
Abstract
The discovery of LRRK2 mutations as a cause of Parkinson’s disease (PD), including the sporadic late-onset form, established the decisive role of genetics in the field of PD research. Among LRRK2 mutations, the G2019S, mostly lying in a haplotype originating from a common Middle Eastern ancestor, has been identified in different populations worldwide. The G2385R and R1628P variants represent validated risk factors for PD in Asian populations. Here, we describe in detail the origin, the present worldwide epidemiology, and the penetrance of LRRK2 mutations. Furthermore, this chapter aims to characterize other definitely/probably pathogenic mutations and risk variants of LRRK2. Finally, we provide some general guidelines for a LRRK2 genetic testing and counseling. In summary, LRRK2 discovery revolutionized the understanding of PD etiology and laid the foundation for a promising future of genetics in PD research.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Leucine-rich repeat kinase 2
- LRRK2
- Dardarin
- Parkinson’s disease
- PARK8
- Parkinson’s disease genetics
- Familial Parkinson’s disease
- LRRK2 mutations
Until the discovery of leucine-rich repeat kinase 2 (LRRK2) mutations as a genetic cause of Parkinson’s disease (PD), the hereditary influences on PD were limited to observation of rare autosomal dominant familial cases harboring highly penetrant SNCA (alpha-synuclein) mutations and juvenile or young onset autosomal recessive forms carrying PRKN, PINK1, and DJ-1 mutations . This scenario was more suggestive of a minor role played by genetic factors in PD, especially considering the common sporadic late-onset form. The innovative finding of LRRK2 low penetrant mutations in common forms of PD revolutionized this outdated view.
Genetic Contribution in Etiology of PD
Epidemiological studies reveal that 10–15% of PD have a positive familial history for the disease, while the majority of cases are sporadic. Through linkage analysis and positional cloning approaches, five genes have been definitely implicated in the etiology of PD. Mutations in the SNCA [1, 2], LRRK2 [3, 4], and VPS35 [5, 6] genes cause autosomal dominant forms, whereas mutations in the PRKN [7], DJ-1 [8], and PINK1 [9] genes cause autosomal recessive forms of PD. Furthermore, mutations in the ATP13A2 [10], PLA2G6 [11], FBXO7 [12, 13], DNAJC6 [14], and SYNJ1 [15] have been reported as rare causes of early-onset parkinsonism with atypical clinical features which might be mechanistically distinct from classical PD. Finally, mutations in UCH-L1 [16], Omi/HtrA2 [17], GIGYF2 [18], EIF4G1 [19], and DNAJC13 [20] genes have also been described in PD cases, but their role in the disease remains uncertain. Another three PD loci have also been mapped (PARK3, PARK10, PARK12, PARK16) [21–23], but the defective genes remain unknown (Table 1.1).
In addition to the Mendelian forms of PD, genetic risk factors for the disease have been investigated in several candidate genes and, more recently, in genome-wide association studies [24]. With the exceptions of SNCA, microtubule-associated protein tau ( MAPT ), and HLA region [25–32], none of the loci reported have so far been convincingly replicated in independent studies.
Another exception is represented by the glucocerebrosidase gene ( GBA ) involved in a recessive neurometabolic disease (Gaucher’s disease). Screening of PD patients for GBA mutations showed a higher number of heterozygous mutations carriers as compared to healthy controls. Mutations have been found in about 2–4% of Caucasian PD patients and less than 1% of controls [33].
The LRRK2 Gene: Mapping and Cloning
Although the discovery of mutations in the SNCA, PRKN, PINK1, and DJ-1 genes clearly contributed to our understanding of the pathogenesis of PD, they were identified in a limited number of PD cases, often with early-onset or pathologically atypical features.
A new locus for PD, termed PARK8, was identified in a large family with autosomal dominant PD, known as the “Sagamihara family” from the region in Japan where the family originated from [34]. The clinical features in affected individuals of the kindred were reported to resemble very closely classical PD, with an average of symptoms onset at 51 ± 6 years. A pattern of “pure nigral degeneration” without Lewy bodies (LB ) was found at autopsy in six PD patients examined, another carrier of the disease haplotype developed multiple system atrophy type P-like pathology, and one showed classical LB pathology [35]. In this family, a genome-wide linkage scan yielded significant evidence for linkage of PD to the centromeric region of chromosome 12 (12p11.2-q13.1). The haplotype analysis suggested an incomplete penetrance of the mutation [34, 35]. In 2004 the linkage to PARK8 was confirmed in two Caucasian families , “family A” (a German–Canadian kindred) and “family D” (from Western Nebraska) with dominantly inherited neurodegeneration [36], and thereafter in several Basque PD [37] families suggesting PARK8 to be a relatively common locus and refining the critical region. A wide clinical–pathological spectrum was shown in these families, including typical PD but also dementia and amyotrophy, diffuse LB and tau pathology, nigral degeneration without inclusions, and atypical, ubiquitin-positive inclusions [38].
In 2004 two independent groups, by positional cloning, identified mutations in a gene at that time annotated as DKFZp434H2111, which cosegregated with PD in several PARK8-linked pedigrees [3, 4]. The gene was renamed LRRK2 (leucine-rich repeat kinase 2) and the encoded protein LRRK2 or dardarin (from the Basque term dardara, meaning tremor, since resting tremor was a consistent clinical feature of the Basque patients who carried LRRK2 mutations).
Subsequently, early in 2005, several groups identified a single LRRK2 mutation (c.G6055A) leading to a G2019S substitution in the encoded protein, which was present in familial and sporadic PD with unprecedented high frequency [39–42]. The following years have seen an explosion of research into the LRRK2 gene in PD and related disorders. The I2020T mutation was detected as the cause of disease in the original “Sagamihara family ” [43].
The G2019S Mutation
Prevalence of G2019S Across Populations
G2019S is particularly important among the PD-causing mutations in LRRK2. This mutation was identified by several groups as a common cause of the disease, being detected initially in ~5–6% of large cohorts of familial PD in Europe and the USA [39, 41] and in ~1–2% of sporadic PD from the UK [40]. Due to the unprecedented high frequency in familial and late-onset classical parkinsonism, which in the past would have been identified as “idiopathic PD,” this specific mutation has been extensively studied worldwide (Fig. 1.1).
So far, large screenings revealed that the frequency of G2019S is population specific. The G2019S mutation has been reported at the highest frequency (up to 37%) among familial PD cases of North African descent and in familial Ashkenazi Jewish patients (23%) [44, 45]. Similar frequencies were replicated in independent studies on PD cases from Tunisia [46–48] and in Ashkenazi Jews [48–51]. Remarkably, the frequency of this mutation was considerably high among sporadic cases (41% North Africans and 13% Ashkenazi Jews) and rarely identified in healthy controls too (3% North Africans and 1.3% Ashkenazi Jews). Other studies reported the presence of the G2019S among 1–2% of healthy North Africans, Ashkenazi, and Sephardic Jewish subjects [49, 50, 52, 53]. So far the G2019S mutation has not been found in sub-Saharan Africans, with exceptions of South Africans where the mutation was present in subjects with European and Jewish ancestry only [54–57]. Little is known about the prevalence in Middle Eastern populations. G2019S is rare in Turkey [58] and has not been identified so far in Yemenite Jews [51] and in Iran [59].
In Western Europe there is a south–north gradient of frequency. The G2019S is found in 9–16% of familial and 3–4% of sporadic PD patients in Portugal [60, 61]; it accounts for 6–16% of familial and 2–6% of sporadic PD in different regions of Spain: Catalonia [62], Cantabria [63, 64], Asturias [65], Galicia [63], and Basque regions (patients without Basque ancestry) [66], while it is less common in patients of Basque origin (1–2%) [66].
In Italy, the G2019S mutation has been reported up to 6–7% of familial and ~1–2% of sporadic cases [39, 67–71]. Similarly, in France the mutation accounts for ~3.5% of familial and ~1.9% of sporadic cases [44, 72–75]. Two independent screening in Sardinians, an isolated population, reported a frequency of ~1.5% in both familial and sporadic cases [76, 77]. Interestingly, the mutation that appeared to be common in the western Mediterranean basin is instead very rare in Greece and Crete [78–81].
