
Abstract
Monocyclic aromatic compounds (MAC) comprise the second most abundant class of natural compounds, many of which are hazardous for the environment and human health. MAC can readily be degraded by many aerobic microorganisms by the extensive using of oxygenases for aromatic ring hydroxylation and cleavage. However, under anoxic conditions, this strategy is not an option and MAC degrading anaerobic prokaryotes employ a totally different enzyme inventory for attacking the resonance-stabilized aromatic ring system or the C–H bond of alky chains from aromatic hydrocarbons. The anaerobic degradation of MAC has become a treasure trove for the discovery of unprecedented enzymatic principles; many involve metalloenzymes catalyzing radical-based reactions. Characteristic enzymatic reactions involved in anaerobic MAC degradation comprise: (i) the addition of alkylated aromatics to fumarate by glycyl-radical enzymes, (ii) the water-dependent hydroxylation or transhydroxylation of MAC by Mo- or flavin-dependent enzymes, (iii) the carboxylation/decarboxylation of aromatic rings by UbiD-/UbiX-like enzyme systems, and (iv) the dearomatization of aromatics rings by ATP-dependent FeS-enzymes or ATP-independent W-enzymes. The multitude of MAC is converted via peripheral channeling pathways to only a few central intermediates that serve as substrates for dearomatizing ring reductases. Depending on the nature of these central intermediates, we divide the anaerobic MAC degradation pathways into five subgroups and highlight the individual characteristic enzymatic steps involved.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

1 Introduction
Only surpassed by the glycosyl moiety, the aromatic benzene nucleus is the second most abundant biogenic structural unit in nature. Aromatic rings mainly derive from plant secondary metabolism (e.g., lignin) where they are synthesized via the shikimate pathway, but they are also abundant in all other living beings in the form of aromatic amino acids or quinones. Aromatic hydrocarbons represent a significant portion of fossil oil reservoirs and serve as source for solvents, dyes, resins, plasticizers, polymers, flame retardants, pesticides, insecticides, and many other synthetic chemicals. Many aromatic hydrocarbons such as BTEX (benzene, toluene, ethylbenzene, xylenes) or polycyclic aromatic hydrocarbons (PAH) are toxic and/or cancerogenic and affect human health and the environment. Due to the chemical inertness of the resonance stabilized ring system, and, in case of aromatic hydrocarbons, low solubility, many aromatic compounds are considered recalcitrant.
The complete degradation of aromatic compounds to CO2 is predominantly performed by microorganisms and has been studied in aerobic bacteria since more than 50 years (Stanier and Ornston 1973; Harayama et al. 1992; Harwood and Parales 1996; Díaz et al. 2013). Aromatic compound degrading aerobes attack aromatic ring systems by the aid of ring-hydroxylating mono- or dioxygenases. A few dihydroxylated central aromatic intermediates (e.g., catechol, protocatechuate, or gentisate) are then dearomatized by ring-cleaving oxygenases. Considering the massive use of the dioxygen molecule in the degradation pathways of aromatic compounds in aerobic microorganisms, their degradation at anoxic sites such as marine/freshwater sediments, oils reservoirs, or aquifers remained obscure until the early 1990s.
Meanwhile, the number of anaerobic microorganisms that are known to use aromatic compounds as growth substrates is steadily growing. They comprise facultative anaerobes, such as denitrifiers or bacteria with anoxygenic photosynthesis (α-, β-Proteobacteria), and obligate anaerobes including sulfate- and metal oxide-reducing or fermenting bacteria in methanogenic co-culture (δ-Proteobacteria and Firmicutes) (Boll et al. 2014). To date, the only known archaeon and hyperthermophilic organism capable of completely degrading aromatic growth substrates to CO2 is the Fe(III)-respiring, strictly anaerobic Ferroglobus placidus (Tor and Lovley 2001).
Today, most of the anaerobic degradation pathways of monocyclic aromatic compounds (MAC) have been elucidated. Characteristic key reactions such as C–H bond activation of aromatic hydrocarbons or the dearomatization of the benzene ring system are typically catalyzed by metalloenzymes, many of which catalyze unprecedented radical-based reactions. In analogy to the aerobic degradation pathways, anaerobic microorganisms channel the multitude of monocyclic aromatic compounds into only a few central intermediates that still contain the aromatic moiety such as benzoyl-CoA and analogues of it, or di-/trihydroxybenzenes with hydroxyl groups in meta-positions (Fuchs et al. 2011). They serve as substrates for dearomatizing ring reductases that, depending on the stability of the aromatic ring, use different electron donors and may couple ring reduction to an exergonic reaction. Dearomatizing ring reductases acting on aryl-CoA-thioesters are also employed in the anaerobic degradation of PAH. However, the pathways of anaerobic PAH degradation differ fundamentally from those of anaerobic MAC degradation. For this reason, anaerobic PAH degradation is the topic of a separate chapter of this volume (Chap. 5, “Catabolic Pathways and Enzymes Involved in the Anaerobic Degradation of Polycyclic Aromatic Hydrocarbons”).
In this contribution, the current knowledge of experimentally verified degradation pathways of monocyclic aromatic compounds with a focus on the function of characteristic key enzymes is summarized and discussed. There are many excellent recent reviews about anaerobic MAC degradation with other focuses than this compendium (Carmona et al. 2009; Fuchs et al. 2011; Philipp and Schink 2012; Heider and Schühle 2013; Boll et al. 2014; Rabus et al. 2016b). Here, we divide anaerobic MAC degradation pathways into five groups with respect to the nature of the substrates for central dearomatizing reductases: (1) benzoyl-CoA, (2–4) ortho-, meta-, or para-substituted benzoyl-CoA derivatives, and (5) di-/trihydroxybenzenes with meta-positioned hydroxy substituents. In all cases dearomatization is accomplished by transferring two electrons to the aromatic ring system yielding cyclic diene products; the latter may undergo tautomerization and/or other spontaneous reactions.
2 Degradation via Dearomatization of Benzoyl-CoA
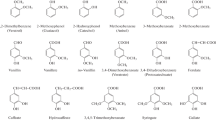
In anaerobic microorganisms, most MAC are channeled into benzoyl-CoA, including the abundant BTEX, phenol, benzoates, phthalates, phenylpropane-derived compounds, phenylacetic acid, phenylalanine, tyrosine, and many more (Fig. 1).

Degradation of selected MAC via benzoyl-CoA. The central intermediate is dearomatized by different classes of benzoyl-CoA reductases yielding the same cyclohexa-1,5-diene-1-carboxyl-CoA (1,5-dienoyl-CoA) product
2.1 General Benzoyl-CoA Degradation Pathway
The so-called benzoyl-CoA degradation pathway is initiated by the key reaction of MAC degradation, the reductive dearomatization of the benzene moiety of benzoyl-CoA. There are two nonrelated classes of dearomatizing benzoyl-CoA reductases (BCRs, class I and II), both yielding the same cyclohexa-1,5-diene-1-carboxyl-CoA (1,5-dienoyl-CoA) product (Fig. 2) (Boll and Fuchs 1995; Kung et al. 2009). An exception was found during benzoate degradation in Rhodopseudomonas palustris, where the 1,5-dienoyl-CoA formed by a class I BCR is further reduced to a cyclic monoenoyl-CoA – probably by the same BCR (Koch et al. 1993; Egland et al. 1997). The redox potential of the benzoyl-CoA/1,5-dienoyl-CoA redox couple is E°′ = −622 mV, more negative than that of any conventional cellular electron donor (Kung et al. 2010). Consequently, BCRs have to couple benzene ring reduction to an exergonic reaction, which is accomplished differently in the two BCR classes. Consequently, the architectures and cofactor contents of both BCR classes differ fundamentally.

Enzymatic reactions involved in the benzoyl-CoA degradation pathway. Benzoyl-CoA is reduced to 1,5-dienoyl-CoA either by ATP-dependent class I (upper reaction) or ATP-independent class II BCR (lower reaction); the latter may drive endergonic electron transfer by electron bifurcation, using an electron donor of insufficient redox potential to reduce the aromatic ring directly (DH2) by coupling its oxidation to the exergonic reduction of a second acceptor (A)
The further catabolism of 1,5-dienoyl-CoA involves a series of β-oxidation-like reactions (Fig. 2). The 1,5-dienoyl-CoA is first hydrated by a specific enoyl-CoA hydratase catalyzing a rather unusual 3,6-addition instead of the typical 2,3-addition of water with respect to the C1-carbonyl of the thioester (Laempe et al. 1998; Peters et al. 2007). In the next step, the hydroxyl group is dehydrogenated to a ketone, yielding 6-oxocyclohex-1-ene-1-carboxyl-CoA that serves as substrate for a ring-cleaving, 3-hydroxypimeloyl-CoA forming hydrolase (Laempe et al. 1999; Kuntze et al. 2008). The encoding gene of this enzyme, referred to as bamA (Geobacter metallireducens notification), is highly conserved and serves as a specific functional marker for monitoring MAC degradation by PCR-, microarray-, or sequencing-based tools (Kuntze et al. 2011a; Porter and Young 2013). The subsequent degradation of the aliphatic 3-hydroxypimeloyl-CoA proceeds again via β-oxidation reactions and yields acetyl-CoA and glutaryl-CoA. After decarboxylation of the latter by a soluble glutaryl-CoA dehydrogenase or by a membrane-bound, sodium-pumping glutaconyl-CoA decarboxylase, crotonyl-CoA, and finally two acetyl-CoA are formed. The benzoyl-CoA degradation pathway can be summarized by the following equation (without considering ATP hydrolysis by class I BCR):
2.2 Benzoyl-CoA Reduction
2.2.1 Class I BCRs
The first BCR was isolated and characterized from the denitrifying β-Proteobacterium Thauera aromatica; to date, it still represents the only characterized enzyme of the class I BCR (Boll and Fuchs 1995). The enzyme uses a reduced ferredoxin as electron donor and couples endergonic transfer of two single electrons to the aromatic ring to a stoichiometric hydrolysis of 2 ATP to 2 ADP + 2 Pi (Fig. 3a) (Boll et al. 1997; Boll and Fuchs 1998). A Birch-like mechanism via reactive radical species has been proposed, which proceeds via alternate single electron transfer and protonation steps; some indirect evidence for such a scenario has been obtained (Möbitz and Boll 2002; Thiele et al. 2008).

Molecular architectures of class I (a) and class II BCRs (b). In class I BCR (BcrABCD subunits) endergonic electron transfer from reduced ferredoxin (E′ ≈ −500 mV) to benzoyl-CoA (E°′ = −622 mV) is driven by stoichiometric ATP hydrolysis. In class II BCR (BamBCDEFGHI), a flavin-based electron bifurcation is hypothesized in which endergonic electron transfer from an unknown donor (Xred−) to benzoyl-CoA is driven by the exergonic electron transfer from the same donor to a high-potential acceptor. Due to similarities of BamH with NAD-binding components of NADH:quinone oxidoreductase, NAD+ (E°′ = −320 mV) has been proposed as a potential second electron acceptor
The 170 kDa class I BCR from T. aromatica binds three [4Fe-4S]+1/+2 clusters and has a BcrABCD-architecture (Fig. 3a) (Buckel et al. 2014). The BcrAD subunits bind a [4Fe-4S] cluster and two ATP molecules; ATP hydrolysis causes conformational changes promoting the electron transfer to the two other [4Fe-4S] clusters of the BcrBC subunits. Class I BCRs are present in aromatic compound degrading facultative anaerobes belonging to genera of the α-Proteobacteria (e.g., Magnetospirillum, Rhodopseudomonas, Rhodomicrobium) or β-Proteobacteria (e.g., Aromatoleum, Thauera, Azoarcus, Georgfuchsia). Surprisingly, a class I BCR has also been identified in the strictly anaerobic archaeon Ferroglobus placidus (Schmid et al. 2015). Based on the amino acid sequence similarities and subunit sizes of the BcrAD-modules, two subclasses of ATP-dependent BCRs have been identified: the Thauera- and the Azoarcus-type. A BCR of the latter subclass has not been isolated and characterized, yet (Buckel et al. 2014). A third BCR subclass recently identified in a 4-methylbenzoate degrading Magnetospirillum species may be specifically involved in the reduction of benzoyl-CoA analogues (Rabus et al. 2016a) (see Sect. 5.1).
2.2.2 Class II BCRs
The active site BamBC components (bam = benzoic acid metabolism) of a class II BCR were isolated and characterized from the Fe(III)-respiring δ-ProteobacteriumG. metallireducens (Kung et al. 2009). The BamB subunit contains an active site W-bis-tungstopterin (bisWPT) cofactor and shows similarities to the W-containing aldehyde:ferredoxin oxidoreductases. The crystal structures of a Bam(BC)2 complex in the presence of the substrate, product, and inhibitor revealed that the tungsten atom is coordinated by four sulfur atoms from the dithiolene groups of the cofactor, by a cysteine-sulfhydryl group and a sixth inorganic ligand, probably a sulfido or cyano group (Fig. 4) (Weinert et al. 2015). Remarkably, the aromatic ring of benzoyl-CoA does not bind directly to the cofactor; instead electron transfer from the reduced bisWPT to the aromatic ring appears to proceed via the sixth inorganic ligand, probably via an outer shell electron transfer mechanism.

