified T cells and CAR T cells include the myriad of mechanisms that cancers (particularly solid tumors) and the tumor microenvironment deploy to suppress effector immune responses including surface expression of PDL-1, PDL-2, and CTLA-4 and elaboration of indoleamine-2,3-dioxygenase (IDO) which depletes tryptophan, increases kynurenine, and thereby promotes formation of Tregs and myeloid-derived suppressor cells (MDSCs). In this regard, the advent of the checkpoint inhibitors including PD-1, PDL-1, and CTLA-4 antibodies and the small molecule inhibitors of the IDO enzyme may provide a logical pathway for developing novel combination approaches.
References
Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK (2012) Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol 42(12):3174–3179
Atanackovic D, Arfsten J, Cao Y, Gnjatic S, Schnieders F, Bartels K, Schilling G, Faltz C, Wolschke C, Dierlamm J, Ritter G, Eiermann T, Hossfeld DK, Zander AR, Jungbluth AA, Old LJ, Bokemeyer C, Kroger N (2007) Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood 109(3):1103–1112
Barrett DM, Teachey DT, Grupp SA (2014) Toxicity management for patients receiving novel T-cell engaging therapies. Curr Opin Pediatr 26(1):43–49
Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, Uckert W, Song JY, Haanen JB, Schumacher TN (2010) Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med 16(5):565–570
Boussiotis VA, Freeman GJ, Taylor PA, Berezovskaya A, Grass I, Blazar BR, Nadler LM (2000) p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat Med 6(3):290–297
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 373(2):123–135
Bridgeman JS, Sewell AK, Miles JJ, Price DA, Cole DK (2012) Structural and biophysical determinants of alphabeta T-cell antigen recognition. Immunology 135(1):9–18
Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, Bossi G, Vuidepot A, Powlesland AS, Legg A, Adams KJ, Bennett AD, Pumphrey NJ, Williams DD, Binder-Scholl G, Kulikovskaya I, Levine BL, Riley JL, Varela-Rohena A, Stadtmauer EA, Rapoport AP, Linette GP, June CH, Hassan NJ, Kalos M, Jakobsen BK (2013) Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5(197):197ra103
Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ (1997) A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A 94(5):1914–1918
Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA (2006) Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66(17):8878–8886
Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA (2007) Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67(8):3898–3903
Condomines M, Hose D, Raynaud P, Hundemer M, De Vos J, Baudard M, Moehler T, Pantesco V, Moos M, Schved JF, Rossi JF, Reme T, Goldschmidt H, Klein B (2007) Cancer/testis genes in multiple myeloma: expression patterns and prognosis value determined by microarray analysis. J Immunol 178(5):3307–3315
Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y (1998) Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol 16:523–544
Dhodapkar MV, Krasovsky J, Olson K (2002) T cells from the tumor microenvironment of patients with progressive myeloma can generate strong, tumor-specific cytolytic responses to autologous, tumor-loaded dendritic cells. Proc Natl Acad Sci U S A 99(20):13009–13013
Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, Gao F, Sami M, Boulter J, Li Y, Jakobsen BK (2006) Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci 15(4):710–721
Ellis JM, Henson V, Slack R, Ng J, Hartzman RJ, Katovich Hurley C (2000) Frequencies of HLA-A2 alleles in five U.S. population groups. Predominance of A*02011 and identification of HLA-A*0231. Hum Immunol 61(3):334–340
Furler S, Paterna JC, Weibel M, Bueler H (2001) Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Ther 8(11):864–873
Gonzalez-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MH, da Silva AL, Teles e Silva AL, Ghattaoraya GS, Alfirevic A, Jones AR, Middleton D (2015) Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 43:D784–D788
Groettrup M, Kirk CJ, Basler M (2010) Proteasomes in immune cells: more than peptide producers? Nat Rev Immunol 10(1):73–78
Gross G, Eshhar Z (1992) Endowing T cells with antibody specificity using chimeric T cell receptors. FASEB J 6(15):3370–3378