A slightly lower frequency was reported in UK screenings of PD patients of Caucasian ethnicity (2.5% familial and 0.3–1.6% sporadic) [40, 82, 83] and also in populations of Celtic and Baltic origin (Ireland 1.1% of familial PD [42, 84], Norway ~1.5% of familial PD [85], and Sweden 1.4% of sporadic cases [86]). Mutation analyses in more than 300 familial and 1200 sporadic PD in Germany suggested a very low frequency of this mutation (0.8% of familial cases [87, 88] 0.2–0.9% of sporadic) [87–89], as well as in Belgium [90], the Netherlands [91], Denmark [92], and Austria [93].
In Poland, Serbia, Hungary, Czech Republic, and Slovakia, the G2019S appeared to be rare (or found in a single subject) [87, 94–98]. On the contrary, four studies have been performed in Russia, where the mutation accounts for 4–7% of familial and 1% of sporadic cases [99–102]. However, subjects were included from a mixed ethnic background, since at least two PD families and one sporadic case reported their ethnic origin as Ashkenazi Jews [101].
An analogous observation can be done when analyzing patients from the USA, where the frequency of the G2019S in Caucasian PD reaches 2–3.5% in familial and 0.5–1.6% in sporadic cases [46, 49, 103–109]; it seems to be rare among American Indians and Afro-Americans (but the sample size for these two ethnic groups is still insufficient to make firm conclusions) [108, 109], whereas it was reported to be higher when patients of Ashkenazi Jewish ancestry were included [49]. In Canadian PD patients, the G2019S is rare/absent [110, 111].
Four different populations of South America, where the Spanish, Portuguese, and Italian ethnic backgrounds are strong, have been studied for this mutation. In Uruguay [112], Chile [113], and Argentina [114], G2019S accounts for 3.5–5.5% of familial and 2.9–4.2% of sporadic cases. The 5.45% of a PD cohort from Argentina was found to carry the mutation, all of them being of Jewish ancestry. While in Peru, the G2019S appears to be rare [112]. Controversial results came from large screening in Brazil (from 3 to 6.8% in familial and 0 to 1.7% in sporadic cases), probably due to the high degree of ethnic heterogeneity within the study cohorts [115–117].
G2019S is rare/absent among Chinese patients with familial and sporadic PD [118–121], as well as in Korea [122, 123] and in India [121, 124]. So far, only three patients have been reported with this mutation in Japan (0.7% of sporadic cases) [48, 125].
Finally, the mutation is present in Australia, among PD patients with European ancestry (2–6% of familial, 0.4% of sporadic PD) [126, 127], while it has not been identified in Australian Aboriginal.
Taken together, these data show that a single LRRK2 mutation represents the most frequent known genetic determinant of PD. The frequency of the G2019S mutation varies widely across populations, indicating that ethnicity is an important factor. For some populations, independent studies on the prevalence of the mutation are already available, and often, the reported results are consistent. These observations imply that most neurologists who treat patients with movement disorders will see patients with LRRK2-related PD that may be addressed to genetic testing. This estimation could be even higher if we include the other LRRK2 definitely pathogenic mutations.
Origins of the G2019S Mutation
So far three different haplotypes have been identified in patients carrying the G2019S mutation.
Haplotype 1
The first studies on unrelated carriers of the G2019S of European or Middle Eastern–North African origin revealed that all shared the same haplotype, consistent with a common founder [42, 44, 45, 70, 128, 129].
Subsequently, the same haplotype has been identified among subjects carrying the G2019S mutation from Italy (independent subset) [71], France [47], Germany [87], Russia [99] Sardinia [76, 77], Spain [65], Portugal [61, 70], Brazil [70], Chile [113], Uruguay and Peru [112], and Australia [126].
According to a general rule in population genetics, the geographic center of the origin of a mutation corresponds to the area where that mutation is most frequent [130]. The highest prevalence of the G2019S mutation has been reported in Berbers [52], followed by North African Arabs, Ashkenazi, and Sephardic Jews. The frequency data combined with the identification of a common haplotype among these populations support the hypothesis that the mutation of haplotype I originated in North Africa or in the Middle East and then spread to other countries following the patterns of migration.
Further studies provided important insights on the estimated age of the common founder for the haplotype 1 carriers. Analyzing the haplotypes of European and Ashkenazi Jews [129] and Tunisian G2019S carriers [131], the age estimated of the common ancestor (using the 30-year intergeneration interval) was 2250 (95% CI 1650–3120) and 3120 (95% CI 2340–4620) years ago, respectively. A third study, on Ashkenazi Jews only, estimated a more recent founder approximately 1525–1830 years ago (150–450 A.D.) [132]. This estimation would fit with the absence of the G2019S in Yemenite Jews [51]. The Yemenite Jews evolved completely separate from all of the other Jewish populations. Most of them arrived in Yemen in the early second century A.D. (~160 A.D.). Finally, a multicentric study proposed a consensus of haplotype 1 origin, estimating the founding mutational event in Ashkenazi Jews ancestors in a period ranging from 4500 to 9100 years. In this scenario, being the Ashkenazi Jews history more recent (at most 2000 years old), it is possible that the G2019S have arisen at least 4000 years ago in the Near East and then Ashkenazi ancestors may have kept the mutation through the different diasporas. Thereafter, the mutation may have been reintroduced by gene flow from Ashkenazi Jews to other European and North African populations [133].
Haplotype 2
A different G2019S haplotype was identified in three families from Western Europe, which appeared to share a more recent founder than haplotype 1. The geographic origin of this haplotype is less certain [129].
Haplotype 3
The third haplotype has been found in Japanese patients carrying the G2019S mutation [125]. This haplotype differs across the markers closest to the mutation, which would suggest an independent origin of the mutation in Japanese and European populations rather than a single ancient founder. Interestingly, the haplotype 3 has also been observed in a single sporadic Turkish patient [134]. This may be the result of a common ancestry (plausibly explained by the large centuries-long migration of the Turkic people across Central Asia) or coincidental presence of Japanese ancestors.
Incomplete Penetrance of G2019S
Incomplete penetrance was already suspected for the mutations underlying the PARK8 locus at the time of the linkage studies. Most of the penetrance analyses have been performed on the frequent G2019S mutation.
Analyses performed on Ashkenazi Jews from the USA revealed a lifetime penetrance of 31.8% [45]. A slighter lower penetrance (24–26% at 80 years) was estimated in independent groups of US Ashkenazi Jews [49, 135].
The International LRRK2 Consortium performed a penetrance study on the largest dataset of G2019S carriers. By analyzing a large sample of PD patients, they calculated a 28% risk of PD at 59 years, 51% at 69 years, and 74% at 79 years for LRRK2 G2019S carriers without differences in penetrance by sex or ethnic group [136]. Interestingly a penetrance study in Tunisian G2019S PD cases, after stratifying by homozygous (n = 23) and heterozygous carriers, reported a penetrance consistently higher in homozygotes in each age group. Considering possible biases in estimating penetrance only from families, this finding, if true, would indicate a gene dosage effect, although the age of onset was not dissimilar between the two groups [46]. However, subsequent studies collecting clinical data of homozygous carriers showed no phenotype differences between heterozygous and homozygous carriers ruling out a gene dosage effect [44, 137, 138].
The reduced penetrance of this frequent mutation is in keeping with the LRRK2 G2019S being the most important genetic determinant, known so far, of sporadic PD. Penetrance can also be expressed in terms of risk (calculated as odd ratio) to develop the disease. For an Ashkenazi Jew who carries the G2019S, the risk of developing PD increases ~18-fold [45]. By analyzing the G2019S in North Africans, a lifetime odds ratio for developing PD of 48.6 (CI 11.2–211.0) [44] has been calculated.
Nevertheless, additional studies in different populations are warranted before G2019S genetic counseling can be implemented, since the precise estimation of the penetrance in some countries is still controversial.
Dissimilar results across the abovementioned Ashkenazi Jews from the USA and other G2019S carriers might be influenced by different methodological approaches (e.g. including only patients with both parents genotyped, excluding patients with GBA mutations, etc.) or by additional genetic or nongenetic factors that can act as modifiers.
The analysis of candidate genes involved in neurodegeneration as potential genetic modifiers of LRRK2 has been reported. The first to be explored was PRKN, since patients who simultaneously harbored PRKN mutations and LRRK2 G2019S have been mentioned in several studies [61, 73, 99, 139–141]. However, the clinical and cosegregation analysis of patients carrying heterozygous PRKN mutations and the G2019S revealed that the combination of the two does not influence the symptoms or the age at disease onset [142].
