
Abstract
The nomenclature of atypical hemolytic uremic syndrome (aHUS) has undergone an evolution as rapid as the scientific understanding of the field during the last decade. Identification of many underlying genetic causes has increased understanding of the major mechanisms of disease. These defects principally, but not exclusively, involve the alternative pathway of complement. Important differences among the specific defects have impact on disease management, and clinical genetics plays a key role in that process. Untreated, aHUS frequently leads to end stage renal disease or death. Fortunately, understanding the major disease mechanisms has allowed development of effective treatment options.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Hemolytic uremic syndrome
- Complement
- Alternative pathway
- Hemolytic anemia
- Thrombocytopenia
- Thrombotic microangiopathy
- Factor H
- Eculizmuab
- Plasmapheresis
- Plasma exchange
Introduction
Hemolytic uremic syndrome (HUS) is form of thrombotic microangiopathy (TMA) characterized by microangiopathic hemolytic anemia, thrombocytopenia, and organ injury. As understood from “uremic” in its designation, the most common organ to be injured by HUS is the kidney but other organs are also frequently involved and in fact sometimes the kidney is relatively spared. The hemolytic anemia is characterized by intravascular red blood cell fragmentation producing schistocytes and by a parallel process leading to thrombocytopenia which can be detected by peripheral blood smear examination. Other forms of TMA such as thrombotic thrombocytopenic purpura (TTP) can present in a very similar clinical manner, and these require careful consideration as the underlying etiology of the process and treatment may differ (Loirat et al. 2016; Nester et al. 2015; George and Nester 2014).
The several types of TMA can be classified in several ways, including by clinical syndrome, as secondary to a coexisting disease or condition, or related to certain infections (Fig. 1) (Loirat et al. 2016). The subjects of this chapter are those forms of HUS in which specific genetic abnormalities predispose to HUS. Commonly referred to as “atypical HUS (aHUS),” the majority of identified individual cases are due to defects that allow for increased activity in the alternative pathway of complement. The typical form of HUS, secondary to STEC infection, is discussed in this chapter.

Basic diagnostic flow chart: TMA thrombotic microangiopathy, TTP thrombotic thrombocytopenic purpura, HUS hemolytic uremic syndrome, ST Shiga-toxin, STEC ST enteropathogenic E. coli, DGKE diacylglycerol kinase epsilon
Epidemiology and Clinical and Manifestations
Both typical and atypical HUS can present at any age. Both will manifest the cardinal laboratory features of HUS (hemolytic anemia, thrombocytopenia, uremia) though the degree of each is variable. Hypertension is very frequent in both except when hemodynamic shock prevails. Therefore, basic hematological and biochemical evaluations as well as a careful physical examination with evaluation of blood pressure are essential for diagnosis. Either typical or atypical HUS can present with multisystem disease involving the CNS, intestine, and other organs; arteriolar and arterial thrombosis may result from progressive microvascular disease and cause large scale ischemia in any organ.
Though exact figures are unclear, aHUS is an “ultra-rare” disease, its incidence estimated between 0.1 and 2 per million population (Loirat and Fremeaux-Bacchi 2011). Re-emphasizing that aHUS can occur at any age, the most frequent age of onset is in very young children, usually under age 2 years. Cumulatively, however, most cases actually manifest in adulthood (Fremeaux-Bacchi et al. 2013). While there is no predominance by sex in childhood, it has been reported and widely accepted that adult cases occur in a female:male ratio of 3:1 perhaps because pregnancy is a potent trigger in susceptible individuals (Fremeaux-Bacchi et al. 2013; Greenbaum 2016).
Clinical features including disease occurring in non-summer months, non-synchronous HUS in a family member, recurrent disease, a prolonged relapsing/remitting or indolent course of illness, or onset prior to age 6 months are strongly suggestive of aHUS. Diarrhea, or indeed any other infectious trigger, is not an infrequent finding and therefore the terminology “diarrhea positive” or “diarrhea negative” to denote typical versus atypical HUS is no longer considered a useful approach to classify this disease.
Unlike typical HUS, in cases of aHUS, the platelet count may present at greater than 30 G/L, and in some cases greater than 150 G/L. In fact, up to 15–20% of cases have subclinical and fluctuating or relapsing/remitting hematological parameters over a prolonged period of time such that the presenting sign may be unexpected such as hypertension, proteinuria, or reduced renal function. In some patients with disorders of the AP of complement regulation, serum C3 or CH50 may be low, but these tests should not be considered sensitive enough to diagnose or exclude aHUS. Many patients have depressed ADAMSTS13 concentrations, but generally not to below 10% (which is characteristic of TTP). One study has documented that these partial deficiencies in aHUS patients are commonly associated with genetic variation in the gene encoding ADAMTS13, contributing to a permissive background to manifest TMA (Feng et al. 2013).
