
Abstract
Over the past two decades, the discovery of various antibodies related to autoimmune encephalitis has given us a new insight into pathogenic mechanisms and treatment of these syndromes. That is important since many patients with autoimmune encephalopathy are children and young adults. These patients often respond favorably to immunotherapy. Delay in diagnosis and treatment has been associated with a worse prognosis. In this chapter, we review the epidemiology, pathophysiology, clinical characteristics, diagnosis, and treatment of different syndromes associated with autoimmune encephalitis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Autoimmune encephalitis represents a group of disorders characterized by various immunologic mechanisms, clinical manifestations, and therapeutic outcomes. They can be associated with paraneoplastic syndromes or nonneoplastic autoimmune processes. Autoimmune encephalitis is usually associated with antibodies that can acutely or subacutely affect any part of the central or peripheral nervous system including neuromuscular junctions and muscles. Antibody-associated encephalitis can be divided into two main categories: (1) encephalopathy associated with antibodies against intracellular antigens and (2) encephalopathy associated with antibodies directed against the neuronal surface and synaptic antigens [1, 2]. The discovery of various antibodies related to autoimmune encephalitis has given us a new insight into pathogenic mechanisms and treatment of these syndromes. That is important since many patients with autoimmune encephalopathy are children and young adults, and they may respond well to immunosuppressive treatment if diagnosed without delay. In this chapter, we review the epidemiology, pathophysiology, clinical characteristics, diagnosis, and treatment of different syndromes associated with autoimmune encephalitis.
History
In 1934, Greenfield initially described two cases of subacute cerebellar degeneration occurring with carcinoma outside the nervous system [3]. Thirteen years later in 1947, Denny-Brown reported two patients with primary sensory neuropathy and muscular changes associated with bronchial carcinoma [4].
In 1954, Henson et al. [5] published a series of 19 cases with various types of carcinomatous neuropathy and myopathy. Eight of these patients had proximal atrophic weakness of limbs and involvement of ocular and bulbar muscles. Four patients also exhibited myasthenic features with a favorable response to neostigmine in some cases. Later in 1956, Chartan et al. [6] described episodes of severe mental disturbance in three male patients with bronchial carcinoma. In all of those, the mental disorder either preceded or overshadowed the presence of cancer.
In 1968, Corsellis et al. [7] reported autoimmune limbic encephalitis associated with small-cell lung cancer. For years, it was believed that “limbic encephalitis” was almost always associated with a form of neoplasia mainly lung, thymic, or testicular tumors. In 2001, it was shown that voltage-gated potassium channels (VGKC) were associated with reversible limbic encephalitis [8]. Four years later, other antibodies to the cell surface or synaptic proteins were detected in six patients with subacute limbic encephalitis and involvement of additional brain regions [9]. Further studies of those patients with immunotherapy-responsive encephalitis resulted in the characterization of the antigen as the NR1 subunit of the N-methyl-D-aspartic acid receptor (NMDA receptor) and the definition of its clinical characteristics [10–12], since many other neuronal cell surface antigens have been detected and introduced in patients with autoimmune encephalitis.
Epidemiology
The California Encephalitis Project was established in 1998 to identify the etiologic agents and to study epidemiology and clinical characteristics of encephalitis. In 2009, they reported ten cases of NMDA receptor antibodies and concluded that unlike classic paraneoplastic encephalitis, anti-NMDA receptor encephalitis affects younger patients [13]. Since, an increasing number of cases have been reported to the California Encephalitis Project, making NMDA receptor antibodies a significant cause of encephalitis among young patients. Between 2007 and 2011, 761 cases of encephalitis of uncertain etiology in individuals aged ≤30 years were reported to the California Encephalitis Project. Of these, 32 patients were tested positive for anti-NMDAR encephalitis; however, viral encephalitis was diagnosed in only 42 patients [14]. Although anti-NMDAR encephalitis was initially thought to affect young women, often with teratomas, it can affect men and children, with or without any identifiable tumor [15]. Overall, 75% of patients with anti-NMDAR encephalitis can significantly recover when diagnosed promptly [10].
