
Abstract
Classically, multiple sclerosis (MS) is characterized by relapses of neurological deficits followed by remission. At present mechanisms of progressive aspects of the disease are currently undergoing increasing attention. Updated diagnostic criteria and improved diagnostic techniques, particularly in the field of magnetic resonance imaging (MRI), have facilitated diagnosis and monitoring of MS patients. Advances in immunology, particularly regarding the role of Th17 helper cells and B cells, together with increasing insight into genetics of MS, continue to improve our knowledge of the disease mechanisms underlying MS.
Symptomatic treatment of MS represents an important aspect of comprehensive care. These include a multitude of symptoms ranging from fatigue, cognitive, and mood disorders to disorders of gait, spasticity, pain, and bladder and bowel abnormalities, as well as less common paroxysmal manifestations. These symptoms necessitate a myriad of treatment approaches tailored to individual patient needs. A boom in the development of disease-modifying therapies for MS has resulted in 14 currently FDA-approved medications for relapsing-remitting MS in 2016 compared to just a handful in the early 2000s.
The pivotal clinical trials are reviewed in regard to safety and efficacy for each of these medications. Of particular concern is the risk of progressive multifocal leukoencephalopathy (PML) with increasingly effective therapies. Such cases have been most commonly been associated with natalizumab (Tysabri®), but a very low incidence of this disorder has been associated with two new drugs, dimethyl fumarate (Tecfidera®) and fingolimod (Gilenya®).
Available data for medications in development is also briefly discussed, with a focus on B-cell therapy disability measures, progressive disease, and repair including remyelination. Development of these new targeted therapies represents an exciting next frontier in MS treatment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Multiple sclerosis
- Epidemiology
- Pathogenesis
- Diagnostic criteria
- Magnetic resonance imaging
- Treatment
- B cells
- Progressive multifocal leukoencephalopathy
Introduction
Great strides in understanding multiple sclerosis (MS) have been made in the areas of immunology, genetics, and most importantly treatment since the first publication of this volume. Advances in drug treatment of MS continue to provide newer, more convenient oral therapies, and potentially more effective options for patients. These areas have been given greater attention for students of this disorder.
History
Charcot first described MS as a unique disorder in the mid nineteenth century in Paris. He attributed the original recognition of this disorder to Cruveillier, the famed professor of anatomy. Others also described the pathological anatomy of the disease in remarkable detail, but it was Charcot who characterized the clinical illness and correlated the illness with its unique neuropathology [1]. From the first descriptions of the illness, it was recognized that MS differed clinically from one patient to another, with the majority of patients experiencing a relapsing-remitting multiple sclerosis (RRMS) [1, 2]. Charcot recognized the illness in a minority of patients was fundamentally different and described them as having an “incomplete” form of illness [1, 2]. From their first symptoms, these patients manifest signs of a progressive spinal cord disease without relapses. They are now designated as having primary progressive (PPMS) [2].
The first person documented to clearly have suffered from MS was a grandson of King George III of England, Sir August D’Este [3]. The course of his illness recorded in his diary was edited and published by Douglas Firth in 1947. While MS is an illness that is more common in the higher socioeconomic strata of society, it is not limited to the well to do by any means [2, 4, 5]. The disease does, however, occur predominantly in persons of European descent [2, 4, 5]. African-Americans have MS diagnosed at approximately half the rate of Caucasians in the United States [4, 5].
Clinical Features of Multiple Sclerosis
Multiple sclerosis is an illness characterized by relapses of neurological deficits followed by remissions with varying degrees of recovery [1–6]. The occurrence and severity of the exacerbations are unpredictable, although several factors are recognized as increasing the risk of attacks. Patients experiencing their initial attacks of MS are more likely to recover “fully,” but an experienced neurologist can virtually always find residual evidence of the previous neurological deficit, no matter how complete the recovery seems to have been. For example, retrobulbar neuritis heralds the onset of illness in 10–15% of MS patients. The severity of the visual impairment varies greatly, with a very small percentage of patients suffering complete loss of light perception. Recovery of vision generally occurs, but occasionally, especially if complete loss of vision occurs, there may be little or no recovery. A skilled examiner can find neurological deficits such as an afferent pupillary defect (Marcus Gunn pupil) and color desaturation (impaired color vision) in the vast majority of patients with a history of retrobulbar neuritis who seem to have recovered normal visual acuity.
Multiple sclerosis is typically manifest by recurrent acute onset of neurological difficulties reflecting damage to multiple areas of the brain and spinal cord, defined clinically as “attacks” or “relapses” [1, 2, 4]. Symptoms associated with these events typically remit, but subsequent relapses occur unpredictably and may become more obviously associated with residual disability [1, 3, 4]. It is this dissemination in time and space that is so characteristic of multiple sclerosis and its principal diagnostic feature [6–9]. Interval progression between, or in the absence of attacks of illness, signifies the onset of secondary progressive multiple sclerosis (SPMS) [2]. However, approximately 10–15% of the overall patient population will develop a progressive form of illness without relapses, usually appearing in midlife, termed primary progressive multiple sclerosis, PPMS [2, 10]. This form of illness is slightly more common in men. This progressive form of MS is approximately three times more common in Irish and Ashkenazi Jewish populations [2, 10]. Should one or more exacerbations occur after onset of primary progressive illness at outset, patients may be designated as having “relapsing progressive MS” [2]. Although in the past, there has been no agreement that SPMS and relapsing progressive patients differ in any fundamental way; evidence from new studies shows differences in the microscopic neuropathology of RRMS, SPMS, and PPMS. Lesions associated with acute relapse in early disease are cellular with abundant CD3+ T cells and do not show smoldering microglial disease activity. In contrast, in PPMS the central nervous system (CNS) is largely devoid of focal cellular collections and smoldering lesions and markers of microglial activation predominate. Secondary progressive patients have a mixture of four types of microscopic lesions with the presence of CD3+ T cells, antibody in plaques, and microglial activation as well as inactive plaques. The majority of the MS population will experience relapsing-remitting illness, but residual persistent disability may variably follow despite remission [11–13]. The presence of residual disability following exacerbations does not signify the onset of secondary progressive illness, however.
Increases in body temperature, or illness, in MS may result in the transient reappearance of neurological symptoms (Uhthoff phenomenon). Despite a previous remission of clinical manifestations of MS, those same symptoms may appear with overheating [2]. Although the Uhthoff phenomenon is not an exacerbation, these phenomena in MS patients are commonly misinterpreted as such. Occasionally heat exposure appears to acutely worsen the severity of an exacerbation and, in other circumstances, worsens a minimal or subclinical event making it more clearly apparent clinically [14]. These events probably reflect the ability of heat to impair the blood-brain barrier, allowing activated lymphocytes and immunoglobulins to enter the brain and spinal cord [14].
The most common initial symptoms of MS are sensory disturbances and fatigue but are often ignored by patients and physicians alike. Perceptions of numbness and tingling by the patient may not be accompanied by obvious abnormalities on initial examination, especially if the patient is not examined completely by a neurologist at the onset of their symptoms. Almost half of initially recognized exacerbations principally affect ambulation. Acute paraparesis varies greatly in degree and in symmetry of the weakness. In many MS patients with motor weakness found by examination, they describe their difficulty as a “heaviness” in their “leg(s).” Alternatively, they may seem only to stumble when their foot catches an uneven area on a sidewalk. The difficulty is often initially recognized only by a family member or a friend during ambulation. Gait problems may be due to motor difficulties and/or, ataxia. Ataxia may occur as a result of vestibular, cerebellar, or sensory impairments. Thus, gait difficulty may reflect motor deficits or ataxia due to one or more problems within the brainstem or spinal cord.
About one out of five or six MS patients will have unilateral retrobulbar (optic) neuritis as their initial clinical difficulty [2, 11]. Other common symptoms at onset include diplopia, facial weakness and/or facial myokymia, vertigo, bladder, and bowel symptoms. Seizures will eventually occur in 10% during the clinical illness but rarely (about 1%) are a presenting sign of illness [2]. Some symptoms, such as hearing loss and impaired night vision, can be seen in MS and also acute disseminated encephalomyelitis (ADEM). The speed of recovery is variable and may be slow over several months or may not occur at all. Other less commonly recognized symptoms include extrapyramidal symptoms and a family of paroxysmal manifestations [15].
Recurrent brief (paroxysmal) stereotyped manifestations in MS include paroxysmal dystonia or “tonic seizures,” paroxysmal dysarthria, paroxysmal akinesis (“paroxysmal falling”), pains (including trigeminal neuralgia and glossopharyngeal neuralgia), and other difficulties [2, 16]. Lhermitte’s sign is precipitated by neck flexion and typically consists of transient shocklike sensations radiating down the neck and back, often into the limbs. It is commonly recognized as a sign of MS especially when it occurs in the young, although it may occur with compressive cervical disc disease or spinal tumors. Except for Lhermitte’s sign, these paroxysmal symptoms seem to occur in a minority of patients and are often not recognized as part of the spectrum of illness. When recognized, these paroxysmal phenomena are of great diagnostic value since they are rarely associated with other illness. When viewed in a cross section of a patient population, they are evident in only about 3% of patients. We have found, however, that with long-term follow-up that paroxysmal phenomena will eventually occur in up to a quarter of patients. Occasionally paroxysmal dystonia involves all four limbs and the truncal muscles as well and may be accompanied by severe pain. Fortunately there is usually a prompt and complete response to carbamazepine in a 400 mg per day dosage, but a course of parenteral corticotrophin may be needed. Unfortunately, many such patients are incorrectly diagnosed as having an acute psychiatric problem. These paroxysmal symptoms are commonly attributed to ephaptic transmission (cross talk between damaged/demyelinated axons), but we suspect that they may be due to inflammatory mediators such as leukotriene C, and other leukotrienes, produced by macrophages. Leukotrienes are extremely potent depolarizing agents. Often the time course of these paroxysmal events approximates that of an exacerbation and, if so, should be considered to be exacerbations.
Although fatigue and fatigability become more prominent with time, especially during periods of disease activity, they may be prominent presenting signs of MS. Anxiety, depression, and cognitive issues, also, may dominate the presentation of illness and may delay disease recognition. In our experience cognitive problems and accompanying emotional reaction occurring early in the course of illness are more important than physical disability as reasons for social dislocation and patients leaving studies or their workplace. A substantial proportion of patients are dismissed as “functional” early in the course of their illness due to their observed emotional status. A recent oral presentation reported the association of MS with schizophrenia and bipolar disorder, with a rate ratio of 1.42 for schizophrenia and 1.73 for bipolar disorder [17].
A bewildering variety of manifestations may occur in MS, singly or in combination with other difficulties. These include limb weakness, “useless limb” syndrome due to severe proprioceptive loss, memory impairment, word-finding difficulty, acalculia, tremor, unusual nonphysiological patterns of sensory loss, and sexual impotence, among others [2, 11]. Motor impersistence is common in the MS population and accompanies proprioceptive impairment. Geschwind also suggested that frontal lobe involvement was a likely contributing factor (Norman Geschwind – personal communication).
Diagnosis of Multiple Sclerosis
Diagnosis of MS is dependent upon the recognition of symptoms and neurological findings typically accompanying exacerbations of MS and affecting different parts of the nervous system over time [7–9]. The importance of an accurate history and physical examination cannot be overemphasized. The senior author’s own observation is that a relative’s recognition of early manifestations of MS is likely to lead to the diagnosis of MS in a family member, rather than the contrary as is commonly believed.
Diagnostic Criteria
The recognition of MS was easy for experienced neurologists in the past. However, long delays in diagnosis were common and many patients were incorrectly diagnosed. The need for standardized criteria for patients entering treatment studies led to the formation of an NIH committee headed by Dr. George Schumacher. Diagnostic criteria have evolved from the 1965 Schumacher criteria [7], that were established primarily for the selection of research subjects for MS studies, to the 1983 Poser criteria [8] which for the first time included laboratory support (magnetic resonance imaging [MRI], evoked response testing, as well as spinal fluid examination). The 2001, 2006, and now 2010 McDonald criteria are based on the original criteria but include validated specific MRI features [9, 10]. These new criteria (Table 2.1) allow the identification of “clinically isolated syndromes” (optic neuritis and brain stem or acute myelitis) with very high (80%) probability of MS. Imaging provides the additional evidence required to establish the presence of dissemination of lesions both in time and space. Early diagnosis of MS with earlier introduction of treatment portends a better outcome in the short-term and prolonged survival, at least for interferon-beta-1a [18, 19]. Consensus definitions of the clinical subtypes of MS were released by the US National Multiple Sclerosis Society Advisory Committee on Clinical Trials in Multiple Sclerosis in 1996 and revised in 2013 [20, 21].
Relapsing MS is characterized by clearly defined relapses with either full recovery or residual deficit, representing about 85% of patients at the outset. Progressive MS is characterized clinically by the gradual accrual of disability independent of relapses and can occur with disease onset (primary progressive) or can be preceded by a relapsing disease course (secondary progressive). In most cases, SPMS is diagnosed retrospectively after several years of gradual worsening after a period of clinical relapses. Currently, there are no clear criteria to mark the transition from RRMS to SPMS. The basis of separating the primary versus secondary progressive forms of MS was derived from a meta-analysis of the COP1 trial in progressive MS as an antecedent of the PROMISE trial [22]. The criteria formulated by Thompson et al. grouped suspected PPMS patients into “definite,” “probable,” and “possible” [21, 23–25]. Multiple sclerosis may be seen as a spectrum with an intense focal inflammatory component in RRMS and more neurodegenerative features with concomitant chronic inflammation and axon loss in progressive forms of MS [26]. Currently, clinical diagnostic criteria exist for both forms. A recent publication provides clear differences in the neuropathological findings separating RRMS, SPMS, and PPMS [27].
Another issue impacting on early diagnosis of MS is the quality of spinal fluid examinations. Importantly, the FDA laboratory standard for oligoclonal banding testing – isoelectric focusing on agarose gel followed by immunoblotting or immunofixation for IgG with paired spinal fluid and serum – avoids technically inadequate studies. The quality of antihuman antibody used in the testing has a major impact on the results. Evoked response testing is relied upon less, but can be helpful, especially visual evoked responses [9].
Diagnostic criteria for PPMS were also updated in 2010 and include (1) a minimum of 1 year of disease progression plus two of three of the following: dissemination in space in the brain or spinal cord or positive CSF, defined as the presence of OCBs, and/or elevated IgG index [10].
Differential Diagnosis
There is a large differential diagnosis, outlined in Table 2.2. In the past meningovascular syphilis was the “great imitator” and topped the list. Today a variety of granulomatous diseases and other diseases are considered in the differential diagnosis, but sarcoidosis and systemic lupus erythematosus (SLE) are the major differential diagnosis considered. The retroviruses human immunodeficiency virus (HIV) and HTLV-I/II can rarely present as a granulomatous disease or mimic MS.
Central nervous system lymphoma may require brain biopsy to establish a diagnosis, but a positive test for HIV ordinarily rules out the diagnosis of MS. Biopsy is ordinarily required to make a diagnosis of primary central nervous system vasculitis (CNS vasculitis). The disorder “CNS vasculitis” is rare and like progressive multifocal leukoencephalopathy (PML) is associated with MS-like attacks resulting in increasing neurological deficit progressing in a stepwise fashion. Unlike PML there may be at least temporary partial resolution of neurological deficit with high-dose steroids or pulse cyclophosphamide therapy in patients with CNS vasculitis. Despite its rarity, establishing a diagnosis of CNS vasculitis is important because it is regularly fatal if not treated aggressively with chronic systemic immunosuppression.
Multiple sclerosis may occasionally present with prominent sensory complaints and marked, symmetrical weakness of the lower extremities and be mistakenly diagnosed as an acute demyelinating polyneuropathy (Guillain-Barré syndrome). Albumino-cytological dissociation, however, is rarely found in MS.
Symptoms of MS must last 24 hours at a minimum. To be considered a new relapse, a new symptom or a relapse of a prior symptom must occur at least 1 month after the previous exacerbation. The symptoms and findings should be of a type recognized as associated with multiple sclerosis. The diagnosis of multiple sclerosis is accepted only if it is established by a neurologist [7–10].