Crystal structure of the active site BamB component fromG. metallireducens class II BCR. The ring of a benzoyl-CoA analogue (a cyclic monoenoyl-CoA) is bound in an aprotic cavity. Electrons for aromatic ring reduction are transferred from the reduced W-atom, coordinated by five sulfur ligands (green), via an unknown inorganic ligand (X). Protons are transferred from His260 and probably Glu251. The structure promotes a Birch-like mechanism via spatially separated single electron transfer and protonation steps; modified from Weinert et al. (2015)
The putative other components of the class II BCR complex are encoded by the bamDEFGHI genes that share similarities to genes encoding soluble heterodisulfide reductase and NADH-binding components of NADH:quinone oxidoreductases (Wischgoll et al. 2005). In silico analysis and preliminary experimental work suggest that class II BCRs form a large BamBCDEFGHI complex harboring bisWPT, >20 FeS clusters, three FAD, and one selenocysteine as cofactors. The BamBCDEGHI complex is considered to drive endergonic benzoyl-CoA reduction via a flavin-based electron bifurcation: the endergonic electron transfer from an unknown donor, probably a reduced ferredoxin, to the aromatic ring is coupled to electron transfer from the same donor to a high-potential second acceptor, probably NAD+. Notably, such an electron bifurcation process has previously been demonstrated for a soluble heterodisulfide reductase from methanogenic archaea which exhibits significant similarities to the BamDE components (Kaster et al. 2011).
The bamBCDEFGHI genes encoding class II BCRs are found in all obligately anaerobic bacteria with the capacity to degrade MAC, but never in any facultatively anaerobic MAC-degrading organism (Löffler et al. 2011). The occurrence of class I BCRs in facultative anaerobes and class II BCRs in obligate anaerobes may be rationalized by the energetic costs for benzoyl-CoA dearomatization. The energy yield of facultatively anaerobic bacteria (e.g., denitrifiers) is relatively high which allows the use of an ATP-dependent, essentially irreversibly operating BCR. A flavin-based electron bifurcation, however, is considered to operate closer to equilibrium and consequently seems to be less energy demanding (Buckel and Thauer 2013). However, it has to be taken into account that the synthesis of a class II BCR depends on efficient metal uptake systems and cofactor synthesis/insertion machineries (tungstopterin, flavins, selenocysteine, FeS clusters).
2.3 Degradation of Phenolic Compounds
2.3.1 Phenol Carboxylation
The pathway of anaerobic phenol degradation has mostly been studied in facultative anaerobes, particularly in the denitrifying bacterium Thauera aromatica. This bacterial species metabolizes phenol via a two-step, ATP-dependent carboxylation of the aromatic ring in para-position to the phenolic hydroxyl group, catalyzed by two distinct enzymes (Fig. 5) (Lack and Fuchs 1994; Breinig et al. 2000; Schühle and Fuchs 2004). In the initial step, phenylphosphate is formed from phenol as a stable activated intermediate (Lack and Fuchs 1994; Narmandakh et al. 2006). This reaction is catalyzed by a phenylphosphate synthase complex (PPS) in an ATP-dependent reaction. Subsequently, phenylphosphate is dephosphorylated and simultaneously carboxylated to 4-hydroxybenzoate by a second multisubunit enzyme complex, phenylphosphate carboxylase (PPC) (Breinig et al. 2000; Schühle and Fuchs 2004).

Enzymatic reactions involved in the conversion of phenol to benzoyl-CoA. Phenylphosphate synthase catalyzes the first step, followed by a carboxylation reaction by phenylphosphate carboxylase. The generated product 4-hydroxybenzoate is then activated to 4-hydroxybenzoyl-CoA, which is reductively dehydroxylated to benzoyl-CoA
Both PPS and PPC have been purified and biochemically characterized (Schühle and Fuchs 2004; Schmeling et al. 2004; Narmandakh et al. 2006), and putative reaction schemes have been derived for both reactions from the biochemical properties of the enzymes. PPS consists of three different subunits, two of which are sufficient to catalyze the reaction. These subunits, PpsA and PpsB, show significant sequence similarity to phospho-enol-pyruvate (PEP) synthetases (Breinig et al. 2000; Schühle and Fuchs 2004; Schmeling et al. 2004), while the third subunit increases the enzyme activity severalfold via an unknown mechanism. The proposed reaction mechanism of PPS involves the transfer of pyrophosphate to a conserved histidine, hydrolysis of the bound pyrophosphate, and the transfer of the remaining phosphoryl group to phenol, yielding phenylphosphate (Schmeling et al. 2004; Schmeling and Fuchs 2009).
The PPC complex consists of four subunits, one of which is proposed to be involved in dephosphorylation of phenylphosphate. This generates phenol (or phenolate) in a tightly enzyme-bound state which seems to be more capable of being carboxylated than free phenol. The second partial reaction consists of the actual carboxylation of the enzyme-bound “phenolate” by subunits affiliated to the UbiD/UbiX family of carboxylases/decarboxylases, constituting a biochemical analog of the chemical Kolbe-Schmitt process (Payne et al. 2015). This partial reaction is freely reversible, causing the extensive exchange between free CO2 and the carboxyl group of 4-hydroxybenzoate as side reaction. In accordance with this proposal, the phosphatase subunit of PPC is only necessary for phenylphosphate carboxylation, but not for CO2 exchange (Schmeling and Fuchs 2009). In contrast to PPS, PPC is highly oxygen sensitive. Oxygen-mediated inactivation of the enzyme is reversible and PPC activity can (at least partially) be restored by removal of oxygen and addition of reducing agents (Schmeling and Fuchs 2009).
Remarkably, strictly anaerobic bacteria, for example, Fe(III)-reducing Geobacter species or several species of sulfate-reducing bacteria, appear to use slightly different biochemical strategies for phenol degradation (Schleinitz et al. 2009; Ahn et al. 2009; Wöhlbrand et al. 2013). Phenol metabolism in these organisms is proposed to proceed via a similar pathway as outlined for T. aromatica, and cell extracts of G. metallireducens grown anaerobically on phenol actually exhibit low activities of both PPS and PPC (Schleinitz et al. 2009). Strictly anaerobic phenol-degrading species contains genes for orthologs of all three PPS subunits, but genes coding for two of the four subunits of PPC are lacking. Further details on the biochemistry of these processes are as yet unknown.
2.3.2 para-Cresol Hydroxylation with Water
The initial steps in the degradation of para-cresol (p-cresol, 4-methylphenol) are identical in aerobic and anaerobic bacteria and comprise the hydroxylation of the methyl functionality to 4-hydroxybenzoyl-CoA via 4-hydroxybenzyl alcohol/aldehyde (Fig. 6) (Hopper and Taylor 1977; Hopper et al. 1991; Rudolphi et al. 1991). The initial hydroxylation with water is catalyzed by p-cresol methylhydroxylase, a flavocytochrome c. The enzyme is composed of the active site subunit in which FAD is covalently attached to a tyrosine residue, and an electron transferring cytochrome c subunit (McIntire et al. 1985; Cunane et al. 2000). The relatively low C–H bond dissociation energy of p-cresol (335 kJ mol−1) allows a hydride abstraction from the methyl group by the FAD cofactor generating a relatively stable quinone-methide intermediate; addition of water yields then the product 4-hydroxybenzyl alcohol, which may be further oxidized to the aldehyde by the same enzyme. In aerobic and facultatively anaerobic p-cresol degrading organisms, electrons are transferred to cytochrome c. In contrast, p-cresol methylhydroxylase was found to be strongly membrane bound in the obligately anaerobic G. metallireducens, probably due to complex formation with cytochrome bc1 complex-like components that contain two heme b and a 2[Fe-2S] cluster (Johannes et al. 2008). In such a complex, electrons derived from p-cresol oxidation may not be transferred to cytochrome c but rather to menaquinone. During the oxidation of 4-hydroxybenzyl alcohol to the corresponding aldehyde (E°′ ≈ −200 mV) by p-cresol methylhydroxylase, electron transfer to menaquinone (E°′ ≈ −75 mV) would be highly exergonic and may be coupled to proton transport across the membrane. Experimental evidence for such an energy conserving process is still lacking.

Enzymatic reactions involved in the conversion of p-cresol to 4-hydroxybenzoate in aerobic and facultatively anaerobic bacteria. The p-cresol methylhydroxylase catalyzes hydride abstraction from p-cresol by the covalently bound FAD cofactor, yielding an enzyme-bound quinone methide; addition of water then yields the product 4-hydroxybenzyl alcohol. A similar reaction mechanism is proposed for the analogous conversion of 4-ethylphenol to 4-hydroxyphenylethanol. The alcohol may be further oxidized to the aldehyde by the same enzyme or by a NAD+-dependent, separate dehydrogenase. Soluble p-cresol methylhydroxylase from aerobic and facultatively organisms transfers electrons to cytochrome c, whereas the membrane-bound enzyme from the obligately anaerobic G. metallireducens is suggested to transfer electrons to menaquinone (not shown)
In sulfate-reducing bacteria, the redox potential of the terminal electron acceptors adenosine-5′-phosphosulfate (E°′ = −60 mV) or sulfite (E°′ = −116 mV) are too negative to accept electrons from p-cresol hydroxylation to 4-hydroxybenzyl alcohol (E°′ = +80 mV). For this reason, a different strategy exists for complete p-cresol oxidation coupled to sulfate reduction. In the p-cresol-degrading bacterium Desulfobacterium cetonicum, in vitro evidence for an addition of p-cresol to fumarate was provided, yielding 4-hydroxybenzylsuccinate as intermediate (Müller et al. 2001). The latter was then further degraded via 4-hydroxybenzoyl-CoA by a reaction sequence similar to that involved in toluene degradation (Fig. 7) (see Sect. 2.8.1).

Enzymatic reactions involved in the conversion of p-cresol to 4-hydroxybenzoyl-CoA in sulfate-reducing bacteria. The reactions leading from 4-hydroxybenzylsucinate to 4-hydroxybenzoyl-CoA are similar to those of benzylsuccinate to benzoyl-CoA involved in toluene degradation (Sect. 2.8.1)
2.3.3 4-Hydroxybenzoyl-CoA Dehydroxylation
Phenol and p-cresol are both metabolized via 4-hydroxybenzoate, which is subsequently activated by a specific AMP-forming CoA ligase to 4-hydroxybenzoyl-CoA (Biegert et al. 1993). This activation is essential for the following reductive dehydroxylation to benzoyl-CoA and water, using reduced ferredoxin as electron donor (Fig. 5) (Brackmann and Fuchs 1993; Breese and Fuchs 1998). The reaction is catalyzed by a 4-hydroxybenzoyl-CoA reductase that belongs to the xanthine oxidase family of molybdenum-cofactor (MPT) containing enzymes. It has so far only been isolated and studied in the denitrifying T. aromatica. The 275 kDa enzyme has an Hcr(ABC)2 composition with the large HcrA-subunit harboring the MPT cofactor, while the HcrB-subunit contains two [2Fe-2S] clusters, and the C-subunit contains an FAD cofactor and an unusual [4Fe-4S] cluster. The latter are usually absent from members of the xanthine oxidase family of Mo enzymes, so this cofactor represents a distinguishing feature of 4-hydroxybenzyol-CoA reductases (Unciuleac et al. 2004). The redox potentials of the Mo cofactor of 4-hydroxybenzoyl-CoA reductase are unusually low (E°′ = −500 mV for the MoV/MoIV transition) reflecting the mechanistically demanding reaction of the enzyme (Boll et al. 2001). A radical-based mechanism has been proposed in which the first electron transfer yields a radical anion that is largely stabilized by the CoA thioester functionality. It also explains why 4-hydroxybenzoate needs to be activated prior to dehydroxylation.
Homologs of the encoding hcrABC genes are present in all phenol, p-cresol or 4-hydroxybenzoate degrading organisms. Due to the relatively high amino acid sequence similarities to other xanthine oxidase-related enzymes, an unambiguous assignment based on the amino acid sequence is not always feasible, although the reaction mechanism of 4-hydroxybenzoyl-CoA reductase fundamentally differs from all other MPT-containing enzymes.
2.3.4 Degradation of Other Para-Hydroxylated Compounds
The degradation of 4-ethylphenol is initiated by hydroxylation to 4-hydroxyphenylethanol with water. The enzyme catalyzing this reaction in A. aromaticum and most probably other 4-ethyphenol degrading organisms appears to be highly similar to p-cresol methylhydroxylase, and identical mechanisms via a quinone methide have been proposed (Fig. 6) (Wöhlbrand et al. 2008; Muhr et al. 2015). The proposed subsequent steps resemble those of the ethylbenzene degradation pathway, involving dehydrogenation of the alcohol to 4-hydroxyacetophenone, and carboxylation of the latter followed by CoA-ester formation. Thiolytic cleavage yields acetyl-CoA and 4-hydroxybenzoyl-CoA. These reactions are discussed in more detail below (see Sect. 2.8.2 ethylbenzene degradation).
Tyrosine and 4-hydroxyphenylacetate are both converted to 4-hydroxybenzoyl-CoA by enzymes similar to those involved in the conversion of the non-phenolic analogues phenylalanine and phenylacetate and will be discussed in Sect. 2.6. Hydroquinone degradation in the fermenting strain HQGö1 is initiated by ortho-carboxylation to gentisate, followed by activation to gentisyl-CoA by an AMP forming CoA ligase (Gorny and Schink 1994a, c). In vitro assays demonstrated the subsequent reductive dehydroxylation to benzoyl-CoA without the accumulation of a mono-hydroxylated intermediate. The genes and enzymes involved in hydroquinone degradation are still unknown.
2.4 Reductive Dehalogenation by Class I BCRs
The major strategy for the anaerobic dehalogenation of haloaromatics is usually associated with organohalide respiration involving vitamin B12-dependent reductive dehalogenases. In this process, halogenated MAC serve only as terminal electron acceptors and the dehalogenated aromatic intermediates are generally not further degraded (Hug et al. 2013). However, there are a number of facultative and obligate anaerobes that completely degrade halobenzoates to CO2 plus the halide anion as only carbon sources (Schennen et al. 1985; Kazumi et al. 1995; Song et al. 2000, 2001; Egland et al. 2001). The position and elemental nature of the halogen substituents in particular halobenzoates seems to determine whether degradation is possible or not. For example, the complete degradation of 3-Cl-benzoate is frequently observed in anaerobic bacteria, whereas anaerobic 3-F-benzoate degradation has never been reported. In contrast, a number of 2- and 4-F-benzoate degrading cultures are known, whereas 2-Cl-/4-Cl-benzoate degradation appears to be rather rare under anoxic conditions.
The complete degradation of 3-Cl-, 3-Br-, and 4-F-benzoates has recently been studied in detail in halobenzoate degrading Thauera strains (Kuntze et al. 2011b; Tiedt et al. 2016). The halobenzoates are first activated by AMP-forming CoA ligases followed by reductive, ATP-dependent dehalogenation by a class I BCR (Fig. 8). The transfer of two single electrons and a proton to 3-Cl-benzoyl-CoA or 4-F-benzoyl-CoA leads to the formation of an anionic transition state, as also proposed for benzoyl-CoA reduction. In case of 3-Cl- or 3-Br-benzoyl-CoA, the anion is irreversibly protonated yielding a 3-Cl-/3-Br-1,5-dienoyl-CoA intermediate, which spontaneously eliminates HCl/HBr in an E2 mechanism. In the case of 4-F-benzoyl-CoA, protonation of the anionic transition state is less favored than fluoride release; the latter event results in re-aromatization to benzoyl-CoA (Tiedt et al. 2016). Notably, this ATP-dependent defluorination of 4-F-benzoyl-CoA to date is the only known oxygen-independent C–F-bond cleavage reaction of any fluoroaromatic in biology (C-F dissociation energy 530 kJ mol−1). In summary, 4-F-benzoyl-CoA defluorination formally represents a nucleophilic aromatic substitution in contrast to the elimination of HCl/HBr from the 3-Cl-/3-Br-1,5-dienoyl-CoA product. Whether complete degradation of halogenated MAC is restricted to halobenzoates needs to be further studied. It is likely that at least some halogenated benzenes, toluenes, phenols, or other MAC may be degraded by promiscuous enzymes to the corresponding halogenated benzoyl-CoA intermediates.