Polymorphic variations in the microtubule-associated protein tau ( MAPT ) have been proposed to be significantly associated with age of disease onset in individuals with LRRK2 mutations [143]. Moreover, SNCA variants have been found as determinant of age of onset in G2019S carriers [144]. It is a common observation among neurologists of the different penetrance of LRRK2 mutations in affected families, implying the great importance of genetic modifiers. Further analyses, especially on large samples and families carrying the G2019S, are warranted to identify genetic factors that can act as modifiers of LRRK2 mutations.
The R1441 Mutational Hot Spot
The LRRK2 R1441 residue is the second most common spot of pathogenic LRRK2 mutations, after G2019S. Three non-synonymous substitutions (R1441C, R1441G, and R1441H) and the synonymous R1441R have been reported in several patients.
R1441C : The Second Most Frequent Pathogenic LRRK2 Mutation
This mutation (c. 4321C > T) represents the second known most common mutation of the LRRK2 gene. The R1441C was identified as causative mutation of the PARK8-linked “family D” (Western Nebraska) [4]. Cosegregation was reported also in smaller PD families from Germany [4, 87], Italy [69], Belgium [90], the USA [42, 145], and Iran [59]. The mutation has also been reported in a few other families, but additional affected relatives were not available for cosegregation analysis [62, 69, 90, 146]. The R1441C is also found among sporadic cases and has been reported in patients from Italy [70], Sardinia [77], Russia (Slavic origin) [100], China [147], and Belgium [90]. The variant was absent in large cohorts of ethnically matched controls (>1000 German, 530 Italian, 208 Sardinian, 400 Chinese, 178 Belgian, and 300 American). Interestingly, the R1441C has been found to be more common than G2019S in southern Italy [148].
Haplotype analysis of LRRK2 R1441C carriers from 20 families of different geographical areas revealed in total four classes of haplotypes. Only for the two major haplotypes, the phase could be established [149]. A first haplotype was identified in the Italian carriers, as well as in German, Spanish, and American patients. A second haplotype was present in the American family D (Western Nebraska) and in Belgian R1441C families. A German and an Irish patient shared a third haplotype for which phase could not be unambiguously determined. Finally, a Chinese proband carried alleles that could not be assigned to any of three previous haplotype classes.
The phenotype associated with this mutation is similar to that of classic PD [149]. The mutation exhibits incomplete penetrance, which could explain its presence in sporadic cases, but calculations performed so far must be interpreted with caution as only a small number of R1441C mutation carriers have been identified until now.
R1441G : A Founder Pathogenic Mutation in the Basques
The LRRK2 R1441G (c. 4321C > G) was initially described in patients with autosomal dominant late-onset PD in PARK8-linked families of Basque ethnicity [3]. The Basques are a homogeneous ethnic group who historically were isolated by linguistic and geographical barriers. The first report on the frequency of this mutation in Basque PD (~8% of familial cases) [3] and the absence in other large populations screened (except for a US patient reported to be of Hispanic descent [50]) suggested that this variant was population specific. Further studies investigated the prevalence of this mutation in Basque. One group detected the R1441G in 16.4% and 4.0% of familial and sporadic Basque PD , respectively [150], while a more recent study reported a prevalence of 46% in familial Basque patients and 2.5% of sporadic cases [66]. It has also been identified at lower frequencies in patients from nearby provinces in Spain who did not report Basque ancestry (6% of non-Basques living in the Basque countries [66], 2.7% in Asturias [151], 0.7% in Catalonia [62], two families from the neighboring region of Navarre, and one from La Rioja [63]), while it is rare in Cantabria [64]. Haplotype analysis on R1441G carriers from Basque and neighborhood regions [63, 150, 152] indicates that this mutation occurred in a single common ancestor, which in one study was estimated to have lived 1350 (95% CI, 1020–1740) years ago [152]. Since the Basque population has a history of emigration to Europe and North, Central, and South Americas, it would not be surprising to find isolated cases in those countries. However, a single case from Uruguay and a family from Japan carrying the R1441G have been reported with a different haplotype than the Basque, suggesting in these cases independent mutational events [112, 153].
R1441H and R1441R : Uncommon but also Likely Pathogenic
This variant, c.G4322A on LRRK2 cDNA, occurs immediately adjacent to the two previously reported pathogenic mutations, c.C4321T (R1441C) and c.C4321G (R1441G), resulting in a different substitution of the same amino acid residue (R1441H).
R1441H has been described in a US PD family, but only the proband and an unaffected sibling were available for testing [146]. It was also reported in PD families from Crete [81], Portugal [61], and Taiwan [84], all not large enough to demonstrate definitive cosegregation with the disease.
Haplotype analysis of the abovementioned R1441H carriers showed diversity suggesting a number of independent founders [154]. Subsequently, the R1441H mutation has been identified in two cases from Australia, both of British origin and with a possible common haplotype, although in these cases the phase was not assessed [126]. A further proof in favor of a pathogenic role of this variant came from the identification of R1441H in two slightly larger French families [72].
R1441H was not found in 281 Americans, 300 Cretans, 200 Portugueses, 174 Europeans, and a set of 1000 control samples (600 North Americans, 200 Taiwaneses, 200 Norwegians, 200 Irish, and 200 Spanish). Moreover several studies screened by sequence the LRRK2 exon 31 in a large sample of healthy controls (>3000 Caucasian [3, 4, 69, 90]) in order to check for the R1441C and R1441G, and none reported mutation in the adjacent nucleotide.
The clinical presentation of affected R1441H carriers appears to be similar to typical Parkinson’s disease with an age at onset range of 32–66 years. All display levodopa-responsive parkinsonism; however, the disease in one of the siblings from the Greek R1441H family appeared to transition into a progressive supranuclear palsy-like disorder [81].
To further highlight the nature of codon 1441 as a mutational hot spot, two groups reported a R1441R (c.C4323T) in a sporadic PD patient [101] from Russia and a PD patient with ascertained LB pathology who additionally developed dementia and dysautonomia (PDD) [155]. As for the R1441H, we can indirectly assume that the variant is rare in the Caucasian population, since sequencing controls for the other mutations at the same codon did not reveal any R1441R carriers. This variant is predicted to lead to a synonymous substitution, which would suggest a nonpathogenic role. Moreover, being the nucleotide change close to the splice site, cDNA analysis from the brain of the PDD patient was performed and did not reveal any aberrations on the LRRK2 transcript [155]. Taken together these results suggest that R1441R is likely to represent a rare but nonpathogenic polymorphism.
Mutations in LRRK2 are associated with pleomorphic pathology, although the Lewy bodies (LB )-positive pathology is the most common pattern, particularly for the G2019S mutation [38, 40, 82, 156, 157].
In a large screen of 405 LB -positive brains, eight (~2%) have been found to be carriers of the G2019S mutation, including four with brainstem type, three with transitional type, and one with diffuse LB pathology. In two G2019S-positive brains , Alzheimer-type pathology was also present, and it was of enough severity to make a concomitant pathological diagnosis of Alzheimer’s disease [157].
A further study on 80 brains with PD or LB dementia screened for the G2019S mutation, and three were found to be carriers. Typical brainstem-type LB-positive pathology was found in one, while the Lewy body variant of Alzheimer’s disease was diagnosed in the second. The third brain showed only cell loss in the substantia nigra and locus coeruleus, but no α-synuclein inclusions were detected. There were only rare tau-positive tangles and occasional plaques. No other ubiquitin-positive inclusions were present either [156].
In family D (Western Nebraska), all R1441C carriers examined showed substantia nigra neuronal loss. Two cases had LB pathology, one brainstem type, and the other one diffuse type. The third case had “nonspecific” substantia nigra degeneration with ubiquitin-positive neuronal inclusions. The final case had PSP-like changes with tau-immunoreactive neuronal and glial lesions [4].
The neuropathological examination of R1441G Basque carriers displayed “nonspecific” nigral degeneration in the substantia nigra without α-synuclein, tau, or ubiquitin inclusions [158].
Japanese cases with the I2020T mutation were found to display hyperphosphorylated tau aggregates [159]. Therefore, despite LBs represent the predominant feature in neuropathological studies, the overall LRRK2-associated pathology has revealed great variability, probably recapitulating the heterogeneity of PD itself, which can be a more complex disease than what we thought until now.