Among paraneoplastic syndrome, Lambert-Eaton myasthenic syndrome, which affects approximately 3% of patients with small-cell lung cancer, and myasthenia gravis, which affects 15% of patients with thymoma, are common [16]. Up to 9% of patients with small-cell lung cancer have at least one form of paraneoplastic syndrome (commonly Lambert-Eaton myasthenic syndrome, sensory neuronopathy, or limbic encephalitis) [16]. γ-Aminobutyric acid (GABA-B) receptor antibodies are also responsible for paraneoplastic limbic encephalitis in patients with small-cell lung cancer [17].
Pathophysiology and Clinical Presentation
Antibodies in autoimmune encephalitis can target intracellular antigens or antigens on neuronal surface/synaptic space. Among those, antibodies which target intracellular antigens are usually associated with paraneoplastic syndromes and a poor prognosis. Antibodies to intracellular antigens include anti-Hu (also known as antineuronal nuclear antibody, type 1, ANNA-1), anti-Ma2 (also called anti-Ta), collapsin-responsive mediator protein-3, protein-4, and protein-5 (CRMP3–5), anti-amphiphysin, anti-Yo, anti-Ri, adenylate kinase 5, and BR serine/threonine kinase (BRSK2) antibodies. Table 8.1 summarizes the antibodies to intracellular antigens and their clinical presentation.
Antibodies to neuronal surface/synaptic antigens can also be associated with cancer; however, they are more responsive to immunotherapy. Antibodies to neuronal surface/synaptic antigens are often related to limbic encephalitis. In this group, anti-N-methyl-D-aspartate (NMDA) receptor encephalitis and anti-leucine-rich glioma-inactivated 1 (LGI1) comprise 85% of patients [1]. Anti-NMDA receptor encephalitis has become one of the most frequently recognized autoimmune encephalitides since its discovery. The disease is more frequent among women (80%) and adults younger than 45 years old [39]. Almost half of the patients initially present with a headache and a viral-like process, followed by psychiatric manifestations, altered mental status, in addition to language and memory dysfunction [15, 40]. Seizure is frequent among pediatric patients [39]. More than two-third of the patients suffer from seizures [39]. Table 8.2 summarizes the antibodies to intracellular antigens, their associated syndromes.
More than half of patients with autoimmune encephalitis present with symptoms of limbic encephalopathy including memory deficits, altered mental status, seizures, and neuropsychiatric syndrome. Refractory seizures and status epilepticus have also been reported [59, 60, 68]. Other common features of autoimmune encephalitis include headache, tremor, language difficulties, ataxia, and sleep disorders.
Diagnostic Approach
The diagnosis of autoimmune encephalitis can be challenging because symptoms usually precede the diagnosis of cancer or resemble other neurological or psychological disorders. An international panel of experts has identified diagnostic criteria for paraneoplastic neurological syndromes (Table 8.3) [69].
Patients with clinical presentations of encephalitis should have a full workup including neuroimaging, cerebrospinal fluid (CSF) examination, electroencephalography (EEG), pertinent laboratory and serological studies, and, in some cases, electromyography (EMG). Many other conditions (Table 8.4) are more frequent than autoimmune etiologies of encephalopathies. They should be considered and excluded.
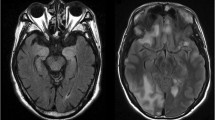
Magnetic resonance imaging (MRI) of the brain is neither sensitive nor specific for the diagnosis of autoimmune encephalitis. However, it is essential to exclude other conditions such as ischemic infarction or tumors. Among patients with encephalitis, signal hyperintensities on fluid-attenuated inversion recovery (FLAIR) and T2-weighted images can be seen in the mesiotemporal lobe, cortical and subcortical regions, or brain stem. Contrast enhancement can be variable, and leptomeningeal enhancement has been reported [70]. The extent of abnormal findings on the MRI is different for each syndrome. For instance, MRI in GABA-A receptor encephalitis often shows multifocal and widespread FLAIR and T2 signal abnormalities [56]. Encephalitic syndromes associated with LGI1 and AMPA receptor antibodies also always cause FLAIR hyperintensity in the mesiotemporal lobe. In a study on 50 patients with paraneoplastic limbic encephalitis, researchers observed that 57% of patients with MRI studies had signal abnormalities in the limbic system [20]. There is also a report of cortical ribboning similar to that seen in Creutzfeldt-Jakob disease (CJD) among patients with voltage-gated potassium channel (VGKC) autoantibody-associated encephalopathy [71]. Brain MRI is often normal or shows transient FLAIR hyperintensity with or without contrast enhancement in anti-NMDAR encephalitis [10, 72].