Untreated, the outcomes of aHUS are historically poor, with the majority developing ESRD and with significant mortality. Improved recognition and treatment options developed recently, and described below, have drastically changed these outcomes for the better. While the presenting episode remains potentially very severe and some cases respond less, the majority of patients can now expect to achieve remission and preserved renal function.
Pathological Findings of aHUS
Atypical HUS is characterized by glomerular capillary wall thickening from endothelial cell swelling and accumulation of material between the endothelial cells and the basement membrane. There is direct endothelial damage resulting in capillary thrombosis from platelet and fibrin thrombi occlusion. In the early stages there is mesangial lysis that later is replaced by sclerotic changes. Fibrin and fibrinogen deposits form in the glomeruli, mesangium, and vessel walls. Complement and immunoglobulin deposits may also be found in the capillaries (Kavanagh et al. 2013).
Extra-renal involvement may affect any organ system, including the CNS, pancreas, colon, and heart leading to severe clinical sequelae in some cases (Hirt-Minkowski et al. 2010). There is essentially no discernable difference in histopathology between aHUS and typical HUS.
Pathogenesis of aHUS
Disorders of the Alternative Pathway of Complement
As noted, the majority of patients with aHUS have a defect in the regulation of the alternative pathway (AP) of complement. The complement system forms part of the innate immunity and consists of plasma proteins, membrane-bound receptors, and membrane-deposited components. These interact with one another for a primary purpose of fighting infection, particularly encapsulated bacteria such as N. Meningitidis.
For the purpose of discussing aHUS, a discussion of complement must center on the AP. There are three pathways for complement activation: the AP, the classical pathway triggered largely by antibody, and the lectin pathway triggered by molecules expressing mannose residues. These pathways converge and overlap, principally at the level of the C3 convertases. C3 convertases are formed in different ways by each pathway but ultimately lead to activation of C5 and onward to formation of the membrane attack complex (MAC). This molecular assembly mediates the toxic end action of the system upon its target. Many so-called intermediates generated as fragments during sequential proteolytic activation of factors have important function. For example C3a and C5a are potent inflammatory mediators and C3b helps with opsonization of pathogens and removal of immune complexes (Murphy 2008; Noris and Remuzzi 2013; Picard et al. 2015).
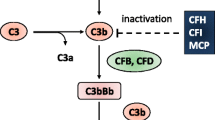
The AP is continuously active at low levels, termed “tickover” which results from spontaneous hydrolysis of C3 by water and Factor B (Fig. 2). Following an additional “triggering” event (such as infection) the AP amplification loop can increase the activity of this system tremendously. This highlights the need for tight regulation of the pathway to avoid promiscuous tissue injury by terminating the additional complement activity when it is no longer required.

A simplified diagram of the AP of complement: The AP pathway is normally active at a continuously low level termed “tickover” and is amplified in the presence of certain microbes and other triggers. The cascade undergoes amplification at several points and leads to formation of the membrane attack complex (MAC) as well as multiple intermediates with anaphylactic and other activities in the fluid phase as well as on native and exogenous cell membranes. (Figure reproduced from Nester et al., “Atypical aHUS: State of the art” with permission).
The initiated and spontaneous activation of the complement system is controlled by regulated proteins at different levels of the cascade in order to limit the response and terminate the process after the triggering event is resolved. Many cases of aHUS (Table 1) result from defects in these regulatory molecules or defects in effector complement proteins that reduce their ability to be regulated. In addition, there is also interaction between the complement system and the intrinsic coagulation/anticoagulation system and there are cases of aHUS that stem from defects at the level of this interaction (Table 1).
Noncomplement-Related Pathogenesis of aHUS
Other genetic causes of aHUS are related to mutation of diacylglycerol kinase epsilon (DGKE) and deficiencies in Cobalamin C, discussed below. The mechanisms by which these defects lead to HUS is less clear than for the defects in the AP of complement.