PPMS is a more difficult diagnosis to establish. This form of MS presents most commonly in midlife (about 40±5 years on average), and distinguishing this form of MS from other potentially treatable illness may be extremely difficult [11, 28]. Manifestations of neurological disease should be observed for at least 6 months before acceptance as evidence supporting a diagnosis of PPMS. Multiple other disorders must be ruled out of the differential diagnosis. Syphilis, vitamin B-12 deficiency (subacute combined myelopathy), and retrovirus-associated myelopathy (HIV-associated myelopathy and human T-cell leukemia-associated myelopathy (TSP/HAM)) [2, 11, 29] can be easily ruled out by laboratory testing. Antibody testing by Western blot for HTLV-I/II, if indeterminate, may not be sufficient [30]. Genetic (“PCR,” polymerase chain reaction) testing in a reliable laboratory test is the most sensitive and specific test for this purpose. In our experience this test is positive in up to 20% of patients who are Western blot indeterminate but who are infected with either HTLV-I/II virus [31]. Radiation myelopathy continues to be an important differential diagnosis in patients with a history of radiation therapy to the head and neck.
Neuroimaging should be carried out to eliminate spinal cord compression, congenital abnormalities, and intraparenchymal tumors from consideration. At times, imaging will not reveal the presence of one or more intraparenchymal spinal cord lesions that are evidenced by clinical examination, however. The finding of hypothyroidism is common in MS, and myelopathy should not be attributed to thyroid disease alone. Adrenocortical leukodystrophy and hereditary spastic paraplegia are easily distinguished from primary progressive multiple sclerosis by the patient’s infantile age of presentation and presence of a family history [2, 32].
It cannot be overemphasized that repeated clinical visits and examinations over time, as well as repeated imaging, may clarify the nature of the illness in difficult cases. This is particularly important when cognitive and emotional issues dominate and obscure the presentation [3, 11]. The McDonald criteria, however, greatly assist early diagnosis and justify the institution of treatment. It should be noted that in using the criteria for a clinically isolated syndrome (CIS), the majority will be correctly diagnosed as having MS, but about 20% of patients may never meet criteria for clinically definite MS. On the other hand, we regularly document relapses within weeks to months in many patients with CIS who initially had no evidence of brain lesions in their MRI scans at clinical presentation. Multiple sclerosis remains a clinical diagnosis [9, 10].
Prognosis
Exacerbation rates in MS patients vary greatly but tend to diminish with increasing duration of illness [13, 14, 18, 33]. When a patient has established disability, exacerbations do not appear to correlate with increasing disability [13]. Pregnancy has long been thought to decrease the risk of relapse in the third trimester, as shown in a large prospective study [34]. This is thought, at least in part, to be secondary to high concentrations of estrogen and progesterone, and phase II clinical trials have shown a potential role for estriol in treatment of MS [35]. The risk of relapse in the first trimester, however, is increased. The French study also confirmed a long recognized phenomenon that the risk of exacerbation of MS is markedly increased for 3 months postpartum. This study also showed this risk continued at a somewhat lower level for the 33 months of follow-up in the study. The importance of infection as a precipitating factor for exacerbations has long been recognized [36].
Emotional stress and its impact on MS has been the subject of a number of excellent studies [37–40]. All of these studies have consistently shown a correlation between major life stress and a significantly increased risk of exacerbation of MS. In a remarkable more recent study, Mohr et al. have demonstrated a correlation between stress, including “hassles” and the appearance of new active gadolinium-enhancing brain lesions [40]. The perception of stress, rather than a particular life event, is related to an increased risk of exacerbation [37–40]. While other factors are thought to influence prognosis in MS patients, no similar studies of risk factors has addressed them adequately.
A large number of neurologists at academic centers in the United States and elsewhere have concluded that the majority of MS patients develop secondary progressive disease and then progress rapidly to disability. Confavreux et al. have published their studies of the natural history of a large population of French patients [13]. The French workers have concluded that there is no relationship between relapses and progression, once disability is established. They have further concluded that only 30% of their relapsing-remitting patients had secondary progressive MS. Pittock et al. at the Mayo clinic published important observations of a 10-year follow-up of their MS population from Olmsted County, Minnesota [14]. They too found that disability in the majority of their patients did not progress measurably during the 10-year period of observation. Only 30% of their patients progressed to needing a cane or a wheel chair, but most patients remained stable despite the fact that only 15% had received immunomodulatory therapy. It is obvious that the perception that the vast majority of MS patients develop secondary progressive disease with rapid progression to serious disability is incorrect. The group in Lyon, France, has also found that longer periods of follow-up show that patients thought to have “benign MS” do develop some neurological impairment over 20–30 years of follow-up. Please see Table 2.3 for a list of proposed prognostic indicators.
Neuroimaging in Multiple Sclerosis
Computerized tomography (CT) neuroimaging for the first time revealed areas of decreased radiodensity in the brain as well as occasional enhancing brain and spinal cord lesions in MS. Interestingly, increasing brain atrophy, although reported early, was largely ignored by the MS community [43–45]. Comparative studies of CT and MRI revealed the relative strength of MRI in visualizing plaques as well as brain atrophy in MS [46–48]. In contrast to the limitations encountered with the use of CT, MRI has had an important impact on both the diagnosis and subsequent management of MS because of the relative ease which it can detect white matter lesions in the brain and spinal cord.
Investigators have sought brain MRI correlations with clinical symptoms of MS, prognosis of the illness, other laboratory findings, as well as with central nervous system pathology. Increased T2 signal, reflecting increases in water content of lesions in hemispheric white matter, was emphasized in earlier studies, but their presence correlates poorly with symptoms and neurological findings (Fig. 2.1a). In our initial experience with this imaging modality, we found that very early in the course of clinical disease, only half of patients with clinically definite MS did have cerebral white matter lesions [47, 49]. However, almost half of those that did not have plaques in their brains exhibited spinal cord lesions that were clearly evident [50]. While, not all cerebrospinal fluids (CSF) had “diagnostic” abnormalities, only 5% of patients did not have either brain MRI abnormality or significant CSF abnormality. In part, the difficulty with the MRI findings in these early studies was related to technical issues such as image slice thickness, noncontiguous sections, etc. Use of fluid-attenuated inversion recovery (FLAIR) sequences, which are easier to visualize, has been made practicable by advances in the hardware and software (Fig. 2.1b). Newer acquisition paradigms and the use of gadolinium to identify “active” inflammatory lesions, in particular, as well as continued hardware improvements have remarkably improved the quality and utility of MRI. However, not all patients with MS, particularly those with PPMS, exhibit white matter lesions in their cerebral hemispheres. The absence of MRI abnormality does not negate the diagnosis of MS [9]. We found that after 9–12 years, the same proportion of MS patients will have white matter lesions evidence by MRI and by pathology, however [47, 49]. In a recent presentation from the Cleveland Clinic, Dr. Robert Fox revealed that approximately 20% of their well-documented patients with progressive MS did not have hemispheric white matter lesions at necropsy [50]. They do, however, have cortical as well as spinal cord, i.e., “corticospinal” involvement. Cortical involvement in MS is rarely evident with standard imaging parameters. Double inversion recovery is capable of documenting about 40% of the cortical lesions found in pathological study [51].

MRI scans of the brain of a 19-year-old woman with relapsing-remitting multiple sclerosis. Axial T2-weighted (a) and fluid-attenuated inversion recovery (b) views show hyperintense lesions in subcortical white matter. Axial T1-weighted postcontrast (c) of the same patient reveals an enhancing lesion, indicating the breakdown of the blood-brain barrier
A strong correlation between increased volume of cerebral MRI T2 signal and long-term disability in MS has been reported in patients followed for 5 years after the onset of a clinically isolated syndrome. However, further follow-up of this cohort of patients has shown only a moderate correlation at 10 years [52]. A number of short-term correlations between stabilization, or reduction, of T2 volumes and clinical stabilization in patients treated with each of the immunomodulatory drugs are currently approved. After the initial 5 years of illness, with some notable exceptions, changes from 1 year to the next are difficult to see in brain MRI scans. Clearly, there must be some reservation about the use of T2 lesion volumes for assessment of longer-term treatment of any kind.
Gadolinium enhancement of white matter lesions is an accepted indicator of active disease, but enhancing lesions are seen several times more often than acute exacerbations of illness in multiple sclerosis (Fig. 2.1c). This surrogate measure of disease activity has been used effectively in preliminary drug efficacy studies to detect a treatment effect. Despite the earlier negative reports, Leist et al. reported a correlation between gadolinium-enhancing lesions and the subsequent appearance of cerebral atrophy [53]. Unlike the earlier studies reporting on correlation, this NIH study was based on frequent (monthly) gadolinium-enhanced brain MRI studies.
Although T1 hypointensities have been reported to correlate with cerebral atrophy, other studies have shown that this type of MRI lesion does not correlate well with either the amount of demyelination or gliosis in tissue lesions. The lack of correlation with tissue changes makes it difficult to understand and accept these observations at face value [54, 55]. Importantly, De Stefano et al. have reported data supporting a role between early axonal damage and subsequent development of disability in multiple sclerosis [66].
Brain atrophy progresses at a rate of 0.5–1.0% per year in patients with MS, considerably higher than the typical rate seen with normal aging at 0.1–0.3% per year. Once thought to be largely a disease of white matter, MS is now recognized to have significant manifestations in the gray matter [56]. The volumetric changes seen on MRI during the course of MS have been correlated with disability progression and cognitive impairment; however, the quantitative cutoffs to determine physiologic versus pathological brain atrophy in MS remain to be determined.
No evidence of disease activity (NEDA) has been proposed as a potential treatment goal for treatment trials in MS. Elimination of relapses and prevention of disease progression, including cognitive loss and impaired ambulation, are the clinical goals (Fig. 2.2).

Disease-free concept: NEDA-4
NEDA-3 includes (1) no sustained increase in disability lasting 3 months, (2) no relapses, and (3) no MRI activity, defined as no new or enlarging T2 and Gad+ lesions. NEDA-4 includes similar parameters, with the addition of no annual brain volume loss >0.4%. NEDA-3 status appears to correlate with subsequent relapse and focal inflammatory MRI activity. NEDA-4, in utilizing measures for tissue destruction at both the focal inflammatory and diffuse level, may be a more comprehensive predictor for subsequent disability-related outcomes. NEDA-4 data has been collected using post hoc analyses of the FREEDOMS and FREEDOMS-II trials [57, 58].
More advanced imaging methods continue to be explored. Double inversion recovery (DIR) can be used to demonstrate cortical inflammatory lesions, although its use is limited by inadequate resolution and inability to identify purely intracortical, versus juxtacortical or leukocortical, lesions [51]. Diffusion tensor imaging (DTI) is used to evaluate the structural integrity of the white matter tracts. DTI can be used for diffusivity measures including mean diffusivity and fractional anisotropy, which may provide even closer evaluation of tissue integrity and axonal damage [56, 59–61]. The value of proton magnetic resonance spectroscopy continues to be investigated and has resulted in many claims that are not entirely consistent. The advent of higher Tesla field strengths, up to ultrahigh-field 7–8 Tesla, has improved characterization of cortical demyelination, with good pathologic correlation but is restricted to research studies for safety reasons [62].
It is obvious that MRI is especially helpful in the evaluation of patients early in the course of their illness. Unfortunately, the question as to the utility of using MRI or other surrogate measures to evaluate the long-term response to treatment remains essentially unanswered. Cerebral atrophy may very well be the most valuable measure.
Other Laboratory Measures
CSF
CSF analysis can be helpful if performed in a specialty laboratory. Increased intrathecal IgG synthesis, measurement of the increase in the proportion of gamma globulin by CSF electrophoresis, and the presence of CSF oligoclonal bands increase the likelihood of a diagnosis of MS [7–9, 11]. Neurofilament chains are potential markers for axonal injury as seen in gadolinium-enhancing lesions in RRMS and progressive forms of MS [63, 64].
Glial Fibrillary Acidic Protein (GFAP) Concentration
CSF GFAP is raised in SPMS and associated with expanded disability status scale (EDSS) scores [65].
Evoked Response Testing
Visual evoked responses carried out in an established laboratory too can be helpful in making a diagnosis [9]. Other evoked responses, brain stem and somatosensory, can be abnormal in other diseases as well as MS, and the studies are technically more difficult. Spinocerebellar degenerations are often associated with markedly abnormal auditory evoked potentials, for example.
Epidemiology
To yield useful data epidemiological studies must be carried out by trained personnel in large populations with good access to good medical care. A number of good studies have been performed, and there is evidence indicating that incidence rates for MS may be increasing.
Age and Sex Distribution
Multiple sclerosis of the relapsing-remitting type is more common in women, about 70% of all patients in most recently studied populations, including our large southern population, with onset of illness in both sexes by the age of 30 in two-thirds [11]. Primary progressive MS is slightly more common in men and typically begins in midlife.
Incidence of MS
Incidence is the rate of occurrence of newly diagnosed (MS) cases per unit of population (usually described per million) per time period, usually reported on an annual basis. The incidence of MS is relatively low (1–5 per million) but seems to have increased over the last century [11]. In the United States the most useful current data comes from Olmsted County, Minnesota, where the incidence rate increased during the last century from two per million to three times that incidence [11].
A number of confounding factors influence incidence figures. Over the last half century, there has been a dramatic increase in the number of trained neurologists. With the advent of effective therapies, more neurologists are interested in MS and many trained in this subspecialty. Consistent easily interpreted diagnostic criteria, and improved diagnostic testing (especially MRI), have greatly facilitated making the diagnosis. Undoubtedly, these factors partly account for the apparent increased incidence of multiple sclerosis. If we can extrapolate from the experience of neuropathologists, and as reported from Stanford, 1–2% of postmortem examinations reveal tissue evidence of “demyelinating disease” in the absence of a clinical history [66, 67]. It is possible that now, given the availability of neurologists, the increasing awareness of MS, and the diagnostic facilities available, many clinically undiagnosed cases in the past would be labeled as having MS.
Despite the low incidence of MS, this illness is the most common cause of chronic disability in young adults because of the minimal impact on the longevity currently. The observations in Olmsted County, Minnesota, clearly indicate a real increase in the incidence, as well as its prevalence, of MS [9].
It is often stated that there are 250,000–350,000 MS patients in the United States [11]. Figures currently used, however, are not based on any current national epidemiological studies. When prevalence figures were reported to be low for the Southern United States, except for California, there were no neurologists in the South. In Florida, for example, the first neurologist established a practice in Florida in 1953 but then entered the military service, a situation similar to many other areas in the South. The appearance of neurologists in the South since that time, as in virtually all under-serviced communities in the United States, is bound to have had a dramatic impact on the recognition and diagnosis of nervous system disease, especially MS. The impact of MRI on the recognition of neurological disease has been dramatic, especially for MS. Considering the increased availability of neurological consultation, improved diagnostic criteria and the availability to MRI, and improved CSF examination, that larger numbers of MS patients will be recognized in life. The quoted prevalence of MS appears to be unrealistically low.
Environmental Factors
Myriad environmental risk factors for MS have been studied with varying degrees of validation. The most robust data supports the association of prior Epstein-Barr virus infection and smoking and development of MS [68]. The significant detrimental effect of smoking has been identified in numerous studies, with a dose-response relationship [69, 70]. Previous infection with EBV and high antibody titers to Epstein-Barr early nuclear antigen are well-established risk factors for MS, especially when contracted as an adolescent or young adult [71, 72].
Other epidemiological factors, which may be associated with an increased risk of MS, include increased salt intake. Kleinewietfeld et al. demonstrated that elevated sodium chloride concentrations in human (dietary) and mouse (tissue culture followed by studies of dietary intake) models increase proinflammatory Th17 cells [73, 74]. Vitamin D may be an early predictor MS activity and progression, though identification of the optimal Vitamin D supplementation strategies remains undetermined [75]. Unpublished follow-up data beyond 10 years of Aschiero’s study group of vitamin D shows maintenance of long-term benefit with vitamin D levels greater than 50 nmol/L. High-dose supplementation with 10,400 IU cholecalciferol daily has been reported as safe [76]. Adolescent obesity, defined as a BMI of > 27 kg/m2 at age 20, is associated with a twofold increased risk of developing MS. Further study has indicated an interaction between adolescent obesity and HLA risk genes in MS [77, 78].