Several autoimmune encephalitis syndromes are associated with seizure or status epilepticus [59, 60]. Diffuse slowing or epileptiform abnormalities in the temporal lobe on EEG are the most common findings in patients with encephalitis. EEG is also important to exclude other etiologies for encephalopathy such as subclinical seizures.
Although CSF examination can be normal especially in the initial phase, a mild elevation of protein (<100 mg/dL) and lymphocytic pleocytosis or oligoclonal bands can be an indicator of autoimmune encephalitis [10, 13, 15, 17, 46, 73]. More than 90% of patients with antibodies against NMDA, AMPA, and GABA-B receptors have pleocytosis or oligoclonal bands on CSF examination [10, 53, 56, 57]. CSF analysis is also essential to exclude other etiologies of encephalopathy including infectious and neoplastic causes.
Pertinent antibody testing should be performed in both serum and CSF. Antibodies to cell surface/ synaptic proteins can be detected primarily in CSF. In a multiinstitutional observational study, detection of NMDA receptor antibodies was compared in 250 paired serum and CSF samples. It showed that the screening test is significantly more sensitive in CSF than serum (100% vs. 85%) [39]. A positive serum antibody testing, when CSF is negative for the antibody, raises the possibility of a false positive diagnosis. Although many tests for autoimmune encephalitis are commercially available, a number of autoimmune encephalitis cases can be caused by other, still unavailable or unknown antibodies. Therefore, a negative test result does not rule out autoimmune encephalitis.
All patients with autoimmune encephalitis should be screened for the presence of a tumor. The detected antibody type can also guide the type and extent of screening. On the other hand, detection of a tumor could also assist in the diagnosis of paraneoplastic encephalitis variants and guide the antibody screening plan.
Treatment and Outcome
Autoimmune encephalitis is often associated with a favorable outcome after tumor removal and antineoplastic treatment (if applicable), as well as immunotherapy. In general, steroids, intravenous immunoglobulin, and plasmapheresis are the first line of immunotherapy especially when a tumor is detected and treated [9, 39]. Rituximab and cyclophosphamide comprise the second-line immunotherapy when the first-line treatment fails. Although seizures must be addressed aggressively during the acute phase of the disease, patients often do not require long-term antiepileptic medication.
In a large multiinstitutional observational study, over 500 patients with anti-NMDA receptor encephalitis were treated and monitored up to 2 years. Out of 501 patients, 94% received first-line immunotherapy (steroids, intravenous immunoglobulin, plasmapheresis) or tumor removal, resulting in improvement within 4 weeks in 53% of patients. More than half of patients who failed first-line therapy received second-line immunotherapy (rituximab, cyclophosphamide), resulting in better outcome than those who did not. During the first 24 months, almost 80% of patients reached a good outcome, where relapses occurred in approximately 12% of the patients. About 6% of patients died [39].
Predictors of poor outcome in anti-NMDA receptor encephalitis are a delay in diagnosis and treatment, the need for intensive care, high titer of antibody in CSF and serum, and the presence of teratoma [39, 74]. The overall prognosis for patients with autoimmune encephalitis is variable. Some patients have a complete recovery, while others die or develop a permanent neurologic disability.