Rough estimates of worldwide G2019S prevalence in PD patients (familial and sporadic)
The Other LRRK2 Variants: Which Are Pathogenic?
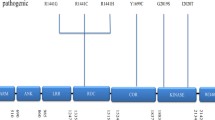
Besides the most recurrent G2019S and R1441C/R1441G/R1441H, more than 50 different LRRK2 sequence variants have been reported in familial and sporadic PD cases so far; moreover, few novel LRRK2 substitutions have been found in healthy control subjects only (Fig. 1.2).

Schematic representation of the LRRK2 gene, the dardarin protein, and its known functional domains. LRRK2 83 coding variants and three putative splice variants are grouped according to evidence of pathogenicity (see main text)
The Y1699C and I2020T mutations are considered as definitely pathogenic. The Y1669C was identified in two independent large families, the Lincolnshire kindred [3, 82] (family PL) of European ancestry, and “family A” (German–Canadian) [4, 38]. The I2020T was identified in “family 32” [4] and “T10738” [89], both of German ancestry. Additionally the same mutation was identified segregating in the large PARK8-linked Sagamihara kindred [43] and in two smaller Japanese families coming from the neighborhood of the Sagamihara region [48].
The role of several other variants remains unclear, since often no family members were available to assess cosegregation and a limited number of ethnically matched controls were screened. Overall, the criteria that may be applied to consider the pathogenicity of the LRRK2 variants should consider several standpoints: frequency in healthy subjects, cosegregation in families, confirmation in independent studies, and pathogenic consequences on cellular and animal models.
Association Studies on LRRK2
In the past few years, many groups put special effort in search of common risk factors for complex diseases. Among these, PD and other neurodegenerative disorders have been extensively studied. However, even using high-throughput techniques allowing to genotype hundreds of thousands of SNPs and covering the whole genome in cases and controls (genome-wide association studies, GWA ), no reproducible risk loci have been reported so far.
One caveat is that the GWA approach can be problematic because the massive number of statistical tests performed presents an unprecedented potential for false-positive results.
After the discovering of mutations in the LRRK2 gene, several studies aimed to explore whether common variant of this gene could represent a risk factor for PD.
Two association studies on LRRK2 have been performed in Caucasians. The first enrolled 340 PD patients and 608 controls from Germany. 121 SNPs (81 tagging SNPs) were genotyped attempting to represent the complete DNA variation of the LRRK2 gene [160]. The second study analyzed four common coding SNPs (L953L, R1398H, G1624G, and T2397M) in 250 controls and 121 unrelated PD, mostly with early-onset and positive family history [141]. Neither of these studies revealed any evidences of association between PD and the LRRK2 SNPs at both allelic and genotypic levels.
In 2005, one study performed in Singapore yielded a significant association. A set of 21 tagging SNPs covering the LRRK2 gene were genotyped in 466 sporadic PD and 374 control individuals all of Chinese ancestry. The authors identified a common haplotype that was highly overrepresented within cases (p = 0.005) and, when present in two copies, significantly increased the risk of PD (OR = 5.5, 95% C.I. = 2.1–14.0, P = 0.0001) [161]. However, no LRRK2 variants within the risk haplotype were reported as the biologically relevant factors.
The G2385R Variant
The LRRK2 G2385R represents the first common genetic risk factor for PD in the Asian population. This variant was first reported in a small PD family from Taiwan [84]. Evidence for cosegregation with PD in that family was limited due to the small pedigree size; however, the mutation was reported to be absent in 200 ethnically matched controls and, therefore, interpreted as putatively pathogenic. At that time very limited data were available on the nature and frequency of LRRK2 mutations and on the polymorphism content of the gene in patients from Asia.
Several groups conducted a mutational screening of three known PD-causing mutations (I2012T, G2019S, and I2020T) which appeared to be very rare or absent in Asian PD patients [43, 118, 120, 121]. A sequence of the whole LRRK2 in Chinese Han patients revealed four coding variants (A419V, P755L, M1869V which were novel substitutions, and the G2385R) that were tested for association with PD in 608 Chinese Han cases and 373 ethnically matched controls.
The heterozygosity for the G2385R variant was significantly higher among PD cases than controls (10% vs 4% p 0012). This suggested that the G2385R variant, or another variant in linkage disequilibrium, is associated with PD in the Taiwanese population.
Since then, several association studies on Asian populations from Taiwan, Singapore, Mainland China, Korea, and Malaysia replicated this finding with a similar size effect. Interestingly the association was also reported in Japanese PD patients and controls, giving a risk of developing PD increased of ~twofold [125, 166] (Table 1.2).
Two groups performed a haplotype analysis of G2385R carriers in a cohort of Chinese Han from Taiwan [165, 168]. A single common haplotype shared by carriers has been identified, likely originated from a single ancestor who lived approximately 4800 years ago. Also all Japanese G2385R carriers shared the same haplotype, with a set of markers (D12S2516, D12S2519, and D12S2521) which overlapped with the Chinese haplotype. This might suggest that the G2385R of Chinese Han and Japanese ancestry has arisen from a common ancestor [125].
The R1628P Variant
The LRRK2 R1628P has been identified in a multicentric study which combined 1986 Chinese individuals from three independent centers in Taiwan and Singapore and so far represents the second most frequent genetic risk factor for PD in Asia [184]. This variant was approximately twice as frequent in affected individuals as control subjects (~6% of PD and ~3.5% of controls, odds ratio 1.84, 95% C.I.: 1.20–2.83, nominal p value = 0.006) [184].
This finding was replicated in two independent Chinese Han cohorts from Singapore [185] and Taiwan [186]. On the contrary, the R1628P is rare in Japan and in non-Chinese Asians [170, 184, 187].
Haplotype analysis strongly indicates that carriers of the R1628P variant share an extended haplotype, indicative of a founder effect [184]. The mutation has been estimated to arise ~2500 years ago and, in contrast to the older G2385R, has remained confined to subjects of Chinese Han ethnicity.
Like for the G2385R, the clinical phenotype of the affected R1628P carriers is that of typical late-onset L-dopa-responsive PD [184, 186, 187].
Taken together, these studies indicate for the first time that common population specific genetic risk factors for PD exist. The association of both LRRK2 variants with PD in Asia has been extensively confirmed in independent dataset of patients. These findings open several opportunities of studies for researchers and clinicians. Discovering how those variants can increase the risk of death of dopaminergic neurons might provide important insight into the pathogenesis of the disease. Other interesting prospects can be provided in clinical practice, for example, studying the effect of neuroprotective drugs in large cohorts of asymptomatic carriers of these two LRRK2 variants, in order to explore whether the risk of developing PD would decrease in the treated subjects.
In conclusion, the LRRK2 gene displays a high polymorphic content in terms of single nucleotide substitutions. No deletions or duplications have been identified until now. Variants identified in patients are located in almost all exons. However, most of them still lack a definite proof of pathogenicity (Tables 1.3). This has direct practical consequences for the genetic studies. LRRK2 is a large gene containing 51 exons. A time-/cost-saving strategy to perform the mutational analysis could be to first screen for the frequent G2019S mutation. If negative, other validated mutations (R1441G/R1441C/R1441H, I2020T, and Y1699C) can be tested next (Tables 1.4). Where a considerable number of affected family members are available for testing, an option is to screen the entire LRRK2 gene, which raises the possibility of discovering one of the above-reported doubtful variants, or even a novel mutation that could be tested for cosegregation in order to verify its pathogenic role. Concerning the significance of these data for the genetic counseling, it is worth to consider that screening the whole coding region or single variants of uncertain role in unselected cases is still a matter of debate, since the identification of any variant would result in more questions than answers for both clinicians and patients. LRRK2 mutations penetrance is a key piece of information for a proper genetic counseling. Only a minority of LRRK2 mutation carriers will develop the disease, making the predictive genetic testing more similar to BRCA test for breast cancer than to presymptomatic test in Huntington’s disease . Dominant transmission involves more subjects and generations inside the family. The involvement of the offspring and the absence of neuroprotective therapy make the offer of predictive/presymptomatic genetic tests in neurodegenerative disease controversial. However, whenever presymptomatic testing is offered, detailed information and counseling at a center with expertise in this area are required [188, 189].