Genetic Basis of aHUS
Genetic variants can be classified as benign, likely benign, a variant of uncertain significance (VUS), likely pathogenic, or pathogenic (Richards et al. 2015). Dysregulation of the AP was the first recognized and the most common etiology of aHUS; in turn, genetic abnormalities affecting this pathway were the first to be elucidated. Inactivating (“loss-of-function”) mutation in genes encoding for Complement Factor H, Factor I, and membrane cofactor protein as well as activating (“gain-of-function”) mutation in C3 and factor B are all described and permit inappropriately intense or prolonged activation of the APC. In addition, acquired antibodies to complement factor H are well described and cumulatively account for a significant proportion of cases. These are almost always linked to mutation in CHFR proteins though the mechanism by which these genetic changes lead to the production of antibodies remains unclear (Durey et al. 2016; Gurjar et al. 2018). Defects in thrombomodulin also lead to abnormal AP activity. Mutations in DGKE and deficient cobalamin C are also reported but are quite infrequent (Berger 2016). It is also relevant to note that while specific genes are discussed separately, a certain proportion of patients manifest more than one pathogenic mutation, and a larger number of patients manifest combinations of pathogenic mutation and permissive common risk variants, and genetic alterations of uncertain significance (Goodship et al. 2017; Bresin et al. 2013). The above noted high frequency of common risk variants of ADAMSTS13 encountered in patients with aHUS is a good example of this issue (Feng et al. 2013).
Atypical HUS and C3 glomerulopathy have much in common and their clinical presentations can overlap. Over time, however, most cases are defined as one or the other largely on the basis of differing hematological parameters. Not surprisingly, there is also overlap in the genetic drivers of disease in these two disease groups. This remains a very active area of research, with recent studies continuing to document both the similarities and differences in the patterns of genetic defects, their location within individual genes, and how these in turn differentially affect complement regulation on surfaces, in fluid phase, and with regard to binding sites (Osborne et al. 2018).
Gene Mutations in Complement Regulatory Proteins (Nester et al. 2015; Loirat and Fremeaux-Bacchi 2011; Noris and Remuzzi 2013; Kavanagh and Goodship 2010; Malina et al. 2012)
Mutations in Factor H (FH)
Factor H is circulating serum protein produced mainly in the liver, though also produced in quantitatively much smaller amounts by circulating lymphocytes, monocytes, dendritic cells, and in glomerular tissue on a “local” basis. It is formed of 20 short consensus repeat (SCR) domains, with an N-terminal SCR that forms the regulatory domain and a C-terminal SCR that forms the recognition terminal. The N-terminal competes with Factor B and accelerates the decay of the C3 convertase. FH also acts as a cofactor for Factor I-mediated inactivation of C3b. The gene encoding for FH is located in the RCA (regulators of complement activation) locus in chromosome in 1q32 and is the most frequently mutated gene in aHUS, identified in ~25% of all cases and in 40% of familial cases. Majority of the mutations are heterozygous and are located in the exons encoding the C-terminal domain of the protein. These mutations may not result in a quantitative deficiency of factor H, but instead normal levels of abnormal protein. In other cases, such as stop or nonsense mutations, FH levels may be drastically reduced.
Mutations in Factor I (FI)
Factor I is a regulatory plasma serine protease also synthesized mainly in the liver. FH has a modular structure consisting of a heavy chain that contains two low density lipoprotein receptor domains, a CD5 domain, and a module only found in FI and complement proteins C6 and C7. FI inactivates C3b to iC3b and this into further fragments in the presence of cofactors including FH, and complement receptor 1 (CR1), MCP or von Willebrand factor. The gene encoding for FI is located on chromosome 4q25. Approximately 40 heterozygous mutations in FI have been reported corresponding to about 12% of aHUS cases. Depending on the nature of the mutation, plasma C3 concentration is decreased in 20–30% and CFI concentration in one third of affected individuals.
Mutations in Factor B (FB)
Factor B is a zymogen with a major role for amplification of the alternative pathway. FB is cleaved into factors Ba and Bb; and facilitates binding of Bb with C3 to form the active C3 convertase. With its formation, C3 convertase achieves a key threshold in the pathway of AP activation; regulation (inactivation) of C3 convertase is promoted by decay accelerating factor, CRI, and FH. The gene encoding FB is located in chromosome 6p21.3. Mutations in FB responsible for aHUS result in a gain of function by increasing the stability (and therefore activity) of C3 convertase via resistance to decay. FB mutations are uncommon, accounting only up to 4% of aHUS cases. Affected individuals have a permanent activation of AP with very low C3 and normal or low FB concentration. It is likely some of the mutations of FB in aHUS patients are either not pathogenic or only permissive, based on functional studies, acting in concert with associated mutations in the few cases reported (Osborne et al. 2018).