There is a geographical pattern distribution of MS, with higher disease incidence in higher latitudes, though this has become less apparent in recent years in the setting of globalization [79]. In this context, the “hygiene hypothesis” was introduced by Strachan in the 1980s. It proposes that persons with less exposure to microbes early in life are more likely to develop autoimmune disorders, including MS [80]. This hypothesis has fallen out of favor, however, as a result of several studies evaluating MS incidence and helminthic infection, and the role of the gut microbiome in MS has become a focus of research. Nonpathogenic intestinal microflora may be mediators of autoimmunity in MS [81–85]. There is no longer evidence for a north-south gradient for MS in the United States.
Pathology of Multiple Sclerosis
Charcot recognized multiple areas of discoloration and hardness (sclerosis) scattered throughout the brain and spinal cord which he termed plaques (plate like) as the cardinal features of MS: hence, the diagnosis of sclerose en plaque, or “multiple sclerosis” [1]. By microscopy, Charcot found that plaques exhibited loss of myelin with relative sparing of axons and varying amounts of gliotic scarring. He also described the presence of inflammatory cells, including large numbers of fat-laden cells. The demyelinated plaque remains the pathological hallmark of this disease [85].
Early in the disease small plaques are prominent in subcortical white matter [42], but in the usual necropsy material obtained after many years of disease, large coalesced plaques are predominantly periventricular [85–89]. No regular association between MS plaques and blood vessels was observed by Adams and Kubik [87] and Zimmerman and Netsky [88]. Subsequently, however, Lampert [89], and others, performed whole brain serial sections of a number of cases, including those previously studied and reported that brain plaques were invariably perivenular [89]. Although oligodendrocyte loss had earlier been reported as a major feature of MS [87, 88], study of whole brain serial sections did not reveal this to be a consistent feature [89]. Another important finding is that so-called shadow plaques seen at the white matter cortical junction are areas of remyelination, rather than areas of incomplete demyelination, as had previously thought [85].
In recent years, the neuropathology of MS has been revisited [90–92], and a new view of the histopathology of MS has emerged based on a study of 51 biopsies and 37 autopsies. A central role for CD4+ T cells and macrophages in the immunopathogenesis of the multiple sclerosis lesions seemed to have been well established (Fig. 2.3) [91]. Lucchinetti et al., however, have suggested four different types of neuropathology in MS, pointing to a predominant role for CD3+ cells and macrophages in type 1, with antibody-mediated demyelination added in type 2, and to loss of oligodendrocytes in others [93].

Biopsy of a large left frontal lobe plaque from a 29-year-old woman with new onset multiple sclerosis with recurrent right hemiparesis over 3 months and new mild speech difficulty. (a) Specimen is stained with Luxol fast blue counterstained with eosin. A new active plaque is shown which is not sharply demarcated but exhibits prominent perivascular cellularity with varying myelin damage and relative sparing of axons. The inflammatory infiltrate is composed of lymphocytes (predominantly CD4 Th1 cells) and a large number of macrophages. These cells are predominantly of hematogenous origin and are considered the perpetrators of tissue damage. These features are in contrast to chronic or inactive plaques which exhibit relatively few or no inflammatory cells but contain prominent myelin damage and gliosis. Axonal loss may be prominent. (b) Frontal lobe biopsy: Luxol fast blue counterstained with eosin. Higher power view showing loss of axons and more prominent myelin loss. Note that axons that are preserved exhibit variable loss of myelin
In type 1, in patients where tissue samples were obtained very early, prominent perivascular infiltrates composed of CD3+ cells and macrophages were present without IgG or complement. In type 2, a similar perivascular picture was seen, except that antibody (IgG) and complement, without cells, were seen at the edge of active demyelination. While prominent loss of myelin basic protein and myelin-associated glycoprotein was found, remyelination was reported to be prominent in types 1 and 2. In type 3 and 4, oligodendrocyte loss was prominent, raising the question of primary oligodendrocyte pathology. Plaques were poorly defined and not related to vessels. However, the authors reported that CD3+ (T) cells and macrophages were present in all four types of multiple sclerosis pathology included in their classification contain, a finding in keeping with other recent analysis of lesions [93]. Their findings that tissue obtained from a small number of patients studied shortly after onset of their illness revealed prominent CD3+ (T) cells and macrophage cellular infiltrates but lacked antibody (type 1) are reminiscent of the findings of patients who died early in the course of their illness, reported by Lumsden [86]. Type 2, where antibody is present in the lesions, is seen at necropsy with some frequency and resembles changes seem in chronic relapsing forms of EAE. In EAE the initial cellular infiltrate is composed primarily of CD4+ cells initially, but this is followed by the appearance of much large numbers of macrophages that induce the damage to myelin and oligodendrocytes [94].
Despite the impressive amount of work their report encompasses [93], the observations that in a proportion of cases the pathology of MS may consist of oligodendrocyte loss, with pathology not associated with blood vessels, raises questions. The numbers of cases are relatively small and many were biopsy specimens, where sampling necessarily was limited and most importantly not based on study of whole brain serial sections. Poser had raised other questions about type 1 pathology [95]. Recently, in 20 patients of a subset of well-documented subset of 150 progressive MS patients without cerebral white matter lesions, pathological evaluation revealed the presence of cortical pathology with an inflammatory component extending from the meninges into the cortex [50]. Spinal cord root entry zone pathology can lead to debilitating pain in MS patients and are rarely identified by neuroimaging [96, 97].
Pathogenesis of Multiple Sclerosis
Genetics
In the past few years, our understanding of the genetic underpinnings of MS has exploded due to the advent of large genome-wide association studies (GWAS). Clustering within families is a well-known phenomenon. Prior to the recent advances, it was found that in a large MS database in Vancouver and our large database in South Florida, a 20% familial incidence was present in both data sets. The Canadian twin study shows a concordance of 31%, similar to other twin studies [98]. Mothers confer a 20–40 times increased risk to their children, greater for girls than boys. Other first-degree relatives also have a much-increased risk of MS [99].
As of press time, more than 159 genetic variants have been associated with an increased risk of developing MS [100, 101]. For several decades, the major histocompatibility (MHC) gene locus located on chromosome 6 has been implicated, and it is clear that the HLA-DRB1 gene in the class II region of the MHC explains up to 10.5% of the genetic variance underlying risk of MS. A monumental linkage study, conducted by the International Multiple Sclerosis Consortium, evaluated 730 families with multiple cases of MS, further emphasized the role of the major histocompatibility (MHC) class II HLA-DRB1*15:01 allele, as the only variant of several genetic loci to achieve statistical significance [102]. Mouse studies also implicate a strong genetic susceptibility for experimental allergic encephalomyelitis (EAE) localized to the region of DQBq*602 [103]. The more complete characterization of MHC contribution to MS and identification of variants outside the MHC region were not appreciated until the advent of the era of GWAS. Using large sample sizes, the largest of which numbered 80,095 subjects, this technique identified 110 non-MHC risk variants in 103 loci. Interestingly, 78% of predicted MS heritability remains undetermined [104]. Improving whole-genome sequencing technologies hold promise to identify rare genetic variants.
A limited number of causative gene variants have been identified. The MS-associated SNP rs6897932, located in the alternatively spliced exon 6 of IL-7Rα, alters the ratio between the soluble and membrane-bound isoforms of the protein by disrupting an exonic splicing enhancer [105]. The risk variant rs1800693 in the tumor necrosis factor (TNF) 1A gene that drives the expression of a novel soluble form of the receptor that can inhibit TNF signaling mimics the effects of TNF-blocking drugs that are known to exacerbate MS pathology [106]. Other variants include rs3453644, acting at the tyrosine kinase 2 protein, and rs12487066 associated with decreased levels of human endogenous retrovirus Casitas B-lineage lymphoma proto-oncogene B in CD4+ T cells [107, 108]. The underlying pathogenic mechanisms for these variants remain unclear. The current collaborative studies arose from early findings by Jersild et al. who found that the alleles A3, B7, and DR2 [109] occurred twice as commonly in MS as compared with the unaffected population. They observed that in patients that possessed both HLA-B7 and DR2, that disease was particularly severe [109]. Many genes important in normal immune function and in immune-mediated tissue damage, such as tumor necrosis factor, are located in the region between HLA-B7 and the DR locus. Several mutations of genes resident in this area are currently being studied. An important study looking for single nucleotide polymorphisms (SNP), modeled on the Crohn’s disease study, is currently under way as part of the human genome project. As yet there is no single gene, or combination of genes, implicated in the risk or causation of MS.
Once disease-causing gene variants are identified, the next step is to identify biomarkers that can predict disease progression. Our understanding of the factors leading to neurodegeneration and increased disability in progressive MS remains limited, and genetics may shed significant light on this process.
Several reports have described familial clustering of MS phenotype. The presence of the HLA-B*44 allele is thought to be associated with better neuroimaging outcomes [110]. Variants associated with age of onset and a range of radiologic outlooks include HLA-DRB1*15:01, HLA-DRB1*07:01 and HLA-DRB1*11:04, and HLA-DRB1*01:03 [111–114]. The absence of HLA-B5 independently associates with a marked increase in the severity of MS, as in the Afro-American population [110]. Future directions for pharmacogenetics research in MS include identification of specific genetic variants associated with treatment response, leading to a tailored therapy approach. SNP genotype data led to the discovery of several HLA genes and may be used to identify IFN-β super-responders. An important recent study found an association between the rs9828519 variants, which is intronic to SLC9A9 and implicated as a regulator of proinflammatory lymphocyte activation and MS disease response and nonresponse to IFN-β [115, 116].
Studies of migrant populations have suggested the presence of an environmental factor. Although generally interpreted as evidence that a viral infection is playing a role in multiple sclerosis, no conclusive evidence of a specific virus playing a role in multiple sclerosis has been produced [11, 71, 117, 118].
Myelin Biochemistry
The genetic basis of a number of leukodystrophies has been firmly established. Of these disorders, the most common are adrenocortical leukodystrophy and metachromatic leukodystrophy. At one time both were considered to have some relationship to MS [2, 11]. Of some importance is Marburg’s disease, sometimes referred to as “acute multiple sclerosis,” which has been attributed to a defect in myelin basic protein (MBP) synthesis and structure [119]. Work on alterations of the 3D structure of MBP and relationship to various demyelinating disease continues. Interestingly, several mutations of the proteolipid of myelin are causative of Pelizaeus-Merzbacher disease, another leukodystrophy, as well as several types of hereditary spastic paraparesis. These disorders ordinarily should not be confused with MS because of early age of presentation of the leukodystrophies, their inexorably progressive course, and their familial setting.
Immunology
Multiple sclerosis is now generally accepted as an immune-mediated illness although its pathogenesis is incompletely understood. The occurrence of MS following about a third of cases of acute disseminated encephalomyelitis complicating infections [120–122] as well as after immunizations, including Semple vaccine (containing spinal cord and killed virus), suggested an autoimmune origin. Although EAE has been studied in animal models for decades, the primary impetus was to elucidate the nature of the immune response [123]. These studies have also provided insight into the pathogenesis of MS as well. Transfer of EAE from immunized to naive animals was first successfully accomplished using lymph node cells but not antibody, thus pointing to a central role for lymphocytes [123]. Nevertheless, antibody from immunized animals, and patients with MS, can induce demyelination in vitro [60, 61].
T cells play a primary role in the pathogenesis of EAE, irrespective of the nervous system antigen used to induce disease [124–127]. A consensus has developed that T cells are the primary effectors both in MS and in EAE [127]. Nevertheless, B cells, plasma cells, and antibody can be found both in EAE pathology and in MS plaques [92, 93]. Despite their emphasis on other findings, these recent studies of pathology in MS show that the predominant cells in active lesions are lymphocytes, in particular CD3+ T cells, and macrophages [93].
Multiple injections of the whole spinal cord were used to induce EAE in early studies, but single immunizations of equivalent amounts of purified myelin or MBP combined with adjuvants were shown to be very effective in disease induction [127]. Myelin proteins other than MBP have also been investigated, notably proteolipid and myelin oligodendrocyte glycoprotein (MOG). Proteolipid protein can induce forms of experimental disease in animal models and, although antibody as well as T cells reactive to this antigen may be present in plaques, no role for sensitization to this antigen has been established [127]. However, an interesting EAE model in marmosets induced using MOG indicates that antibody may mediate demyelination [128, 129]. Passive transfer of the disease by serum from MOG-sensitized animals has been accomplished [129]. However, T cells (CD4+ Th2, rather than CD4+ Th1 cells) may be the primary mediators of myelin damage in MOG-sensitized marmosets [129]. The situation is complicated by the fact that CD4+ cells reactive to MBP, capable of inducing EAE, are present in naive animals as well as in these immunized animals coincidently with anti-MOG antibody [129]. Anti-MOG antibody has been reported at the outset of MS and is common in RRMS [130, 131]. In contrast to anti-MOG antibody being limited to MS relapse, CD4+ cells reactive to MOG are ubiquitous [132].
Antigen presentation by MHC class I or MHC class II by antigen-presenting cells (APC) to T cells results in the initiation of immune responses: antibody production or a cellular immune response. Activated CD4+ T helper (Th) cells fall into three functionally distinct classes, Th1 and Th2, and Th17 with distinctive profiles of lymphokine production. Following antigenic stimulation CD4+ Th1 cells produce interleukin-1 (IL-1), IL-2, IFN-γ, and tumor necrosis factor α (TNF-α) are postulated to mediate inflammatory pathological processes in immune-mediated tissue damage seen in MS and EAE [133]. In contrast, Th2 cells produce IL-4, IL-5, IL-6, and IL-10 and induce upregulation of antibody production and downregulation of Th1 cellular responses (Fig. 2.4) [133]. The observed failure of increased production of the regulatory cytokine IL-10, by myelin-reactive T cells in MS by Ozenci et al. in Sweden, has recently been confirmed by Cao et al. at MIT [134, 135]. More recently a role for Th17 helper cells in a large subpopulation of MS patients has been identified and characterized. Sera from interferon-β-1a treatment failure patients from Denmark were shown to contain IL-17F. Naive patients that had IL-17F and elevated levels of endogenous INF-β failed to respond to IFN-β-1a subsequently also. These IFN-β failure MS patients resemble EAE animals induced by Th17-polarized cells [136, 137].

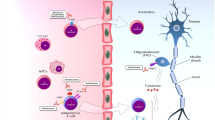
A model of immunopathogenesis of multiple sclerosis. Following exposure to certain environmental antigen(s) in genetically susceptible individuals, myelin-reactive T cells migrate from peripheral circulation to the central nervous system. Interaction between activated T cell and cerebral endothelial cells leads to upregulation of the adhesion molecules (E-selectin, vascular cell adhesion molecule, intercellular adhesion molecule, mucosal addressin cell adhesion molecule, and platelet endothelial cells adhesion molecule). Transendothelial migration of reactive T cells is heralded by the disruption of the blood-brain barrier, which is in part mediated by the activities of the matrix metalloproteinases. Matrix metalloproteinases digest the activated T cells (such as TNF-α and IFN-γ) and upregulate the expression of cell surface molecules on antigen-presenting cells (in this figure, glial cell). Binding of putative multiple sclerosis antigen (e.g., myelin basic protein and myelin oligodendrocyte glycoprotein) by the trimolecular complex T-cell receptor and class II major histocompatibility molecules on the antigen-presenting cells precipitates a massive inflammatory cascade, which leads to production of both pro- and anti-inflammatory cytokines. This inflammatory reaction ultimately results in loss of myelin-oligodendrocyte complexes
Macrophages are the principal sources of IL-1, IL-12, and TNF-α, driven by IL-2 production from antigen-activated CD4+ cells. Importantly, IL-12 production is IFN-γ dependent and TNF-α production is IL-12 dependent [138]. Traditionally the macrophage was considered to be the principal APC, but B cells are now recognized as important in this task. However, macrophages are central effector cells in cell-mediated immunity. After antigen presentation, CD4+ cells respond by clonal proliferation and recruitment of other CD4+ cells to participate in the initiation of cellular immune responses. Cytotoxic CD8+ cells, driven by IL-12, may exert their effect directly or target antibody complexed with antigen on target tissue, i.e., antibody-dependent cytotoxicity [127, 139]. Macrophages may also target these complexes. The spectrum of CD4+ Th2 responses includes a regulatory role in switching of CD8+ cell cytotoxic function to active suppression of CD4 Th1 responses, suppressor T cells. In the CNS microglial cells can function as APC and exhibit certain other macrophage behaviors including an anti-inflammatory response.