References
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276(5321):2045–2047
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44(4):595–600
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44(4):601–607
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN et al (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89(1):168–175
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89(1):162–167
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392(6676):605–608
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299(5604):256–259
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304(5674):1158–1160
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38(10):1184–1191
Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW et al (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65(1):19–23
Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L et al (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72(3):240–245
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi GA et al (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82(6):1375–1384
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S et al (2012) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7(5), e36458
Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J et al (2013) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34(9):1208–1215
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E et al (1998) The ubiquitin pathway in Parkinson’s disease. Nature 395(6701):451–452
Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D et al (2005) Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet 14(15):2099–2111
Lautier C, Goldwurm S, Durr A, Giovannone B, Tsiaras WG, Pezzoli G et al (2008) Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet 82(4):822–833
Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, Lincoln SJ, Lepretre F, Hulihan MM et al (2011) Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 89(3):398–406
Vilarino-Guell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J et al (2014) DNAJC13 mutations in Parkinson disease. Hum Mol Genet 23(7):1794–1801
Gasser T, Muller-Myhsok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V et al (1998) A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet 18(3):262–265
Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA et al (2002) Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet 70(4):985–993
Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C et al (2002) Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet 71(1):124–135
Singleton AB, Farrer MJ, Bonifati V (2013) The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 28(1):14–23
Gonzalez-Perez A, Gayan J, Marin J, Galan JJ, Saez ME, Real LM et al (2009) Whole-genome conditional two-locus analysis identifies novel candidate genes for late-onset Parkinson’s disease. Neurogenetics 10(3):173–181
Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW et al (2009) Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124(6):593–605
Sutherland GT, Halliday GM, Silburn PA, Mastaglia FL, Rowe DB, Boyle RS et al (2009) Do polymorphisms in the familial Parkinsonism genes contribute to risk for sporadic Parkinson’s disease? Mov Disord 24(6):833–838
Skipper L, Wilkes K, Toft M, Baker M, Lincoln S, Hulihan M et al (2004) Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am J Hum Genet 75(4):669–677
Tobin JE, Latourelle JC, Lew MF, Klein C, Suchowersky O, Shill HA et al (2008) Haplotypes and gene expression implicate the MAPT region for Parkinson disease: the GenePD study. Neurology 71(1):28–34
Zabetian CP, Hutter CM, Factor SA, Nutt JG, Higgins DS, Griffith A et al (2007) Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson’s disease. Ann Neurol 62(2):137–144
Saiki M, Baker A, Williams-Gray CH, Foltynie T, Goodman RS, Taylor CJ et al (2010) Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. J Neurol Neurosurg Psychiatry 81(8):890–891
Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D et al (2010) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42(9):781–785
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351(19):1972–1977
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51(3):296–301
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S (2009) Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15(4):300–306
Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M et al (2004) The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet 74(1):11–19
Paisan-Ruiz C, Saenz A, Lopez de Munain A, Marti I, Martinez Gil A, Marti-Masso JF et al (2005) Familial Parkinson’s disease: clinical and genetic analysis of four Basque families. Ann Neurol 57(3):365–372
Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ et al (2004) Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 62(9):1619–1622
Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F et al (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365(9457):412–415
Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ et al (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365(9457):415–416
Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA et al (2005) Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365(9457):410–412
Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J et al (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76(4):672–680
Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H et al (2005) An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 57(6):918–921
Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 354(4):422–423
Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354(4):424–425
Ishihara L, Gibson RA, Warren L, Amouri R, Lyons K, Wielinski C et al (2007) Screening for Lrrk2 G2019S and clinical comparison of Tunisian and North American Caucasian Parkinson’s disease families. Mov Disord 22(1):55–61
Lesage S, Ibanez P, Lohmann E, Pollak P, Tison F, Tazir M et al (2005) G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol 58(5):784–787
Tomiyama H, Li Y, Funayama M, Hasegawa K, Yoshino H, Kubo S et al (2006) Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov Disord 21(8):1102–1108
Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J et al (2006) Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 67(10):1786–1791
Deng H, Le W, Guo Y, Hunter CB, Xie W, Huang M et al (2006) Genetic analysis of LRRK2 mutations in patients with Parkinson disease. J Neurol Sci 251(1–2):102–106
Djaldetti R, Hassin-Baer S, Farrer MJ, Vilarino-Guell C, Ross OA, Kolianov V et al (2008) Clinical characteristics of Parkinson’s disease among Jewish Ethnic groups in Israel. J Neural Transm 115(9):1279–1284
Change N, Mercier G, Lucotte G (2008) Genetic screening of the G2019S mutation of the LRRK2 gene in Southwest European, North African, and Sephardic Jewish subjects. Genet Test 12(3):333–339
Saunders-Pullman R, Lipton RB, Senthil G, Katz M, Costan-Toth C, Derby C et al (2006) Increased frequency of the LRRK2 G2019S mutation in an elderly Ashkenazi Jewish population is not associated with dementia. Neurosci Lett 402(1–2):92–96
Bardien S, Marsberg A, Keyser R, Lombard D, Lesage S, Brice A et al (2010) LRRK2 G2019S mutation: frequency and haplotype data in South African Parkinson’s disease patients. J Neural Transm 117(7):847–853
Yonova-Doing E, Atadzhanov M, Quadri M, Kelly P, Shawa N, Musonda ST et al (2012) Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ-1 in Zambian patients with Parkinson’s disease. Parkinsonism Relat Disord 18(5):567–571
Cilia R, Sironi F, Akpalu A, Cham M, Sarfo FS, Brambilla T et al (2012) Screening LRRK2 gene mutations in patients with Parkinson’s disease in Ghana. J Neurol 259(3):569–570
Okubadejo N, Britton A, Crews C, Akinyemi R, Hardy J, Singleton A et al (2008) Analysis of Nigerians with apparently sporadic Parkinson disease for mutations in LRRK2, PRKN and ATXN3. PLoS One 3(10), e3421
Hanagasi HA, Lohmann E, Dursun B, Honore A, Lesage S, Dogu O et al (2011) LRRK2 mutations are uncommon in Turkey. Eur J Neurol 18(10), e137