Mutations in Membrane Cofactor Protein (MCP/CD46)
MCP is a membrane cofactor protein expressed by most cells including leukocytes which serves as a cofactor for FI to degrade C3b and C4b. The MCP gene is located in within the RCA gene cluster on chromosome 1q32 and account for up to 15% of aHUS. MCP mutation is characterized by decreased MCP cell surface expression and less frequently the expression is normal but functionally inactive. Affected patients may present with normal C3 levels but about 1/4 to 1/3 of cases do have decreased levels; this often occurs when another complement-related mutation coexists. When MCP is defective, the most relevant cell surface involved in aHUS is the endothelial lining of the microvasculature of the kidneys. When affected individuals undergo kidney transplant, the graft and its endothelium is protected by expression of its intrinsic MCP. While posttransplant recurrence of aHUS is therefore unlikely in cases due to MCP mutation (in the absence of additional pathogenic defects in fluid-phase proteins), it is believed that recipient vascular cells do migrate into the graft, rendering the endothelium partially chimeric and variably susceptible to the effects of deficient MCP (Fremeaux-Bacchi et al. 2007).
C3 Mutations
C3 is abundant in plasma and its hydrolysis causes activation of the AP leading to the generation of C3 convertase. This reactions are regulated by cofactors including FH and MCP increasing the rate of dissociation of the convertase or serving as cofactor for FI to cleave C3b. The gene encoding for C3 is located on chromosome 19 and mutations in this gene account up to 10% of aHUS. In such cases, there is indirect gain of function by decreasing the ability of C3 to bind to the regulator MCP, with enhanced capacity of FB to bind C3b, resulting in greater formation of C3 convertase. Such patients present with low C3 levels in 70–80% of cases.
Antibodies Against Complement Factor H
Antibodies against FH (anti-FH) is an acquired dysfunction of FH but in a sense may be considered a complement mutation because it is highly associated with rearrangements or deletions of FH-related proteins (typically homozygosity for delCFHR3-CFHR1) (Loirat and Fremeaux-Bacchi 2014). Interestingly, the deletion itself is not uncommon, found in up to 9% of healthy individuals in several populations studied; the mechanism leading to the association remains unclear. Anti FH Ab accounts for approximately 5–10% of cases of aHUS.
Thrombomodulin Mutations
Thrombomodulin (THBD) is a transmembrane glycoprotein expressed on vascular endothelial cells. Interacting with both the coagulation cascade and complement system, this protein facilitates activation of protein C and enhances thrombin-mediated activation of plasma procarboxypeptidase B, which inactivates C3a and C5a. THBD downregulates the AP by accelerating FI-mediated activation of C3b in the presence of cofactors. THBD mutations result in decreased function and account for 3% of cases of aHUS.
Noncomplement-Related Genetic Causes of aHUS
DGKE Mutation
The DGKE gene encodes diacylglycerol kinase-epsilon, an intracellular lipid kinase that phosphorylates diacylglycerol (DAG) to phosphatidic acid. DGKE is expressed in endothelium, platelets, and podocytes, and its mutation has been associated with aHUS in infants.
It is believed that the mechanism stems from loss of DGKE in endothelial cells, leading to endothelial cell damage and death, with a resulting microvascular prothrombotic consequence (Nester et al. 2015; Vieira-Martins et al. 2016). DGKE mutation is particularly frequent among infants (age < 1 year) with aHUS; it accounts for 27% of sporadic cases and up to 50% of familial cases in that age group (Lemaire et al. 2013). While patients do not respond to anti-complement therapy, posttransplant recurrence risk appears to be very low. Recently, as with defects in the APC, the spectrum of renal disease due to DGKE has been extended beyond aHUS to include proliferative forms of GN, suggesting heterogeneity of effects (Azukaitis et al. 2017).
Cobalamin Deficiency Associated HUS
Cobalamin C disease is caused by a mutation in the gene encoding for the methylmalonic aciduria and homocystinuria type C protein (MMACHC). It is suggested that in Cobalamin deficiency–associated TMA, the hyperhomocysteinemia induces endothelial damage, leading to platelet aggregation and an induced procoagulant state with formation of microthrombi. Affected patients most commonly present in the newborn period with vomiting, poor sucking, failure to thrive, lethargy, hypotonia, and laboratory features of HUS/TMA (Beck et al. 2017).