Enzymatic reactions involved in the conversion of halobenzoates to benzoyl-CoA. After activation of 3-Cl- or 4-F-benzoate to the corresponding CoA-thioesters by AMP-forming CoA ligases, the 3-Cl-/4-F-benzoyl-CoA formed are converted by promiscuous class I BCR to the common, enzyme-bound anionic transition states (in brackets). In the case of 3-Cl-benzoyl-CoA, irreversible protonation yields 3-Cl-1,5-dienoyl-CoA that spontaneously eliminates HCl. In the case of 4-F-benzoyl-CoA conversion, fluoride release from the anionic transition state is favored over protonation, resulting in a formal nucleophilic aromatic substitution at C4
2.5 Decarboxylation of Phthalates
Phthalate (1,2-dicarboxybenzene) and its isomers isophthalate (1,3-dicarboxybenzene) and terephthalate (1,4-dicarboxybenzene) are annually produced in the million ton scale due to their massive use of their esters as plasticizers or as precursors for polymers. In this context, it is surprising that the degradation pathways of phthalates remained obscure for a long time. In recent studies with phthalate-degrading species of the genera Aromatoleum, Azoarcus, and Thauera, phthalate degradation was shown to be initiated by a highly specific phthaloyl-CoA forming, succinyl-CoA-dependent CoA transferase (Ebenau-Jehle et al. 2016; Junghare et al. 2016). The phthaloyl-CoA formed is then decarboxylated by an oxygen-sensitive enzyme belonging to the UbiD-family of decarboxylases (ubiD encodes 3-octaprenyl-4-hydroxybenzoate carboxy-lyase involved in ubiquinone biosynthesis) (Fig. 9). The UbiD-like protein involved in phthaloyl-CoA decarboxylation shows significant similarities to phenylphosphate carboxylase or putative carboxylases involved in aromatic hydrocarbon carboxylation. Thioesterification is considered as an essential step for stabilizing an anionic transition state during phthaloyl-CoA decarboxylation. The extreme instability of phthaloyl-CoA requires highly balanced phthaloyl-CoA forming/decarboxylating enzyme activities to avoid its cellular accumulation that would result in spontaneous hydrolysis.

Enzymatic reactions involved in the conversion of phthalate to benzoyl-CoA. The phthaloyl-CoA formed by a specific, succinyl-CoA dependent CoA transferase is decarboxylated by the oxygen-sensitive UbiD-like phthaloyl-CoA decarboxylase possibly carrying a prenylated FMN cofactor (prFMN). The prenylated cofactor is suggested to be formed by the UbiX-like prenyl transferase using dimethylallyl monophosphate (DMAP) as co-substrate
In the genomes of all investigated phthalate-degrading organisms, the ubiD-like genes coding for phthaloyl-CoA decarboxylase are flanked by ubiX-like genes (Nobu et al. 2015; Ebenau-Jehle et al. 2016; Junghare et al. 2016). A UbiX-like protein was recently reported to act as a prenyltransferase that forms a UbiX-bound prenylated FMN cofactor. The latter is then transferred to the apo-form of an UbiD-like enzyme catalyzing the decarboxylation of cinnamate to styrene; dimethylallyl monophosphate serves as prenyl-donor for UbiX (Payne et al. 2015; White et al. 2015). In analogy, phthaloyl-CoA decarboxylase and other UbiD-like carboxylases/decarboxylases, whose genes are often flanked by ubiX-like genes, are all suggested to contain a prenylated flavin cofactor that is formed by an UbiX-like flavin prenyltransferase (Fig. 9).
Only little is known about the anaerobic degradation of isophthalate and terephthalate, though the complete degradation has been reported in strictly anaerobic Pelotomaculum and Syntrophorhabdus species (Qiu et al. 2004, 2006, 2008). In case of terephthalate, a similar strategy as for phthalate degradation via thioesterification followed by UbiX/UbiD-dependent decarboxylation is conceivable, and UbiD-like candidate genes have been identified by genomic/proteomic studies (Nobu et al. 2015). Nothing is known about the genes and enzymes involved in isophthalate degradation.
2.6 Degradation of Phenylalanine, Tyrosine, Phenylacetates, and Phenylpropionates
All three aromatic amino acids, phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp), are readily degraded in the absence of oxygen (Heider and Fuchs 1997a, b). Many organisms capable of anaerobic respiration mineralize these compounds completely in pure or mixed cultures, and many fermentative bacteria or archaea are transforming aromatic amino acids to simpler aromatic or even aliphatic fermentation products (Barker 1981).
Fermentative transformation of aromatic amino acids is well known via the pathway of Stickland fermentation in strictly anaerobic bacteria affiliated to the Clostridia or related families of Firmicutes (Barker 1981; Schink and Stams 2013). Stickland fermentation involves the transformation of amino acid pairs, one of which is oxidized and decarboxylated to yield an organic acid lacking one C-atom, whereas the other is reduced to the organic acid of the same size as the amino acid. The oxidative pathway can be coupled to energy conservation via substrate-level phosphorylation from the acyl-CoA intermediate, while the reductive pathway is usually only required for equilibrating the redox balance and does not lead to energy conservation (with the exception of glycine reduction). All three aromatic amino acids can principally be channeled either into oxidative or reductive pathways. Both are initiated by the conversion of the amino acids to the respective arylpyruvates via transaminases, using 2-oxoglutararate as acceptor molecule. The respective arylpyruvate intermediates are then either oxidized to the arylacetates plus CO2 or reduced to the arylpropionates.
In the case of phenylpyruvate (Fig. 10), the reductive pathway involves an initial reduction to phenyllactate, which is then activated to the CoA-thioester by a specific enzyme affiliated to class III of CoA-transferases (Heider 2001) and dehydrated to cinnamoyl-CoA by a complex phenyllactyl-CoA dehydratase complex in a radical-type reaction mechanism (Dickert et al. 2002; Buckel et al. 2012). Cinnamoyl-CoA serves as CoA-donor for phenyllactate activation, generating cinnamate, which is finally reduced to phenylpropionate by an enoate reductase (Buckel et al. 2012). Because of the involvement of a CoA-transferase, the pathway proceeds without energy consumption despite the required activation of phenyllactate for the mechanistically difficult elimination of the α-hydroxy group (Dickert et al. 2002; Buckel et al. 2012).

Anaerobic degradation of aromatic amino acids. Reactions in green: reductive branch during fermentative transformations via Stickland fermentation; reactions in red: oxidative branch during fermentative Stickland reactions or during oxidative transformations in hyperthermophilic archaea; reactions in blue: complete degradation in denitrifying bacteria
The oxidative part of arylpyruvate metabolism is common to all known degradation pathways, but occurs in two variants. The first variant involves simultaneous oxidation and decarboxylation of the arylpyruvates by a ferredoxin-dependent oxidoreductase. The best characterized of these enzymes is indolepyruvate oxidoreductase from the hyperthermophilic archaeum Pyrococcus furiosus, which converts any arylpyruvate derived from the aromatic amino acids to the corresponding acylacetyl-CoA derivative (Mai and Adams 1994). The same type of enzyme is known from other archaea degrading aromatic amino acids (Parthasarathy et al. 2013; Aklujkar et al. 2014), and genes coding for this type of enzymes regularly occur in anaerobic amino-acid-degrading bacteria, although there are no reports on their expression patterns available. The thioesters generated by this reaction provide a means of energy conservation via substrate level phosphorylation (Fig. 10), either by CoA transfer to succinate and succinyl-CoA synthetase or by ADP-coupled acetyl-CoA synthetases with broad substrate specificities that include the arylacetyl-CoAs (Mai and Adams 1996; Bräsen and Schönheit 2004). Therefore, this variant of the pathway appears to be particularly required for fermentative microorganisms which excrete the corresponding arylacetates as fermentation products and use these reactions as their main energy conserving pathway during growth on aromatic amino acids.
The second variant of arylpyruvate oxidation has mostly been documented in denitrifying microorganisms capable of fully degrading phenylalanine. It involves an initial decarboxylation of phenylpyruvate to phenylacetaldehyde, which is then oxidized to phenylacetate (Heider and Fuchs 1997a, b; Debnar-Daumler et al. 2014). Phenylpyruvate decarboxylases are very common enzymes in microorganisms and have been identified in the phenylalanine degradation pathways of denitrifying bacteria affiliated to the β-Proteobacteria (Heider and Fuchs 1997a, b; Debnar-Daumler et al. 2014). The subsequent oxidation of phenylacetaldehyde to phenylacetate is then either catalyzed by NAD- or NADP-coupled aldehyde oxidoreductases or by aldehyde:ferredoxin oxidoreductases containing a tungsten cofactor (Debnar-Daumler et al. 2014). The organic acids generated from the aromatic amino acids are further degraded via separate metabolic modules. We describe here the pathways of anaerobic phenylacetate and phenylpropionate metabolism, which are probably shared with the 4-hydroxyphenyl acids derived from tyrosine (Fig. 1). For indoleacetate degradation see Sect. 3.3.
Phenylacetate metabolism is initiated by its activation to the CoA-thioester by an AMP-generating CoA ligase, followed by a four-electron oxidation to phenylgloxylate, as catalyzed by a membrane-bound molybdenum enzyme, phenylacetyl-CoA:acceptor oxidoreductase (Rhee and Fuchs 1999). Finally, phenylgloxylate is oxidized and decarboxylated by phenylgloxylate:ferredoxin oxidoreductase, yielding the common intermediate benzoyl-CoA (Hirsch et al. 1998) (Fig. 10). In contrast, anaerobic phenylpropionate degradation follows a classical β-oxidation pathway, as inferred by a proteomic analysis in the denitrifying species Aromatoleum aromaticum (Trautwein et al. 2012) and recent biochemical characterization of some of the enzymes (J. Heider, unpublished data). Phenylpropionate is activated by a AMP-producing CoA ligase which also accepts several substrate analogs such as cinnamate or several hydroxyphenylpropionates (J. Heider, unpublished data), indicating that the same enzymes are involved in metabolizing intermediates of tyrosine or lignin degradation. Phenylpropionyl-CoA then seems to be oxidized to cinnamoyl-CoA, hydrated to 3′-hydroxyphenylpropionyl-CoA, further oxidized to benzoylacetyl-CoA and finally cleaved to benzoyl-CoA and acetyl-CoA by a specific thiolase. The genes for all proteins involved in this pathway appear to form a common substrate-induced operon in A. aromaticum (Trautwein et al. 2012).
2.7 Fermentative Formation of Toluene, p-Cresol, or Skatole
Some anaerobic fermentative organisms affiliated to the Clostridia are able to decarboxylate the arylacetates formed from the three aromatic amino acids to produce toluene, p-cresol, or skatole as final fermentation products. In the case of 4-hydroxyphenylacetate conversion to p-cresol, the enzyme responsible has been intensively studied and biochemically characterized. 4-Hydroxyphenylacetate decarboxylase (4Hpad) from the pathogenic gut bacterium Clostridium difficile has been identified as a glycyl radical enzyme and its structure and mechanism have been identified (Selmer and Andrei 2001; Selmer et al. 2005; Selvaraj et al. 2016). While C. difficile is resistant against very high concentrations of p-cresol, the presence of 4Hpad allows it to accumulate p-cresol at toxic concentrations for most other bacteria or the cells of the gut wall. Therefore, this metabolic pathway may be considered as one of the pathogenicity factors of this bacterium. Very similar enzymes seem to be involved in producing skatole from indoleacetate (Yu et al. 2006) or even toluene from phenylacetate (Zargar et al. 2016).
2.8 Degradation of Alkylbenzenes
2.8.1 Fumarate Addition to Toluene
Toluene is one of the most widely occurring aromatic hydrocarbons and appears to be readily degraded under anaerobic conditions. Anaerobic toluene degraders are known among different phylogenetic groups of denitrifying, metal-ion or sulfate-reducing bacteria, as well as among anoxygenic phototrophic bacteria and syntrophic proton-reducing bacteria in co-culture with methanogenic archaea or other hydrogen-consuming anaerobes (Evans et al. 1992; Heider et al. 1998; Heider and Schühle 2013; Rabus et al. 2016a, b). The most prevalent genera of anaerobic toluene degraders are affiliated to the betaproteobacterial genera Thauera and Aromatoleum/Azoarcus or the deltaproteobacterial genera Geobacter, Desulfobacula, and Desulfobulbus. Despite the wide range of phylogenetic distribution, the pathway of anaerobic toluene degradation is completely conserved in all known cases, starting with the formation of (R)-benzylsuccinate from toluene and the fumarate co-substrate by the glycyl-radical enzyme benzylsuccinate synthase (BSS ; Leuthner et al. 1998; Heider et al. 2016b). BSS exhibits a heterohexameric (αβγ)2 structure with two large α subunits of ca. 100 kDa harboring a single glycyl radical at the active center of one of the subunits and two very small β- and γ- subunits containing unusual Fe4S4 clusters (Leuthner et al. 1998; Funk et al. 2015; Heider et al. 2016b). As known for all glycyl radical enzymes, BSS needs to be activated to the active radical-containing form by an activating enzyme, which belongs to the family of “S-adenosylmethione radical” enzymes. In the activated state, BSS is proposed to catalyze the reaction by binding both substrates into a shielded active site pocket, followed by abstracting a hydrogen from the methyl group of toluene, radical addition of the transiently formed benzyl radical to the double bond of fumarate, and donating the hydrogen back to the generated benzylsuccinyl (product) radical, forming the product benzylsuccinate and regenerating the radical form of BSS (Heider et al. 2016b; Szaleniec and Heider 2016).
The intermediate (R)-benzylsuccinate is further degraded by β-oxidation via a conserved specific pathway consisting of a benzylsuccinate CoA-transferase, benzylsuccinyl-CoA dehydrogenase, phenylitaconyl-CoA hydratase, (hydroxybenzyl)succinyl-CoA dehydrogenase, and benzoylsuccinyl-CoA thiolase (Leuthner and Heider 2000; Heider et al. 2016b; Fig. 11). In all investigated bacteria capable of anaerobic toluene degradation, all enzymes needed for benzylsuccinate formation and β-oxidation are encoded in two separate but coordinately induced genetic units, namely the bss and bbs operons (Heider et al. 2016b).