The blood-brain barrier (BBB) is a physical barrier that prevents intravascular cellular elements, antibodies, and other proteins free access to the brain and spinal cord [138]. The endothelial cells in the brain and spinal cord possess tight junctions that are impervious to intravascular fluids as well as nonactivated cells. These endothelial cells are also surrounded by astrocytic foot processes that further support and maintain the integrity of the BBB. However, activated CD4+ cells do cross the BBB [140–145]. However, the BBB is an actual physical barrier which may be breached only in an organized and well-orchestrated fashion [140, 145, 146]. The mechanisms of cellular transmigration across the blood-brain barrier are now well understood [140–146].
Interleukin-17 and Type 17 Helper T Cells
T cells were found to produce cytokines that could not be classified into either the Th1 or Th2 scheme detailed above. Primary among these cytokines is interleukin-17 (IL-17), and the cells that produce IL-17A have been named Th17 cells. Other cytokines produced include IL-17F, IL-21 and IL-22, IL-26, and TNFα. Their important role in the pathogenesis of MS is increasingly recognized [147, 148]. In vitro studies have suggested that Th17 cells can permeate the blood-brain barrier, and elevated levels of IL-17 have been detected both in serum and CSF in some patients with MS [149]. In addition, an increase in IL-17 mRNA has been detected in MS plaques at autopsy [150, 151]. Th17 cells can induce and regulate tissue inflammation. In the setting of chronic inflammation and autoimmunity, initially studied in rheumatoid arthritis, signaling through Th17 receptors induces production of inflammatory cytokines such as IL-6, IL-1, TNF, IL-8, and matrix metalloproteinases [147]. A recent study has implicated glutamate excitotoxicity as a possible effector mechanism for inflammation in MS [152]. Studies to elucidate the role of Th17 cells in MS are ongoing. Secukinumab, a selective anti-IL-17A monoclonal antibody, is being studied as a potential treatment for MS [153].
Adhesion Molecules
Venules control CD4+ and other cell migration from blood into the nervous system. Attachment requires cellular adhesion molecules and endothelial counter receptors to overcome the considerable shear stresses produced by blood flow. Adhesion molecules on CD4+ cells and macrophages act as functional anchors forming stable bonds with their ligands on the vascular wall. In addition to functioning as mechanical anchors, adhesion molecules function as tissue-specific recognition molecules [140–146].
Entry of CD4+ cells and macrophages into the CNS is accomplished by a series of steps including tethering or rolling, adhesion (binding), and finally transendothelial migration across the BBB [141–146]. Subsequent to their egress, they migrate through the extracellular matrix in the CNS. Selectins mediate the initial step of tethering leading to rolling [146, 154, 155] but selectin-mediated bonds are reversible. To arrest these cells on the endothelium, these low-affinity interactions must be supplemented by high-affinity adhesion molecules, the integrins [153, 154]. The integrins, including α4β1-integrin (VLA-4), are members of the endothelial immunoglobulin superfamily [156, 157]. The predominant function of the β2-integrin leukocyte function antigen-1 (LFA-1) and α4-integrins (integrin-α4β1/VLA-4) is to bind the cells to their ligands intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM-1) [155–157]. Blocking of attachment of the α4 moiety on lymphocytes by natalizumab is highly effective treatment in MS but is complicated by a risk of progressive multifocal leukoencephalopathy (PML) [158].
Selectins expressed on leukocytes (P-selectin and L-selectin) and endothelium (E-selectin) result in rolling and slowing of the cells. P-selectin and its ligand PECAM-1 appear to play a special role in EAE and MS [159, 160]. As cells roll and are slowed by the interaction of selectins and their ligands, they respond to endothelial cell chemokines. Specific chemokines are fixed on the endothelial surface and are molecular signals that direct cells to tissues and with specific adhesion molecules confer organ specificity [145]. Chemokines are divided into four families that are specific for different T-cell subgroups [145]. Distinctive chemokine receptors on Th1 cells include CCR5 and CXCR3. In MS, all of the infiltrating Th1 cells express these chemokine receptors [161]. They play a central role in the egress of specific lymphocyte subgroups into specific target organs. Selectin binding to ligand is an activating signal that induces rapid activation of α4-integrins and β2-integrins [155–157].
From the first availability of IFN-β, about half of the population placed on this drug did not appear to benefit from it. In a prospective study, Byun and coworkers found that half of MS patients placed on IFN-β were “super-responders” [162]. They found that a number of genes were expressed in this super-responder subpopulation following their first dosage, and this predicted the clinical response. Interestingly, these genes included heparan proteoglycans [160]. Further support for the identification of IFN-β responder/nonresponder populations followed with a report by Axtell et al. in 2010 [136]. They reported that serum from Danish IFN-β-1a nonresponders contained IL-17. Most recently the evidence correlating response or nonresponse to IFN-β to polymorphisms of a specific gene rs9828519, a sodium-hydrogen channel, has been published [115]. Apart from illuminating the mechanisms of the drug response, these observations hopefully will help identify potential “super-responders” and assist in advising them in regard to their therapeutic choices for MS. This should reduce the human and financial cost of treatment failure in managing MS.
T-cell vaccine studies are continuing. The initial approach was to remove immunocompetent cells from patients by immunizing them with antigen analogous to V-beta chains of T-cell receptors that are capable recognizing encephalitogenic fragments of MBP. More recent studies have focused on using CNS antigen-stimulated cells from the patient’s own T-cell repertoire and, following irradiation, infusing these autoreactive cells back into the donors. There has been a remarkable impact on reducing sustained progression of disability patients with RRMS, and the current study is hoping to replicate these findings in patients with SPMS. A preliminary report in RRMS was encouraging for progressive MS [163].
Treatment of Multiple Sclerosis
Treatment issues in MS generally fall into four categories. These are (1) symptomatic treatment; (2) treatment of acute MS exacerbations; (3) reducing the risk (“prevention”) of future exacerbations and, more importantly, reducing the risk of sustained increases in disability; and (4) neurological rehabilitation. In recent years there have been advances in each of these four areas.
In the past, treatment of MS was limited to empirical management of symptoms, i.e., symptomatic treatment. Most treatments were untested and were of questionable value, at best. Interested readers are referred to the Diary of Augustus D’Este where descriptions of treatments employed are recounted [3]. Treatments were really generic, ineffective, and sometimes dangerous remedies such as cathartics, enemas, and bloodletting. Many ineffective empirical treatments continue to be offered by misguided individuals and quacks.
Symptomatic Treatment
Symptomatic treatment covers many areas, but only a few specific issues will be dealt with in this review. Fatigue, spasticity, and bladder symptoms are among the most important areas. Also important is the management of the paroxysmal disorders: paroxysmal dystonia, paroxysmal akinesia, paroxysmal dysarthria, trigeminal neuralgia, facial myokymia, and hemifacial spasm. Treatment can be dramatically effective.
Fatigue is a prominent complaint in the majority of patients. In reality, the fatigue of which patients complain is predominantly fatigability, although the occasional patients with severe exacerbations may awaken with overwhelming fatigue. The first drug for fatigue to be evaluated in double-blind trials (and shown to effective) was amantadine HCl (Symmetrel®) [164]. A dose of 100 mg twice daily is an effective antiviral, initially virtually preventing all influenza type A infections and 90% of type B infections and a lower but important risk reduction for other paramyxovirus infections. The sustained reduction of fatigue observed in the majority of patients is presumably due to its weak dopamine agonist properties, rather than an antiviral effect. In addition, a variety of adrenergic drugs have been used to treat fatigue, but tolerance tends to develop quickly and habituation is also problem [165]. Modafinil (Provigil®), a more selective member of this family of drugs appears safe and tolerated in small (200 mg) daily doses [166]. Unfortunately, in our experience, tolerance seems to develop quickly too. A matter of concern is that in vitro adrenergic drugs appear to promote cellular immune mechanisms, calling into question their use in fatigue management. Fatigue and depression commonly coexist, and fluoxetine (Prozac®) is commonly used to manage these patients. Interestingly, fluoxetine has immunomodulatory properties, with resultant increases in the Th2 lymphokines, IL-4, and TGFβ [167]. Fatigue lessens in patients who stabilize clinically, spontaneously, or in conjunction with immunomodulatory therapy.
Mobility
Dalfampridine (Ampyra®) was approved in 2010 for the improvement of walking ability. It is a nonspecific potassium channel blocker that is thought to improve conduction in focally demyelinated axons by delaying repolarization and prolonging duration of action potentials. Enhanced neuronal conduction is thought to strengthen skeletal muscle fiber twitch activity, resulting in improved motor function [168–170].
Spasticity continues to be a major problem in MS patients [2]. Diazepam (Valium®) was the first drug to be proven to reduce spasticity in MS, and it continues to be a very helpful drug. The use of single oral dose of 5 mg at bedtime is convenient and cost-effective treatment in a large proportion of patients with mild-to-moderate spasticity. Occasionally, a small additional dose can be added in the morning, but the long half-life of the drug usually makes that unnecessary or undesirable. Baclofen (Lioresal®) is an important and useful drug that is less frequently associated with sedation than diazepam, even at high doses. The oral form of the drug, which is a racemic mixture, does not seem to have a predictable dose response in many patients, however. In contrast, those patients with severe refractory spasticity predictably respond to intrathecal baclofen [171]. This, in part, reflects the addition of l-baclofen to the racemic forms of baclofen for intrathecal use. Use of the intrathecal drug requires the implantation of a pump to deliver the drug, however [171]. Tizanidine (Zanaflex®), an alpha-2-adrenergic agonist, has good dose-response characteristics [122]. On the negative side, tizanidine has a short half-life and 40% of patients experience prominent fatigue and dry mouth as side effects. In some patients use of tizanidine avoids the necessity of pump implantation and therefore is a welcome alternative [172]. Hopefully, in the future an oral formulation of l-baclofen will advance to phase III studies and become a clinical option.
Bladder dysfunction occurs in the majority of patients, largely due to hyperreflexia of the detrusor muscle. However, dyssynergia accompanies this in 90% of cases. Managing urinary frequency is usually attempted with the use of low doses of anticholinergic and oral baclofen, but is often unsatisfactory. Often a single dosage of an anticholinergic drug before retiring at night and prior to occasional social outings is more satisfactory than a multiple doses. Incomplete emptying is usually best handed by intermittent catheterization. The management of infections is very important. Avoidance of antibiotics for unproven infections, and obtaining bacterial sensitivities for each infection, is crucial to avoid pseudomonas infections. Often chronic use of oral ascorbic acid 2–4 g daily with hippuric acid 2 g daily to acidify the urine together with six to eight glasses of water successfully prevents recurrent infections. Mirabegron (Myrbetriq®) is a remarkable new adrenergic drug for hyperreflexic bladder with incontinence [173].
More extensively studied in spinal cord injury, botulinum toxin A has recently been approved as an effective alternative for uncontrolled neurogenic detrusor overactivity resulting in incontinence in patients with MS [174, 175]. It is clear that good bladder management significantly contributes to quality of life [176].
Management of the paroxysmal disorders is relatively simple in most patients once they are recognized and identified by physicians [2]. Paroxysmal dystonia (or tonic spasms), paroxysmal akinesia, trigeminal neuralgia, facial myokymia, and hemifacial spasm are often successfully managed with modest doses of anticonvulsant drugs. However, the response in patients with paroxysmal dysarthria tends is less predictable. For patients requiring treatment, carbamazepine in doses of 100 mg orally three times daily controls about 70% of these disorders and 400 mg daily increases the response rate to 80–85%. Higher doses sometimes are helpful but the addition of a second anticonvulsant is often more effective. Some patients require two or more drugs, including gabapentin and topiramate, to control these symptoms, but often carbamazepine can be withdrawn if the second drug is effective [177]. The use of corticotrophin (ACTH) intravenously or intramuscularly, but not steroids, is sometimes necessary to gain control of the situation [178].
Treatment of Acute Exacerbations
In the past management of MS exacerbations consisted principally of continuous enforced rest [2]. At the onset of an exacerbation, rest relieves (or prevents) fatigue. Thankfully, the injudicious use of extended periods of rest has given way to the enthusiastic use of physical rehabilitation.
The senior author’s career has spanned the era of validation and FDA approval of corticotrophin (adrenocorticotropic hormone/ACTH) [122] and the subsequent introduction and use of high-dose intravenous steroids for the management of exacerbations of multiple sclerosis. Dr. Leo Alexander, Harvard Medical School, initially used corticotrophin because steroids (that he hypothesized should be helpful) were not available (personal communication). The effectiveness of corticotrophin was established by multiple controlled trials, the first for any MS treatment [178]. The pivotal trial was a multicenter double-blind placebo-controlled trial was published in Neurology 1970 and became the basis of the FDA approval in 1978. No other drug has been validated as an effective treatment for exacerbations of MS. However, 40 years ago neurologists at the Montreal Neurological Institute, including the senior author with other MS physicians, first employed high-dose intravenous steroids in patients diagnosed with MS. The use of high-dose parenteral steroids was limited to patients who had lost vision, in one or both eyes due to optic neuritis, or who were acutely paraplegic due to acute myelitis. In retrospect, these patients probably had neuromyelitis optica rather than MS. On the basis of the analogy with trauma and tumor management, it was hypothesized that that acute severe edematous swelling of the optic nerve or spinal cord resulted in complicating ischemia due to the limited capacity to expand within the dura spaces. Although patients often improved rapidly, frequent complications of high-dose therapy problems were encountered. Gastrointestinal complications are now rare, but psychiatric disturbances, infectious complications, osteoporosis, and aseptic necrosis of the hip and other bones which are side effects are not rare. Despite weak evidence of benefit from the single-blind (intravenous) optic neuritis treatment trial indicating short-term benefit [178, 179], no well-organized appropriate sized, double-blind trials have been carried out to date. The double-blind oral steroid use portion of the optic neuritis trial showed clearly that oral steroids were deleterious to patients with optic neuritis (most of whom would develop clinically definite multiple sclerosis). Patients receiving oral steroids subsequently experienced a doubled relapse rate of optic neuritis, apart from other manifestations of MS compared with oral placebo recipients. A German trial has confirmed experimental observations of increased damage from the use of steroids equivalent to doses used in human [180]. In patient with optic neuritis treated with steroids, treatment is associated with damage to the affected optic nerve that can be reduced by the concomitant administration of erythropoietin [181]. We interpret these results as evidence that oral steroids, alone, should not be used in the management of MS. It is important to note that a neuroprotective effect for neurons from corticotrophin is well established [182–184]. Methylprednisolone, however, has recently been shown to induce programmed cell death (apoptosis) of neurons [180]. Because of the effectiveness, and the neuroprotective effect, of corticotrophin, we continue to favor its use.
A trial of natalizumab for the management of acute exacerbations failed to influence the outcome of such clinical exacerbations [185]. The drug, however, did reduce the risk of new MRI brain lesions over the subsequent 12 weeks following a single infusion. Despite its failure to induce a more rapid recovery from exacerbations, natalizumab did improve the sense of well-being of the drug recipients, also. Benefit was observed in subsequent studies aimed at reducing the risk of MS exacerbations and/or sustained increase in disability also.
Reduction of Multiple Sclerosis Exacerbations and Disability
For more than a decade and a half, there has been intensive study of several drugs and their potential value in reducing the risk of exacerbations in MS. As a corollary to this outcome, there has been increasing emphasis on their potential impact on reducing the risk of disability due to this disease. At press time, there are ten FDA-approved disease-modifying therapies for relapsing MS (see Table 2.4).
The first drug to be approved (1993) to reduce the frequency of MS exacerbations of (33% reduction) was IFN-β-1b (Betaseron®) [186, 187]. The drug also had a remarkable effect, significantly reducing the burden of disease as measured by brain MRI T2 lesion volumes [187]. Unfortunately, use of IFN-β-1b is consistently associated with flu-like symptoms and local inflammatory reaction at the injection site.
The drug IFN-β-1a is produced using mammalian cell lines and the authentic human genetic sequence, unlike IFN-β-1b that has two genetic alterations and which is made using coliform bacteria. IFN-β-1a is rapidly absorbed from the injection site and local reactions as well as neutralizing antibody formation are less. Avonex® brand of IFN-β-1a was approved in 1996 as a result of a study using 30 micrograms intramuscularly once weekly [188]. Risk of sustained disability for 24 weeks, the primary outcome measure, was reduced for drug recipients to 21.9 vs. 39.7% for placebo recipients in the study. Relapse risk was also reduced, 0.61 vs. 0.90 for those who completed the 104 weeks of the trial. However, data analysis employing “intent-to-treat analysis” showed a reduction in the risk of relapses with active drug treatment of 0.61 vs. 0.82 for placebo. The latter results reflect the fact that 40% of the patients did not complete the study because study drug was not available. Subsequently, the benefits on disability prevention were shown to be sustained [189].