Diagnostic Approach
The age, clinical presentation, and baseline laboratory should guide the diagnostic approach. It is critical to rule out infectious etiology (STEC-HUS or pneumonia-associated HUS) with bacterial and stool cultures. Cobalamin deficiency should be suspected in neonates and infants with supporting systemic findings including homocystinuria and methylmalonic aciduria. ADAMTS13 activity below 10% is consistent with primary or acquired TTP; testing for anti-ADAMTS13 antibodies can be helpful but it is important not to rule out aHUS with ADAMTS13 levels which are reduced, but greater than 10%. In many cases, it will not be clear whether a patient has typical HUS, aHUS, or another type of TMA during the acute presentation of the illness; regardless, in such cases empiric treatment must not be delayed pending clarification. When possible, it behooves clinicians to save plasma from the acute (as yet untreated) setting for future measurements that may clarify the diagnosis.
In suspected aHUS, thorough evaluation of the AP system is warranted. This can include plasma C3 which is widely available; measurement of FH, FI, FB levels may also be done but as noted earlier, normal levels do not rule out aHUS and abnormal levels can be found in other conditions. Assessment for antibodies to FH and genetic testing, where available, should be performed for affected individuals. While these results do not inform initial diagnosis or guide acute therapy, the findings may be important for longer-term treatment, prognostication, and genetic counseling.
Treatment
Often, the first episode of aHUS an individual experiences will require treatment while the diagnosis remains suspected but not proven. Typical HUS, TTP, and other conditions like hemophagocytic syndromes, disseminated intravascular coagulation, and acute hemolytic anemia of other causes can remain in the differential diagnosis without impeding empiric treatment of aHUS while further diagnostic testing continues. Indeed, many of these conditions share some common treatment approaches.
The treatment of aHUS requires an experienced clinical team and supportive capacity typically available in tertiary care hospitals. The potential for rapid AKI with need for dialysis, as well as multiple organ pathology including CNS, cardiac, pulmonary, and GI systems must be respected. Untreated, the mortality rate and rate of renal failure is very high. In the immediate support of patients, red blood cell transfusion is often required, while platelet transfusion is generally reserved for those about to undergo a procedure with a high risk of bleeding, as there remains a clinical perception, somewhat controversial, that platelets may worsen the TMA by providing further substrate to create microthrombi.
Plasma Therapy
Historically, this was considered first line treatment; despite a lack of definitive studies, there was considerable observational evidence of benefit in many cases. Retrospective analysis supports the fact that plasma exchange (PE) in patients with aHUS can remove mutant FH, FI, FB, C3, and anti-FH antibodies, while plasma product replacement (in conjunction with PE or in isolation) can restore more normal protein levels. Following its definition as a cause of aHUS, patients with MCP gene mutation proved exceptions to this necessity with up to 90% of episodes resolving without the need for such treatment; this difference is believed to be the case because MCP is membrane-bound not in the fluid phase (Loirat et al. 2016; Loirat and Fremeaux-Bacchi 2011; Loirat et al. 2012).
Because plasma therapy is available relatively widely and quickly, and especially when eculizumab (discussed below) is not available or its administration is unavoidably delayed, plasma therapy can still be recommended. This treatment should begin as quickly as possible, at least within 24 h of presentation or when the empiric (“working”) diagnosis of aHUS is recognized. Plasma exchange, rather than plasma infusion, is often recommended so as to enable larger volumes of plasma to be provided while reducing the risk of volume overload and severe hypertension especially in patients with AKI. Among infants or others in whom placing appropriate vascular access can itself present significant risk, plasma infusion may be used as a temporizing measure.
Plasma therapy should be performed daily until the platelet count, hemoglobin, and LDH normalize and renal function improves. Some patients may respond in less than a week and then tapering the frequency of treatment may be possible at that point. Maintenance therapy should be guided by frequency of relapses and the identified complement anomaly. While rapid institution of plasma therapy may be of benefit, it is also very important to consider the acute risks of PE including infusion reactions, hypotension or hypertension, altered coagulation profile, hypocalcemia, and anemia with additional requirement for blood transfusion, as well as catheter-related thrombosis and infection. As noted, placement and use of extracorporeal circuits can be life-threatening in hemodynamically unstable infants and children.
Chronic plasma therapy may help to gain control the acute episodes of aHUS, as well as to prevent (as maintenance therapy) future episodes. Nonetheless, retrospective study has demonstrated that there is incomplete protection and that over time, a significant proportion of individuals with aHUS treated in this manner will continue to experience some amount of ongoing aHUS activity, accompanied by declining renal function and elevated risk of end stage kidney disease.