Anaerobic degradation of toluene. Reactions are shown that are catalyzed by enzymes encoded by genes of the bss and bbs operons
The strategy of adding fumarate to activate nonreactive alkyl groups is used for a surprisingly large number of other substrates in addition to toluene. The enzymes involved in these reactions are all highly similar to BSS and apply the same apparent mechanism starting with a glycyl radical in their active sites. For example, fumarate addition has been reported in anaerobic degradation pathways of m-xylene (Rabus et al. 2016; see Sect. 4.1), m- or p-cresol (see Sects. 4.2 and 2.3.2), 2-methylnaphthalene (see Chap. 5, “Catabolic Pathways and Enzymes Involved in the Anaerobic Degradation of Polycyclic Aromatic Hydrocarbons” about PAH degradation of this volume on the degradation of polycyclic aromatic hydrocarbons in this volume) p-cymene (see Sect. 5.2), ethylbenzene (see Sect. 2.8.2), or even alkanes or cycloalkanes (Wilkes et al. 2016; see Chap. 3, “Catabolic Pathways Involved in the Anaerobic Degradation of Saturated Hydrocarbons” on the degradation alkanes in this volume). In the case of p-cresol, p-cymene, ethylbenzene, and possibly long-chain alkanes, the degradation pathways initiated by fumarate addition are not unique, and alternative pathways initiated by oxygen-independent hydroxylation are known. At least in some of these cases, the distribution of these different pathways appears to be dependent on the physiological type of the respective bacterial strains: p-cresol or ethylbenzene seem to be preferentially degraded via hydroxylation in facultatively anaerobic denitrifying bacteria and via fumarate addition in strictly anaerobic sulfate-reducers. However, both types of initial reactions have been observed for p-cymene in closely related species of denitrifying bacteria (Strijkstra et al. 2014; Rabus et al. 2016a), and alkanes seem to be degraded by either initial reaction in different sulfate-reducing bacteria (Heider and Schühle 2013).
2.8.2 Hydroxylation of Ethylbenzene or Propylbenzene by Water
The first indications of an oxygen-independent hydroxylation reaction initiating anaerobic hydrocarbon degradation arose with the isolation of denitrifying bacteria capable of degrading ethylbenzene or propylbenzene (Rabus and Widdel 1995; Rabus and Heider 1998). The initial reaction is catalyzed by ethylbenzene dehydrogenase (EBDH), a new type of soluble periplasmic molybdenum enzyme, which hydroxylates ethylbenzene or propylbenzene stereospecifically to the respective 1-(S)-alcohols (Kniemeyer and Heider 2001a). EBDH belongs to subfamily 2 of the dimethylsulfoxide reductase enzyme family of Mo-enzymes. It is a trimer comprising a large catalytic subunit, which contains a Mo-bis-molybdopterin guanine dinucleotide (MGD) cofactor and an Fe4S4 cluster, a medium subunit containing four additional FeS-clusters, and a small subunit containing a heme b cofactor (Kloer et al. 2006). Hydroxylation of the substrate occurs at the Mo cofactor with water as hydroxyl donor (Ball et al. 1996), and the probable mechanism of this reaction has been modeled by computational methods (Szaleniec et al. 2010, 2014). The electrons released from oxidation of the substrate are then transferred through the enzyme from the Mo-bis-MGD cofactor via the FeS-clusters toward the heme b, where they are transferred to an external acceptor such as cytochrome c (Kloer et al. 2006).
The generated alcohol enters the cytoplasm and is further oxidized to the ketone (acetophenone or propiophenone, respectively) by a short-chain alcohol dehydrogenase (Kniemeyer and Heider 2001b). The genes for EBDH and the alcohol dehydrogenase are encoded in a common substrate-induced operon (Rabus et al. 2005). The further metabolism of the aromatic ketones is initiated by an ATP-dependent carboxylation to benzoylacetate or 2-benzoylpropionate, respectively, by acetophenone carboxylase, which belongs to a special family of carboxylases together with acetone carboxylases from many microorganisms (Jobst et al. 2010; Heider et al. 2016a). Benzoylacetate is finally activated to the CoA-thioester by a specific CoA ligase, which is then cleaved to benzoyl-CoA and acetyl-CoA by a thiolase (Fig. 12). The genes coding for the subunits of acetophenone carboxylase and benzoylacetate-CoA ligase form a second operon, which is specifically induced by the presence of acetophenone or a few very similar substrates (Heider et al. 2016a; Muhr et al. 2016).

Anaerobic degradation of ethylbenzene. The degradation of p-ethylphenol (4-hydroxyethylbenzene) proceeds in analogous reactions in an induced pathway to 4-hydroxybenzoyl-CoA
Molybdenum enzymes with high similarities to EBDH have also been identified in the anaerobic degradation of the aromatic hydrocarbon p-cymene (see Sect. 5.2) and the isoprenoid side chains of cholesterol or other steroids (Dermer and Fuchs 2012; Heider et al. 2016c, see Chap. 7, “Anaerobic Biodegradation of Steroids” on the degradation of steroids in this volume) and may also be involved in an alternative pathway of anaerobic alkane degradation (Heider et al. 2016c).
2.9 Degradation of Benzene
The C–H bond dissociation energy of benzene is the strongest among all hydrocarbons (473 kJ mol−1), and the oxygen-independent attack on the benzene ring has been considered to afford an unprecedented mechanism. To date numerous benzene-degrading pure/enrichment cultures have been obtained under denitrifying, sulfate-reducing, Fe(III)-reducing conditions and in methanogenic consortia [for a recent review see (Meckenstock et al. 2016)]. Recently, even the genetically tractable G. metallireducens was reported to completely degrade benzene (Zhang et al. 2013). Considering the number of well-characterized benzene-degrading cultures available, it is surprising that benzene degradation is still controversially discussed. The identification of benzene-induced UbiD-like enzymes in conjunction with a number of 13C-labeling studies in benzene degrading Fe(III)-reducing or methanogenic enrichment cultures (Kunapuli et al. 2008; Abu Laban et al. 2009, 2010; Luo et al. 2014), as well as in the hyperthermophilic, benzene-degrading Ferroglobus placidus (Holmes et al. 2011), convincingly suggested that benzene is initially carboxylated to benzoate (Fig. 13). However, a recent study with G. metallireducens reported the anoxic hydroxylation of benzene to phenol with water (Zhang et al. 2013) (Fig. 13). In this study, H218O labeling experiments indicated that the hydroxyl functionality of phenol derived from water and not from reactive oxygen species that may be formed by reduced media components during sample preparation. In contrast to the carboxylation scenario, no plausible genes were identified that could be assigned to water-dependent benzene hydroxylation (Zhang et al. 2014). In summary, anaerobic benzene degradation still remains obscure, but recent work suggested that anaerobic benzene activation may be accomplished by different strategies in different organisms thriving at different habitats.

Scenarios for the initial conversion of benzene without oxygen. The two alternative reactions in benzene degrading Fe(III)-reducing/methanogenic enrichment cultures (carboxylation) or G. metallireducens (hydroxylation) are shown. In the case of benzene carboxylation, the induction of ubiD-like genes putatively coding for a benzene carboxylase is in favor for the carboxylation scenario
3 Degradation via Dearomatization of ortho-Substituted Benzoyl-CoAs
A number of aromatic compounds are anaerobically degraded via ortho-substituted benzoyl-CoA analogues including (acetyl-)salicylate, anthranilate, indoleacetate, ortho-xylene, or 2-F-/2-benzoates. Ortho-positioned substituents cannot be removed by specific reductases, but hydroxy- or amino-substituented benzoyl-CoA analogues may be directly converted to intermediates of the benzoyl-CoA degradation pathway after dearomatization by benzoyl-CoA reductases (Fig. 14).

Degradation of MAC via ortho-substituted benzoyl-CoA intermediates
3.1 Degradation of Salicylate and Anthranilate
Growth with salicylate (2-hydroxybenzoate) and anthranilate (2-aminobenzoate) has been reported for denitrifying bacteria and was always initiated by the activation to the corresponding CoA esters by specific and growth substrate-induced AMP forming CoA ligases (Bonting and Fuchs 1996; Lochmeyer et al. 1992). The anthranoyl-CoA and salicyl-CoA formed serve both as direct substrates for class I BCRs that form the corresponding instable ortho-substituted 1,5-dienoyl-CoAs. In the case of 2-hydroxy-1,5-dienoyl-CoA, tautomerization yields 2-oxocylohex-1-ene-1-carboxyl-CoA, which is the substrate for the ring-cleaving hydrolase of the general benzoyl-CoA degradation pathway (Fig. 14). In case of 2-amino-1,5-dienoyl-CoA, the imine-tautomer will spontaneously hydrolyze to 2-oxocylohex-1-en-1-carboxyl-CoA. In summary, both salicylate and anthranilate degradation requires only a specific CoA ligase for channeling the growth substrates into the benzoyl-CoA degradation pathway.
3.2 Degradation of o-Xylene, 2-Methyl-, and 2-Fluorobenzoate
The anaerobic degradation of o-xylene has been demonstrated for several anaerobic cultures and is considered to be initiated by addition to fumarate followed by the oxidation to 2-methylbenzoyl-CoA in analogy to the benzylsuccinate degradation pathway involved in toluene degradation. Reaction of BSS isoenzymes with all three xylene isomers has previously been demonstrated, although it is unclear how the generated methylbenzylsuccinates are further metabolized to the corresponding methylbenzoyl-CoAs (Verfürth et al. 2004). The further degradation of 2-methylbenzoyl-CoA via a modified benzoyl-CoA degradation pathways has not been studied yet. In case of 2-F-benzoate, complete degradation has frequently been reported for denitrifying bacteria that are capable of growing with benzoate. Though standard benzoate-CoA ligase and class I BCRs generally show a high activity with the 2-F-analogues, the enzymes involved in C–F-bond cleavage are unknown. Mechanisms as reported for C-halide cleavage during 3-Cl- or 4-F-benzoyl-CoA reduction by class I BCR are not applicable in case of 2-F-benzoyl-CoA. Thus, C–F-bond cleave has to occur in so far unknown downstream reactions of the benzoyl-CoA degradation pathway.
3.3 Degradation of Indoleacetate via Anthranoyl-CoA
Indoleacetate is a common degradation intermediate of the amino acid tryptophan and serves as an important plant hormone, auxin. Therefore, it occurs in large amounts and serves as growth substrate for bacteria such as Aromatoleum or Azoarcus species, even under anaerobic conditions. Recently, the pathway of anaerobic indoleacetate metabolism in these bacteria was discovered and the enzymes and genes involved were identified (Fig. 15; Ebenau-Jehle et al. 2012; Schühle et al. 2016). The pathway consists of ten successive enzymatic reactions, starting with uptake and activation of indoleacetate to the CoA-thioester using a highly specific CoA ligase (Schühle et al. 2016), followed by hydroxylation at the pyrrole ring to 2-oxoindoleactyl-CoA by a molybdenum enzyme of the xanthine dehydrogenase family and hydrolytic ring opening to yield (2-aminophenyl)succinyl-CoA (Fig. 15). The CoA moiety is then intramolecularly transferred to the other carboxy group by a CoA-transferase (Schühle et al. 2016), allowing the reconfiguration of the molecule to (2-aminobenzyl)malonyl-CoA via a coenzyme B12-dependent mutase (Fig. 15). Finally, the remaining pathway consists of a simultaneous oxidation and decarboxylation to 2-aminocinnamoyl-CoA, followed by β-oxidation steps generating anthranoyl-CoA and acetyl-CoA (Fig. 15).