A large three-arm pivotal (PRISMS) trial was reported in 2002, showing results resembling those reported for IFN-β-1b [190]. Subsequently, after additional studies, a head-to-head trial of Rebif® vs. Avonex® was undertaken [191]. The 16-month trial benefit favored Rebif® at each time point in the study. However, the “survival” curve of Avonex® appeared to approach that of Rebif® as the study progressed, however. The PRISM trial extension did show more benefit for patients at the higher dose who initially had received placebo and who were switched to either 22 or 44 micrograms three times weekly [192, 193].
Pegylated IFN-β-1a (Plegridy®) was approved by the FDA in 2014 and is administered subcutaneously at 2-week intervals at a maintenance dose of 125 μcg /0.5 mL, available both as a pen injector and prefilled syringe. It is an IFN-β-1a to which a single, linear 20,000-dalton methoxy poly(ethyleneglycol)-O-2-methylpropionaldehyde molecular is covalently attached to the alpha amino group of the N-terminal amino acid residue. The efficacy of Plegridy® was demonstrated in the ADVANCE study, a randomized, double-blind, placebo-controlled study of RRMS that examined clinical and MRI outcomes at 48 weeks, comparing the treatment group against placebo. The primary outcome of related reduction of annualized relapse rate over 1 year was met, with statistically significant (p=0.0007) relative reduction of 36%. MRI outcomes at 48 weeks showed a 67% relative reduction of mean number of new or newly enlarging T2 hyperintense lesions and 86% relative reduction in the mean number of Gd-enhancing lesions (p≤0.0001) [194]. The side-effect profile is quite similar to that of Rebif®, including flu-like symptoms, injection site reactions, hepatic injury, and depression. The dose-frequency blinded extension study (ATTAIN) is ongoing.
Glatiramer acetate (Copaxone®) was approved in 1997 as a result of a double-blind placebo-controlled trial [195]. The outcome of the trial was a 30% reduction in the risk of relapse for glatiramer, compared with placebo, similar to the IFN-β studies. A follow-up of a subset of patients by the original investigators has shown apparent robust long-term benefits with the majority of the study subjects stabilized [196]. This information has become part of the package insert. More recently in the Glatiramer Acetate Low-Frequency Administration (GALA) study, glatiramer acetate at a dose of 40 mg/mL administered subcutaneously thrice weekly compared to placebo showed a 34.0% reduction in risk of confirmed relapses, and this new dosing regimen is now approved for use [197].
A marked reduction of gadolinium lesion enhancement has been found following initiation of IFN-β-1b [198] and IFN-β-1a [188] and for glatiramer acetate [199]. Similar results for natalizumab have been reported [200]. Interestingly, the serially studied placebo patients showed that while enhancement disappears with steroid administration, enhancement returns, finally disappearing about 2 months after its first appearance [185]. In recent years, increasing emphasis has been placed on techniques of measuring brain atrophy [201–203].
Natalizumab (Tysabri®) is a humanized monoclonal antibody that binds α4-integrin and blocks interaction of α4β1-integrin on leukocytes with vascular cell adhesion molecules (VCAM) and connects segment-1 on fibronectin sites on vascular endothelial cells [204]. Two phase III clinical trials demonstrated the efficacy of natalizumab, administered at a dose of 300 mg intravenously every 4 weeks. The AFFIRM trial showed that natalizumab reduced ARR by 68% over 2 years, disability progression by 42% over 12 weeks and 54% over 24 weeks, an 83% decrease in new or enlarging T2 hyperintense lesions, and decrease in gadolinium-enhancing lesions on MRI by 92% compared to placebo. The SENTINEL trial examined natalizumab in combination with IM IFN-β-1α is more effective than IM IFN-β-1α alone [205–207]. Natalizumab is generally tolerated well. Side effects include infusion-related symptoms, allergic hypersensitivity reactions, anxiety, fatigue, pharyngitis, bladder and respiratory infections, sinus congestion, and peripheral edema. The primary safety concern is the increased risk of PML, the risk of which increases with duration of therapy and serum JCV Ab status and index [208, 209]. Approximately 6% of patients develop persistent anti-natalizumab-neutralizing antibodies [210]. Switching of natalizumab to alternative agents like fingolimod more than 8 weeks after cessation of natalizumab may be associated with lower risk of MRI and clinical disease reactivation [211].
In 2010, Fingolimod (Gilenya®) was the first oral disease-modifying drug to be approved by the Food and Drug Administration for MS. Fingolimod is a sphingosine-1-phosphate receptor (S1P1) modulator, initially acting as an agonist of the S1P1 receptor, and then becomes a potent functional antagonist, leading to internalization of S1P1 receptors on lymph node T cells, resulting in sequestration of lymphocytes in the lymph node. Uniquely, circulating naive T cells and central memory cells are reduced by fingolimod, since both express the chemokine receptor lymph node homing CCR7. Fingolimod does not affect effector memory cells, but some of its mechanisms of action may be explained by the enhancement of function of potent circulating regulatory T cells. Other effects include the modulation of human oligodendrocyte progenitor cells, which potentially could affect myelin repair, astrocyte proliferation, migration and gliosis, and neuroprotection. The clinical efficacy of fingolimod was demonstrated in two large, phase III, double-blind, randomized trials: (1) FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis (FREEDOMS) and (2) Trial Assessing Injectable Interferon Versus FTY720 Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS). The FREEDOMS trial enrolled 1272 patients who were assigned either oral fingolimod 0.5 mg or 1.25 mg daily versus placebo for 2 years. The primary end point, ARR, was 0.18 in the 0.5 mg dose group, 0.16 in the 1.25 mg dose group, and 0.40 in the placebo group. There was also a statistically significant effect on reduction of sustained disability progression. After 12 weeks progression was seen in 17.7% in the 0.5 mg dose group and 16.6% in the 1.25 mg dose group versus 24.1% in the placebo group. Fingolimod also showed a reduction in the number of new or enlarging lesions on T2-weighted imaged, gadolinium-enhancing lesions at year 2. Importantly, reductions in whole brain volume were less at both 12 and 24 months in the fingolimod group [212, 213]. The TRANSFORMS trial included 1292 patients randomly assigned to the 0.5 mg dose and 1.25 mg dose, but this time a comparator of 30 μg weekly IM interferon-beta-1a. Orally administered fingolimod at a dose of 0.5 mg daily was found to be superior to IFN-β-1a at reducing ARR and MRI activity, although the sustained use of IFN in patients prior to the initiation of the trial is considered a confounder of this data [214]. Fingolimod is generally well tolerated; however, low-frequency specific safety issues including first-dose bradycardia, herpes virus dissemination, macular edema, and elevated blood pressure require screening and regular monitoring. Of note, four cases of PML have now been reported with fingolimod use, without prior exposure to natalizumab.
Teriflunomide (Aubagio®) is an oral medication that interferes with the de novo synthesis of pyrimidines via inhibition of the mitochondrial enzyme dihydroorotate dehydrogenase, resulting in blocking cell replication in rapidly dividing cells. The precise mechanism for its effect in RRMS is unknown. Teriflunomide is a derivative of leflunomide, used for many years in the management of rheumatoid arthritis. Two clinical trials examined the efficacy of teriflunomide: (1) TEMSO and (2) TOWER. The TEMSO study evaluated both 7 mg and 14 mg doses versus placebo in 1088 patients with active relapsing MS. Both doses showed a significant reduction in the primary outcome measure, ARR, compared to placebo by 31.2% (7 mg) and 31.5% (14 mg). Both the 7 mg and 14 mg dose reduced MRI outcomes, slightly more in favor of the14 mg dose. In the TEMSO extension study, adjusted ARR remained low 5 years after initial randomization [215–217]. In the TOWER study, 1169 were randomly assigned to a 7 mg dose, 14 mg dose, and placebo group. The ARR was higher in the placebo group (0.50) compared to the 14 mg (0.32) and 7 mg dose groups (0.39). Teriflunomide at the 14 mg dose reduced the risk of sustained accumulation of disability at 48 weeks; however, the 7 mg dose did not show this effect [218, 219]. A third head-to-head study compared the effectiveness and safety of teriflunomide and subcutaneous interferon-β-1a (44 μg three times per week) in patients with relapsing multiple sclerosis (TENERE) over a 2-year period. The primary end point was time to failure, defined as the first occurrence of confirmed relapse or permanent treatment discontinuation for any reason, and no statistical superiority between IFN-β-1a and the 14 mg dose of teriflunomide was found, although IFN-β-1a was superior to the 7 mg dose of teriflunomide [220]. The ongoing phase III TERACLES trial is examining the clinical usefulness of combination teriflunomide with IFN-β. (ClinicalTrials.gov identifier: NCT01252355)
The most common adverse effects of teriflunomide are mild-moderate, including elevation in transaminases, hair thinning, GI upset, and headache. We have had two apparent allergic reactions to this drug. The greatest concern is the potential for teratogenicity based on animal data, and teriflunomide is contraindicated in women in childbearing potential not using reliable contraception, and men with the potential to father a child are also advised to utilize contraception. As teriflunomide may remain in the serum for up to 2 years, an enhanced drug elimination procedure using cholestyramine or activated charcoal powder is used for patients planning on becoming pregnant or who already are pregnant [221]. Despite these precautions, as of 2013 the AUBAGIO Pregnancy Registry data indicated that 12 newborns have been conceived while on teriflunomide, with no structural or functional deficits reported [222].
Dimethyl fumarate (DMF) (BG-12, Tecfidera®) is the third oral therapeutic option. It is a fumaric acid ester in an enteric-coated microtablet. When it enters the CNS is immediately hydrolyzed by esterases to its metabolite monomethyl fumarate. DMF is associated with decreased GI side effects compared to MMF. It acts on nuclear factor erythroid2-related factor 2 (Nrf-2), which upregulates various antioxidative pathways and inhibits the translocation of nuclear factor-ĸB into the nucleus, therefore avoiding the expression of a cascade of inflammatory cytokines, chemokines, and adhesion molecules. While the forgoing mechanism is thought to be responsible to it clinical effect, the exact mechanism of action in RRMS, however, is unknown [223].
Two clinical trials have evaluated the efficacy of BG-12 for RRMS: (1) determination of the efficacy and safety of oral fumarate in relapsing-remitting multiple sclerosis (DEFINE) and (2) comparator and an oral fumarate in relapsing-remitting multiple sclerosis (CONFIRM). The DEFINE study evaluated 1234 patients with RRMS and EDSS scores of ≤5 who were randomized to a 240 mg twice-a-day dosing regimen, 240 mg three-times-a-day dosing regimen, or placebo. The primary outcome measure was the proportion of patients relapsing at 2 years, whereas unlike other clinical trials, the ARR and risk for disability progression were secondary outcomes. Both doses of BG-12 met the primary outcome measure, with a reduction in the proportion of patients relapsing by almost 50%. Twenty-seven percent of patients on the twice-a-day dosing and 26% of patients on the three-times-a-day regimen had at least one relapse at 2 years, versus 46% of patients on placebo. ARR in both doses of BG-12 was reduced by 53% relative to placebo. EDSS progression was also reduced at 12 weeks in both dosing regimens, with 16% (twice-a-day regimen) and 18% (three-times-a-day regimen) progressing versus 27% of patients on placebo. Other measures, including new or enlarging MRI lesions were significantly lower in the BG-12-treated patients as well. The CONFIRM trial evaluated 1430 patients randomized to one of the two BG-12 dosing regimens or an active comparator glatiramer acetate (GA) 20 mg/d subcutaneously. The primary end point, difference in ARR over a 2-year period, was 44% lower with BG-12 at the twice-a-day regimen, 51% lower with the three-times-a-day regimen, and 29% lower with GA. There was no significant reduction in sustained increase in disability, but a preplanned analysis of the combined outcomes of the DEFINE and CONFIRM studies did reveal a significant reduction in the risk of sustained increase in disability. Of note, the study was powered to evaluate the doses against placebo, but not against GA. The most common adverse effects include abdominal pain, flushing, nausea, and diarrhea. These effects can be ameliorated with the administration of the medication with food and/or regular aspirin at a dose of ≤325 mg 30 minutes prior to administration. Severe lymphopenia may occur, and PML has been reported in four patients. It is recommended that a CBC with differential be obtained at least at 6-month intervals. Reduction of CD8+ T cells is more pronounced than that of CD4+ T cells, and this can be serially monitored with lymphocyte subset panels [224–226].
Despite hopes that oral therapy would lead to increased compliance, it has been shown that oral medications, particularly dimethyl fumarate which is dosed twice daily, is associated with poorer compliance, especially in the young population [227–229]. Alemtuzumab (Lemtrada®) is a humanized anti-CD52 monoclonal antibody. The exact mechanism by which alemtuzumab exerts its therapeutic effects in RRMS is unknown, but is thought to work via depletion and subsequent repopulation of both circulating T and B lymphocytes. These cell populations recover at variable rates, with CD4+ T lymphocytes being the slowest, leading to long-term adaptive immunity. The CARE-MS I trial was a phase III randomized clinical trial of 581 treatment-naive patients comparing alemtuzumab (12 mg/d over a 5-day IV administration with a second 3-day IV administration 1 year later) to subcutaneous IFN-β-1a administered three times a week at a ratio of 2:1. Two primary end points were identified: reduction in relapse rate and 6-month sustained accumulation of disability. Alemtuzumab reduced risk for relapse by 55% compared to IFN-β-1a, with a yearly relapse rate of 0.39 in the IFN-β-1a group compared to 0.18 in the alemtuzumab group, monitored over a period of 2 years. A secondary outcome measure, maintenance of relapse-free status for 2 years, was met in 77.6% of alemtuzumab-treated patients and 58.7% of IFN-β-1a-treated patients. Multiple MRI outcomes also favored alemtuzumab. These included a reduction in the percentage of new and enlarging T2 lesions, new gadolinium-positive lesions, or persistent gadolinium-positive lesions at 24 months and new T1-hypointense lesions. The alemtuzumab group had slower progression of brain atrophy as compared to IFN-β-1a (0.87 versus -1.49 median percent change at year 2) [230]. CARE-MS II evaluated 840 patients who, unlike CARE-MS I, had recently relapsed while taking a standard disease-modifying therapy. Randomization was performed in a 2:2:1 ratio of high-dose (24 mg) alemtuzumab, low-dose (12 mg) alemtuzumab, and IFN-β-1a. Yearly rate of relapse was significantly reduced in the low-dose alemtuzumab group (0.26) compared to the IFN-β-1a group (0.52) over 2 years. A 42% reduction in the risk for sustained accumulation of disability over 6 months was seen in the low-dose alemtuzumab group (12.7%) versus the IFN-β-1a group (21.1%). Of the low-dose alemtuzumab group, 28.8% had sustained improvement in their EDSS score compared to the IFN-β-1a group (12.9%). There was no significant change in total T2 burden, but fewer patients had new or enlarging T2 lesions or new gadolinium-positive lesions over 24 months in the alemtuzumab group. There was less reduction in mean brain parenchymal fraction in the alemtuzumab group (−0.615% versus −0.81%). No advantage of the 24 mg over 12 mg dose of alemtuzumab was seen [231].
Alemtuzumab is associated with several safety issues. Mild-moderate infusion-related reactions are seen in 90%. The incidence of infections is higher, most commonly upper respiratory tract infections, urinary tract infections, and oral herpes. The development of secondary autoimmune disorders is of primary concern, with 16–19% of alemtuzumab-treated patients developing thyroid-related problems and 1% developing immune thrombocytopenia. There is concern for development of antiglomerular basement membrane disease as well. Monthly CBC with differential, serum creatinine levels, and urinalysis with urine cell counts are recommended for 48 months after the last dose of alemtuzumab. Prophylactic medications for pneumocystis pneumonia and herpes viral infections must be administered during treatment and for at least 2 months following the last dose or until CD4+ counts recover to ≥200 cells/mm3 [232].