Eculizumab
Eculizumab is a humanized monoclonal antibody that inhibits terminal complement activity Eculizumab prevents C5 cleavage and formation of C5a, reducing the C5a pro-inflammatory and the toxic consequences of uncontrolled formation of the MAC. In the USA, the FDA approved eculizumab for the treatment of aHUS in 2011. Initial studies were widely viewed as demonstrating that eculizumab resulted in definitive remission of most cases of aHUS as well as superiority to plasma therapy (Legendre et al. 2013). Currently, eculizumab remains the only agent approved for aHUS, though newer generations of this treatment and alternative agents are in active development.
Eculizumab has been shown to be effective in patients with aHUS with or without detectable complement mutation, although it is not effective in cases due to DGKE mutation. Where early use of eculizumab was reserved for cases resistant to plasmatherapy, it should be considered standard of care in all phases of illness. Prompt empiric use in suspected cases of aHUS is indicated since outcomes after prompt treatment are superior than with delayed treatment (Yuksel et al. 2016). Even in cases where prompt diagnosis or treatment was delayed or not possible, administration of eculizumab has been shown to improve renal function over chronic periods of use, with some chronically dialysis-dependent patients regaining a degree of renal function. Treatment is also demonstrated to be effective in preventing or treating posttransplant aHUS recurrence, depending on the etiology. Patients with MCP mutation do not require and those with DGKE mutations do not benefit from eculizumab therapy. Certain uncommon variations in C5 complement reduce efficacy as well (Nishimura et al. 2014).
Due to inhibition of complement activity, eculizumab increases the risk of serious and fatal infection by encapsulated bacteria principally including N. meningitidis, but also S. pneumoniae and H. influenza. Other infections like respiratory tract infections, urinary tract infections, as well as viral syndromes have been reported in 23% to up to 42% of patients taking eculizumab. Anti-meningococcal vaccination must be used, and vaccination for the other mentioned pathogens is extremely important as well. Because vaccination takes at least 2–4 weeks to achieve protective titers, prophylaxis with penicillin or other appropriate agent is required when eculizumab is required acutely (Benamu and Montoya 2016). Some clinicians will choose to use both vaccination as well as chronic antibiotic prophylaxis in all patients for concern of waning immunity on in particular for fear of impaired vaccine response. This concern may be particularly valid for example in a patient with a kidney transplant under immunosuppression or for infants whose vaccine responses may be incomplete.
Transplantation Considerations
Ideally, treatment of aHUS should prevent end-stage kidney disease. However, there are patients who suffered kidney failure before effective treatment was available or in whom it could not be delivered effectively. In other patients, the diagnosis of aHUS was not recognized and may only come to light after kidney failure or after transplantation when the disease may recur. Thus, the need to manage kidney transplantation for patients with aHUS remains highly relevant.
Achieving a good outcome after any of type of transplantation requires the best practices of “routine” surgical and perioperative management like dialysis, plasma exchange, central catheter management, posttransplant anticoagulation, and immunosuppression. Historically, many individuals suffering from aHUS and kidney failure remained dialysis-dependent for very long periods, in some cases many years. Severe TMA with medium-sized and large vessel involvement is also not uncommon in aHUS even in cases where dialysis is not required. Prior to transplant surgery in such individuals, it is important to use imaging to carefully assess the central vasculature for unsuspected occlusions in order to identify both occluded as well as appropriate sites for transplant vessel anastomosis.
Like other inheritable causes of renal disease, living-related donor evaluations for individuals suffering from aHUS also require additional care. The individual genetic evaluation of each case is therefore of high importance and should be required before a related donor be accepted. In some cases, the potential donor may have risk of aHUS in addition to transmission of risk to the recipient.
Posttransplant recurrence and less frequently, de novo occurrence, of aHUS may occur. In a patient with recognized aHUS undergoing transplantation, the optimal approach requires an assessment of whether the patient’s condition had been or will be responsive to eculizumab. In cases where recurrence is most likely, such as mutation in CFH, CFB, or CFI, the transplantation protocol must include preoperative eculizumab followed by regular treatment for prevention of aHUS. The surgical procedure of transplantation, along with the relative ischemia of the graft and its reperfusion are themselves potential triggers for aHUS. Unavailability of chronic eculizumab in any case should be considered a relative contraindication to transplant because other options such as observation alone or use of plasma therapy are more likely to result in graft loss and morbidity.