Shojaee S, Sina F, Farboodi N, Fazlali Z, Ghazavi F, Ghorashi SA et al (2009) A clinic-based screening of mutations in exons 31, 34, 35, 41, and 48 of LRRK2 in Iranian Parkinson’s disease patients. Mov Disord 24(7):1023–1027
Bras JM, Guerreiro RJ, Ribeiro MH, Januario C, Morgadinho A, Oliveira CR et al (2005) G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord 20(12):1653–1655
Ferreira JJ, Guedes LC, Rosa MM, Coelho M, van Doeselaar M, Schweiger D et al (2007) High prevalence of LRRK2 mutations in familial and sporadic Parkinson’s disease in Portugal. Mov Disord 22(8):1194–1201
Gaig C, Ezquerra M, Marti MJ, Munoz E, Valldeoriola F, Tolosa E (2006) LRRK2 mutations in Spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol 63(3):377–382
Gonzalez-Fernandez MC, Lezcano E, Ross OA, Gomez-Esteban JC, Gomez-Busto F, Velasco F et al (2007) Lrrk2-associated parkinsonism is a major cause of disease in Northern Spain. Parkinsonism Relat Disord 13(8):509–515
Infante J, Rodriguez E, Combarros O, Mateo I, Fontalba A, Pascual J et al (2006) LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci Lett 395(3):224–226
Mata IF, Ross OA, Kachergus J, Huerta C, Ribacoba R, Moris G et al (2006) LRRK2 mutations are a common cause of Parkinson’s disease in Spain. Eur J Neurol 13(4):391–394
Gorostidi A, Ruiz-Martinez J, Lopez de Munain A, Alzualde A, Marti Masso JF (2009) LRRK2 G2019S and R1441G mutations associated with Parkinson’s disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics 10(2):157–159
Civitelli D, Tarantino P, Nicoletti G, Ciro Candiano IC, Annesi F, De Marco EV et al (2007) LRRK2 G6055A mutation in Italian patients with familial or sporadic Parkinson’s disease. Clin Genet 71(4):367–370
De Rosa A, Criscuolo C, Mancini P, De Martino M, Giordano IA, Pappata S et al (2009) Genetic screening for LRRK2 gene G2019S mutation in Parkinson’s disease patients from Southern Italy. Parkinsonism Relat Disord 15(3):242–244
Di Fonzo A, Tassorelli C, De Mari M, Chien HF, Ferreira J, Rohe CF et al (2006) Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson’s disease. Eur J Hum Genet 14(3):322–331
Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M et al (2005) The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet 42(11), e65
Marongiu R, Ghezzi D, Ialongo T, Soleti F, Elia A, Cavone S et al (2006) Frequency and phenotypes of LRRK2 G2019S mutation in Italian patients with Parkinson’s disease. Mov Disord 21(8):1232–1235
Lesage S, Condroyer C, Lannuzel A, Lohmann E, Troiano A, Tison F et al (2009) Molecular analyses of the LRRK2 gene in European and North-African autosomal dominant Parkinson’s disease. J Med Genet 46(7):458–464
Lesage S, Janin S, Lohmann E, Leutenegger AL, Leclere L, Viallet F et al (2007) LRRK2 exon 41 mutations in sporadic Parkinson disease in Europeans. Arch Neurol 64(3):425–430
Lesage S, Leclere L, Lohmann E, Borg M, Ruberg M, Durr A et al (2007) Frequency of the LRRK2 G2019S mutation in siblings with Parkinson’s disease. Neurodegener Dis 4(2–3):195–198
Funalot B, Nichols WC, Perez-Tur J, Mercier G, Lucotte G (2006) Genetic screening for two LRRK2 mutations in French patients with idiopathic Parkinson’s disease. Genet Test 10(4):290–293
Cossu G, van Doeselaar M, Deriu M, Melis M, Molari A, Di Fonzo A et al (2007) LRRK2 mutations and Parkinson’s disease in Sardinia: a Mediterranean genetic isolate. Parkinsonism Relat Disord 13(1):17–21
Floris G, Cannas A, Solla P, Murru MR, Tranquilli S, Corongiu D et al (2009) Genetic analysis for five LRRK2 mutations in a Sardinian parkinsonian population: importance of G2019S and R1441C mutations in sporadic Parkinson’s disease patients. Parkinsonism Relat Disord 15(4):277–280
Kalinderi K, Fidani L, Bostantjopoulou S, Katsarou Z, Kotsis A (2007) The G2019S LRRK2 mutation is uncommon amongst Greek patients with sporadic Parkinson’s disease. Eur J Neurol 14(10):1088–1090
Papapetropoulos S, Adi N, Shehadeh L, Bishopric N, Singer C, Argyriou AA et al (2008) Is the G2019S LRRK2 mutation common in all southern European populations? J Clin Neurosci 15(9):1027–1030
Papapetropoulos S, Argyriou AA, Mitsi G, Chroni E (2007) Re: the G2019S LRRK2 mutation is uncommon amongst Greek patients with familial Parkinson’s disease. Eur J Neurol 14(11), e6
Spanaki C, Latsoudis H, Plaitakis A (2006) LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology 67(8):1518–1519
Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL et al (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128(Pt 12):2786–2796
Williams-Gray CH, Goris A, Foltynie T, Brown J, Maranian M, Walton A et al (2006) Prevalence of the LRRK2 G2019S mutation in a UK community based idiopathic Parkinson’s disease cohort. J Neurol Neurosurg Psychiatry 77(5):665–667
Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T et al (2005) Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics 6(4):171–177
Toft M, Haugarvoll K, Ross OA, Farrer MJ, Aasly JO (2007) LRRK2 and Parkinson’s disease in Norway. Acta Neurol Scand Suppl 187:72–75
Carmine Belin A, Westerlund M, Sydow O, Lundstromer K, Hakansson A, Nissbrandt H et al (2006) Leucine-rich repeat kinase 2 (LRRK2) mutations in a Swedish Parkinson cohort and a healthy nonagenarian. Mov Disord 21(10):1731–1734
Hedrich K, Winkler S, Hagenah J, Kabakci K, Kasten M, Schwinger E et al (2006) Recurrent LRRK2 (Park8) mutations in early-onset Parkinson’s disease. Mov Disord 21(9):1506–1510
Moller JC, Rissling I, Mylius V, Hoft C, Eggert KM, Oertel WH (2008) The prevalence of the G2019S and R1441C/G/H mutations in LRRK2 in German patients with Parkinson’s disease. Eur J Neurol 15(7):743–745
Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P et al (2005) Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain 128(Pt 12):3000–3011
Nuytemans K, Rademakers R, Theuns J, Pals P, Engelborghs S, Pickut B et al (2008) Founder mutation p.R1441C in the leucine-rich repeat kinase 2 gene in Belgian Parkinson’s disease patients. Eur J Hum Genet 16(4):471–479
Macedo MG, Verbaan D, Fang Y, van Rooden SM, Visser M, Anar B et al (2009) Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson’s disease. Mov Disord 24(2):196–203
Bech S, Norremolle A, Winge K, Hasholt L, Tommerup N, Svenstrup K et al (2011) The lrrk2 p.Gly2019Ser mutation is uncommon in a Danish cohort with various neurodegenerative disorders. Parkinsonism Relat Disord 17(5):398–399
Haubenberger D, Bonelli S, Hotzy C, Leitner P, Lichtner P, Samal D et al (2007) A novel LRRK2 mutation in an Austrian cohort of patients with Parkinson’s disease. Mov Disord 22(11):1640–1643
Bialecka M, Hui S, Klodowska-Duda G, Opala G, Tan EK, Drozdzik M (2005) Analysis of LRRK 2 G 2019 S and I 2020 T mutations in Parkinson’s disease. Neurosci Lett 390(1):1–3
Jankovic MZ, Kresojevic ND, Dobricic VS, Markovic VV, Petrovic IN, Novakovic IV et al (2015) Identification of novel variants in LRRK2 gene in patients with Parkinson’s disease in Serbian population. J Neurol Sci 353(1–2):59–62
Bognar C, Baldovic M, Benetin J, Kadasi L, Zatkova A (2013) Analysis of Leucine-rich repeat kinase 2 (LRRK2) and Parkinson protein 2 (parkin, PARK2) genes mutations in Slovak Parkinson disease patients. Gen Physiol Biophys 32(1):55–66
Balicza P, Bereznai B, Takats A, Klivenyi P, Dibo G, Hidasi E et al (2012) The absence of the common LRRK2 G2019S mutation in 120 young onset Hungarian Parkinon’s disease patients. Ideggyogy Sz 65(7–8):239–242
Fiala O, Pospisilova L, Prochazkova J, Matejckova M, Martasek P, Novakova L et al (2010) Parkin mutations and phenotypic features in Czech patients with early-onset Parkinson’s disease. Neuro Endocrinol Lett 31(2):187–192
Illarioshkin SN, Shadrina MI, Slominsky PA, Bespalova EV, Zagorovskaya TB, Bagyeva G et al (2007) A common leucine-rich repeat kinase 2 gene mutation in familial and sporadic Parkinson’s disease in Russia. Eur J Neurol 14(4):413–417
Pchelina SN, Yakimovskii AF, Emelyanov AK, Ivanova ON, Schwarzman AL, Singleton AB (2008) Screening for LRRK2 mutations in patients with Parkinson’s disease in Russia: identification of a novel LRRK2 variant. Eur J Neurol 15(7):692–696