Summary
Autoimmune encephalitis has different immunologic mechanisms, clinical manifestations, and therapeutic outcomes. It can be divided into two categories: antibodies against intracellular antigens or antibodies against neuronal surface/synaptic antigens. More than half of patients with autoimmune encephalitis present with symptoms of limbic encephalopathy including memory deficits, altered mental status, seizures, and neuropsychiatric syndrome. Patients with the clinical presentation of encephalitis should have a complete workup including neuroimaging, EEG, lumbar puncture, and serologic testing. Other etiologies of encephalitis are more common and should be excluded. Patients often respond favorably to immunotherapy. Delay in diagnosis and treatment has been associated with a worse prognosis.
References
Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77:179–89.
Tüzün E, Dalmau J. Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13:261–71.
Greenfield JG. Subacute spino-cerebellar degeneration occurring in elderly patients. Brain. 1934;57:161–76.
Denny-Brown D. Primary sensory neuropathy with muscular changes associated with carcinoma. J Neurol Neurosurg Psychiatry. 1948;11:73–87.
Henson RA, Russell DS, Wilkinson M. Carcinomatous neuropathy and myopathy a clinical and pathological study. Brain. 1954;77:82–121.
Charatan FB, Brierley JB. Mental disorder associated with primary lung carcinoma. Br Med J. 1956;1:765–8.
Corsellis JA, Goldberg GJ, Norton AR. ‘Limbic encephalitis’ and its association with carcinoma. Brain. 1968;91:481–96.
Buckley C et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50:73–8.
Ances BM et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764–77.
Dalmau J et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–8.
Dalmau J et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–26.
Gable MS et al. Anti-NMDA receptor encephalitis: report of ten cases and comparison with viral encephalitis. Eur J Clin Microbiol Infect Dis. 2009;28:1421–9.
Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California encephalitis project. Clin Infect Dis. 2012;54:899–904.
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
Gozzard P et al. Paraneoplastic neurologic disorders in small cell lung carcinoma: a prospective study. Neurology. 2015;85:235–9.
Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology. 2011;76:795–800.
Graus F et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–48.
Sillevis Smitt P et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745–53.
Gultekin SH et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123(Pt 7):1481–94.
Honnorat J et al. Autoimmune limbic encephalopathy and anti-Hu antibodies in children without cancer. Neurology. 2013;80:2226–32.
Dalmau J, Graus F, Rosenblum MK, Posner JB. Anti-Hu--associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (Baltimore). 1992;71:59–72.
Shams’ili S et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126:1409–18.
Saiz A et al. Anti-Hu-associated brainstem encephalitis. J Neurol Neurosurg Psychiatry. 2009;80:404–7.
Luque FA et al. Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol. 1991;29:241–51.
Kim H, Lim Y, Kim K-K. Anti-ri-antibody-associated paraneoplastic syndrome in a man with breast cancer showing a reversible pontine lesion on MRI. J Clin Neurol. 2009;5:151–2.
Greenlee JE et al. Association of anti-Yo (type I) antibody with paraneoplastic cerebellar degeneration in the setting of transitional cell carcinoma of the bladder: detection of Yo antigen in tumor tissue and fall in antibody titers following tumor removal. Ann Neurol. 1999;45:805–9.
Voltz R et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999;340:1788–95.
Dalmau J et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127:1831–44.
Waragai M et al. Anti-Ma2 associated paraneoplastic neurological syndrome presenting as encephalitis and progressive muscular atrophy. J Neurol Neurosurg Psychiatry. 2006;77:111–3.
Saiz A et al. Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry. 1999;66:214–7.
Antoine JC et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol. 1999;56:172–7.
David C, McPherson PS, Mundigl O, de Camilli P. A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci U S A. 1996;93:331–5.
Yu Z et al. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49:146–54.
Vernino S et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol. 2002;51:625–30.
Knudsen A et al. Antibodies to CRMP3-4 associated with limbic encephalitis and thymoma. Clin Exp Immunol. 2007;149:16–22.
Tüzün E, Rossi JE, Karner SF, Centurion AF, Dalmau J. Adenylate kinase 5 autoimmunity in treatment refractory limbic encephalitis. J Neuroimmunol. 2007;186:177–80.