Unexpected aHUS following transplantation should be evaluated and treated in the same way as disease in the native kidneys. Such an occurrence usually indicates the original cause of ESRD was aHUS though there are exceptions. Posttransplantation aHUS in individuals with transplantation of other solid organs should also be evaluated as with aHUS in nontransplanted individuals. In addition to the simple fact that liver failure and transplantation surgery with reperfusion injury are potentially large triggers of aHUS, the possibility of “inheriting” a specific risk factor is evident. In one case, a donor had an asymptomatic CFH mutation and the recipient an unknown MCP defect which together permitted aHUS at a later date (Brown et al. 2012). The de novo occurrence of aHUS following stem cell or marrow transplantation is more common than for solid organs and may be unrelated to the mechanisms of aHUS discussed in this chapter. However, it is now well-described that TMA in such patients can and often does result from the same pathophysiological pathways described above. Thus, a similar, though modified, diagnostic and therapeutic approach will apply; importantly, clinicians may need to evaluate the genetic factors of both the graft and the recipient host.
As described earlier in the chapter, FH, FI, FB, and C3 are circulating proteins mainly produced in the liver, explaining the high recurrence rate of disease after isolated kidney transplant in patients with mutations leading to defects in these proteins. On the other hand, MCP and DGKE mutations are widely expressed, so a kidney transplant from a healthy donor not only restores kidney function but also represents local gene therapy by allowing expression of normal protein in that kidney. Also discussed above, individuals with the latter defects should not be considered totally immune from relapse following transplantation. Not only are multiple simultaneous genetic defects possible but exceptions do occur, for example, via evolution of recipient microchimerism in grafts of patients with MCP mutation which render the recipient susceptible to recurrence (Fremeaux-Bacchi et al. 2007).
Among patients with mutations in genes encoding for hepatically synthesized and circulating complement proteins, combined liver-kidney to cure patients with aHUS and end-stage kidney disease, or isolated liver transplantation to cure patients with aHUS with preserved renal function, may be considered (Saland et al. 2009; Saland 2014). This approach carries some important risks and also some potential benefits, which must be considered carefully for each patient. A perioperative strategy to approach transplant in the same manner as isolated kidney transplant in order to prevent aHUS recurrence is required, namely, using perioperative eculizumab or intensive plasma exchange. While this option predated eculizumab and has largely been supplanted, given its potential for cure, it should not be discarded as an option, but given its risks, it should also be reserved for cases where eculizumab is not available, strong patient preference, or in the uncommon scenario in which eculizumab is not effective (Coppo et al. 2016). Use of liver transplantation to cure aHUS should not be considered for patients with no evidence of gene mutations, patients with isolated MCP or DGKE mutation, anti FH-autoantibodies, or in the small minority of patients with mutations in a hepatically synthesized product (like FH or FI) but in whom affected family members (or themselves) have undergone successful isolated kidney transplantation (Table 2).
References
Azukaitis K, Simkova E, Majid MA, Galiano M, Benz K, Amann K et al (2017) The phenotypic spectrum of nephropathies associated with mutations in diacylglycerol kinase epsilon. J Am Soc Nephrol 28(10):3066–3075
Beck BB, van Spronsen F, Diepstra A, Berger RM, Komhoff M (2017) Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol 32:733–741. 2016 Jun 11
Benamu E, Montoya JG (2016) Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr Opin Infect Dis 29(4): 319–329
Berger BE (2016) The alternative pathway of complement and the evolving clinical-pathophysiological Spectrum of atypical hemolytic uremic syndrome. Am J Med Sci 352(2):177–190
Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S et al (2013) Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24(3):475–486
Brown JH, Tellez J, Wilson V, Mackie IJ, Scully M, Tredger MM et al (2012) Postpartum aHUS secondary to a genetic abnormality in factor H acquired through liver transplantation. Am J Transplant 12(6): 1632–1636
Coppo R, Bonaudo R, Peruzzi RL, Amore A, Brunati A, Romagnoli R et al (2016) Liver transplantation for aHUS: still needed in the eculizumab era? Pediatr Nephrol 31(5):759–768
Durey MA, Sinha A, Togarsimalemath SK, Bagga A (2016) Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol 12(9):563–578