Pchelina SN, Yakimovskii AF, Ivanova ON, Emelianov AK, Zakharchuk AH, Schwarzman AL (2006) G2019S LRRK2 mutation in familial and sporadic Parkinson’s disease in Russia. Mov Disord 21(12):2234–2236
Shadrina MI, Illarioshkin SN, Bagyeva G, Bespalova EV, Zagorodskaia TB, Slominskii PA et al (2007) A PARK8 form of Parkinson’s disease: a mutational analysis of the LRRK2 gene in Russian population. Zh Nevrol Psikhiatr Im S S Korsakova 107(3):46–50
Chen-Plotkin AS, Yuan W, Anderson C, McCarty Wood E, Hurtig HI, Clark CM et al (2008) Corticobasal syndrome and primary progressive aphasia as manifestations of LRRK2 gene mutations. Neurology 70(7):521–527
Johnson J, Paisan-Ruiz C, Lopez G, Crews C, Britton A, Malkani R et al (2007) Comprehensive screening of a North American Parkinson’s disease cohort for LRRK2 mutation. Neurodegener Dis 4(5):386–391
Kay DM, Zabetian CP, Factor SA, Nutt JG, Samii A, Griffith A et al (2006) Parkinson’s disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov Disord 21(4):519–523
Paisan-Ruiz C, Nath P, Washecka N, Gibbs JR, Singleton AB (2008) Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Hum Mutat 29(4):485–490
Papapetropoulos S, Singer C, Ross OA, Toft M, Johnson JL, Farrer MJ et al (2006) Clinical heterogeneity of the LRRK2 G2019S mutation. Arch Neurol 63(9):1242–1246
Patra B, Parsian AJ, Racette BA, Zhao JH, Perlmutter JS, Parsian A (2009) LRRK2 gene G2019S mutation and SNPs [haplotypes] in subtypes of Parkinson’s disease. Parkinsonism Relat Disord 15(3):175–180
Scholz S, Mandel RJ, Fernandez HH, Foote KD, Rodriguez RL, Barton E et al (2006) LRRK2 mutations in a clinic-based cohort of Parkinson’s disease. Eur J Neurol 13(12):1298–1301
Dupre N, Riviere JB, Myers RH, Provencher P, Pourcher E, Emond F et al (2007) LRRK2 is not a significant cause of Parkinson’s disease in French-Canadians. Can J Neurol Sci 34(3):333–335
Grimes DA, Racacho L, Han F, Panisset M, Bulman DE (2007) LRRK2 screening in a Canadian Parkinson’s disease cohort. Can J Neurol Sci 34(3):336–338
Mata IF, Cosentino C, Marca V, Torres L, Mazzetti P, Ortega O et al (2009) LRRK2 mutations in patients with Parkinson’s disease from Peru and Uruguay. Parkinsonism Relat Disord 15(5):370–373
Perez-Pastene C, Cobb SA, Diaz-Grez F, Hulihan MM, Miranda M, Venegas P et al (2007) Lrrk2 mutations in South America: a study of Chilean Parkinson’s disease. Neurosci Lett 422(3):193–197
Gatto EM, Parisi V, Converso DP, Poderoso JJ, Carreras MC, Marti-Masso JF et al (2013) The LRRK2 G2019S mutation in a series of Argentinean patients with Parkinson’s disease: clinical and demographic characteristics. Neurosci Lett 537:1–5
Camargos ST, Dornas LO, Momeni P, Lees A, Hardy J, Singleton A et al (2009) Familial Parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov Disord 24(5):662–666
Munhoz RP, Wakutani Y, Marras C, Teive HA, Raskin S, Werneck LC et al (2008) The G2019S LRRK2 mutation in Brazilian patients with Parkinson’s disease: phenotype in monozygotic twins. Mov Disord 23(2):290–294
Pimentel MM, Moura KC, Abdalla CB, Pereira JS, de Rosso AL, Nicaretta DH et al (2008) A study of LRRK2 mutations and Parkinson’s disease in Brazil. Neurosci Lett 433(1):17–21
Fung HC, Chen CM, Hardy J, Hernandez D, Singleton A, Wu YR (2006) Lack of G2019S LRRK2 mutation in a cohort of Taiwanese with sporadic Parkinson’s disease. Mov Disord 21(6):880–881
Lin CH, Tzen KY, Yu CY, Tai CH, Farrer MJ, Wu RM (2008) LRRK2 mutation in familial Parkinson’s disease in a Taiwanese population: clinical, PET, and functional studies. J Biomed Sci 15(5):661–667
Lu CS, Simons EJ, Wu-Chou YH, Fonzo AD, Chang HC, Chen RS et al (2005) The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson’s disease. Parkinsonism Relat Disord 11(8):521–522
Tan EK, Shen H, Tan LC, Farrer M, Yew K, Chua E et al (2005) The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson’s disease patients. Neurosci Lett 384(3):327–329
Cho JW, Kim SY, Park SS, Kim HJ, Ahn TB, Kim JM et al (2007) The G2019S LRRK2 mutation is rare in Korean patients with Parkinson’s disease. Can J Neurol Sci 34(1):53–55
Choi JM, Woo MS, Ma HI, Kang SY, Sung YH, Yong SW et al (2008) Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 9(4):263–269
Punia S, Behari M, Govindappa ST, Swaminath PV, Jayaram S, Goyal V et al (2006) Absence/rarity of commonly reported LRRK2 mutations in Indian Parkinson’s disease patients. Neurosci Lett 409(2):83–88
Zabetian CP, Morino H, Ujike H, Yamamoto M, Oda M, Maruyama H et al (2006) Identification and haplotype analysis of LRRK2 G2019S in Japanese patients with Parkinson disease. Neurology 67(4):697–699
Huang Y, Halliday GM, Vandebona H, Mellick GD, Mastaglia F, Stevens J et al (2007) Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson’s disease. Mov Disord 22(7):982–989
Mellick GD, Siebert GA, Funayama M, Buchanan DD, Li Y, Imamichi Y et al (2009) Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat Disord 15(2):105–109
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A, Jain S, Meyer-Lindenberg A, Evans EW et al (2005) Clinical and positron emission tomography of Parkinson’s disease caused by LRRK2. Ann Neurol 57(3):453–456
Zabetian CP, Hutter CM, Yearout D, Lopez AN, Factor SA, Griffith A et al (2006) LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet 79(4):752–758
Watterson GA, Guess HA (1977) Is the most frequent allele the oldest? Theor Popul Biol 11(2):141–160
Warren L, Gibson R, Ishihara L, Elango R, Xue Z, Akkari A et al (2008) A founding LRRK2 haplotype shared by Tunisian, US, European and Middle Eastern families with Parkinson’s disease. Parkinsonism Relat Disord 14(1):77–80
Bar-Shira A, Hutter CM, Giladi N, Zabetian CP, Orr-Urtreger A (2009) Ashkenazi Parkinson’s disease patients with the LRRK2 G2019S mutation share a common founder dating from the second to fifth centuries. Neurogenetics 10(4):355–358
Lesage S, Patin E, Condroyer C, Leutenegger AL, Lohmann E, Giladi N et al (2010) Parkinson’s disease-related LRRK2 G2019S mutation results from independent mutational events in humans. Hum Mol Genet 19(10):1998–2004
Pirkevi C, Lesage S, Condroyer C, Tomiyama H, Hattori N, Ertan S et al (2009) A LRRK2 G2019S mutation carrier from Turkey shares the Japanese haplotype. Neurogenetics 10(3):271–273
Marder K, Wang Y, Alcalay RN, Mejia-Santana H, Tang MX, Lee A et al (2015) Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 85(1):89–95
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol 7(7):583–590
Ishihara L, Warren L, Gibson R, Amouri R, Lesage S, Durr A et al (2006) Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol 63(9):1250–1254
Brice A (2005) Genetics of Parkinson’s disease: LRRK2 on the rise. Brain 128(Pt 12):2760–2762
Marras C, Klein C, Lang AE, Wakutani Y, Moreno D, Sato C et al (2010) LRRK2 and Parkin mutations in a family with parkinsonism-Lack of genotype-phenotype correlation. Neurobiol Aging 31(4):721–722
Dachsel JC, Mata IF, Ross OA, Taylor JP, Lincoln SJ, Hinkle KM et al (2006) Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci Lett 410(2):80–84
Paisan-Ruiz C, Lang AE, Kawarai T, Sato C, Salehi-Rad S, Fisman GK et al (2005) LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology 65(5):696–700
Solla P, Cannas A, Floris G, Murru MR, Corongiu D, Tranquilli S et al (2010) Parkin Exon rearrangements and sequence variants in LRRK2 mutations carriers: analysis on a possible modifier effect on LRRK2 penetrance. Parkinsons Dis 2010:537698
Golub Y, Berg D, Calne DB, Pfeiffer RF, Uitti RJ, Stoessl AJ et al (2009) Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkinsonism Relat Disord 15(7):539–541
Botta-Orfila T, Ezquerra M, Pastor P, Fernandez-Santiago R, Pont-Sunyer C, Compta Y et al (2012) Age at onset in LRRK2-associated PD is modified by SNCA variants. J Mol Neurosci 48(1):245–247
Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI et al (2008) The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med 6:32
Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D et al (2005) A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 65(5):741–744