The management of primary and secondary progressive disease is far from satisfactory but based on prospective studies; two drugs are now approved: mitoxantrone [233, 234] (Novantrone®) and IFN-β-1b [235]. The use of IFN-β-1b varies greatly from one geographic area to another, varying on the impatience and experience of physicians and patients alike. Its use is tempered by the fact that many patients seemingly stabilized initially subsequently begin to progress despite continued use of the drug. In retrospect, this is seen in drug trials that included patients who no longer experienced relapses [235]. This observation is also in keeping with the meta-analysis of the US trial. The use of mitoxantrone resulted in cessation of exacerbations and apparent stabilization in the majority of drug recipients vs. controls in the study. This was accompanied by the realization that the drug is cardiotoxic [233, 234]. The results as published are difficult to under interpret for the non-statistician, and the specter of cardiotoxicity combined with the risk of promyelocytic leukemia has limited its use of this effective drug, despite clear-cut guidelines. It is best used in larger centers with experience with this drug.
High doses of oral biotin (100–300 mg daily) were studied in France for chronic progressive multiple sclerosis [236]. Data in an open-label study of 23 patients showed that 91.3% improved clinically suggested that biotin may have an effect on disability and progression. The results of a randomized, double-blind, multicenter placebo-controlled (2:1) trial of MD1003 (pharmaceutical grade biotin dosed at 300 mg/day) in patients with progressive MS were reported at both the 2015 AAN meeting and 1st Congress of the European Academy of Neurology [237]. A second clinical trial is underway evaluating the effect of biotin in MS patients with permanent visual loss following optic neuritis. A significant reduction in disability progression is preliminarily reported.
Other nonspecific immunosuppressants have been used in the clinical setting. Some were employed in open-label settings, and limited trials of azathioprine, methotrexate, and cyclophosphamide have been carried out. There appears to be a desirable effect from the use of these drugs, but potential infections are real risks, and other problems potentially complicate their use. Hopefully, pivotal trials of one or more of these agents will be organized in the near future. If employed, their use again should be limited or guided by neurologists who are experienced in their use.
Future Directions in Treatment
Though traditionally B cells were not thought to be of central importance in the pathogenesis of MS, and therefore not initially a target for disease-modifying therapy, an anti-B-cell therapy a proof of concept (phase II) study indicated a potential role for rituximab (Rituxan®) in the treatment of RRMS [238]. While a clinical trial evaluating the use of rituximab in primary progressive MS (PPMS) patients did not show a statistically significant difference in time to confirmed disease progression compared to placebo, subgroup analysis revealed a significant difference in patients aged <51 years with gadolinium-enhancing lesions seen on MRI [239].
Data presented at the 2015 ECTRIMS meeting from recently completed pivotal studies of ocrelizumab, a humanized anti-CD20 monoclonal antibody given intravenously, have revealed a highly significant impact on both relapse reduction and reduction in the risk of progression in RRMS. Another anti-CD20 humanized monoclonal antibody under study, ofatumamab, has been successful in a proof of concept studies with either intravenous or subcutaneous preparations. The data of three large pivotal (phase III) clinical trials, two evaluating ocrelizumab in the RRMS population (OPERA I and II), and another in the progressive MS population (ORATORIO) were revealed at the 2015 ECTRIMS annual meeting in Barcelona, Spain. Ocrelizumab showed a significant effect for both relapsing-remitting and progressive MS. Ocrelizumab reduced the ARR at 96 weeks by 46% in OPERA I and 47% in OPERA II compared to IFN-β-1a [240]. In the ORATORIO PPMS study, ocrelizumab met the primary end point of a significant 24% reduction in 12-week confirmed disability progression (CDP) [241]. Key secondary end points including a 25% reduction in risk of CDP at 24 weeks, 17.5% reduction in brain volume loss, and 3.4% decrease in T2 lesion volume. The most common adverse events were mild-to-moderate infusion-related reactions [242]. Official publication of the results is newly released [243], [244].
Daclizumab is yet another humanized monoclonal antibody that binds to the α-subunit (CD25) of the high-affinity interleukin-2 (IL-2) receptor expressed on activated T cells and CD4+CD25+FoxP3+ regulatory T cells. Its mechanism of action in MS is thought to be via blockage of the activation and expansion of autoreactive T cells. An important biological effect of daclizumab is the activation and expansion of immunoregulatory CD56 bright natural killer cells. Two phase III trials are recently completed and the drug has been submitted for approval by the Federal Drug Agency. The DECIDE study, which compared subcutaneous daclizumab high-yield process (HYP), administered at a dose of 150 mg every 4 weeks, with intramuscular IFN-β-1a. The annualized relapse rate was significantly lower with daclizumab HYP than with IFN-β-1a (0.22 vs. 0.39, 45% lower rate with daclizumab HYP). The number of new or newly enlarged hyperintense lesions on T2-weighted magnetic resonance imaging (MRI) over a period of 96 weeks was lower with daclizumab HYP than with IFN-β-1a (4.3 vs. 9.4, 54% lower number of lesions with daclizumab HYP, P<0.001). At week 144, the estimated incidence of disability progression confirmed at 12 weeks was 16% with daclizumab HYP and 20% with IFN-β-1a, but this finding was not statistically significant [245]. The results of the OBSERVE single-arm study, which is evaluating the immunogenicity and pharmacokinetics of daclizumab HYP, have not been published at press time [246].
There is understandably substantial interest in the development of remyelinating agents in MS to repair damage myelin. The anti-LINGO-1 monoclonal antibody BIIB033 has undergone phase I randomized trials, and phase II results from the SYNERGY trial were reported in Barcelona in 2015 [247, 248]. Another monoclonal antibody under consideration for development is GSK1223249 which targets NOGO-A, an inhibitor of neurite outgrowth [249].
Laquinomod is a derivative of linomide, an agent studied in the 1980s for use in MS whose development was halted due to multiple adverse events including myocardial infarction. As with its parent molecule, serious adverse experience including cardiotoxicity has been recognized, and the pivotal study has been halted.
Other treatments in early clinical studies include secukinumab, an anti-IL-17A monoclonal antibody and firategrast, an oral agent acting against anti-α4-integrin (the target for natalizumab) [250, 251]. Second-generation, more specific sphingosine receptor agents being studied include siponimod and ONO-4641 [252, 253]. Ibudilast is a phosphodiesterase-4 inhibitor that reduces microglial inflammation and hopefully neurodegeneration in MS and is a promising option for treatment of progressive MS. The phase IIb trial Secondary and Primary Progressive Ibudilast NeuroNEXT trial in Multiple Sclerosis (SPRINT-MS) is currently under way.
Rehabilitation
There is renewed interest in exercise in MS both here in the United States and in Europe, and strategies employed in rehabilitation have continued to evolve [254, 255]. The recognition and acceptance of the principal of shorter periods of exercise for MS patients repeated after periods of rest has helped many patients greatly. The use of aquatic exercises, where the patient is cooled during exercise and allowed longer periods of sustained effort, also has resulted in more effective rehabilitation. The impact of daily exercise on experimental models of CNS disease is striking [256–258].
The use of more modern orthotics devices, which are lighter and reduce fatigue in the MS patient, is a major advance in patient management. New neuroprosthetic technology in the form of functional electrical stimulation, such as Bioness® and WalkAide®, can be helpful in selected patients. Fitting these devices and monitoring by experienced physicians and therapists increases their effectiveness and is particularly important. Patients require training and encouragement to adapt to these devices. Similarly, simply giving a patient a prescription for a cane is insufficient. Early introduction of stretching, and judicious use of muscle stretching and use of drugs for control of spasticity prevent contractures and simplify management of most patients. The primary role of the therapist is to instruct the patient and caregivers as to what they must do to decrease the risk of contractures and increase mobility. At the same time they must increase self-confidence of the patient avoid making the patient dependent on the therapist.
Conclusions
The age of rational therapy for MS arrived in the early 2000s with natalizumab and therapeutic options continue to expand. Increased efficacy may be associated with complications such as PML as first evidenced with natalizumab. Its continued use is contingent upon improved risk stratification for PML based on JC virus antibody indices with values less than 1.3 indicative of a low risk (less than 1:10,000). There is continuing concern that other effective drugs may share such risks but the jury is still out. Risks for natalizumab vary with duration of treatment, peaking at the end of the third year of use for high JC virus antibody index subjects and subsequently decreasing to levels resembling those observed after 2 years. Prior use of mitoxantrone or methotrexate raises the risk to especially high levels (1:90) in the presence of high index JVC antibody. L-selectin (CD62L) was thought to be a possible useful biomarker, but a recently published prospective study failed to show any utility [209]. From the available data, fingolimod and dimethyl fumarate appear to be associated with a very low risk for PML, far less than the risk for natalizumab with low JVC antibody indices.
Future trials of compounds discussed in the “emerging therapies” section are exciting prospects. Of particular importance are the anti-B-cell therapies. The focus for disease-modifying therapy has been in relapsing-remitting multiple sclerosis, and there is newfound enthusiasm for treatment of progressive MS stimulated by the recently announced ocrelizumab trial results for PPMS. The FDA has just declared this drug as a “breakthrough” in the treatment of progressive MS.
Abbreviations
- ACTH:
-
Corticotrophin
- APC:
-
Antigen-presenting cell
- CIS:
-
Clinically isolated syndrome
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CT:
-
Computerized tomography
- DIR:
-
Double inversion recovery
- DTI:
-
Diffusion tensor imaging
- EAE:
-
Experimental allergic encephalomyelitis
- EDSS:
-
Expanded disability status scale
- GFAP:
-
Glial fibrillary acidic protein
- HIV:
-
Human immunodeficiency virus
- IL:
-
Interleukin
- MBP:
-
Myelin basic protein
- MHC:
-
Major histocompatibility class
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MRI:
-
Magnetic resonance imaging
- MS:
-
Multiple sclerosis
- NEDA:
-
No evidence of disease activity
- PCR:
-
Polymerase chain reaction
- PML:
-
Progressive multifocal leukoencephalopathy
- PPMS:
-
Primary progressive multiple sclerosis
- RRMS:
-
Relapsing-remitting multiple sclerosis
- SLE:
-
Systemic lupus erythematosus
References
Charcot JM. Histologie de la sclerose en plaques. Gaz Hop Paris. 1868;41:554–66.
Compston A, Ebers G, Lassman H, McDonald I, Mathews B, Wekerle H. McAlpine’s multiple sclerosis. 3rd ed. London: Churchill Livingstone; 1988.
Firth D. The case of sir Augustus d’Este. London: Cambridge University Press; 1947.
Kurtzke JF. A reassessment of the distribution of multiple sclerosis. Part one. Acta Neurologica Scand. 1975;51:110–36.
Kurtzke JF. A reassessment of the distribution of multiple sclerosis. Art two. Acta Neurologica Scand. 1975;51:137–57.
Weinshenker BG, Bass B, Rice GPA, et al. The natural history of multiple sclerosis: a geographically based study. 1. Clinical course and disability. Brain. 1989;112:133–46.
Schumacher GA, Beebe G, Kibler RF, Kurland LT, Kurtzke JF, McDowell F, Nagler B, Sibley W, Tourtellotte W, Willmon TL. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann New York Academy of Sciences, NY. 1965;123:552–68.
Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–31.
McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol. 2001;50:121–7.
Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Flippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O’Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Noseworthy JH, Luccinetti C, Rodriguez M, Weinschenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–52.
Leibowitz U, Halpern L, Alter M. Clinical studies of multiple sclerosis in Israel. 5. Progressive spinal syndromes and multiple sclerosis. Neurology. 1967;17:988–92.
Confavreux C, Vukusic S, Moreau T, Adeline P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343:1430–8.
Pittock SJ, Mayr WT, McClelland RL, Jorgensen NW, Weigand SD, Noseworthy JH, Weinshenker BG, Rodriguez M. Change in MS-related disability in a population-based cohort: a 10-year follow-up study. Neurology. 2004;62:51–9.
Berger J, Sheremata WA. Persistent neurological deficit in multiple sclerosis precipitated by hot bath test. JAMA. 1983;133:1224–6.
Berger JR, Sheremata WA, Melmed E. Paroxysmal dystonia as the initial manifestation of multiple sclerosis. Arch Neurol. 1984;41:747–50.
Ramagopalan S, Meier U, Goldacre R, Goldacre M. Co-associations of multiple sclerosis with schizophrenia and bipolar disorder: record linkage studies. Presented at: ACTRIMS-ECTRIMS MS, Boston; 2014.
Jacobs LD, Beck RW, Simon JH, Kinkel P, Brownscheidle CM, Murray TJ, Simonian NA, Slasor PJ, Sandrock AW, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000;343:898–904.
Goodin DS, Reder AT, Ebers GC, et al. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78(17):1315–22.
Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46:907–11.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørenson PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell R, Barkhof F, Bebo Jr B, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O’Connor PW, Petkau J, Pzzilli C, Rudick RA, Sormani MP, Stüve O, Waubant E, Polman CH. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Bornstein MB, Miller A, Slagle S, Weitzman M, Drexler E, Keilson M, Spada V, Weiss W, Appel S, Rolak L, et al. A placebo controlled, double-blind, randomized, two-center, pilot trial of Cop 1 in chronic progressive multiple sclerosis. Neurology. 1991;41:533–9.
Thompson AJ, Montalban X, Barkhof F, Brochet B, Flippi M, Miller DH, Polman CH, Stevenson VL, McDonald WI. Diagnostic criteria for primary progressive multiple sclerosis: a position paper. Ann Neurol. 2000;47(6):831–5.
Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14(2):208–23.
Wolinsky JS, Narayana PA, O’Connor P, Coyle PK, Ford C, Johnson K, Miller A, Pardo L, Kadosh S, Ladkani D. PROMiSe Trial Study Group. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-control trial. Ann Neurol. 2007;61(1):14–24.
Lassmann H. Multiple sclerosis: is there neurodegeneration independent from inflammation? J Neurol Sci. 2007;259(1–2):3–6.
Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, Mandrekar J, Bramow S, Metz I, Brück W, Lassmann H, Lucchinetti CF. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015;78:710–21.
Cottrell DA, Kremenchutzky M, Rice GPA, et al. The natural history of multiple sclerosis: a geographically based study. The clinical features and natural history of primary progressive multiple sclerosis. Brain. 1999;122:625–89.
Sheremata WA, Berger JR, Harrington Jr W, Ayyar R, Stafford JM, Defreitas E. Human lymphotropic (HTLV-I) associated myelopathy: a report of ten cases born in the United States. Arch Neurol. 1992;31:34–8.
Biswas HH, Engstrom JW, Kaidarova Z, Garratty G, Gibble JW, Newman BH, Smith JW, Ziman A, Fridey JL, Sacher RA, Murphy EL. Neurologic abnormalities in HTLV-1 and HTLV-II infected individuals without overt myelopathy. Neurology. 2009;73(10):781–9.
Lowis GW, Sheremata WA, Minagar A. Epidemiologic features of HTLV-II: serological and molecular evidence. Ann Epidemiol. 2002;12:46–66.
Fink JK. Hereditary spastic paraplegia: the pace quickens. Ann Neurol. 2002;51:669–72.
Sadovnick AD, Ebers GC. Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci. 1993;20:17–9.
Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T, et al. Rate of pregnancy-related relapse in multiple sclerosis. N Engl J Med. 1998;339:285–91.
Voskuhl RR, Wang H, Wu TC, Sicotte NL, Nakamura K, Kurth F, Itoh N, BArdens J, Bernard JT, Corboy JR, Cross AH, Dhib-Jalbut S, Ford CC, Frohman EM, Giesser B, Jacobs D, Kasper LH, Lynch S, Parry G, RAcke MK, REder AT, Rose J, Wingerchuk DM, MacKenzie-Graham AJ, Arnold DL, Tseng CH, Elashoff R. Estriol combined with glatiramer acetate for women with relapsing-remitting multiple sclerosis: a randomized, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(1):35–46.
Confavreux C. Infections and the risk of relapse in multiple sclerosis. (Editorial). Brain. 2002;125:933–4.
Warren S, Greenhill S, Warren KG. Emotional stress and the development of multiple sclerosis: case–control evidence of a relationship. J Chronic Dis. 1982;35:821–31.
Grant I, Brown GW, Harris T, McDonald WI, Patterson T, Trimble MR. Severely threatening events and marked life difficulties preceding onset or exacerbation of multiple sclerosis. J Neurol Neurosurg Psychiatry. 1989;52:8–13.
Warren S, Warren KG, Cockerill R. Emotional stress and coping in multiple sclerosis and exacerbations. J Psychosom Res. 1991;35:37–47.
Mohr DC, Goodkin DE, Bacchetti P, Boudewyn AC, Huang L, Marietta P, Cheuk W, Dee B. Psychological stress and he subsequent appearance of new brain MRI lesions in MS. Neurology. 2000;55:55–61.
Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, DAumer M, Ebers GC. The natural history of multiple sclerosis, a geographically based study 10: relapses and long-term disability. Brain. 2010;133:1914–29.
Lublin FD, Baier M, Gutter G. Effect of relapses on development of multiple sclerosis. Neurology. 2003;61:1528–32.
Cala LA, Mastaglia FL, Black JL. Computerized tomography of brain and optic nerve in multiple sclerosis: observation in 100 patients including serial studies in 16. J Neurol Sci. 1978;36:411–26.
Hershey LA, Gado MH, Trotter JL. Computerized tomography in the diagnostic evaluation of multiple sclerosis. Ann Neurol. 1979;5:32–9.
Barrett L, Drayer B, Shin C. High-resolution computerized tomography in the diagnostic evaluation of multiple sclerosis. Ann Neurol. 1985;17:33–8.
Bradley WG, Walauch Y, Yadley RA, Wycoff RR. Comparison of CT and MR in 400 patients with suspected disease of the brain and cervical spinal cord. Radiology. 1984;152:895–702.
Sheldon JJ, Siddharthan R, Tobias J, Sheremata WA, et al. Magnetic resonance imaging of multiple sclerosis: comparison with clinical, paraclinical, laboratory and CT examination. AJNR. 1985;6:683–90.
Jacobs L, Kinkel WR, Polachini I, Kinkel RP. Correlations of nuclear magnetic resonance imaging, computerized tomography, and clinical profiles in multiple sclerosis. Neurology. 1986;36:27–34.
Honig LS, Siddharthan R, Sheremata WA, Sheldon JJ, Sazant A. Multiple sclerosis: correlation of magnetic resonance imaging with cerebrospinal fluid findings. Neurol Neurosurg Psychiatry. 1988;51:27–280.
Fox R et al. Consortium of Multiple Sclerosis Centers annual meeting. Indianapolis; 2015.
Seewann A, Kooi EJ, Pouwels PJ, Wattjes MP, van der Valk P, Barkhof F, Polman CH, Geurts JJ. Postmortem verification of MS cortical lesion detection with 3D DIR. Neurology. 2012;78(5):302–8.
Honig LS, Sheremata WA. Magnetic resonance imaging of spinal cord lesions in multiple sclerosis. Neurol Neurosurg Psychiatry. 1989;52:459–66.
Brex PA, Ciccarelli O, O’Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;348:158–64.
Leist TP, Gobbini MI, Frank JA, McFarland HF. Enhancing magnetic resonance imaging lesions and cerebral atrophy in patients with relapsing multiple sclerosis. Arch Neurol. 2000;57:57–60.
van Walderveen MA, Kamphorst W, Scheltens P, et al. Histopathologic correlate of hypointense lesions on T1-wighted spin-echo magnetic resonance images in multiple sclerosis. Neurology. 1998;50:1282–8.
Bermel RA, Bakshi R. The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol. 2006;5:158–70.
Kappos L et al. Predictive value of NEDA for disease outcomes over 6 years in patients with RRMS. Presented at: 31st ECTRIMS Annual Congress; 7–10 Oct 2015; Barcelona; Abstract 570.
Cree BAC et al. Long-term effects of fingolimod on NEDA by year of treatment. Poster presented at: 31st ECTRIMS Annual Congress; 7–10 Oct 2015; Barcelona. Poster Session 1; P627.
Filippi M, Rocca MA. MR imaging of gray matter involvement in multiple sclerosis: implications for understanding disease pathophysiology and monitoring treatment efficacy. Am J Neuroradiol. 2010;31:1171–7.
Moll NM, Rietsch AM, Thomas S, et al. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. 2011;70(5):764–73.
Fox RJ, Sakaie K, Lee JC, et al. A validation study of multicenter diffusion tensor imaging: reliability of fractional anisotropy and diffusivity values. ANJR. 2012;33(4):695–700.
Pitt D, Boster A, Pei W, et al. Imaging cortical lesions in multiple sclerosis with ultra-high-field magnetic resonance imaging. Arch Neurol. 2010;67(7):812–8.
Burman J, Zetterberg H, Fransson M, Loskog AS, Raininko R, Fagius J. Assessing tissue damage in multiple sclerosis: a biomarker approach. Acta Neurol Scand. 2014;130:81–9.
Kuhle J, Plattner K, Bestwick JP, et al. A comparative study of CSF neurofilament light and heavy chain protein in MS. Mult Scler. 2013;19:1597–603.
Petzold A, Eikelenboom MJ, Gveric D, et al. Markers for different glial cell responses in multiple sclerosis: clinical and pathological correlations. Brain. 2002;125:1462–73.
De Stefano N, Narayanan S, Francis GS, et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;5:65–70.
Sobel RA. The pathology of multiple sclerosis. In: Multiple sclerosis. Antel J, editor. Neurologic clinics. Philadelphia: Sanders; 1995; 13(1):1–22.
Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015;14(3):263–73.
Wingerchuk DM. Smoking: effects on multiple sclerosis susceptibility and disease progression. Ther Adv Neurol Disord. 2012;5:13–22.
Hedström AK, Hiller J, Olsson T, Alfredsson L. Smoking and multiple sclerosis susceptibility. Eur J Epidemiol. 2013;28(11):867–74.
Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288–99.
Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol. 2006;49(3):499–503.
Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic T17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–51.
Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic T17 cells. Nature. 2013;496:518–22.
Ascherio A, Munger KL, White R, et al. Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA Neurol. 2014;71(3):306–14.
Sotirchos ES, Bhargava P, Eckstein C, et al. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology. 2016;86(4):382–90.
Hedström AK, Olsson T, Alfredsson L. High body mass index before age 20 is associated with increased risk for multiple sclerosis in both men and women. Mult Scler J. 2012;18(9):1334–6.
Hedström AK, Bomfirm IL, Barcellos L, Gianfrancesco M, Schaefer C, Kockum I, Olsson T, Alfredsson L. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology. 2014;82(10):867–72.
Simpson Jr S, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:1132–41.
Strachan DP. Family size, infection and atopy: the first decade of the “hygiene hypothesis”. Thorax. 2000;55(Suppl 1):S2–10.
Correale J. Helminth/parasite treatment of multiple sclerosis. Curr Treat Options Neurol. 2014;16:296.
Correale J, Farez MF. The impact of parasite infections on the course of multiple sclerosis. J Neuroimmunol. 2011;233(1–2):6–11.
Fleming JO. Helminth therapy and multiple sclerosis. Int J Parasitol. 2013;43(3–4):259–74.
Mielcarz DW, Kasper LH. The gut microbiome in multiple sclerosis. Curr Treat Options Neurol. 2015;17:18.
Oppenheimer DR. Demyelinating diseases. In: Blackwood W, Corsellis JAN, editors. Greenfield’s neuropathology. 3rd ed. London: Edward Arnold; 1976. p. 470–99.
Lumsden CE. The neuropathology of multiple sclerosis. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology. New York: Elsevier; 1969. p. 217–309.
Adams RD, Kubick CS. The morbid anatomy of the demyelinative disease. Am J Med. 1952;12:510–46.
Zimmerman HM, Netsky HG. The pathology of multiple sclerosis. Res Publ Res Nerv Ment Dis. 1950;28:271–312.
Lampert PW. Fine structure of the demyelinating process. In: Hallpike JF, Adams CWM, Tourtelotte WW, editors. Multiple sclerosis: pathology, diagnosis and management. Baltimore: Williams and Wilkins; 1983. p. 29–46.
Trapp BD, Peterson J, Ransahoff RM, Rudick R, Moerk S, Boe L. Axonal transaction in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85.
Lassmann H, Vass K. Are current immunological concepts of multiple sclerosis reflected by the Immunopathology of its lesions? Springer Semin Immunopathol. 1995;17:77–87.
Lassman H, Raine CS, Antel J, Prineas JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol. 1998;86:213–7.
Lucchinetti C, Brueck W, Paris J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–17.
Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol. 1995;37:424–35.
Poser C. The pathogenesis of multiple sclerosis: a commentary. Clin Neurol Neurosurg. 2000;102:191–204.
Khan N, Smith MT. Multiple sclerosis-induced neuropathic pain: pharmacological management and pathophysiological insights from rodent EAE models. Inflammopharmacology. 2014;22(1):1–22.
Pender MP, Sears TA. Involvement of the dorsal root ganglion in acute experimental allergic encephalomyelitis in the Lewis rat: a histological and electrophysiological study. J Neurol Sci. 1986;72(2–3):231–42.
Sadovnick AD, Armstrong H, Rice GF, et al. A population based study of multiple sclerosis in twins: an update. Ann Neurol. 1993;33:281–5.
Sadovnick AD, Baird PA, Ward RH. Multiple sclerosis: update risks for relatives. Am J Med Genet. 1988;29:533–41.
Didonna A, Oksenberg JR. Genetic determinants of risk and progression in multiple sclerosis. Clin Chim Acta. 2015;449:16–22.
De Jager P et al. ACTRIMS-ECTRIMS. Boston, MA; 2014.
Sawcer S, Ban M, Maranian M, et al. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. 2005;77:454–67.
Kaushansky N, Altmann DM, David CS, Lassmann H, Ben-Nun A. DQB1*0602 rather than DRB1*1501 confers susceptibility to multiple sclerosis-like disease induced by proteolipid protein (PLP). J Neuroinflammation. 2012;9:29.
International Multiple Sclerosis Genetics Consortium. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Brain. 2010;133:2603–11.
Gregory SG, Schmidt S, Seth P, et al. Interleukin 7 receptor alpha chain shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–11.
Gregory AP, Dendrou CA, Attfield KE, et al. TNF receptor 1 genetic risks mirrors outcome of ant-TNF therapy in multiple sclerosis. Nature. 2012;488:508–11.
Couturier N, Bucciarelli F, Nurtdinov RN, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. 2011;134:693–703.
Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J Immunol. 2014;193:4439–47.
Jersild C, Fog T, Hansen GS, Thomsen M, Svejgaard A, Dupont B. Histocompatibility determinants in multiple sclerosis with special reference to clinical course. Lancet. 1973;2:1221–5.
Healy BC, Liguori M, Tran D, et al. HLA B*44: protective effects in MS susceptibility and MRI outcome measures. Neurology. 2010;75:634–40.
Qju W, Raven S, James I, et al. Spinal cord involvement in multiple sclerosis: a correlative MRI and high-resolution HLA-DRB1 genotyping study. J Neurol Sci. 2011;300:114–9.
Okuda DT, Srinivasan R, Oksenberg JR, et al. Genotype-phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain. 2009;132:250–9.
Masterman T, Ligers A, Olsson T, Andersson M, Olerup O, Hillert J. HLA-DR15 is associated with lower age at onset in multiple sclerosis. Ann Neurol. 2000;48:211–9.
Smestad C, Brynedal B, Jonasdottir G, et al. The impact of HLA-A and –DRB1 on age at onset, disease course and severity in Scandinavian multiple sclerosis patients. Eur J Neurol. 2007;14:835–40.
Esposito F, Sorosina M, Ottoboni L, et al. A pharmacogenetics study implicates SLC9A9 in multiple sclerosis disease activity. Ann Neurol. 2015;78:115–27.
Dhib-Jalbut S, Valenzuela RM, Ito K, Kaufman M, Picone AM, Buyske S. HLA DR and DQ alleles and haplotypes associated with clinical response to glatiramer acetate in multiple sclerosis. Mult Scler Relat Disord. 2013;2(4):340–8.
Levin LI, Munger KL, Ruberstone MV, et al. Multiple sclerosis and Epstein-Barr virus. JAMA. 2003;289:1533–6.
DeLorenzo GN, Munger KL, Lennette ET, Orentreich N, Vogelman JH, Ascherio A. Epstein-Barr virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch Neurol. 2006;63(6):839–44.
Woods DD, Bilbao JM, O’Connor P, Moscarello MA. A highly deaminized form of myelin basic protein in Marburg’s disease. Ann Neurol. 1996;40:18–24.
Schwartz S, Mohr A, Knauth M, Wildemann B, Storch-Hagenlocher B. Acute disseminated encephalomyelitis. A follow-up study of 40 adult patients. Neurology. 2001;56:1312–8.
Hartung HP, Grossman RI. ADEM. Distinct disease or part of the MS spectrum? Neurology. 2001;56:1257–60.
Murthy JM, Yangala R, Meena AK, Jaganmohan-Reddy J. Acute disseminated encephalomyelitis: clinical and MRI study from South India. J Neurol Sci. 1999;165:133–6.
Patterson PY. Transfer of allergic encephalomyelitis in rats by means of lymph node cells. J Exp Med. 1960;111:119–36.
Bornstein MB, Appel SH. Application of tissue culture to the study of experimental allergic encephalomyelitis. 1. Patterns of demyelination. J Neuropathol Exp Neurol. 1961;20:141–57.
Bornstein MB, Raine CS. Multiple sclerosis and experimental allergic encephalomyelitis: specific demyelination of CNS in culture. Neuropathol Appl Neurobiol. 1977;3:359–67.
Ben-Nun A, Cohen IR. Genetic control of experimental autoimmune encephalomyelitis at the level of cytotoxic lymphocytes in guinea pigs. Eur J Immunol. 1982;12:709–13.
Owens T, Sriram S. The immunology of multiple sclerosis and its animal model experimental allergic encephalomyelitis. Neurol Clin. 1995;13(1):57–73.
Massacesi L, Genain CP, Lee-Parritz D, Letvin NL, Confield D, Hauser SL, et al. Active and passively induced experimental autoimmune encephalomyelitis in common marmosets: as new model for multiple sclerosis. Ann Neurol. 1995;37:519–30.
Uccelli A, Giunti D, Capello E, Roccatagliata L, Mancardi GL. EAE in the common marmoset Callithrix jacchus. Int MS J. 2003;10:6–12.
Bronstein JM, Lallone RL, Seitz RS, Ellison GW, Myers LW. A humoral response to oligodendrocyte-specific protein in MS. A potential molecular mimic. Neurology. 1999;53:154–61.
Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, Dilitz E, Deisenhammer F, Reindl M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2004;349:139–45.
Yu T, Ellison GW, Mendoza F, Bronstein JM. T-cell responses to oligodendrocyte-specific protein in multiple sclerosis. J Neurosci Res. 2001;66:506–9.
Adorini L, Singaglia F. Pathogenesis and immunotherapy of autoimmune disease. Immunol Today. 1997;18:209–11.
Özenci V, Kouwenhoven M, Huang YM, Xiao BG, Kivisäkk P, Fredrikson S, Link H. Multiple sclerosis: levels of interleukin-10-secreting blood mononuclear cells are low in untreated patients but augmented during interferon-β-1b treatment. Scand J Immunol. 1999;49:554–61.
Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, Hafler DA. Functional inflammatory profiles distinguished myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. 2015;7(287):1–10.
Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal MR, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16(4):406–12.
Wekerle H. Immune pathogenesis of multiple sclerosis. Brain autoimmune reactivity and its control by neuronal function. Mult Scler. 1998;4:136–7.
Yang Y, Tomura M, Ono S, Hamaoka T, Fujiwara H. Requirement for IFN-γ in IL-12 production induced by collaboration between Vα14+NKT cells and antigen-presenting cells. Int Immunol. 2000;12:1669–75.
Liu C-C, Young LHY, Young JD-E. Lymphocyte-mediated cytolysis and disease. N Engl J Med. 2004;335:1651–9.
Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2003;9:540–9.
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–6.
Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–101.
Frenette PS, Wagner DD. Adhesion molecules--Part 1. N Engl J Med. 1996;334:1526–9.
Frenette PS, Wagner DD. Adhesion molecules--Part II: blood vessels and blood cells. N Engl J Med. 1996;335:43–5.
von Andrian UH, MacKay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–34.
von Adrian UH, Engelhardt B. α4 integrins as therapeutic targets in autoimmune disease. N Engl J Med. 2004;348:68–72.
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–98.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature. 2006;441:235–8.
Matusevicius D, Kivisäkk P, He B, Kostulas N, Özenci V, Fredikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mul Scler J. 1999;5(2):101–4.
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172(1):146–55.
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Alalrd J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8.