Feng S, Eyler SJ, Zhang Y, Maga T, Nester CM, Kroll MH et al (2013) Partial ADAMTS13 deficiency in atypical hemolytic uremic syndrome. Blood 122(8):1487–1493
Fremeaux-Bacchi V, Arzouk N, Ferlicot S, Charpentier B, Snanoudj R, Durrbach A (2007) Recurrence of HUS due to CD46/MCP mutation after renal transplantation: a role for endothelial microchimerism. Am J Transplant 7(8):2047–2051
Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaime F, Dragon-Durey MA, Ngo S et al (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8(4):554–562
George JN, Nester CM (2014) Syndromes of thrombotic microangiopathy. N Engl J Med 371(7):654–666
Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Fremeaux-Bacchi V, Kavanagh D et al (2017) Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney disease: improving global outcomes” (KDIGO) controversies conference. Kidney Int 91:539–551. 2016 Dec 15
Greenbaum L (2016) The physician’s guide to atypical hemolytic uremic syndrome (aHUS). In: (NORD) NOfRD, editor. http://www.nordphysicianguidesorg/wp-content/uploads/2015/12/NORD_Physician%E2%80%99s-Guide-to-AHUS.pdf
Gurjar BS, Sriharsha TM, Bhasym A, Prabhu S, Puraswani M, Khandelwal P, et al (2018) Characterization of genetic predisposition and autoantibody profile in atypical hemolytic uremic syndrome. Immunology. https://doi.org/10.1111/imm.12916. [Epub ahead of print]. PMID:29485195
Hirt-Minkowski P, Dickenmann M, Schifferli JA (2010) Atypical hemolytic uremic syndrome: update on the complement system and what is new. Nephron Clin Pract 114(4):c219–c235
Kavanagh D, Goodship T (2010) Genetics and complement in atypical HUS. Pediatr Nephrol 25(12): 2431–2442
Kavanagh D, Goodship TH, Richards A (2013) Atypical hemolytic uremic syndrome. Semin Nephrol 33(6): 508–530
Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C et al (2013) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368(23):2169–2181
Lemaire M, Fremeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M et al (2013) Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45(5):531–536
Loirat C, Fremeaux-Bacchi V (2011) Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 6:60
Loirat C, Fremeaux-Bacchi V (2014) Anti-factor H autoantibody-associated hemolytic uremic syndrome: the earlier diagnosed and treated, the better. Kidney Int 85(5):1019–1022
Loirat C, Saland J, Bitzan M (2012) Management of hemolytic uremic syndrome. Presse Med 41(3 Pt 2):e115–e135
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A et al (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 31(1): 15–39
Malina M, Roumenina LT, Seeman T, Le Quintrec M, Dragon-Durey MA, Schaefer F et al (2012) Genetics of hemolytic uremic syndromes. Presse Med 41(3 Pt 2):e105–e114
Murphy K (2008) Janeway’s immunobiology, 7th edn. Garland Science, New York
Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH et al (2015) Atypical aHUS: state of the art. Mol Immunol 67(1): 31–42
Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL et al (2014) Genetic variants in C5 and poor response to eculizumab. N Engl J Med 370(7):632–639
Noris M, Remuzzi G (2013) Overview of complement activation and regulation. Semin Nephrol 33(6): 479–492
Osborne AJ, Breno M, Borsa NG, Bu F, Fremeaux-Bacchi V, Gale DP et al (2018) Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J Immunol 200:2464–2478. 2018 Mar 2
Picard C, Burtey S, Bornet C, Curti C, Montana M, Vanelle P (2015) Pathophysiology and treatment of typical and atypical hemolytic uremic syndrome. Pathol Biol 63(3):136–143
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Saland J (2014) Liver-kidney transplantation to cure atypical HUS: still an option post-eculizumab? Pediatr Nephrol 29(3):329–332
Saland JM, Ruggenenti P, Remuzzi G, Consensus Study G (2009) Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol 20(5):940–949
Saland J, Satlin L, Zalsos-Johnson J, Cremers S, Ginsberg H (2016) Impaired postprandial lipemic response in chronic kidney disease. Kidney Int 90:172–180. in press
Vieira-Martins P, El Sissy C, Bordereau P, Gruber A, Rosain J, Fremeaux-Bacchi V (2016) Defining the genetics of thrombotic microangiopathies. Transfus Apher Sci 54(2):212–219
Yuksel S, Evrengul H, Ozcakar ZB, Becerir T, Yalcin N, Korkmaz E et al (2016) First-line, early and long-term eculizumab therapy in atypical hemolytic uremic syndrome: a case series in pediatric patients. Paediatr Drugs 18(6):413–420
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
Reyes, L.C., Saland, J.M. (2019). Hemolytic Uremic Syndrome, Genetic. In: Trachtman, H., Herlitz, L., Lerma, E., Hogan, J. (eds) Glomerulonephritis. Springer, Cham. https://doi.org/10.1007/978-3-319-49379-4_43
Download citation
DOI: https://doi.org/10.1007/978-3-319-49379-4_43
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49378-7
Online ISBN: 978-3-319-49379-4
eBook Packages: MedicineReference Module Medicine