Tan EK, Skipper L, Chua E, Wong MC, Pavanni R, Bonnard C et al (2006) Analysis of 14 LRRK2 mutations in Parkinson’s plus syndromes and late-onset Parkinson’s disease. Mov Disord 21(7):997–1001
Criscuolo C, De Rosa A, Guacci A, Simons EJ, Breedveld GJ, Peluso S et al (2011) The LRRK2 R1441C mutation is more frequent than G2019S in Parkinson’s disease patients from southern Italy. Mov Disord 26(9):1733–1736
Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM et al (2008) Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology 70(16 Pt 2):1456–1460
Simon-Sanchez J, Marti-Masso JF, Sanchez-Mut JV, Paisan-Ruiz C, Martinez-Gil A, Ruiz-Martinez J et al (2006) Parkinson’s disease due to the R1441G mutation in Dardarin: a founder effect in the Basques. Mov Disord 21(11):1954–1959
Mata IF, Taylor JP, Kachergus J, Hulihan M, Huerta C, Lahoz C et al (2005) LRRK2 R1441G in Spanish patients with Parkinson’s disease. Neurosci Lett 382(3):309–311
Mata IF, Hutter CM, Gonzalez-Fernandez MC, de Pancorbo MM, Lezcano E, Huerta C et al (2009) Lrrk2 R1441G-related Parkinson’s disease: evidence of a common founding event in the seventh century in Northern Spain. Neurogenetics 10(4):347–353
Hatano T, Funayama M, Kubo S, Mata IF, Oji Y, Mori A et al (2014) Identification of a Japanese family with LRRK2 p.R1441G-related Parkinson’s disease. Neurobiol Aging 35(11):2656.e17–2656.e23
Ross OA, Spanaki C, Griffith A, Lin CH, Kachergus J, Haugarvoll K et al (2009) Haplotype analysis of Lrrk2 R1441H carriers with parkinsonism. Parkinsonism Relat Disord 15(6):466–467
Gaig C, Ezquerra M, Marti MJ, Valldeoriola F, Munoz E, Llado A et al (2008) Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci 270(1–2):94–98
Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ et al (2006) Biochemical and pathological characterization of Lrrk2. Ann Neurol 59(2):315–322
Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC et al (2006) Lrrk2 and Lewy body disease. Ann Neurol 59(2):388–393
Marti-Masso JF, Ruiz-Martinez J, Bolano MJ, Ruiz I, Gorostidi A, Moreno F et al (2009) Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord 24(13):1998–2001
Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW et al (2015) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72(1):100–105
Biskup S, Mueller JC, Sharma M, Lichtner P, Zimprich A, Berg D et al (2005) Common variants of LRRK2 are not associated with sporadic Parkinson’s disease. Ann Neurol 58(6):905–908
Skipper L, Li Y, Bonnard C, Pavanni R, Yih Y, Chua E et al (2005) Comprehensive evaluation of common genetic variation within LRRK2 reveals evidence for association with sporadic Parkinson’s disease. Hum Mol Genet 14(23):3549–3556
Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF et al (2006) A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 7(3):133–138
Tan EK (2006) Identification of a common genetic risk variant (LRRK2 Gly2385Arg) in Parkinson’s disease. Ann Acad Med Singapore 35(11):840–842
Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR (2006) A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol 6:47
Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K et al (2007) Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord 13(2):89–92
Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M et al (2007) Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 18(3):273–275
Tan EK, Zhao Y, Tan L, Lim HQ, Lee J, Yuen Y et al (2007) Analysis of LRRK2 Gly2385Arg genetic variant in non-Chinese Asians. Mov Disord 22(12):1816–1818
Li C, Ting Z, Qin X, Ying W, Li B, Guo Qiang L et al (2007) The prevalence of LRRK2 Gly2385Arg variant in Chinese Han population with Parkinson’s disease. Mov Disord 22(16):2439–2443
An XK, Peng R, Li T, Burgunder JM, Wu Y, Chen WJ et al (2008) LRRK2 Gly2385Arg variant is a risk factor of Parkinson’s disease among Han-Chinese from mainland China. Eur J Neurol 15(3):301–305
Zabetian CP, Yamamoto M, Lopez AN, Ujike H, Mata IF, Izumi Y et al (2009) LRRK2 mutations and risk variants in Japanese patients with Parkinson’s disease. Mov Disord 24(7):1034–1041
Miyake Y, Tsuboi Y, Koyanagi M, Fujimoto T, Shirasawa S, Kiyohara C et al (2010) LRRK2 Gly2385Arg polymorphism, cigarette smoking, and risk of sporadic Parkinson’s disease: a case–control study in Japan. J Neurol Sci 297(1–2):15–18
Kim JM, Lee JY, Kim HJ, Kim JS, Shin ES, Cho JH et al (2010) The LRRK2 G2385R variant is a risk factor for sporadic Parkinson’s disease in the Korean population. Parkinsonism Relat Disord 16(2):85–88
Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G et al (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case–control study. Lancet Neurol 10(10):898–908
Gopalai AA, Lim SY, Chua JY, Tey S, Lim TT, Mohamed Ibrahim N et al (2014) LRRK2 G2385R and R1628P mutations are associated with an increased risk of Parkinson’s disease in the Malaysian population. Biomed Res Int 2014:867321
Nichols WC, Elsaesser VE, Pankratz N, Pauciulo MW, Marek DK, Halter CA et al (2007) LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology 69(18):1737–1744
Xiromerisiou G, Hadjigeorgiou GM, Gourbali V, Johnson J, Papakonstantinou I, Papadimitriou A et al (2007) Screening for SNCA and LRRK2 mutations in Greek sporadic and autosomal dominant Parkinson’s disease: identification of two novel LRRK2 variants. Eur J Neurol 14(1):7–11
Skipper L, Shen H, Chua E, Bonnard C, Kolatkar P, Tan LC et al (2005) Analysis of LRRK2 functional domains in nondominant Parkinson disease. Neurology 65(8):1319–1321
Schlitter AM, Woitalla D, Mueller T, Epplen JT, Dekomien G (2006) The LRRK2 gene in Parkinson’s disease: mutation screening in patients from Germany. J Neurol Neurosurg Psychiatry 77(7):891–892
Covy JP, Yuan W, Waxman EA, Hurtig HI, Van Deerlin VM, Giasson BI (2009) Clinical and pathological characteristics of patients with leucine-rich repeat kinase-2 mutations. Mov Disord 24(1):32–39
Aasly JO, Vilarino-Guell C, Dachsel JC, Webber PJ, West AB, Haugarvoll K et al (2010) Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson’s disease. Mov Disord 25(13):2156–2163
Lorenzo-Betancor O, Samaranch L, Ezquerra M, Tolosa E, Lorenzo E, Irigoyen J et al (2012) LRRK2 haplotype-sharing analysis in Parkinson’s disease reveals a novel p.S1761R mutation. Mov Disord 27(1):146–151
Cilia R, Siri C, Rusconi D, Allegra R, Ghiglietti A, Sacilotto G et al (2014) LRRK2 mutations in Parkinson’s disease: confirmation of a gender effect in the Italian population. Parkinsonism Relat Disord 20(8):911–914
Clarimon J, Pagonabarraga J, Paisan-Ruiz C, Campolongo A, Pascual-Sedano B, Marti-Masso JF et al (2008) Tremor dominant parkinsonism: clinical description and LRRK2 mutation screening. Mov Disord 23(4):518–523
Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI et al (2008) Analysis of Lrrk2 R1628P as a risk factor for Parkinson’s disease. Ann Neurol 64(1):88–92
Tan EK, Tan LC, Lim HQ, Li R, Tang M, Yih Y et al (2008) LRRK2 R1628P increases risk of Parkinson’s disease: replication evidence. Hum Genet 124(3):287–288
Lu CS, Wu-Chou YH, van Doeselaar M, Simons EJ, Chang HC, Breedveld GJ et al (2008) The LRRK2 Arg1628Pro variant is a risk factor for Parkinson’s disease in the Chinese population. Neurogenetics 9(4):271–276
Tan EK, Tang M, Tan LC, Wu YR, Wu RM, Ross OA et al (2008) Lrrk2 R1628P in non-Chinese Asian races. Ann Neurol 64(4):472–473
Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F et al (2007) Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology 68(14):1141–1143
McInerney-Leo A (2005) Genetic testing in Parkinson’s disease. Mov Disord 20(7):908–909
Conflict of Interest
The author declares no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Monfrini, E., Di Fonzo, A. (2017). Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. In: Rideout, H. (eds) Leucine-Rich Repeat Kinase 2 (LRRK2). Advances in Neurobiology, vol 14. Springer, Cham. https://doi.org/10.1007/978-3-319-49969-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-49969-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49967-3
Online ISBN: 978-3-319-49969-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)