Conversion of indoleacetate to 2-aminobenzoyl-CoA. The reactions catalyzed by the Iaa-gene products are indicated
4 Degradation via Dearomatization of meta-Substituted Benzoyl-CoAs
A number of MAC containing meta-positioned functionalities are converted to the corresponding meta-substituted benzoyl-CoA analogues (Fig. 16). Non-halogenic substituents in meta-position generally cannot be easily removed during reductive dearomatization, and stable meta-substituted 1,5-dienoyl-CoA analogues are formed by BCRs. For this reason the degradation via meta-substituted benzoates often requires modifications of the benzoyl-CoA degradation pathway which include the synthesis of isoenzymes acting on the meta-substituted benzoyl-CoA analogues and their metabolites in a modified benzoyl-CoA degradation pathway. An exception of this rule was reported for 3-hydroxybenzoate degradation in the fermenting Sporotomaculum hydroxybenzoicum where a reductive dehalogenation of 3-OH-benzoyl-CoA to benzoyl-CoA was reported (Müller and Schink 2000).

Degradation of MAC via meta-substituted benzoyl-CoA intermediates
4.1 Degradation via Dearomatization of 3-Methylbenzoyl-CoA
3-Methylbenzoyl-CoA is suggested to be an intermediate during the degradation of 3-methlybenzoate, m-xylene and o-cresol (Fig. 16). During 3-methylbenzoate degradation in the denitrifying Azoarcus sp. CIB, a specific AMP-forming CoA ligase forms 3-methylbenzoyl-CoA (Juárez et al. 2013). The anaerobic degradation of m-xylene has been demonstrated in several denitrifying and sulfate-reducing pure/enrichment cultures. The initial conversion of m-xylene to 3-methylbenzylsuccinate was reported either by metabolite analyses of the culture medium or by in vitro assays (Krieger et al. 1999; Elshahed et al. 2001; Morasch et al. 2004; Verfürth et al. 2004); 3-methylbenzylsuccinate is then expected to be converted to 3-methylbenzoyl-CoA via a modified benzylsuccinate degradation pathway (Juárez et al. 2013). In the case of o-cresol, carboxylation to 3-methyl-4-hydroxybenzoyl-CoA is suggested to be catalyzed by a phenyl-phosphate carboxylase-like enzyme system (Bisaillon et al. 1991; Rudolphi et al. 1991) (Fig. 16). Reductive dehydroxylation by a 4-hydroxybenzoyl-CoA reductase-like Mo-enzyme then results in 3-methylbenzoyl-CoA formation. In analogy to p-cresol oxidation to 4-hydroxybenzoate, o-cresol could also be oxidized to 2-hydroxybenzoate; however, experimental evidence for such a degradation pathway is lacking so far (Schink et al. 2000).
The genome of Azoarcus sp. CIB contains the 3-methylbenzoate-induced mbd gene cluster comprising the genes encoding a 3-methylbenzoate-inducible class I BCR. The gene product is specifically involved in the reduction of 3-methylbenzoyl-CoA to a methylated 1,5-dienoyl-CoA (Fig. 17) (Juárez et al. 2013). Other 3-methylbenzoate-induced gene products are involved in modified beta-oxidation of CoA ester substrates carrying the additional methyl group (e.g., the methylated 1,5-dienoyl-CoA) (Fig. 17). It is unknown whether 3-methylbenzoyl-CoA reductase from Azoarcus sp. CIB forms the 3-methyl-1,5-dienoyl-CoA or 5-methyl-1,5-dienoyl-CoA isomer or both. Depending on the regioselectivity of the 3-methylbenzoyl-CoA reductase, 2-methylcrotonyl-CoA and/or 2,3-dehydrovaleryl-CoA (2-pentenoyl-CoA) intermediates would be formed by isoenzymes or promiscuous enzymes of the lower benzoyl-CoA degradation pathway. In any case, 3-methylbenzoyl-CoA is expected to be converted to two acetyl-CoA, propionyl-CoA and CO2 (Juárez et al. 2013).

Enzymatic reactions involved in the degradation of 3-methylbenzoyl-CoA to acetyl-CoA, propionyl-CoA, and CO2. The degradation has been studied in Azoarcus sp. CIB
4.2 Degradation via Dearomatization of 3-Hydroxybenzoyl-CoA
The degradation of 3-hydroxybenzoate via dearomatization of 3-hydroxybenzoyl-CoA was studied in the denitrifying T. aromatica, and shown to be initiated by an AMP-forming, specific 3-hydroxybenzoate CoA ligase (Laempe et al. 2001). The activated thioester was then reduced by ATP-dependent class I benzoyl-CoA reductase to either 3-hydroxy-1,5-dienoyl-CoA or 5-hydroxy-1,5-dienoyl-CoA; the latter is expected to tautomerize to the more stable keto-form (Fig. 18). However, the metabolites and enzymes involved in the further degradation of the products of 3-hydroxybenzoyl-CoA reduction are still unknown. It is obvious that the degradation pathway of 3-hydroxybenzoyl-CoA has to differ substantially from that of benzoyl-CoA.

Initial enzymatic reaction involved in the degradation of 3-hydroxybenzoate via 3-hydroxybenzoyl-CoA dearomatization. The degradation has been studied in T. aromatica; the steps involved in modified β-oxidation after ring reduction are still unknown
In the sulfate-reducing Desulfosarcina cetonica, the formation of 3-hydroxybenzylsuccinate from meta-cresol is catalyzed by a fumarate-adding glycyl-radical enzyme (Müller et al. 1999). The further degradation is then similar to that of toluene and results in the formation of 3-hydroxybenzoyl-CoA.
Protocatechuate (3,4-dihydroxybenzoate) is a central intermediate in the aerobic degradation of many aromatic compounds, but also one in the anaerobic degradation of catechol and 3,4-methoxylated aromatic carboxylic acids that may derive from lignin degradation (vanillate, isovanillate, or veratrate). Anaerobic catechol degradation was studied in Desulfobacterium sp. strain Cat2 and in T. aromatica. It appears to be initiated by promiscuous enzymes of anaerobic phenol degradation and involves the phosphorylation to catechuylphosphate followed by carboxylation to protocatechuate (Gorny and Schink 1994b; Ding et al. 2008). Under anoxic conditions, methoxylated aromatic compounds are substrates for methylotrophic acetogens that use corrinoid-dependent O-demethylases to cleave the phenyl methyl ether bond and to transfer the methyl group to tetrahydrofolate (Engelmann et al. 2001). As a result, protocatechuate may be formed, for example, from vanillate. Complete protocatechuate degradation is initiated by the activation to protocatechuyl-CoA by a promiscuous 3-hydroxybenzoate-CoA ligase, followed by reductive dehydroxylation to 3-hydroxybenzoyl-CoA, possibly catalyzed by a promiscuous 4-hydroxybenzoyl-CoA reductase (Gorny and Schink 1994b; Ding et al. 2008).
5 Degradation via Dearomatization of para-Substituted Benzoyl-CoA Derivatives
Insights into the degradation of MAC via dearomatization of non-halogenic/non-hydroxylated para-substituted benzoyl-CoA derivatives have only recently been obtained in studies of 4-methylbenzoate and p-cymene degradation in Magnetospirillum, Aromatoleum, and Thauera strains (Lahme et al. 2012; Strijkstra et al. 2014) (Fig. 19).

Degradation of MAC via para-substituted benzoyl-CoA intermediates
In Magnetospirillum sp. pMbN1, an ATP-dependent class I BCR catalyzed the dearomatization of 4-methylbenzoyl-CoA to 4-methyl-1,5-dienoyl-CoA (Lahme et al. 2012) (Fig. 19). This finding is remarkable as conventional Thauera- and Azoarcus-type BCRs have not been reported to accept para-substituted benzoyl-CoA analogues other than 4-F-benzoyl-CoA (see Sect. 2.4). The 4-methylbenzoyl-CoA converting BCR differs from the typical class I BCRs with respect to substrate preference and amino acid sequence similarity, and it has been suggested that it represents a member of a new subclass of class I BCRs.
A 4-methylbenzoyl-CoA reductase is also expected to be involved in anaerobic p-xylene degradation (Fig. 19). Metabolite analyses in p-xylene-degrading, sulfate-reducing, and denitrifying enrichment cultures revealed that 4-methylbenzylsuccinate is an intermediate during p-xylene degradation (Morasch and Meckenstock 2005; Rotaru et al. 2010). This finding suggests that the degradation pathway of p-xylene is similar to that of toluene: after initial addition of p-xylene to fumarate by a glycyl-radical enzyme homologous to benzylsuccinate synthase, the 4-methylbenylsuccinate formed is then oxidized to 4-methylbenzoyl-CoA by a set of enzymes catalyzing β-oxidation like reactions similar to those involved in benzylsuccinate conversion to benzoyl-CoA.
5.1 Enzymatic Reactions Involved in 4-Methylbenzoate Degradation
The catabolism of 4-methylbenzoate has been elucidated in recent studies with Magnetospirillum pMbN1 by proteogenomic analyses, metabolite analyses, and in vitro enzyme assays (Lahme et al. 2012) (Fig. 20). After activation of 4-methylbenzoate by an AMP-forming 4-methylbenzoyl-CoA synthetase, a specific 4-methylbenzoyl-CoA reductase (class I BCR) formed 4-methyl-1,5-dienoyl-CoA in an ATP- and electron donor-dependent manner. In the following, a series of β-oxidation-like reactions similar to those of the standard benzoyl-CoA degradation pathway converted 4-methyl-1,5-dienoyl-CoA to central intermediates. Similar to 3-methylbenzoyl-CoA the methyl group is retained during the entire pathway. However, due to the different positions of the methyl-group in the ring, the 4-methylbenzoyl-CoA degradation pathway is suggested to yield acetoacetate and acetyl-CoA, whereas propionyl-CoA, acetyl-CoA, and CO2 are proposed to be formed from 3-methylbenzoyl-CoA.

Enzymatic reactions involved in the anaerobic degradation of 4-methylbenzoate. The degradation has been studied in Magnetospirillum pMbN1
5.2 Enzymatic Reactions Involved in p-Cymene Degradation
The degradation of p-cymene (4-isopropyltoluene) was studied in a denitrifying A. aromaticum and a Thauera sp. strain related to T. terpenica (Strijkstra et al. 2014). Surprisingly, both use different strategies for initiation of complete p-cymene degradation (Fig. 21).

Enzymatic reactions involved in the conversion of p-cymene to 4-isopropylbenzoyl-CoA. The upper pathway was identified in the p-cymene-degrading A. aromaticum strain pCyN1 and proceeds via anoxic hydroxylation to 4-isopropylbenzyl alcohol by a Mo-enzyme related to ethylbenzene dehydrogenase. The alternative lower route is initiated by addition of p-cymene to fumarate similar to addition of toluene to fumarate by benzylsuccinate synthase; it was found in the p-cymene-degrading Thauera strain pCyN2. The enzymes involved have not been studied, yet, and the further degradation of 4-isopropylbenzoyl-CoA is unknown
In the A. aromaticum strain pCyN1, a Mo-enzyme similar to ethylbenzene dehydrogenase catalyzed the hydroxylation of p-cymene to 4-isopropylbenzyl alcohol with water. The presumptive p-cymene dehydrogenase is encoded by the cmdABC genes, and a mechanism similar to that established for ethylbenzene dehydrogenase has been proposed. The further conversion of 4-isopropylbenzyl alcohol is then likely to be accomplished by two dehydrogenases that oxidize the alcohol stepwise to the carboxylic acid; finally, an AMP-forming CoA ligase activates 4-isopropylbenzoate to a thioester (Fig. 21).
An alternative pathway has been identified in the p-cymene degrading Thauera strain pCyN2, where p-cymene was initially converted to 4-isopropylbenzylsuccinate; most likely by addition to fumarate catalyzed by a glycyl radical enzyme. Proteogenomic analyses suggested a pathway in which 4-isopropylbenzylsuccinate is oxidized to 4-isopropylbenzoyl-CoA, similar to the benzylsuccinate degradation pathway involved in toluene degradation. The further degradation of the branched chain containing 4-isopropylbenzoyl-CoA remains to be studied; the presence of a novel modification of the benzoyl-CoA degradation pathway is anticipated (Fig. 21).
6 Degradation via Dearomatization of Di- and Trihydroxybenzenes
Next to benzoyl-CoA and derivatives of it, three di- and trihydroxybenzenes with at least two meta-positioned phenolic hydroxy-functionalities serve as substrates for dearomatizing enzymes: resorcinol, hydroxyhydroquinone, and phloroglucinol. The possibility of these intermediates to tautomerize to 1,3-diketones strongly weakens the aromatic character of these compounds and, consequently, common biological electron donors such as reduced ferredoxin or NAD(P)H serve directly as electron donors without the need for coupling the ring reduction to an exergonic reaction (Schink et al. 2000; Fuchs et al. 2011). In case of hydroxyhydroquinone degradation, even an oxidative dearomatization of the aromatic ring is possible – the only such example under anoxic conditions. The dearomatized cyclic 1,3-diketones can then be cleaved hydrolytically, and the acyclic products are converted to central intermediates (Fig. 22).