Kostic M, Dzopalic T, Zivanovic S, Zivkovic N, Cvetanovic A, Stojanovic I, Vojinovic S, Marjanovic G, Savic V, Colic M. IL-17 and glutamate excitotoxicity in the pathogenesis of multiple sclerosis. Scand J Immunol. 2014;79(3):181–6.
Elain G, Jeanneau K, Rutkowska A, Mir AK, Dev KK. The selective anti-IL17A monoclonal antibody secukinumab (AIN457) attenuates IL17A-induced levels of IL6 in human astrocytes. Glia. 2014;62(5):725–35.
Vestweber D, Blanks JE. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79:181–213.
Takada Y, Elices MJ, Crouse C, Hemler ME. The primary structure of the alpha 4 subunit of VLA-4: homology to other integrins and a possible cell-cell adhesion function. EMBO J. 1989;8:1361–8.
Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48(4):549–54.
Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25.
Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, Frohman EM, Phillips JT, Arendt G, Hemmer B, Monson NL, Racke MK. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2006;59(5):745–7.
Piccio L, Rossi B, Scarpini E, Laudanna C, et al. Molecular mechanisms involved in lymphocyte recruitment in inflamed brain microvessels: critical roles for P-selectin glycoprotein Ligand-1 and heterotrimeric Gi-Linked receptors. J Immunol. 2002;168:1940–849.
Minagar A, Jy W, Jimenez JJ, Mauro LM, Horüman L, Sheremata WA, Ahn YS. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology. 2001;56:1319–24.
Qin S, Rottman JB, Myers P, et al. The chemokine receptors CSCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reaction. J Clin Invest. 1998;101:746–54.
Byun E, Caillier SJ, Montalban X, Villoslada P, Fernandez O, Brassat D, Comabella M, Wang J, Barcellos LF, Baranzini SE, Oksenberg JR. Genome-wide pharmacogenomics analysis of the response to interferon beta therapy in multiple sclerosis. Arch Neurol. 2008;65(3):337–44.
Study of Tecelan (Imilecleucel-T) in secondary progressive multiple sclerosis (Abili-T). ClinicalTrials.gov, Jul 7, 2015. Accessed Feb 27, 2016 from https://clinicaltrials.gov/ct2/show/NCT01684761.
Murray TJ. Amantadine therapy for fatigue in MS. Can J Neurol Sci. 1994;21:9–14.
Krupp LB, Coyle PK, Doscher C, et al. Fatigue therapy in MS: results of a double-blind, randomized, parallel trial of amantadine, pemoline, and placebo. Neurology. 1995;45:1956–61.
Rammohan KW, Rosenberg JH, Lynn DJ, et al. Efficacy and safety of modafinil (Provigil) for the treatment of fatigue in multiple sclerosis: a two centre phase 2 study. J Neurol Neurosurg Psychiatry. 2002;72:150–79.
Traugott U. Detailed analysis of immunomodulatory properties of fluoxetine (Prozac) in chronic experimental allergic encephalomyelitis in SJL/J mice. Neurology. 1998;50:1998. (abstract)
Goodman AD, Brown TR, Edwards KR, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68(4):494–502.
Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373(9665):732–8.
Korenke AR, Rivey MP, Allington DR. Sustained-release fampridine for symptomatic treatment of multiple sclerosis. Ann Pharmacother. 2008;42(10):1458–65.
Penn RD, Savoy SM, Corcos D, et al. Intrathecal baclofen for severe spinal spasticity. N Engl J Med. 1989;320:1517–21.
Nance P, Sheremata WA, Lynch SG, et al. Relationship of the antispasticity effect of tizanidine to plasma concentration in patients with multiple sclerosis. Arch Neurol. 1997;54:731–06.
Rossanese M, Novara G, Challacombe B, Iannetti A, Dasgupta P, Ficarra V. Critical analysis of phase II and III randomized control trials evaluating efficacy and tolerability of a β3-adrenoreceptor agonist for overactive bladder. BJU Int. 2015;115(1):32–40.
Mehnert U, Birzele J, Reueter K, Schurch B. The effect of botulinum toxin type a on overactive bladder symptoms in patients with multiple sclerosis: a pilot study. J Urol. 2010;184(3):1011–116.
Goessaert AS, Everaert KC. Onabotulinum toxin A for the treatment of neurogenic detrusor overactivity due to spinal cord injury or multiple sclerosis. Expert Rev Neurother. 2012;12(7):763–75.
Browne C, Salmon N, Kehoe M. Bladder dysfunction and quality of life for people with multiple sclerosis. Disabil Rehabil. 2015;37:2350–8.
Solaro C, Uccelli MM, Guglieri P, Uccelli A, Mancardi GL. Gabapentin is effective in treating nocturnal painful spasms in multiple sclerosis. Mult Scler. 2000;6(3):192–3.
Rose AS, Kuzma JW, Kurtzke JF, et al. Cooperative study in the evaluation of therapy in multiple sclerosis: ACTH vs. placebo. Final Report. Neurology. 1970;20 Part 2:1–19.
Beck BW, Cleary PA, Anderson MM, et al. A randomized controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581–8.
Diem R, Hobom M, Maier K, Weissert R, Storch MK, Meyer R, Bähr M. Methylprednisolone increases neuronal apoptosis during autoimmune CNS inflammation by inhibition of an endogenous neuroprotective pathway. J Neurosci. 2003;23(18):6993–7000.
Diem R, Sättler MB, Merkler D, et al. Combined therapy with methylprednisolone and erythropoietin in a model of multiple sclerosis. Brain. 2005;129(Pt 2):375–85.
Botticelli LJ, Wurtman RJ. Septo-hippocampal cholinergic neurons are regulated transynaptically by endorphin and corticotrophin neuropeptides. J Neurosci. 1982;2:1316–21.
Spruijt BM, Van Rijzingen I, Masswinkel H. The ACTH 4-9 analog Org2766 modulates the behavioral changes induced by NMDA and the NMDA receptor antagonist AP5. J Neurosci. 1994;14:3225–30.
Hol EM, Mandys V, Sodnar P, Gispen WH, Bar PR. Protection by ACTH4-9 analogue against the toxic effects of cisplatin and taxol on sensory neurons and Glial cells in vitro. J Neurosci Res. 1994;39:178–85.
O’Connor PW, Goodman A, Willmer-Hulme AJ, et al. Randomized. Multicenter trial of intravenous natalizumab in acute MS relapses: clinical and MRI effects. Neurology. 1994;62:2038–43.
The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. 1. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–61.
Paty DW, Li KDB, the UBC MS/MRI Group and the IFN Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Neurology. 1993;42:662–7.
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285–94.
Rudick RA, Goodkin DE, Jacobs LD, et al. Impact of interferon beta-1a on Neurologic disability in relapsing multiple sclerosis. Neurology. 1997;49:358–63.
PRISMS (Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in multiple sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 2002;352:1498–504.
Panitch H, Goodin DS, Francis G, Chang P, Coyle PK, O’Connor P, Monaghan E, Li D, Weinshenker B. Randomized, comparative study of interferon ß-1a treatment regimens in MS: the EVIDENCE Trial. Neurology. 2002;59:1496–506.
The PRISMS Study Group and the University of British Columbia MS/MRI Analysis Group. PRISMS-4: longer term efficacy of interferon-beta-1a in relapsing MS. Neurology. 2001;56:1628–36.
Cohen BA, Rivera VM. PRISMS: the story of a pivotal clinical trial series in multiple sclerosis. Curr Med Res Opin. 2010;26(4):827–38.
Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomized, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657–65.
Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45:1268–76.
Johnson KP, Brooks BR, Ford CC, et al. Sustained clinical benefits of Glatiramer acetate (Copaxone) in multiple sclerosis patients observed for 6 years. Mult Scler. 2000;6:255–66.
Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R, and for the GALA study group. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Ann Neurol. 2013;73(6):705–13.
Stone LA, Frank JA, Albert PS, et al. Characterization of MRI response to treatment with interferon beta-1b: contrast-enhancing MRI lesion frequency as a primary outcome measure. Neurology. 1997;49:862–9.
Mancardi GL, Sardanelli F, Parodi RC, et al. Effect of copolymer-1 on serial gadolinium-enhanced RMI in relapsing remitting multiple sclerosis. Neurology. 1998;50:1127–33.
Dalton CM, Miszkiel KA, Barker GJ, et al. The effect of natalizumab on conversion of T1 gadolinium enhancing lesions to T1 hypodense lesions. Neurology. 2004;60(Supp1):S484.
Rudick RA, Fisher E, Lee J-C, Simon J, Jacobs L, and the Multiple Sclerosis Collaborative Research Group. Us of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS. Neurology. 1999;53:1698–704.
Ge Y, Grossman RI, Udupa JK, et al. Glatiramer acetate (Copaxone) treatment in relapsing-remitting MS. Neurology. 2000;54:813–7.
Frank JA, Richert N, Bash C, et al. Interferon-β-1b slows progression of atrophy in RRMS. Neurology. 2004;62:719–25.
Ransohoff RM. Natalizumab for multiple sclerosis. N Engl J Med. 2007;356(25):2622–9.
Polman CH, O’Connor PW, Hardova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Miller DH, Soon D, Fernando KT, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007;68(17):1390–401.
Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–23.
McGuigan C, Craner M, Guadagno J, et al. Stratification and monitoring of natalizumab-associated progressive multifocal leukoencephalopathy risk: recommendations from an expert group. J Neurol Neurosurg Psychiatry. 2016;87(2):117–25.
Schwab N, Schneider-Hohendorf T, Pignolet B, et al. PML risk stratification using anti-JCV antibody index and L-selectin. Mult Scler. 2016;22:1048–60. doi:10.1177/135245851607651.
Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69(14):1391–403.
Kappos L, Radue EW, Comi G, et al. Switching from natalizumab to fingolimod: a randomized, placebo-controlled study in RRMS. Neurology. 2015;85(1):29–39.
Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. 2012;366(4):339–47.
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–303.
Miller AE, O’Connor P, Wolinsky JS, et al. Pre-specified subgroup analyses of a placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis. Mult Scler. 2012;18(11):1625–32.
O’Connor P, Wolinsky J, Confavreux C, Comi G, Kappos L, Olsson T, et al. Extension of a phase III trial (TEMSO) of oral teriflunomide in multiple sclerosis with relapses: clinical and MRI data 5 years after initial randomisation. Mult Scler. 2011;17(Supp 17):S414. P924
Miller A, Kappos L, Comi G, et al. Teriflunomide efficacy and safety in patients with relapsing multiple sclerosis: results from TOWER, a second, pivotal, phase 3 placebo-controlled study (S01.004). Neurology. 2013;80(meeting abstracts 1):S01.004.
Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–56.
Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomized, controlled phase 3 trial. Mult Scler. 2014;20(6):705–16.
Genzyme Corporation. Aubagio prescribing information. Cambridge, MA; 2012.
Jung Henson L, Stüve O, Kieseier B, Benamor M, Benzerdjeb H. Pregnancy outcomes from the teriflunomide clinical development program: retrospective analysis of the teriflunomide clinical trial database. Neurology. 2013;80:1001–11.
Gold R, Linker RA, Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis with antioxidative mechanism of action. Clin Immunol. 2012;142(1):44–8.
Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098Y1107.
Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087Y1097.
Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BAC, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e76.
Dionne CA, Ganguly R, Camac A, Chaves C. Do oral disease modifying agents improve adherence to MS treatment? A comparison or oral and injectable drugs. CMSC 2015 Indianapolis; Abstract DX19.
Munsell M, Locklear JC, Phillips AL, Frean M, Menzin J. An assessment of adherence among MS patients newly initiating treatment with a self-injectable versus oral disease-modifying drug. CMSC 2015 Indianapolis; Abstract DX43.
Ko JJ, Nazareth TA, Friedman H, Navaratnam P, Herriott DA, Sasane R. Treatment discontinuation after initiation of oral disease-modifying therapies in patients with MS. CMSC 2015 Indianapolis; Abstract DX44.
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomized controlled phase 3 trial. Lancet. 2012;380(9856):1819–28.
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomized controlled phase 3 trial. Lancet. 2012;380(9856):1829–39.
Lemtrada® package insert: http://products.sanofi.us/lemtrada/lemtrada.pdf.
Hartung HP, Gonsette R, Koenig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo controlled, double-blind, randomized, multicentre trial. Lancet. 2002;360:2018–25.
Ghalie RG, Edan G, Laurent M, et al. Cardiac adverse effects associated with mitoxantrone (Novantrone) therapy in patients with MS. Neurology. 2002;59:909–13.
European Study Group on Interferon β-1b in Secondary Progressive MS. Placebo-controlled multicentre randomised trial of interferon β-1b in treatment of secondary progressive multiple sclerosis. Lancet. 1998;352:1491–7.
Sedel F, Papeix C, Bellanger A, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4:159–69.
Tourbah A, Frenay CL, Edan G, et al. Effect of MD10003 [high doses of biotin] in progressive multiple sclerosis: results of a pivotal phase III randomized double blind placebo controlled study. Neurology. 2015;84(14):Supplement PL2.002.
Hauser SL et al. B-cell depletion in rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–71.
Hauser S. Phase III results in relapsing MS (OPERA I and OPERA II studies). ECTRIMS 2015; Barcelona; Abstract #190.
Montalban X. Phase II results of the ORATORIO study. ECTRIMS 2015; Barcelona; Abstract #228.
Kappos L et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomized, placebo-controlled, multicenter trial. Lancet. 2011;378(9805):1779–87.
Hauser SL, Bar-Or A, Comi G et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2016; doi: 10.1056/NEJMoa1601277.
Montalban X, Hauser SL, Kappos L et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2016; doi: 10.1056/NEJMoa1606468.
Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A, Kaufman M, Rose J, Greenberg S, Sweetser M, Riester K, O’Neill G, Elkins J. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2015;373(15):1418–28.
An immunogenicity and pharmacokinetics study of BIIB019 (daclizumab high yield process) prefilled syringe in relapsing remitting multiple sclerosis (OBSERVE). www.clinicaltrials.gov, last updated Dec 23, 2015. Accessed 27 Feb 2016.
Cadavid D, Hupperts R, Dulović et al. Correlation of brain volume and physical measures with cognitive function using baseline data from the anti-LINGO-1 SYNERGY trial in multiple sclerosis. ECTRIMS 2015 Barcelona; Abstract P629.
Tran JQ, Rana J, Barkhof F, et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e18.
Wang T, Xiong JQ, Ren XB, Sun W. The role of Nogo-A in neuroregeneration: a review. Brain Res Bull. 2012;87:499–503.
Hvardova E. Positive proof of concept of AIN457, an antibody against interleukin-17A, in relapsing-remitting multiple sclerosis, in ECTRIMS. Lyons; 2012.
Miller DH et al. Firategrast for relapsing remitting multiple sclerosis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:131–9.
Stuve O, Hartung HP, Freedman M, Li D, Hemmer B, Kappos L, Rieckmann P, Montalban X, Ziemssen T, Selmaj K. Phase 2 BOLD extension study efficacy results for siponimod (BAF312) in patients with relapsing–remitting multiple sclerosis. Mul Scler Relat Disord. 2014;3(6):754–5.
Komiya T et al. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin Exp Immunol. 2013;171:54–62.
Aisen ML. Justifying neurorehabilitation. (Editorial). Neurology. 1999;52:8.
Thompson A. Symptomatic management and rehabilitation in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2001;71(Suppl 11):112–1127.
Heine M, van de Port I, Rietberg MB, van Wegen EE, Kwakkel G. Exercise therapy for fatigue in multiple sclerosis. Cochrane Database Syst Rev. 2015;9:CD009956.
Latimer-Cheung AE, Pilutti LA, Hicks AL, et al. The effects of exercise training on fitness, mobility, fatigue, and health related quality of life among adults with multiple sclerosis: a systematic review to inform guideline development. Arch Phys Med Rehabil. 2013;94:1800–28.
Motl RW, Pilutti LA. The benefits of exercise training in multiple sclerosis. Nat Rev Neurol. 2012;8:487–97.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Lizarraga, A.A., Sheremata, W.A. (2017). Multiple Sclerosis: Clinical Features, Immunopathogenesis, and Treatment. In: Minagar, A., Alexander, J. (eds) Inflammatory Disorders of the Nervous System. Current Clinical Neurology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-51220-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-51220-4_2
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-51218-1
Online ISBN: 978-3-319-51220-4
eBook Packages: MedicineMedicine (R0)