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 86(24):10024–10028
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368(16):1509–1518
Haidar JN, Pierce B, Yu Y, Tong W, Li M, Weng Z (2009) Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins 74(4):948–960
Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, Speiser DE, Rufer N (2013) SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest 123(3):1044–1056
Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N (2015) Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol 6:582
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C (2008) Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358(25):2698–2703
Ilyas S, Yang JC (2015) Landscape of tumor antigens in T cell immunotherapy. J Immunol 195(11):5117–5122
Irving M, Zoete V, Hebeisen M, Schmid D, Baumgartner P, Guillaume P, Romero P, Speiser D, Luescher I, Rufer N, Michielin O (2012) Interplay between T cell receptor binding kinetics and the level of cognate peptide presented by major histocompatibility complexes governs CD8+ T cell responsiveness. J Biol Chem 287(27):23068–23078
Ito R, Katano I, Kawai K, Hirata H, Ogura T, Kamisako T, Eto T, Ito M (2009) Highly sensitive model for xenogenic GvHD using severe immunodeficient NOG mice. Transplantation 87(11):1654–1658
Jager E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jager D, Arand M, Wada H, Noguchi Y, Stockert E, Old LJ, Knuth A (1998) Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 187(2):265–270
Jameson SC, Hogquist KA, Bevan MJ (1995) Positive selection of thymocytes. Annu Rev Immunol 13:93–126
Johnson LA, Heemskerk B, Powell DJ Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA (2006) Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol 177(9):6548–6559
Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, Van Waes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114(3):535–546
Jungbluth AA, Antonescu CR, Busam KJ, Iversen K, Kolb D, Coplan K, Chen YT, Stockert E, Ladanyi M, Old LJ (2001) Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int J Cancer 94(2):252–256
Jungbluth AA, Ely S, DiLiberto M, Niesvizky R, Williamson B, Frosina D, Chen YT, Bhardwaj N, Chen-Kiang S, Old LJ, Cho HJ (2005) The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood 106(1):167–174
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DA, Morton KE, Toomey MA, Rosenberg SA (2015) Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol Off J Am Soc Clin Oncol 33(6):540–549
Kvistborg P, van Buuren MM, Schumacher TN (2013) Human cancer regression antigens. Curr Opin Immunol 25(2):284–290
Li L, Yee C, Beavo JA (1999) CD3- and CD28-dependent induction of PDE7 required for T cell activation. Science 283(5403):848–851
Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM (2005) Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol 23(3):349–354
Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122(6):863–871
Linnemann C, Heemskerk B, Kvistborg P, Kluin RJ, Bolotin DA, Chen X, Bresser K, Nieuwland M, Schotte R, Michels S, Gomez-Eerland R, Jahn L, Hombrink P, Legrand N, Shu CJ, Mamedov IZ, Velds A, Blank CU, Haanen JB, Turchaninova MA, Kerkhoven RM, Spits H, Hadrup SR, Heemskerk MH, Blankenstein T, Chudakov DM, Bendle GM, Schumacher TN (2013) High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med 19(11):1534–1541
Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, Angell HK, Fredriksen T, Elie N, Fauquembergue E, Drouet A, Leprince J, Benichou J, Mauillon J, Le Pessot F, Sesboue R, Tuech JJ, Sabourin JC, Michel P, Frebourg T, Galon J, Latouche JB (2015) Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and Frameshift mutations: a rationale for personalized immunotherapy. Cancer Res 75(17):3446–3455
Malecek K, Grigoryan A, Zhong S, Gu WJ, Johnson LA, Rosenberg SA, Cardozo T, Krogsgaard M (2014) Specific increase in potency via structure-based design of a TCR. J Immunol 193(5):2587–2599
Mallabiabarrena A, Fresno M, Alarcon B (1992) An endoplasmic reticulum retention signal in the CD3 epsilon chain of the T-cell receptor. Nature 357(6379):593–596
Maus MV, Grupp SA, Porter DL, June CH (2014) Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123(17):2625–2635
Miles JJ, Douek DC, Price DA (2011) Bias in the alphabeta T-cell repertoire: implications for disease pathogenesis and vaccination. Immunol Cell Biol 89(3):375–387
Minami Y, Weissman AM, Samelson LE, Klausner RD (1987) Building a multichain receptor: synthesis, degradation, and assembly of the T-cell antigen receptor. Proc Natl Acad Sci U S A 84(9):2688–2692
Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, Rodolfo M, Schneider C (2006) MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A 103(30):11160–11165
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314(5796):126–129
Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36(2):133–151
Nitta T, Murata S, Sasaki K, Fujii H, Ripen AM, Ishimaru N, Koyasu S, Tanaka K, Takahama Y (2010) Thymoproteasome shapes immunocompetent repertoire of CD8+ T cells. Immunity 32(1):29–40
Noonan K, Matsui W, Serafini P, Carbley R, Tan G, Khalili J, Bonyhadi M, Levitsky H, Whartenby K, Borrello I (2005) Activated marrow-infiltrating lymphocytes effectively target plasma cells and their clonogenic precursors. Cancer Res 65(5):2026–2034
Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA (2009) Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res 15(1):169–180
Porrata LF, Markovic SN (2004) Timely reconstitution of immune competence affects clinical outcome following autologous stem cell transplantation. Clin Exp Med 4(2):78–85
Porrata LF, Gertz MA, Inwards DJ, Litzow MR, Lacy MQ, Tefferi A, Gastineau DA, Dispenzieri A, Ansell SM, Micallef IN, Geyer SM, Markovic SN (2001) Early lymphocyte recovery predicts superior survival after autologous hematopoietic stem cell transplantation in multiple myeloma or non-Hodgkin lymphoma. Blood 98(3):579–585
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365(8):725–733
Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, Veloso E, Zheng Z, Westphal S, Mair R, Chi N, Ratterree B, Pochran MF, Natt S, Hinkle J, Sickles C, Sohal A, Ruehle K, Lynch C, Zhang L, Porter DL, Luger S, Guo C, Fang HB, Blackwelder W, Hankey K, Mann D, Edelman R, Frasch C, Levine BL, Cross A, June CH (2005) Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med 11(11):1230–1237
Rapoport AP, Stadtmauer EA, Aqui N, Vogl D, Chew A, Fang HB, Janofsky S, Yager K, Veloso E, Zheng Z, Milliron T, Westphal S, Cotte J, Huynh H, Cannon A, Yanovich S, Akpek G, Tan M, Virts K, Ruehle K, Harris C, Philip S, Vonderheide RH, Levine BL, June CH (2009) Rapid immune recovery and graft-versus-host disease-like engraftment syndrome following adoptive transfer of costimulated autologous T cells. Clin Cancer Res 15(13):4499–4507
Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Fang HB, Cai L, Janofsky S, Chew A, Storek J, Akpek G, Badros A, Yanovich S, Tan MT, Veloso E, Pasetti MF, Cross A, Philip S, Murphy H, Bhagat R, Zheng Z, Milliron T, Cotte J, Cannon A, Levine BL, Vonderheide RH, June CH (2011) Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after AHCT for myeloma. Blood 117(3):788–797
Rapoport AP, Aqui NA, Stadtmauer EA, Vogl DT, Xu YY, Kalos M, Cai L, Fang HB, Weiss BM, Badros A, Yanovich S, Akpek G, Tsao P, Cross A, Mann D, Philip S, Kerr N, Brennan A, Zheng Z, Ruehle K, Milliron T, Strome SE, Salazar AM, Levine BL, June CH (2014) Combination immunotherapy after AHCT for multiple myeloma using MAGE-A3/poly-ICLC immunizations followed by adoptive transfer of vaccine-primed and costimulated autologous T cells. Clin Cancer Res 20(5):1355–1365
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, Kulikovskaya I, Sinha SK, Kronsberg S, Gupta M, Bond S, Melchiori L, Brewer JE, Bennett AD, Gerry AB, Pumphrey NJ, Williams D, Tayton-Martin HK, Ribeiro L, Holdich T, Yanovich S, Hardy N, Yared J, Kerr N, Philip S, Westphal S, Siegel DL, Levine BL, Jakobsen BK, Kalos M, June CH (2015) NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21(8):914–921
Robbins PF (2015) T-cell receptor-transduced T cells: clinical experience. Cancer J 21(6):480–485
Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, Bennett AD, Dunn SM, Mahon TM, Jakobsen BK, Rosenberg SA (2008) Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 180(9):6116–6131
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol Off J Am Soc Clin Oncol 29(7):917–924
Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CC, Li YF, El-Gamil M, Rosenberg SA (2015) A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21(5):1019–1027
Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE (1994) Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 271(12):907–913
Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10(9):909–915
Rudolph MG, Stanfield RL, Wilson IA (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24:419–466
Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, Gomez-Eerland R, Thome M, Schumacher TN, Romero P, Speiser DE, Zoete V, Michielin O, Rufer N (2010) Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol 184(9):4936–4946
Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, Heath K, McClanahan T, Lunceford J, Gause C, Cheng JD, Chow LQ (2016) Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 17(7):956–965
Stadtmauer EA, Vogl DT, Luning Prak E, Boyer J, Aqui NA, Rapoport AP, McDonald KR, Hou X, Murphy H, Bhagat R, Mangan PA, Chew A, Veloso EA, Levine BL, Vonderheide RH, Jawad AF, June CH, Sullivan KE (2011) Transfer of influenza vaccine-primed costimulated autologous T cells after stem cell transplantation for multiple myeloma leads to reconstitution of influenza immunity: results of a randomized clinical trial. Blood 117(1):63–71
Stone JD, Chervin AS, Kranz DM (2009) T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology 126(2):165–176
Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA (2004) Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol 22(5):589–594
Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, Robbins PF, Rosenberg SA, Dudley ME (2008) Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother 31(8):742–751
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F (2015) Proteomics. Tissue-based map of the human proteome. Science 347(6220):1260419
van Baren N, Brasseur F, Godelaine D, Hames G, Ferrant A, Lehmann F, Andre M, Ravoet C, Doyen C, Spagnoli GC, Bakkus M, Thielemans K, Boon T (1999) Genes encoding tumor-specific antigens are expressed in human myeloma cells. Blood 94(4):1156–1164
van den Berg JH, Gomez-Eerland R, van de Wiel B, Hulshoff L, van den Broek D, Bins A, Tan HL, Harper JV, Hassan NJ, Jakobsen BK, Jorritsma A, Blank CU, Schumacher TN, Haanen JB (2015) Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol Ther 23(9):1541–1550
van der Merwe PA, Davis SJ (2003) Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol 21:659–684
van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JH, Heemskerk MH (2010) Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A 107(24):10972–10977
van Rhee F, Szmania SM, Zhan F, Gupta SK, Pomtree M, Lin P, Batchu RB, Moreno A, Spagnoli G, Shaughnessy J, Tricot G (2005) NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood 105(10):3939–3944
Yuan J, Gnjatic S, Li H, Powel S, Gallardo HF, Ritter E, Ku GY, Jungbluth AA, Segal NH, Rasalan TS, Manukian G, Xu Y, Roman RA, Terzulli SL, Heywood M, Pogoriler E, Ritter G, Old LJ, Allison JP, Wolchok JD (2008) CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A 105(51):20410–20415
Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, Rosenberg SA, Morgan RA (2007) High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol 179(9):5845–5854
Zoete V, Irving M, Ferber M, Cuendet MA, Michielin O (2013) Structure-based, rational design of T cell receptors. Front Immunol 4:268
Acknowledgments
The authors warmly thank Gwendolyn Binder-Scholl for critical review of the manuscript and Kris Rifkin for assistance with Fig. 7.1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rapoport, A.P., Yared, J.A. (2019). T Cell Receptors-Gene-Modified T Cells for Cancer: Methods, Data, and Challenges. In: Perales, MA., Abutalib, S., Bollard, C. (eds) Cell and Gene Therapies. Advances and Controversies in Hematopoietic Transplantation and Cell Therapy. Springer, Cham. https://doi.org/10.1007/978-3-319-54368-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-54368-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-54367-3
Online ISBN: 978-3-319-54368-0
eBook Packages: MedicineMedicine (R0)