Degradation of MAC via resorcinol, hydroxyhydroquinone, and phloroglucinol
6.1 Degradation via Dearomatization of Resorcinol
The anaerobic degradation of 2,4- and 2,6-resorcyclic acids (β- and γ-resorcyclic acids) has initially been studied in a fermenting Clostridium co-culture (Tschech and Schink 1985; Kluge et al. 1990). After decarboxylation by specific enzymes, the resorcinol (1,3-dihydroxybenzene) formed was found to be reduced to 1,3-cyclohexadione by resorcinol reductase (Fig. 22). The purified enzyme contains a FAD cofactor and probably uses a reduced ferredoxin as electron donor (Schink et al. 2000). The 1,3-cyclohexadione product is finally hydrolytically cleaved to 2-oxocaproate, which is then finally converted to the fermentation products acetate and butyrate. As an alternative, resorcinol can be converted via hydroxyhydroquinone (see Sect. 6.2).
6.2 Degradation via Dearomatization of Hydroxyhydroquinone
Hydroxyhydroquinone (1,2,4-trihydroxybenzene) serves as a substrate for different dearomatizing enzymes. It may be formed via anoxic hydroxylation of resorcinol (Philipp and Schink 1998), or by hydroxylation of 3,5-dihydroxybenzoate (α−resorcylate) to a trihydroxybenzoate, followed by decarboxylation (Gallus and Schink 1998). Resorcinol hydroxylase was studied in the denitrifying Azoarcus anaerobius strain LuFRes1, and the genes encoding a Mo-enzyme with similarities to pyrogallol-phloroglucinol transhydroxylase of Pelobacter acidigallici were identified (Darley et al. 2007) (see Sect. 6.3).
There are three strategies for hydroxyhydroquinone dearomatization that have been identified in different physiological classes of anaerobic bacteria. The oxidative pathway was identified in the denitrifying Azoarcus anaerobius and involves a membrane bound hydroxybenzoquinone-forming dehydrogenase. Finally, a series of noncharacterized reactions yield acetate and malate (Philipp and Schink 1998) (Fig. 23).

Catabolism of resorcinol via the oxidative hydroxyhydroquinone degradation pathway
Alternatively, hydroxyhydroquinone is dearomatized by reduction to dihydro-hydroxyhydroquinone in the sulfate-reducing Desulfovibrio inopinatus (Reichenbecher et al. 2000). The product is then further oxidized to acetate and to a second nonidentified compound. Finally, a series of at least three consecutive transhydroxylation reactions were suggested to be involved to form phloroglucinol from hydroxyhydroquinone in Pelobacter massiliensis. Mo-containing enzymes, similar to pyrogallol transhydroxylase (see Sect. 6.3), were suggested to be involved (Brune et al. 1992). The phloroglucinol formed is then oxidized to three acetate.
6.3 Degradation via Dearomatization of Phloroglucinol
Phloroglucinol is an important intermediate during the anaerobic degradation of tannins and flavonoids derived from plants. For example, degradation of hydrolysable tannins yields gallate (3,4,5-trihydroxybenzoate), which can easily be decarboxylated to pyrogallol (1,2,3-trihydroxybenzene) (Schink and Pfennig 1982). This compound is not feasible for direct reduction but can be converted to phloroglucinol catalyzed by the molybdenum enzyme pyrogallol transhydroxylase (Fig. 24). Phloroglucinol is then dearomatized by NADPH-dependent reduction to dihydrophloroglucinol. The hydrolytic cleavage of the latter results in the formation of 3-hydroxy-5-oxohexanoic acid; in fermenting bacteria, the latter is converted to three acetate molecules as fermentation end products (Schink et al. 2000).