Sabater L, Gómez-Choco M, Saiz A, Graus F. BR serine/threonine kinase 2: a new autoantigen in paraneoplastic limbic encephalitis. J Neuroimmunol. 2005;170:186–90.
Titulaer MJ et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–65.
Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti–N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70:1133–9.
Hacohen Y et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry. 2013;84:748–55.
Iizuka T et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology. 2008;70:504–11.
Shimazaki H, Ando Y, Nakano I, Dalmau J. Reversible limbic encephalitis with antibodies against the membranes of neurones of the hippocampus. J Neurol Neurosurg Psychiatry. 2007;78:324–5.
Pillay N, Gilbert JJ, Ebers GC, Brown JD. Internuclear ophthalmoplegia and ‘optic neuritis’: paraneoplastic effects of bronchial carcinoma. Neurology. 1984;34:788–91.
Lebas A, Husson B, Didelot A, Honnorat J, Tardieu M. Expanding spectrum of encephalitis with NMDA receptor antibodies in young children. J Child Neurol. 2010;25:742–5.
Wingfield T et al. Autoimmune encephalitis: a case series and comprehensive review of the literature. QJM. 2011;104:921–31.
Andrade DM, Tai P, Dalmau J, Wennberg R. Tonic seizures: a diagnostic clue of anti-LGI1 encephalitis? Neurology. 2011;76:1355–7.
Irani SR et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900.
Sen A et al. Pathognomonic seizures in limbic encephalitis associated with anti-LGI1 antibodies. Lancet (London/England). 2014;383:2018.
Tofaris GK et al. Immunotherapy-responsive chorea as the presenting feature of LGI1-antibody encephalitis. Neurology. 2012;79:195–6.
Fukata Y et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A. 2010;107:3799–804.
Lai M et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776–85.
Höftberger R et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology. 2015;84:2403–12.
Klein CJ et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol. 2013;70:229–34.
Irani SR et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72:241–55.
Petit-Pedrol M et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13:276–86.
Höftberger R et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81:1500–6.
Sabater L et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13:575–86.
Suleiman J et al. VGKC antibodies in pediatric encephalitis presenting with status epilepticus. Neurology. 2011;76:1252–5.
Cornelius JR et al. Sleep manifestations of voltage-gated potassium channel complex autoimmunity. Arch Neurol. 2011;68:733–8.
Hutchinson M et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71:1291–2.
Mas N et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 2011;82:1399–401.
Clerinx K et al. Progressive encephalomyelitis with rigidity and myoclonus: resolution after thymectomy. Neurology. 2011;76:303–4.
Boronat A et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73:120–8.
Tobin WO et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology. 2014;83:1797–803.
Prüss H et al. Limbic encephalitis with mGluR5 antibodies and immunotherapy-responsive prosopagnosia. Neurology. 2014;83:1384–6.
Lancaster E et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77:1698–701.
Johnson N, Henry C, Fessler AJ, Dalmau J. Anti-NMDA receptor encephalitis causing prolonged nonconvulsive status epilepticus. Neurology. 2010;75:1480–2.
Graus F et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135–40.
Flanagan EP et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc. 2010;85:881–97.
Geschwind MD et al. Voltage-gated potassium channel autoimmunity mimicking creutzfeldt-jakob disease. Arch Neurol. 2008;65:1341–6.
Florance NR et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66:11–8.
Lawn ND, Westmoreland BF, Kiely MJ, Lennon VA, Vernino S. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clin Proc. 2003;78:1363–8.
Gresa-Arribas N et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–77.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Zand, R. (2017). Autoimmune Encephalitis: Clinical Features, Pathophysiology, and Treatment. In: Minagar, A., Alexander, J. (eds) Inflammatory Disorders of the Nervous System. Current Clinical Neurology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-51220-4_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-51220-4_8
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-51218-1
Online ISBN: 978-3-319-51220-4
eBook Packages: MedicineMedicine (R0)