Degradation of pyrogallol via the phloroglucinol degradation pathway. Pyrogallol degradation is initiated by intermolecular hydroxyl transfer catalyzed by the Mo-enzyme transhydroxylase. An enzyme-bound 1,3,4,5-tetrahydroxybenzene (in red) serves as cofactor, which after hydroxyl transfer to pyrogallol is released as product. The cofactor is regenerated by the hydroxyl accepting pyrogallol
The isomerization of pyrogallol to phloroglucinol is catalyzed by the Mo-enzyme transhydroxylase that has been isolated and studied in detail in the fermenting Pelobacter acidigallici (Reichenbecher et al. 1994). The transhydroxylase belongs to the dimethylsulfoxid reductase family of Mo-enzymes and requires 1,3,4,5-tetrahydroxybenzene as a co-substrate (Fig. 24). The crystal structure in conjunction with the chemical synthesis of reaction intermediates gave rise to a hexahydroxydiphenyl ether intermediate in the course of catalysis (Messerschmidt et al. 2004; Paizs et al. 2007). The ether is formed by covalently linking the substrate phloroglucinol and the cofactor 1,3,4,5-tetrahydroxybenzene. Its cleavage releases the initial co-substrate as phloroglucinol, whereas the pyrogallol is converted to the co-substrate. In summary, the transhydroxylase catalyzes an intermolecular hydroxyl transfer between two phenolic compounds.
Phloroglucinol reductase has been isolated and characterized in some detail from the fermenting rumen bacterium Eubacterium oxidoreducens (Haddock and Ferry 1989). The monomeric 78 kDa enzyme specifically uses NADPH as electron donor, but does not depend on any other cofactor.
7 Research Needs
In the past 20 years, our knowledge in the field of anaerobic MAC degradation has increased tremendously, and the function of some of the previously enigmatic key enzymes is now understood on the molecular level. The unique, but meanwhile well-characterized processes comprise the anaerobic hydroxylation of alkyl chains from aromatic hydrocarbons with water by Mo- or flavo-enzymes, or the addition of alkyl chains to fumarate by glycyl radical enzymes. In addition, class I BCRs emerged as biocatalysts with a much broader function than previously anticipated, and a number of MAC degradation pathway depend on the catalytic versatility of class I BCRs. Next to benzoyl-CoA, these enzymes accept numerous analogues as substrates for reductive dearomatization, and depending whether substituents can be removed or not by class I BCRs, modified dienoyl-CoA degradation pathways are induced. Moreover, the capacity to dehalogenate fluorinated or chlorinated benzoyl-CoA analogues appears to be a key process for the complete degradation of haloaromatics in anaerobic bacteria. The lack of a crystal structure, however, still hampers detailed knowledge of the function of class I BCRs. Although valuable insights into W-cofactor containing active site of class II BCRs have been obtained, novel crucial questions about these enzymes have emerged. One of the most intriguing questions is the energetic coupling of endergonic benzoyl-CoA reduction in the ATP-independent class II BCRs; experimental evidence for the proposed and plausible flavin-based electron bifurcation is still lacking.
The probably least understood process in anaerobic MAC degradation is the initial reaction involved in anaerobic benzene degradation. While the previously favored methylation is meanwhile rather considered as unlikely, there is evidence for two completely different scenarios for the initial attack of the extremely stable C–H bond of benzene: the carboxylation by an UbiD-like enzyme and the hydroxylation with water by an unknown enzyme. The former scenario appears to be plausible due to the identification of induced UbiD-like genes during anaerobic growth with benzene. In this context, the previously obtained insights obtained into the role of UbiD/UbiX-enzyme system in decarboxylation of aromatic carboxylic acids could open the door to understand the carboxylation of nonsubstituted aromatic compounds by related enzymes. It is possible that either initiation reaction, benzene carboxylation or hydroxylation may occur in different anaerobic benzene degrading organisms, dependent on their phylogenetic positions.
In addition to the importance of anaerobic MAC degradation for the biological removal of environmentally hazardous compounds, a number of the enzymatic reactions involved may be useful for biotechnological applications. These comprise the formation of chiral alcohols, either via stereospecific hydroxylation of alkyl side chains or by reduction of the ketones formed, the carboxylation of phenol and related reactions, enzymatic Birch reductions, and many other reactions. Thus, anaerobic MAC degradation has indeed emerged as a treasure trove for the discovery of new biocatalysts.
References
Abu Laban N, Selesi D, Jobelius C, Meckenstock RU (2009) Anaerobic benzene degradation by gram-positive sulfate-reducing bacteria. FEMS Microbiol Ecol 68:300–311
Abu Laban N, Selesi D, Rattei T, Tischler P, Meckenstock RU (2010) Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ Microbiol 12:2783–2796
Ahn Y, Chae J, Zylstra GJ, Häggblom MM (2009) Degradation of phenol via phenylphosphate and carboxylation to 4-hydroxybenzoate by a newly isolated strain of the sulfate-reducing bacterium Desulfobacterium anilini. Appl Environ Microbiol 75:4248–4253
Aklujkar M, Risso C, Smith J, Beaulieu D, Dubay R, Giloteaux L, DiBurro K, Holmes D (2014) Anaerobic degradation of aromatic amino acids by the hyperthermophilic archaeon Ferroglobus placidus. Microbiology 160:2694–2709
Ball HA, Johnson HA, Reinhard M, Spormann AM (1996) Initial reactions in anaerobic ethylbenzene oxidation by a denitrifying bacterium, strain EB1. J Bacteriol 178:5755–5761
Barker HA (1981) Amino acid degradation by anaerobic bacteria. Annu Rev Biochem 50:23–40
Biegert T, Altenschmidt U, Eckerskorn C, Fuchs G (1993) Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoate-CoA ligase from a denitrifying Pseudomonas species. Eur J Biochem 213:555–561
Bisaillon JG, Lépine F, Beaudet R, Sylvestre M (1991) Carboxylation of o-cresol by an anaerobic consortium under methanogenic conditions. Appl Environ Microbiol 57:2131–2134
Boll M, Fuchs G (1995) Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem 234:921–933
Boll M, Fuchs G (1998) Identification and characterization of the natural electron donor ferredoxin and of FAD as a possible prosthetic group of benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. Eur J Biochem 251:946–954
Boll M, Albracht SS, Fuchs G (1997) Benzoyl-CoA reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. A study of adenosinetriphosphatase activity, ATP stoichiometry of the reaction and EPR properties of the enzyme. Eur J Biochem 244:840–851
Boll M, Fuchs G, Meier C, Trautwein A, El Kasmi A, Ragsdale SW, Buchanan G, Lowe DJ (2001) Redox centers of 4-hydroxybenzoyl-CoA reductase, a member of the xanthine oxidase family of molybdenum-containing enzymes. J Biol Chem 276:47853–47862
Boll M, Löffler C, Morris BE, Kung JW (2014) Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: organisms, strategies and key enzymes. Environ Microbiol 16:612–627
Bonting CF, Fuchs G (1996) Anaerobic metabolism of 2-hydroxybenzoic acid (salicylic acid) by a denitrifying bacterium. Arch Microbiol 165:402–408
Brackmann R, Fuchs G (1993) Enzymes of anaerobic metabolism of phenolic compounds. 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from a denitrifying Pseudomonas species. Eur J Biochem 213:563–571
Bräsen C, Schönheit P (2004) Unusual ADP-forming acetyl-coenzyme A synthetases from the mesophilic halophilic euryarchaeon Haloarcula marismortui and from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Arch Microbiol 182:277–287
Breese K, Fuchs G (1998) 4-Hydroxybenzoyl-CoA reductase (dehydroxylating) from the denitrifying bacterium Thauera aromatica–prosthetic groups, electron donor, and genes of a member of the molybdenum-flavin-iron-sulfur proteins. Eur J Biochem 251:916–923
Breinig S, Schiltz E, Fuchs G (2000) Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J Bacteriol 182:5849–5863
Brune A, Schnell S, Schink B (1992) Sequential transhydroxylations converting hydroxyhydroquinone to phloroglucinol in the strictly anaerobic, fermentative bacterium Pelobacter massiliensis. Appl Environ Microbiol 58:1861–1868
Buckel W, Thauer RK (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113
Buckel W, Zhang J, Friedrich P, Parthasarathy A, Li H, Djurdjevic I, Dobbek H, Martins BM (2012) Enzyme catalyzed radical dehydrations of hydroxy acids. Biochim Biophys Acta 1824:1278–1290
Buckel W, Kung JW, Boll M (2014) The benzoyl-coenzyme a reductase and 2-hydroxyacyl-coenzyme a dehydratase radical enzyme family. Chembiochem 15:2188–2194
Carmona M, Zamarro MT, Blázquez B, Durante-Rodríguez G, Juárez JF, Valderrama JA, Barragán MJL, García JL, Díaz E (2009) Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol Mol Biol Rev 73:71–133
Cunane LM, Chen ZW, Shamala N, Mathews FS, Cronin CN, McIntire WS (2000) Structures of the flavocytochrome p-cresol methylhydroxylase and its enzyme-substrate complex: gated substrate entry and proton relays support the proposed catalytic mechanism. J Mol Biol 295:357–374
Darley PI, Hellstern JA, Medina-Bellver JI, Marqués S, Schink B, Philipp B (2007) Heterologous expression and identification of the genes involved in anaerobic degradation of 1,3-dihydroxybenzene (resorcinol) in Azoarcus anaerobius. J Bacteriol 189:3824–3833
Debnar-Daumler C, Seubert A, Schmitt G, Heider J (2014) Simultaneous involvement of a tungsten-containing aldehyde: ferredoxin oxidoreductase and a phenylacetaldehyde dehydrogenase in anaerobic phenylalanine metabolism. J Bacteriol 196:483–492
Dermer J, Fuchs G (2012) Molybdoenzyme that catalyzes the anaerobic hydroxylation of a tertiary carbon atom in the side chain of cholesterol. J Biol Chem 287:36905–36916
Díaz E, Jiménez JI, Nogales J (2013) Aerobic degradation of aromatic compounds. Curr Opin Biotechnol 24:431–442
Dickert S, Pierik AJ, Buckel W (2002) Molecular characterization of phenyllactate dehydratase and its initiator from Clostridium sporogenes. Mol Microbiol 44:49–60
Ding B, Schmeling S, Fuchs G (2008) Anaerobic metabolism of catechol by the denitrifying bacterium Thauera aromatica–a result of promiscuous enzymes and regulators? J Bacteriol 190:1620–1630
Ebenau-Jehle C, Thomas M, Scharf G, Kockelkorn D, Knapp B, Schühle K, Heider J, Fuchs G (2012) Anaerobic metabolism of indoleacetate. J Bacteriol 194:2894–2903
Ebenau-Jehle C, Mergelsberg M, Fischer S, Brüls T, Jehmlich N, von Bergen M, Boll M (2016) An unusual strategy for the anoxic biodegradation of phthalate. ISME J 11:224. https://doi.org/10.1038/ismej.2016.91
Egland PG, Pelletier DA, Dispensa M, Gibson J, Harwood CS (1997) A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc Natl Acad Sci USA 94:6484–6489
Egland PG, Gibson J, Harwood CS (2001) Reductive, coenzyme A-mediated pathway for 3-chlorobenzoate degradation in the phototrophic bacterium Rhodopseudomonas palustris. Appl Environ Microbiol 67:1396–1399
Elshahed MS, Gieg LM, Mcinerney MJ, Suflita JM (2001) Signature metabolites attesting to the in situ attenuation of alkylbenzenes in anaerobic environments. Environ Sci Technol 35:682–689
Engelmann T, Kaufmann F, Diekert G (2001) Isolation and characterization of a veratrol: corrinoid protein methyl transferase from Acetobacterium dehalogenans. Arch Microbiol 175:376–383
Evans PJ, Ling W, Goldschmidt B, Ritter ER, Young LY (1992) Metabolites formed during anaerobic transformation of toluene and o-xylene and their proposed relationship to the initial steps of toluene mineralization. Appl Environ Microbiol 58:496–501
Fuchs G, Boll M, Heider J (2011) Microbial degradation of aromatic compounds – from one strategy to four. Nat Rev Microbiol 9:803–816
Funk MA, Marsh E, Neil G, Drennan CL (2015) Substrate-bound structures of benzylsuccinate synthase reveal how toluene is activated in anaerobic hydrocarbon degradation. J Biol Chem 290:22398–22408
Gallus C, Schink B (1998) Anaerobic degradation of alpha-resorcylate by Thauera aromatica strain AR-1 proceeds via oxidation and decarboxylation to hydroxyhydroquinone. Arch Microbiol 169:333–338
Gorny N, Schink B (1994a) Complete anaerobic oxidation of hydroquinone by Desulfococcus sp. strain Hy5: indications of hydroquinone carboxylation to gentisate. Arch Microbiol 162:131–135
Gorny N, Schink B (1994b) Anaerobic degradation of catechol by Desulfobacterium sp. strain Cat2 proceeds via carboxylation to protocatechuate. Appl Environ Microbiol 60:3396–3400
Gorny N, Schink B (1994c) Hydroquinone degradation via reductive dehydroxylation of gentisyl-CoA by a strictly anaerobic fermenting bacterium. Arch Microbiol 161:25–32
Haddock JD, Ferry JG (1989) Purification and properties of phloroglucinol reductase from Eubacterium oxidoreducens G-41. J Biol Chem 264:4423–4427
Harayama S, Kok M, Neidle EL (1992) Functional and evolutionary relationships among diverse oxygenases. Annu Rev Microbiol 46:565–601
Harwood CS, Parales RE (1996) The beta-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol 50:553–590
Heider J (2001) A new family of CoA-transferases. FEBS Lett 509:345–349
Heider J, Fuchs G (1997a) Anaerobic metabolism of aromatic compounds. Eur J Biochem 243:577–596
Heider J, Fuchs G (1997b) Microbial anaerobic aromatic metabolism. Anaerobe 3:1–22
Heider J, Schühle K (2013) Anaerobic biodegradation of hydrocarbons including methane. In: Rosenberg E, Delong E, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: prokaryotic physiology and biochemistry. Springer, Heidelberg, pp 601–630
Heider J, Spormann AM, Beller HR, Widdel F (1998) Anaerobic bacterial metabolism of hydrocarbons. FEMS Microbiol Rev 22:459–473
Heider J, Schühle K, Frey J, Schink B (2016a) Activation of acetone and other simple ketones in anaerobic bacteria. J Mol Microbiol Biotechnol 26:152–164
Heider J, Szaleniec M, Martins BM, Seyhan D, Buckel W, Golding BT (2016b) Structure and function of benzylsuccinate synthase and related fumarate-adding glycyl radical enzymes. J Mol Microbiol Biotechnol 26:29–44
Heider J, Szaleniec M, Sünwoldt K, Boll M (2016c) Ethylbenzene dehydrogenase and related molybdenum enzymes involved in oxygen-independent alkyl chain hydroxylation. J Mol Microbiol Biotechnol 26:45–62
Hirsch W, Schägger H, Fuchs G (1998) Phenylglyoxylate:NAD+ oxidoreductase (CoA benzoylating), a new enzyme of anaerobic phenylalanine metabolism in the denitrifying bacterium Azoarcus evansii. Eur J Biochem 251:907–915
Holmes DE, Risso C, Smith JA, Lovley DR (2011) Anaerobic oxidation of benzene by the hyperthermophilic archaeon Ferroglobus placidus. Appl Environ Microbiol 77:5926–5933
Hopper DJ, Taylor DG (1977) The purification and properties of p-cresol-(acceptor) oxidoreductase (hydroxylating), a flavocytochrome from Pseudomonas putida. Biochem J 167:155–162
Hopper DJ, Bossert ID, Rhodes-Roberts ME (1991) p-cresol methylhydroxylase from a denitrifying bacterium involved in anaerobic degradation of p-cresol. J Bacteriol 173:1298–1301
Hug LA, Maphosa F, Leys D, Löffler FE, Smidt H, Edwards EA, Adrian L (2013) Overview of organohalide-respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans R Soc B Biol Sci 368:20120322
Jobst B, Schühle K, Linne U, Heider J (2010) ATP-dependent carboxylation of acetophenone by a novel type of carboxylase. J Bacteriol 192:1387–1394
Johannes J, Bluschke A, Jehmlich N, von Bergen M, Boll M (2008) Purification and characterization of active-site components of the putative p-cresol methylhydroxylase membrane complex from Geobacter metallireducens. J Bacteriol 190:6493–6500
Juárez JF, Zamarro MT, Eberlein C, Boll M, Carmona M, Díaz E (2013) Characterization of the mbd cluster encoding the anaerobic 3-methylbenzoyl-CoA central pathway. Environ Microbiol 15:148–166
Junghare M, Spiteller D, Schink B (2016) Enzymes involved in the anaerobic degradation of ortho-phthalate by the nitrate-reducing bacterium Azoarcus sp. strain PA01. Environ Microbiol 18:3175. https://doi.org/10.1111/1462-2920.13447
Kaster A, Moll J, Parey K, Thauer RK (2011) Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci USA 108:2981–2986
Kazumi J, Häggblom MM, Young LY (1995) Diversity of anaerobic microbial processes in chlorobenzoate degradation: nitrate, iron, sulfate and carbonate as electron acceptors. Appl Microbiol Biotechnol 43:929–936
Kloer DP, Hagel C, Heider J, Schulz GE (2006) Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure 14:1377–1388
Kluge C, Tschech A, Fuchs G (1990) Anaerobic metabolism of resorcylic acids (m-dihydroxybenzoic acids) and resorcinol (1,3-benzenediol) in a fermenting and in a denitrifying bacterium. Arch Microbiol 155:68–74
Kniemeyer O, Heider J (2001a) Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J Biol Chem 276:21381–21386
Kniemeyer O, Heider J (2001b) (S)-1-phenylethanol dehydrogenase of Azoarcus sp. strain EbN1, an enzyme of anaerobic ethylbenzene catabolism. Arch Microbiol 176:129–135
Koch J, Eisenreich W, Bacher A, Fuchs G (1993) Products of enzymatic reduction of benzoyl-CoA, a key reaction in anaerobic aromatic metabolism. Eur J Biochem 211:649–661
Krieger CJ, Beller HR, Reinhard M, Spormann AM (1999) Initial reactions in anaerobic oxidation of m-xylene by the denitrifying bacterium Azoarcus sp. strain T. J Bacteriol 181:6403–6410
Kunapuli U, Griebler C, Beller HR, Meckenstock RU (2008) Identification of intermediates formed during anaerobic benzene degradation by an iron-reducing enrichment culture. Environ Microbiol 10:1703–1712
Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, van Dorsselaer A, Friedrich T, Boll M (2009) Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc Natl Acad Sci USA 106:17687–17692
Kung JW, Baumann S, von Bergen M, Müller M, Hagedoorn P, Hagen WR, Boll M (2010) Reversible biological Birch reduction at an extremely low redox potential. J Am Chem Soc 132:9850–9856
Kuntze K, Shinoda Y, Moutakki H, McInerney MJ, Vogt C, Richnow H, Boll M (2008) 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ Microbiol 10:1547–1556
Kuntze K, Vogt C, Richnow H, Boll M (2011a) Combined application of PCR-based functional assays for the detection of aromatic-compound-degrading anaerobes. Appl Environ Microbiol 77:5056–5061
Kuntze K, Kiefer P, Baumann S, Seifert J, von Bergen M, Vorholt JA, Boll M (2011b) Enzymes involved in the anaerobic degradation of meta-substituted halobenzoates. Mol Microbiol 82:758–769
Lack A, Fuchs G (1994) Evidence that phenol phosphorylation to phenylphosphate is the first step in anaerobic phenol metabolism in a denitrifying Pseudomonas sp. Arch Microbiol 161:132–139
Laempe D, Eisenreich W, Bacher A, Fuchs G (1998) Cyclohexa-1,5-diene-1-carbonyl-CoA hydratase [corrected], an enzyme involved in anaerobic metabolism of benzoyl-CoA in the denitrifying bacterium Thauera aromatica. Eur J Biochem 255:618–627
Laempe D, Jahn M, Fuchs G (1999) 6-Hydroxycyclohex-1-ene-1-carbonyl-CoA dehydrogenase and 6-oxocyclohex-1-ene-1-carbonyl-CoA hydrolase, enzymes of the benzoyl-CoA pathway of anaerobic aromatic metabolism in the denitrifying bacterium Thauera aromatica. Eur J Biochem 263:420–429
Laempe D, Jahn M, Breese K, Schägger H, Fuchs G (2001) Anaerobic metabolism of 3-hydroxybenzoate by the denitrifying bacterium Thauera aromatica. J Bacteriol 183:968–979
Lahme S, Eberlein C, Jarling R, Kube M, Boll M, Wilkes H, Reinhardt R, Rabus R (2012) Anaerobic degradation of 4-methylbenzoate via a specific 4-methylbenzoyl-CoA pathway. Environ Microbiol 14:1118–1132
Leuthner B, Heider J (2000) Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of β-oxidation of the intermediate benzylsuccinate. J Bacteriol 182:272–277
Leuthner B, Leutwein C, Schulz H, Hörth P, Haehnel W, Schiltz E, Schägger H, Heider J (1998) Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol Microbiol 28:615–628
Lochmeyer C, Koch J, Fuchs G (1992) Anaerobic degradation of 2-aminobenzoic acid (anthranilic acid) via benzoyl-coenzyme A (CoA) and cyclohex-1-enecarboxyl-CoA in a denitrifying bacterium. J Bacteriol 174:3621–3628
Löffler C, Kuntze K, Vazquez JR, Rugor A, Kung JW, Böttcher A, Boll M (2011) Occurrence, genes and expression of the W/Se-containing class II benzoyl-coenzyme A reductases in anaerobic bacteria. Environ Microbiol 13:696–709
Luo F, Gitiafroz R, Devine CE, Gong Y, Hug LA, Raskin L, Edwards EA (2014) Metatranscriptome of an anaerobic benzene-degrading, nitrate-reducing enrichment culture reveals involvement of carboxylation in benzene ring activation. Appl Environ Microbiol 80:4095–4107
Mai X, Adams MW (1994) Indolepyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus. A new enzyme involved in peptide fermentation. J Biol Chem 269:16726–16732
Mai X, Adams MW (1996) Purification and characterization of two reversible and ADP-dependent acetyl coenzyme A synthetases from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 178:5897–5903
McIntire W, Hopper DJ, Singer TP (1985) p-cresol methylhydroxylase. Assay and general properties. Biochem J 228:325–335
Meckenstock RU, Boll M, Mouttaki H, Koelschbach JS, Cunha Tarouco P, Weyrauch P, Dong X, Himmelberg AM (2016) Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J Mol Microbiol Biotechnol 26:92–118
Messerschmidt A, Niessen H, Abt D, Einsle O, Schink B, Kroneck PM (2004) Crystal structure of pyrogallol-phloroglucinol transhydroxylase, an Mo enzyme capable of intermolecular hydroxyl transfer between phenols. Proc Natl Acad Sci USA 101:11571–11576
Möbitz H, Boll M (2002) A Birch-like mechanism in enzymatic benzoyl-CoA reduction: a kinetic study of substrate analogues combined with an ab initio model. Biochemistry 41:1752–1758
Morasch B, Meckenstock RU (2005) Anaerobic degradation of p-xylene by a sulfate-reducing enrichment culture. Curr Microbiol 51:127–130
Morasch B, Schink B, Tebbe CC, Meckenstock RU (2004) Degradation of o-xylene and m-xylene by a novel sulfate-reducer belonging to the genus Desulfotomaculum. Arch Microbiol 181:407–417
Muhr E, Schühle K, Clermont L, Sünwoldt K, Kleinsorge D, Seyhan D, Kahnt J, Schall I, Cordero PR, Schmitt G, Heider J (2015) Enzymes of anaerobic ethylbenzene and p-ethylphenol catabolism in ‘Aromatoleum aromaticum’: differentiation and differential induction. Arch Microbiol 197:1051–1062
Muhr E, Leicht O, González Sierra S, Thanbichler M, Heider J (2016) A fluorescent bioreporter for acetophenone and 1-phenylethanol derived from a specifically induced catabolic operon. Front Microbiol 6:1561
Müller JA, Schink B (2000) Initial steps in the fermentation of 3-hydroxybenzoate by Sporotomaculum hydroxybenzoicum. Arch Microbiol 173:288–295
Müller JA, Galushko AS, Kappler A, Schink B (1999) Anaerobic degradation of m-cresol by Desulfobacterium cetonicum is initiated by formation of 3-hydroxybenzylsuccinate. Arch Microbiol 172:287–294
Müller JA, Galushko AS, Kappler A, Schink B (2001) Initiation of anaerobic degradation of p-cresol by formation of 4-hydroxybenzylsuccinate in Desulfobacterium cetonicum. J Bacteriol 183:752–757
Narmandakh A, Gad’on N, Drepper F, Knapp B, Haehnel W, Fuchs G (2006) Phosphorylation of phenol by phenylphosphate synthase: role of histidine phosphate in catalysis. J Bacteriol 188:7815–7822
Nobu MK, Narihiro T, Hideyuki T, Qiu Y, Sekiguchi Y, Woyke T, Goodwin L, Davenport KW, Kamagata Y, Liu W (2015) The genome of Syntrophorhabdus aromaticivorans strain UI provides new insights for syntrophic aromatic compound metabolism and electron flow. Environ Microbiol 17:4861–4872
Paizs C, Bartlewski-Hof U, Rétey J (2007) Investigation of the mechanism of action of pyrogallol-phloroglucinol transhydroxylase by using putative intermediates. Chemistry 13:2805–2811
Parthasarathy A, Kahnt J, Chowdhury NP, Buckel W (2013) Phenylalanine catabolism in Archaeoglobus fulgidus VC-16. Arch Microbiol 195:781–797
Payne KA, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SE, Scrutton NS, Hay S, Leys D (2015) New cofactor supports α,β-unsaturated acid decarboxylation via 1,3-dipolar cycloaddition. Nature 522:497–501
Peters F, Shinoda Y, McInerney MJ, Boll M (2007) Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J Bacteriol 189:1055–1060
Philipp B, Schink B (1998) Evidence of two oxidative reaction steps initiating anaerobic degradation of resorcinol (1,3-dihydroxybenzene) by the denitrifying bacterium Azoarcus anaerobius. J Bacteriol 180:3644–3649
Philipp B, Schink B (2012) Different strategies in anaerobic biodegradation of aromatic compounds: nitrate reducers versus strict anaerobes. Environ Microbiol Rep 4:469–478
Porter AW, Young LY (2013) The bamA gene for anaerobic ring fission is widely distributed in the environment. Front Microbiol 4:302
Qiu Y, Sekiguchi Y, Imachi H, Kamagata Y, Tseng I, Cheng S, Ohashi A, Harada H (2004) Identification and isolation of anaerobic, syntrophic phthalate isomer-degrading microbes from methanogenic sludges treating wastewater from terephthalate manufacturing. Appl Environ Microbiol 70:1617–1626
Qiu Y, Sekiguchi Y, Hanada S, Imachi H, Tseng I, Cheng S, Ohashi A, Harada H, Kamagata Y (2006) Pelotomaculum terephthalicum sp. nov. and Pelotomaculum isophthalicum sp. nov.: two anaerobic bacteria that degrade phthalate isomers in syntrophic association with hydrogenotrophic methanogens. Arch Microbiol 185:172–182
Qiu Y, Hanada S, Ohashi A, Harada H, Kamagata Y, Sekiguchi Y (2008) Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl Environ Microbiol 74:2051–2058
Rabus R, Heider J (1998) Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate-reducing bacteria. Arch Microbiol 170:377–384
Rabus R, Widdel F (1995) Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch Microbiol 163:96–103
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rabus R, Boll M, Golding B, Wilkes H (2016a) Anaerobic degradation of p-alkylated benzoates and toluenes. J Mol Microbiol Biotechnol 26:63–75
Rabus R, Boll M, Heider J, Meckenstock RU, Buckel W, Einsle O, Ermler U, Golding BT, Gunsalus RP, Kroneck PM, Krüger M, Lueders T, Martins BM, Musat F, Richnow HH, Schink B, Seifert J, Szaleniec M, Treude T, Ullmann GM, Vogt C, von Bergen M, Wilkes H (2016b) Anaerobic microbial degradation of hydrocarbons: from enzymatic reactions to the environment. J Mol Microbiol Biotechnol 26:5–28
Reichenbecher W, Brune A, Schink B (1994) Transhydroxylase of Pelobacter acidigallici: a molybdoenzyme catalyzing the conversion of pyrogallol to phloroglucinol. Biochim Biophys Acta 1204:217–224
Reichenbecher W, Philipp B, Suter MJ, Schink B (2000) Hydroxyhydroquinone reductase, the initial enzyme involved in the degradation of hydroxyhydroquinone (1,2,4-trihydroxybenzene) by Desulfovibrio inopinatus. Arch Microbiol 173:206–212
Rhee SK, Fuchs G (1999) Phenylacetyl-CoA: acceptor oxidoreductase, a membrane-bound molybdenum-iron-sulfur enzyme involved in anaerobic metabolism of phenylalanine in the denitrifying bacterium Thauera aromatica. Eur J Biochem 262:507–515
Rotaru A, Probian C, Wilkes H, Harder J (2010) Highly enriched betaproteobacteria growing anaerobically with p-xylene and nitrate. FEMS Microbiol Ecol 71:460–468
Rudolphi A, Tschech A, Fuchs G (1991) Anaerobic degradation of cresols by denitrifying bacteria. Arch Microbiol 155:238–248
Schennen U, Braun K, Knackmuss HJ (1985) Anaerobic degradation of 2-fluorobenzoate by benzoate-degrading, denitrifying bacteria. J Bacteriol 161:321–325
Schink B, Pfennig N (1982) Fermentation of trihydroxybenzenes by Pelobacter acidigallici gen. nov. sp. nov., a new strictly anaerobic non-sporeforming bacterium. Arch Microbiol 133:195–201
Schink B, Stams AJ (2013) Syntrophism among prokaryotes. In: Rosenberg E, Delong E, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes. Vol 2, Ecophysiology and biochemistry. Springer, Berlin, pp 471–493
Schink B, Philipp B, Müller J (2000) Anaerobic degradation of phenolic compounds. Naturwissenschaften 87:12–23
Schleinitz KM, Schmeling S, Jehmlich N, von Bergen M, Harms H, Kleinsteuber S, Vogt C, Fuchs G (2009) Phenol degradation in the strictly anaerobic iron-reducing bacterium Geobacter metallireducens GS-15. Appl Environ Microbiol 75:3912–3919
Schmeling S, Fuchs G (2009) Anaerobic metabolism of phenol in proteobacteria and further studies of phenylphosphate carboxylase. Arch Microbiol 191:869–878
Schmeling S, Narmandakh A, Schmitt O, Gad'on N, Schühle K, Fuchs G (2004) Phenylphosphate synthase: a new phosphotransferase catalyzing the first step in anaerobic phenol metabolism in Thauera aromatica. J Bacteriol 186:8044–8057
Schmid G, René SB, Boll M (2015) Enzymes of the benzoyl-coenzyme A degradation pathway in the hyperthermophilic archaeon Ferroglobus placidus. Environ Microbiol 17:3289–3300
Schühle K, Fuchs G (2004) Phenylphosphate carboxylase: a new C-C lyase involved in anaerobic phenol metabolism in Thauera aromatica. J Bacteriol 186:4556–4567
Schühle K, Nies J, Heider J (2016) An indolacetate-CoA ligase and a phenylsuccinyl-CoA transferase involved in anaerobic metabolism of auxin. Environ Microbiol 18:3120. https://doi.org/10.1111/1462-2920.13347
Selmer T, Andrei PI (2001) p-hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol. Eur J Biochem 268:1363–1372
Selmer T, Pierik AJ, Heider J (2005) New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol Chem 386:981–988
Selvaraj B, Buckel W, Golding BT, Ullmann GM, Martins BM (2016) Structure and function of 4-Hydroxyphenylacetate decarboxylase and its cognate activating enzyme. J Mol Microbiol Biotechnol 26:76–91
Song B, Palleroni NJ, Häggblom MM (2000) Isolation and characterization of diverse halobenzoate-degrading denitrifying bacteria from soils and sediments. Appl Environ Microbiol 66:3446–3453
Song B, Palleroni NJ, Kerkhof LJ, Häggblom MM (2001) Characterization of halobenzoate-degrading, denitrifying Azoarcus and Thauera isolates and description of Thauera chlorobenzoica sp. nov. Int J Syst Evol Microbiol 51:589–602
Stanier RY, Ornston LN (1973) The beta-ketoadipate pathway. Adv Microb Physiol 9:89–151
Strijkstra A, Trautwein K, Jarling R, Wöhlbrand L, Dörries M, Reinhardt R, Drozdowska M, Golding BT, Wilkes H, Rabus R (2014) Anaerobic activation of p-cymene in denitrifying betaproteobacteria: methyl group hydroxylation versus addition to fumarate. Appl Environ Microbiol 80:7592–7603
Szaleniec M, Heider J (2016) Modeling of the reaction mechanism of enzymatic radical C-C coupling by benzylsuccinate synthase. Int J Mol Sci 17:514
Szaleniec M, Borowski T, Schühle K, Witko M, Heider J (2010) Ab initio modeling of ethylbenzene dehydrogenase reaction mechanism. J Am Chem Soc 132:6014–6024
Szaleniec M, Dudzik A, Kozik B, Borowski T, Heider J, Witko M (2014) Mechanistic basis for the enantioselectivity of the anaerobic hydroxylation of alkylaromatic compounds by ethylbenzene dehydrogenase. J Inorg Biochem 139:9–20
Thiele B, Rieder O, Golding BT, Müller M, Boll M (2008) Mechanism of enzymatic Birch reduction: stereochemical course and exchange reactions of benzoyl-CoA reductase. J Am Chem Soc 130:14050–14051
Tiedt O, Mergelsberg M, Boll K, Müller M, Adrian L, Jehmlich N, von Bergen M, Boll M (2016) ATP-dependent C-F bond cleavage allows the complete degradation of 4-fluoroaromatics without oxygen. MBio 7:e00990–e00916
Tor JM, Lovley DR (2001) Anaerobic degradation of aromatic compounds coupled to Fe(III) reduction by Ferroglobus placidus. Environ Microbiol 3:281–287
Trautwein K, Wilkes H, Rabus R (2012) Proteogenomic evidence for β-oxidation of plant-derived 3-phenylpropanoids in “Aromatoleum aromaticum” EbN1. Proteomics 12:1402–1413
Tschech A, Schink B (1985) Fermentative degradation of resorcinol and resorcylic acids. Arch Microbiol 143:52–59
Unciuleac M, Warkentin E, Page CC, Boll M, Ermler U (2004) Structure of a xanthine oxidase-related 4-hydroxybenzoyl-CoA reductase with an additional [4Fe-4S] cluster and an inverted electron flow. Structure 12:2249–2256
Verfürth K, Pierik AJ, Leutwein C, Zorn S, Heider J (2004) Substrate specificities and electron paramagnetic resonance properties of benzylsuccinate synthases in anaerobic toluene and m-xylene metabolism. Arch Microbiol 181:155–162
Weinert T, Huwiler SG, Kung JW, Weidenweber S, Hellwig P, Stärk H, Biskup T, Weber S, Cotelesage JJ, George GN, Ermler U, Boll M (2015) Structural basis of enzymatic benzene ring reduction. Nat Chem Biol 11:586–591
White MD, Payne KA, Fisher K, Marshall SA, Parker D, Rattray NJ, Trivedi DK, Goodacre R, Rigby SE, Scrutton NS, Hay S, Leys D (2015) UbiX is a flavin prenyltransferase required for bacterial ubiquinone biosynthesis. Nature 522:502–506
Wilkes H, Buckel W, Golding BT, Rabus R (2016) Metabolism of hydrocarbons in n-alkane-utilizing anaerobic bacteria. J Mol Microbiol Biotechnol 26:138–151
Wischgoll S, Heintz D, Peters F, Erxleben A, Sarnighausen E, Reski R, van Dorsselaer A, Boll M (2005) Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol Microbiol 58:1238–1252
Wöhlbrand L, Wilkes H, Halder T, Rabus R (2008) Anaerobic degradation of p-ethylphenol by “Aromatoleum aromaticum” strain EbN1: pathway, regulation, and involved proteins. J Bacteriol 190:5699–5709
Wöhlbrand L, Jacob JH, Kube M, Mussmann M, Jarling R, Beck A, Amann R, Wilkes H, Reinhardt R, Rabus R (2013) Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ Microbiol 15:1334–1355
Yu L, Blaser M, Andrei PI, Pierik AJ, Selmer T (2006) 4-Hydroxyphenylacetate decarboxylases: properties of a novel subclass of glycyl radical enzyme systems. Biochemistry 45:9584–9592
Zargar K, Saville R, Phelan RM, Tringe SG, Petzold CJ, Keasling JD, Beller HR (2016) In vitro characterization of phenylacetate decarboxylase, a novel enzyme catalyzing toluene biosynthesis in an anaerobic microbial community. Sci Rep 6:31362
Zhang T, Tremblay P, Chaurasia AK, Smith JA, Bain TS, Lovley DR (2013) Anaerobic benzene oxidation via phenol in Geobacter metallireducens. Appl Environ Microbiol 79:7800–7806
Zhang T, Tremblay P, Chaurasia AK, Smith JA, Bain TS, Lovley DR (2014) Identification of genes specifically required for the anaerobic metabolism of benzene in Geobacter metallireducens. Front Microbiol 5:245
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this entry

Cite this entry
Boll, M., Estelmann, S., Heider, J. (2020). Catabolic Pathways and Enzymes Involved in the Anaerobic Degradation of Monocyclic Aromatic Compounds. In: Boll, M. (eds) Anaerobic Utilization of Hydrocarbons, Oils, and Lipids. Handbook of Hydrocarbon and Lipid Microbiology . Springer, Cham. https://doi.org/10.1007/978-3-319-50391-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-50391-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50390-5
Online ISBN: 978-3-319-50391-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences